

Abstract
NMDA receptors (NMDARs) are ionotropic glutamate receptors that are crucial for neuronal development and higher cognitive processes. NMDAR dysfunction is involved in a variety of neurological and psychiatric diseases; however, the mechanistic link between the human pathology and NMDAR dysfunction is poorly understood. Rare missense variants within NMDAR subunits have been identified in numerous patients with mental or neurological disorders. We specifically focused on the GluN2B NMDAR subunit, which is highly expressed in the hippocampus and cortex throughout development. We analyzed several variants located in the GluN2B C terminus and found that three variants in patients with autism (S1415L) or schizophrenia (L1424F and S1452F) (S1413L, L1422F, and S1450F in rodents, respectively) displayed impaired binding to membrane-associated guanylate kinase (MAGUK) proteins. In addition, we observed a deficit in surface expression for GluN2B S1413L. Furthermore, there were fewer dendritic spines in GluN2B S1413L-expressing neurons. Importantly, synaptic NMDAR currents in neurons transfected with GluN2B S1413L in GluN2A/B-deficient mouse brain slices revealed only partial rescue of synaptic current amplitude. Functional properties of GluN2B S1413L in recombinant systems revealed no change in receptor properties, consistent with synaptic defects being the result of reduced trafficking and targeting of GluN2B S1413L to the synapse. Therefore, we find that GluN2B S1413L displays deficits in NMDAR trafficking, synaptic currents, and spine density, raising the possibility that this mutation may contribute to the phenotype in this autism patient. More broadly, our research demonstrates that the targeted study of certain residues in NMDARs based on rare variants identified in patients is a powerful approach to studying receptor function.
SIGNIFICANCE STATEMENT We have used a “bedside-to-bench” approach to investigate the functional regulation of NMDA receptors (NMDARs). Using information from deep sequencing of patients with neurological or psychiatric disorders, we investigated missense variants identified in the intracellular C-terminal domain of the GluN2B NMDAR subunit. We found several variants that displayed altered properties. In particular, one variant identified in a patient with autism, human GluN2B S1415L, displayed reduced surface expression and binding to PSD-95. Furthermore expression of GluN2B S1415L (S1413L in mouse) showed a deficit in rescue of synaptic NMDAR currents and fewer dendritic spines, consistent with other reports of spine abnormalities being associated with autism. More broadly, we demonstrate that using patient data is an effective approach to probing the structure/function relationship of NMDARs.
Keywords: dendritic morphology, human diseases, MAGUK binding, NMDA receptor, surface expression, synaptic function
Introduction
NMDA receptors (NMDARs) are ionotropic glutamate receptors that regulate neurotransmission, neuronal development, and plasticity in the brain (Traynelis et al., 2010). NMDARs are heterotetramers composed of two GluN1 and two GluN2/3 subunits and are primarily located at the postsynaptic density (PSD) at excitatory synapses in the CNS. The C-terminal domains of GluN2 subunits play an important role in regulating surface expression, synaptic stability, and removal of NMDARs from the synapse. In particular, GluN2B has many residues within the C terminus that modulate GluN2B trafficking and synaptic targeting. The last 4 aa (1479–1482; ESDV) of GluN2B constitute a well characterized PDZ-binding domain that interacts with PSD-95 family members, which are membrane-associated guanylate kinase proteins (MAGUKs) (Kim and Sheng, 2004). GluN2B is phosphorylated by casein kinase 2 (CK2) on serine 1480 within the PDZ ligand, which eliminates MAGUK binding via that domain (Chung et al., 2004). Phosphorylation of GluN2B S1480 first increases during development and upon neuronal activity and mediates the lateral movement of NMDARs from the PSD to extrasynaptic areas, where they undergo endocytosis (Chen et al., 2012). Once GluN2B-containing NMDARs are removed, GluN2A-containing NMDARs are inserted to the PSD and become a major NMDAR subtype at mature synapses, resulting in faster NMDAR kinetics. Reduction of S1480 phosphorylation is observed at later developmental stages (Sanz-Clemente et al., 2010). Several regions of the GluN2B C terminus upstream of the PDZ ligand have also been identified as being important for protein binding and synaptic expression, including a CaMKII-binding site. When CaMKII is activated, it binds to GluN2B and recruits CK2, resulting in phosphorylation of GluN2B S1480 and destabilization of synaptic GluN2B (Sanz-Clemente et al., 2013b). In addition, secondary non-PDZ-binding sites have been identified for PSD-95 between aa residues 1157 and 1458 on GluN2B (Cousins and Stephenson, 2012) and for another MAGUK protein, SAP102, in a region including aa residues 1391 and 1392 (Chen et al., 2012).
Loss of GluN2B-containing NMDARs leads to early neonatal mortality in mice (Kutsuwada et al., 1996). Conditional deletion of GluN2B at the bed nucleus of the stria terminalis eliminates LTP and actions of ethanol on NMDAR function (Wills et al., 2012). In addition to null mutations, there are multiple studies reporting GluN2B missense variants in patients with schizophrenia, autism, epilepsy, mental retardation, and other neurological or psychiatric diseases (Ohtsuki et al., 2001; Arning et al., 2007; Allen et al., 2008; Freunscht et al., 2013; Epi et al., 2013; Lemke et al., 2014; Hamdan et al., 2014; Andreoli et al., 2014; Zhu et al., 2015; Swanger et al., 2016; for review, see Yuan et al., 2015 and Hu et al., 2016). However, pathological and biological consequences of these missense variants have not been well-characterized in humans or animal models.
Third-generation deep sequencing is a high-throughput technique that enables the detection of single novel variants from individual patients. From the existing literature and the public COSMIC database, we obtained a list of rare variants in NMDAR subunits and specifically focused on those identified within the GluN2B C-terminal domain (Parsons et al., 2008; Tarabeux et al., 2011; Myers et al., 2011). We tested several GluN2B variants with the most dramatic amino acid changes in regard to charge, size, and polarity of the side chain. Three GluN2B variants, S1415L (S1413L in rat and mouse), L1424F (L1422F in rat and mouse), and S1452F (S1450F in rat and mouse), exhibited impaired binding affinity to PSD-95 and SAP102. Moreover, GluN2B S1413L showed reduced surface expression in hippocampal neurons. GluN2B S1413L-expressing neurons also had reduced dendritic spine density compared with neurons expressing GluN2B WT. Because GluN2B S1413L displayed abnormal phenotypes in all of the assays we performed, we further characterized this variant to investigate functional properties. We expressed GluN2B WT and S1413L in slices prepared from GluN2A/B conditional knock-out mice and found that GluN2B S1413L only partially rescued synaptic NMDAR currents compared with full recovery with GluN2B WT. Our results demonstrate that serine 1413 is important in maintaining normal GluN2B function and subcellular localization and in regulating synaptic activity. These findings also provide mechanistic insight into neurological diseases caused by missense variants in the GluN2B C terminus.
Materials and Methods
Neuronal cultures, antibodies, and reagents.
We adhered to the guidelines of the National Institutes of Health's Animal Research Advisory Committee regarding the care and use of animals for this study. For immunocytochemistry experiments, we used primary cultured hippocampal neurons from embryonic day 18 (E18) Sprague Dawley rats of either sex as described previously (Roche and Huganir, 1995). C57BL/6 GluN2A/GluN2B floxed mice (Gray et al., 2011) of either sex were bred in pairs and used to prepare neuronal cultures for recording NMDAR currents. Brain tissue from WT adult C57BL/6 mice of either sex was used for GST-pull down assays.
PSD-95 and SAP102 antibodies were from NeuroMab. Anti-GFP and secondary antibodies for immunofluorescence were from Invitrogen. GFP-tagged rat GluN2B cDNA was kindly provided by Dr. Stefano Vicini (Georgetown University). Point mutations were made by PCR with targeted primers. Portions of the GluN2B C terminus was amplified from rat GluN2B cDNA using PCR and the resulting product was subcloned in frame into the pGEX-4T1 vector at the EcoRI and XhoI sites.
Preparation of synaptosome fractions from mouse brain and GST pull-down assays.
We followed previously published procedures (Sanz-Clemente et al., 2010) to perform membrane fractionation of forebrain collected from 3- to 6-month-old rats. Tissue was homogenized in cold TEVP buffer (10 mm Tris pH 7.5, 1 mm EDTA, 1 mm EGTA, 1 mm Na3VO4) supplemented with 0.32 m sucrose. Buffer was supplemented with phosphatase inhibitors (P5726 and P0044; Sigma-Aldrich) and protease inhibitors (cOmplete EDTA-free, 05056489001; Roche). The homogenate was centrifuged at 1000 × g for 10 min at 4°C to remove nuclei. The P2 fraction was isolated by taking the supernatant and centrifuging at 10,000 × g for 20 min at 4°C. The P2 fraction was then solubilized in TEVP buffer with 35.6 mm sucrose and 1% SDS for 30 min at 37°C. Solubilized P2 fraction was neutralized with 3 volumes of TEVP buffer containing 35.6 mm sucrose and 1% Triton X-100 for 30 min on ice and sonicated on ice for 15 s to shear DNA. The solubilized P2 solution was centrifuged at 30,000 × g for 10 min to exclude insoluble debris and the supernatant was incubated with GST-GluN2B.
A plasmid (pGEX-4T-1) containing the GST-rat GluN2B (residues 1315–1482) WT, S1413L, L1422F, or S1450F mutants was transformed into BL-21 E. coli. GluN2B ΔSTOP is the same construct described (V1482Stop) in Sanz-Clemente et al. (2010). Protein expression was induced by IPTG; the bacteria were collected, lysed, and GST-GluN2B fusion proteins present in the supernatant were enriched by isolating the fusion proteins on glutathione Sepharose-4B beads (GE Healthcare). The P2 fraction from mouse brain was incubated with GST-GluN2B (WT or mutant) Sepharose beads for 2 h at 4°C. After three washes with PBS, bound proteins were eluted and immunoblotted for PSD-95 or SAP102 to evaluate the protein interaction. The amount of GST-GluN2B fusion protein was determined by staining with GelCode Blue (Thermo Fisher).
Immunofluorescence microscopy.
Surface receptors were analyzed using a fluorescence-based antibody binding assay as described previously (Sanz-Clemente et al., 2010). Briefly, hippocampal neurons were transfected with GFP-GluN2B at d in vitro (DIV) 14 with Lipofectamine 2000 (Invitrogen). At DIV 17, surface receptors were labeled with anti-GFP antibody for 15 min at room temperature before fixation with 4% PFA in PBS (containing 4% sucrose). Surface receptors were detected by Alexa Fluor 555-conjugated secondary antibody (shown in red in merged picture). After permeabilization with 0.25% Triton X-100 in PBS, the intracellular receptors were labeled with anti-GFP antibody followed by Alexa Fluor 633 or Alexa Fluor 647-conjugated secondary antibody (shown in blue in merged picture). We measured the fluorescence intensity using MetaMorph 6.0 software (Universal Imaging). We analyzed three independent areas per neuron for the quantitation. Intensity is presented as mean ± SEM. We calculated the ratio of the intensity of surface/intracellular in our analyses of surface expression. The number of dendritic spines was measured by cotransfecting neurons with pCAG-EGFP and GFP-tagged GluN2B to visualize spines. Spine number was counted in 20 μm secondary dendritic regions of pyramidal neurons. Three regions were selected and averaged for every neuron.
Images were collected using a Zeiss LSM 510 confocal microscope. Serial optical sections at 0.35 μm intervals were used to create maximum projection images. Three to five independent experiments were conducted and significance was analyzed using one-way ANOVA analysis (n = number of cells).
Two-electrode voltage-clamp (TEVC) current recording and whole-cell voltage-clamp recording.
For this study, we transiently transfected HEK cells with cDNA that encoded a fusion protein of WT or mutant rat GluN2B with a GFP tag (GenBank: NP_036706.1) in the pRK5 vector and WT rat GluN1-1a (hereafter GluN1; GenBank: NP_058706.1). Synthesis and injection of cRNA from WT GluN1 and GluN2B into Xenopus laevis oocytes (Ecocyte), as well as TEVC recordings from oocytes were performed as described previously at 23°C (Hansen et al., 2013). The recording solution contained the following (in mm): 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2, and 0.01 EDTA, pH 7.4. The membrane potential was held at −40 mV for all TEVC recordings from oocytes unless otherwise stated. The concentration–response curves were fitted with two equations:
 |
where EC50 is the concentration of the agonist that induces a half-maximal effect, N is the Hill slope, IC50 is the concentration of the negative modulators that produces a half-maximal effect, and “minimum” is the degree of residual inhibition at a saturating concentration of the negative modulators.
To evaluate whether the mutants influence the NMDAR response time course, the whole-cell voltage-clamp recordings were performed on transiently transfected HEK 293 cells at −60 mV (23°C). Recording electrodes (3–5 ΜΩ) were pulled from thin wall glass micropipettes (TW150F-4; World Precision Instruments) using a vertical puller (Narishige P-10) and filled with solution containing the following (in mm): 110 D-gluconate, 110 CsOH, 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 NaATP, and 0.3 NaGTP, pH 7.35. The external solution contained the following (in mm): 150 NaCl, 10 HEPES, 30 D-mannitol, 3 KCl, 1.0 CaCl2, and 0.01 EDTA, pH 7.4. Rapid solution exchange was performed by computer-controlled piezoelectric solution switching system and data were analyzed by ChanneLab (Synaptosoft) (Yuan et al., 2009). Deactivation or response time course after the rapid removal of glutamate was fitted with the following equation:
 |
NMDAR-mediated EPSCs were recorded in acute hippocampal slices. Briefly, 300 μm transverse hippocampal slices from postnatal day 13 (P13)–P15 mice were cut on a DTK Microslicervibratome (Ted Pella) in chilled sucrose cutting solution containing the following (in mm): 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, 7 glucose, and 210 sucrose. Hippocampal slices were subsequently placed in a chamber with artificial CSF (ACSF) containing the following (in mm) 119 NaCl, 2.5 KCl, 26 NaHCO3, 1 Na2PO4, 11 glucose, 2.5 CaCl2, and 1.3 MgCl2 at 32°C for ∼30–60 min. Slices were then maintained at room temperature for 0.5–1 h in the same solution before recording. NMDA EPSCs were recorded from identified CA1 pyramidal cells using whole-cell patch-clamp methods in the presence of picrotoxin (100 μm) and NBQX (10 μm). Membrane voltage was held at +40 mV and the Schaffer collateral pathway was stimulated at 0.05 Hz with a monopolar glass electrodes filled with ACSF. The internal solution in the patch pipette contained the following (in mm): 135 CsMeSO4, 8 NaCl, 10 HEPES, 0.3 Na3GTP 4 MgATP, 0.3 EGTA, 5 QX-314, and 0.1 spermine; 3–5 MΩ borosilicate glass pipettes were used for recording. A single weighted decay measure of NMDA EPSCs (referred in the text as decay time; see Cathala et al., 2005) was calculated from the area under the peak-normalized current for 1.3 s after the peak. Series resistance was monitored and not compensated and neurons in which series resistance varied by 25% during a recording session were discarded. Synaptic responses were recorded with a Multiclamp 700B amplifier (Molecular Devices), filtered at 2 kHz, and digitized at 10 Hz. All recordings were performed at room temperature.
In utero electroporation.
Pregnant GRIN2Afl/flGRIN2Bfl/fl mice were anesthetized with isofluorane at E14.5. Each lateral ventricle of the embryo was injected with 2 μl of plasmid DNA with 0.05% fast green (Sigma-Aldrich 68724). After injection, voltage steps (45 V for 5 pulses at 1 Hz with 50 ms) were applied across the uterus to target hippocampal progenitor neurons. Warmed PBS (32°C) was given to moisten the embryos. Buprenex (0.1 mg/kg) was applied and the wound was subsequently sutured.
Results
Surface expression of GluN2B variants in hippocampal neurons
Because GluN2B is critical for development throughout the CNS, it is not surprising that de novo mutations in GluN2B have been identified in a variety of psychiatric and neurological diseases (Hu et al., 2016). We focused on variants within the C terminus because the C termini of the GluN2 subunits are important for the subunit specificity of NMDAR trafficking and signaling (Myers et al., 2011; Tarabeux et al., 2011; Parsons et al., 2008; and COSMIC database). We selected variants within the last 68 aa of the GluN2B C-terminal domain, which were identified in patients with schizophrenia, autism spectrum disorders (ASDs), and mental retardation (Fig. 1A,B). The relevant residues in GluN2B are shown in Figure 1A. To determine whether these variants in the GluN2B C terminus alter GluN2B trafficking to the surface, we generated three rat mutant GluN2B subunits using site-directed mutagenesis to introduce the following missense changes: S1413L, L1422F, and S1450F (S1415L, L1424F, and S1452F in human, respectively). All GluN2B cDNAs were engineered to produce a fusion protein that contained a GFP tag appended in-frame to the N-terminal domain to allow for detection of surface NMDARs using immunofluorescence microscopy. We expressed each GluN2B mutant transiently in cultured rat hippocampal neurons and labeled surface receptors with GFP antibody followed by fixation. Subsequently, we applied GFP primary antibody again to label the intracellular pool and used a secondary antibody conjugated to a different chromophore to visualize intracellular immunoreactivity (Fig. 1C). We compared the ratio of surface GluN2B versus intracellular receptors. Among the mutants tested, only GluN2B S1413L displayed a statistically significant deficit in surface expression (30% reduction) compared with GluN2B WT (Fig. 1D). The other GluN2B mutants displayed no significant change in surface expression.
Figure 1.

Differential surface expression of GluN2B C-terminal variants expressed in cultured rat hippocampal neurons. A, Three rare variants in the GluN2B C terminus were identified from human patients via third-generation deep sequencing. Human S1415 is analogous to S1413 in rat; L1424 is analogous to L1422 in rat; and S1452 is analogous to S1450 in rat. B, Reference and minor allele frequency of three C-terminal variants in human diseases. C, D, Three rat GluN2B mutants are expressed after transient transfection in DIV 14 hippocampal neurons. Surface expression of GluN2B S1413L is reduced, whereas other GluN2B variants do not alter surface expression in hippocampal neurons. C, Representative images of surface (red channel) and intracellular (blue channel) expression of rat GluN2B mutants. Scale bar, 10 μm. D, Quantitive analysis of C. **p < 0.01, n = 5, one-way ANOVA.
Several GluN2B C-terminal variants disrupt binding to MAGUKs
The GluN2B subunit binds to a variety of MAGUK proteins via a direct interaction between the last 4 aa (ESDV), often referred to as the PDZ ligand, and the PDZ domains of MAGUKs. The PDZ ligand on GluN2B stabilizes NMDARs on the cell surface. Moreover, the synaptic expression of NMDARs is stabilized via binding to PSD-95. To test whether the three variants affect PSD-95 or SAP102 binding to GluN2B, we generated GST-GluN2B C-terminal fusion proteins with single variants in the GluN2B tail. We used a fusion protein containing the last 175 aa of GluN2B, as described previously (Sanz-Clemente et al., 2010), to compare WT GST-GluN2B (including aa 1315–1482) to the GluN2B harboring S1413L, L1422F, or S1450F variants. We conducted pull-down assays with the WT or mutant GST-GluN2B fusion proteins using lysates from rat brain P2 fractions and probed for PSD-95 or SAP102. Interestingly, all three GluN2B mutant GST fusion proteins showed impaired binding to both PSD-95 and SAP102 (Fig. 2A,B). The GST-GluN2B S1413L, L1422F, and S1450F binding was reduced by ∼50% (Fig. 2C,D). As has been reported previously, GST-GluN2B truncated to exclude the PDZ ligand by the introduction of a stop codon at Ser1482 (ΔSTOP) disrupts MAGUK binding. Therefore, we used this truncated version of GluN2B as a comparison for impaired binding (Fig. 2C,D).
Figure 2.

Binding to MAGUKs is impaired in three rat GluN2B mutants. The GST-GluN2B C terminus fusion protein was used for a pull-down assay of PSD-95 (A, C) and SAP102 (B, D) from mouse brain synaptosomal fractions. *p < 0.05; **p < 0.01, p < 0.001, n = 5, one-way ANOVA.
Reduced spine number in GluN2B S1413L-expressing neurons
Because reduced surface expression of GluN2B is often associated with other physiological changes, we next investigated the dendritic morphology of neurons expressing the three GluN2B mutants (S1413L, L1422F, or S1450F) that had shown a clear phenotype in our biochemical assays. EGFP was coexpressed with GluN2B, WT or mutant, in hippocampal cultures (DIV 14). Dendritic spines were visualized with GFP immunostaining 3 d after transfection (DIV 17). Representative images of dendritic spines in GluN2B-GFP-expressing neurons are shown in Figure 3A. We calculated the number of spines in 20 μm dendrites in three areas of each transfected neuron. Only GluN2B S1413L-expressing neurons showed a statistically significant reduction in dendritic spine number compared with GluN2B WT-expressing neurons (20% decrease; Fig. 3B). The other two mutants did not alter the spine density detectably in rat hippocampal neurons.
Figure 3.

Dendritic spine number is decreased in hippocampal pyramidal neurons expressing GluN2B S1413L. A, DIV 14 hippocampal neurons were transiently transfected with pCAG-GFP and GluN2B. At DIV 17, neurons were stained for GFP-tagged GluN2B and spines filled with GFP in 20 μm dendrites were analyzed. Three regions were selected and averaged for every neuron. Scale bar, 5 μm. B, Quantification of spine density. ***p < 0.001, n = 3, one-way ANOVA.
NMDAR pharmacological properties and response time course are not affected by the GluN2B C-terminal variants
We next tested whether the three GluN2B mutants (S1413L, L1422F, and S1450F) that had impaired MAGUK binding also displayed altered pharmacological properties. We coexpressed GluN2B (WT, S1413L, L1422F, or S1450F) with GluN1 in X. laevis oocytes and performed TEVC recordings. We determined the agonist potencies (EC50, the agonist concentrations that can produce a half-maximal current response) for glutamate and glycine (Fig. 4A,B), the potency for inhibition by extracellular magnesium (IC50; Fig. 4C), and the proton sensitivity (percentage inhibition by pH 6.8 vs pH 7.6; Fig. 4D). Except for a modest decrease of glutamate potency (increased EC50 value; Table 1) in the mutant L1422F and a mild increase of glycine potency (decreased EC50 value; Table 1) in the mutant S1450F, the data indicated that the major pharmacological properties remain unchanged in NMDARs containing any of these three variants.
Figure 4.

The GluN2B C-teminal variants do not change functional properties of NMDARs. Current responses in A–D were recorded under TEVC from Xenopus oocytes to determine pharmacological properties. A, B, Concentration–response curves (VHOLD = −40 mV) for glutamate in the presence of 30 μm glycine (A) and glycine in the presence of 100 μm glutamate (B) are shown. C, Concentration–response curves for inhibition of mutant and wild-type NMDA receptors by extracellular Mg2+; currents (VHOLD = −60 mV) were activated by 100 μm glutamate and 30 μm glycine. D, Mean ratio (in percentage) of the current response to 100 μm glutamate and 30 μm glycine recorded at pH 6.8 and the current response at pH 7.6 (VHOLD = −40 mV). E, Superimposed whole-cell current recordings in response to 1000 μm glutamate and 30 μm glycine from HEK cells expressing GluN1/GluN2B (VHOLD = −60 mV). F, Weighted mean time constants describing deactivation were not detectably different among the mutations tested. p > 0.05, one-way ANOVA, Bonferroni's multiple-comparisons test.
Table 1.
Summary of pharmacological results of GluN2B WT and mutants in X. laevis oocytes
| Glu, EC50, μM (nH, n) | Gly, EC50, μM (nH, n) | Mg2+, IC50, μM (nH, n) | Proton, IpH6.8/IpH7.6% (n) | |
|---|---|---|---|---|
| WT 2B | 1.3 ± 0.14 (1.1, 15) | 0.36 ± 0.02 (1.2, 12) | 28 ± 3.7 (0.92, 12) | 21 ± 1.6 (12) |
| S1413L | 1.3 ± 0.16 (1.0, 10) | 0.36 ± 0.06 (1.4, 6) | 20 ± 2.2 (0.91, 7) | 19 ± 0.33 (6) |
| L1422F | 2.1 ± 0.15 (1.2, 8)* | 0.43 ± 0.02 (1.4, 9) | 28 ± 3.0 (0.92, 6) | 17 ± 1.2 (6) |
| S1450F | 1.3 ± 0.10 (1.1, 6) | 0.25 ± 0.02 (1.3, 6)* | 31 ± 4.1 (0.86, 8) | 23 ± 1.7 (6) |
Data are shown as mean ± SEM.
nH, Hill slope.
*p < 0.05, one way ANOVA, Bonferroni's multiple-comparisons test.
In addition, we assessed the effect of the three variants within the GluN2B C terminus on the response amplitude and time course of NMDARs. The NMDAR current–response time course after glutamate removal (in the presence of saturating glycine) is thought to determine the time course of the synaptic current (Lester et al., 1990). We therefore recorded the current response under voltage clamp from HEK cells transiently cotransfected with GluN1 plus mutant or WT GluN2B during rapid solution exchange. The deactivation time course of all three NMDARs variants after the removal of glutamate could be described by two exponential components, which were similar to GluN2B WT receptors (Fig. 4E,F, Table 2). No significant differences were detected among any fitted parameters (Table 2). In addition, there were no detectable differences in the response amplitude, response rise time, or charge transfer during brief glutamate application. Together, these data suggest that these three rare variants do not change NMDAR pharmacology, response amplitude, or response time course significantly.
Table 2.
The three GluN2B C-terminal mutants do not change the response properties of NMDARs
| WT GFP-2B | S1413L | L1422F | S1450F | |
|---|---|---|---|---|
| Prolonged application | ||||
| Amplitude (peak, pA/pF) | 77 ± 16 | 93 ± 9.5 | 66 ± 16 | 50 ± 14 |
| Amplitude (SS, pA/pF) | 47 ± 13 | 53 ± 1.9 | 41 ± 7.4 | 32 ± 8.4 |
| ISS/IPEAK | 59 ± 8.1% | 59 ± 4.0% | 65 ± 6.0% | 69 ± 6.0% |
| Rise time (ms) | 9.6 ± 0.5 | 8.9 ± 0.4 | 9.5 ± 0.8 | 8.9 ± 0.7 |
| Desensitization, τ (ms) | 314 ± 50 | 320 ± 28 | 358 ± 47 | 372 ± 95 |
| Deactivation | ||||
| τFAST (ms) | 422 ± 111 | 321 ± 24 | 359 ± 41 | 300 ± 58 |
| τSLOW (ms) | 1081 ± 90 | 1296 ± 143 | 1229 ± 116 | 1069 ± 104 |
| % τFAST | 55 ± 6.2% | 48 ± 4.0% | 50 ± 4.4% | 42 ± 11% |
| τW (ms) | 779 ± 92 | 789 ± 50 | 789 ± 75 | 744 ± 68 |
| Brief application | ||||
| Amplitude (peak, pA/pF) | 63 ± 18 | 58 ± 10 | 52 ± 14 | 40 ± 12 |
| Rise time (ms) | 8.5 ± 0.5 | 8.3 ± 0.5 | 9.7 ± 0.7 | 7.8 ± 0.4 |
| Deactivation | ||||
| τFAST (ms) | 118 ± 18 | 96 ± 16 | 268 ± 102 | 135 ± 23 |
| τSLOW (ms) | 912 ± 73 | 981 ± 132 | 670 ± 87 | 733 ± 60 |
| % τFAST | 78% | 77% | 72% | 72% |
| τW (ms) | 270 ± 27 | 274 ± 32 | 328 ± 42 | 292 ± 71 |
| Charge transfer | 15,615 | 15,754 | 16,505 | 9,726 |
| n | 8–10 | 9–12 | 6–9 | 6–8 |
p > 0.05 compared with WT GFP-2B, one-way ANOVA, Bonferroni's multiple-comparisons test.
NMDAR currents are impaired in GluN2B S1413L-expressing neurons
Our data showed that, compared with GluN2B WT, GluN2B S1413L displays lower surface expression (Fig. 1), impaired binding to MAGUKs (Fig. 2), and decreased dendritic spine density (Fig. 3). From these data, we would predict that synaptic current responses would be reduced if there were not confounding changes in receptor function produced by these amino acid substitutions. Our functional analysis should allow us to interpret any change in synaptic response amplitude or time course as being reflective of the number of receptors because we know that the variants have no effect on NMDAR time course, amplitude, or agonist potency. We therefore designed a series of experiments to assess whether mutant GluN2B would alter the synaptic current response properties. We used GRIN2Afl/flGRIN2Bfl/fl mice to perform molecular replacement experiments (Gray et al., 2011). Because GluN2A and GluN2B are expressed in principal cells of the hippocampus, deletion of these subunits should eliminate NMDAR currents. In agreement with previous reports (Gray et al., 2011), electrophysiological analysis in CA1 pyramidal neurons in acute hippocampal slices showed that the NMDA EPSCs were lost in Cre-mCherry-positive neurons (Fig. 5A). In addition, transient transfection of neurons with WT GluN2B fully rescued NMDA EPSCs in Cre-expressing GRIN2Afl/flGRIN2Bfl/fl cells, as assessed by comparison of EPSC response amplitudes. In contrast, transient transfection of neurons with GluN2B S1413L restored only ∼60% of the NMDA EPSCs in Cre-positive GRIN2Afl/flGRIN2Bfl/fl neurons (Fig. 5B,C). NMDA EPSC decay time was not altered by the GluN2B S1413L variant compared with GluN2B (Fig. 5D). Because GluN2B S1413L has no effect on NMDAR response properties or amplitudes, these deficits in synaptic currents are consistent with deficits in synaptic targeting.
Figure 5.
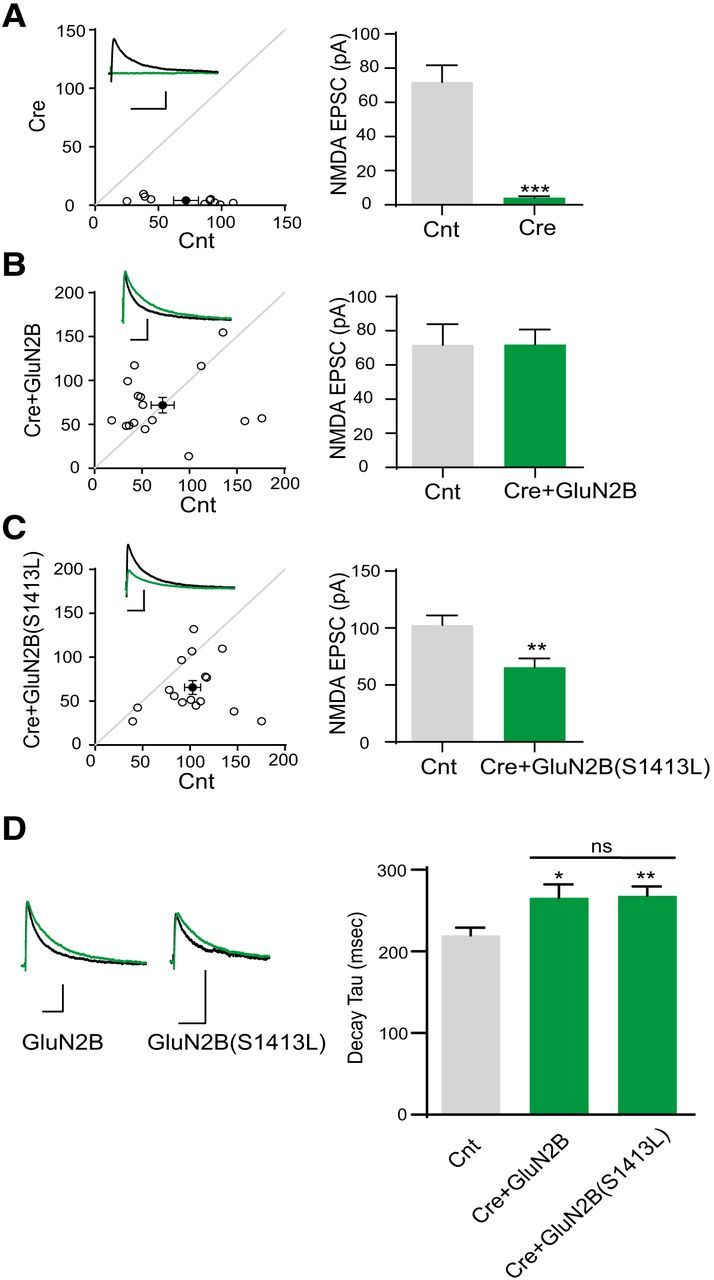
GluN2B S1413L cannot rescue NMDA EPSCs. A, Loss of evoked NMDA EPSCs in Cre-positive GRIN2Afl/flGRIN2Bfl/fl hippocampal CA1 neurons at P13–P15. Amplitude: Control (Cnt), 71.9 ± 9.9 pA; Cre, 4.2 ± 0.9 pA, n = 10. ***p < 0.0001. B, Coexpression of Cre-mCherry with GluN2B-IRES-GFP rescued NMDA EPSCs Amplitude: Cnt, 71.8 ± 12.2 pA; Cre+GluN2B, 71.9 ± 8.8 pA, n = 16. p = 0.99. C, Coexpression of Cre-mCherry with GluN2B S1413L–IRES-GFP did not fully rescue NMDA EPSCs [103 ± 8.5 pA; Cre + GluN2B(S1413L), 66 ± 7.9 pA]. n = 16. **p = 0.0042. D, GluN2B S1413L variant does not change the NMDA EPSC decay time constant compared with GluN2B. Averaged and peak-aligned NMDA EPSCs were recorded from control or transfected cells from GRIN2Afl/flGRIN2Bfl/fl mice [decay time constant: Cnt, 220.0 ± 9.4 ms, n = 32; Cre + GluN2B, 265.9 ± 16.4 ms, n = 16; Cre + GluN2B(S1413L), 268.2 ± 11.3, n = 16. Cnt vs Cre + GluN2B, p = 0.012 < 0.05; Cnt vs Cre + GluN2B(S1413L), p = 0.0033 < 0.01; Cre + GluN2B vs Cre + GluN2B(S1413L), p = 0.91]. Scale bar, 50 pA and 0.02 s. Error bars indicate SEM.
Discussion
Many lines of evidence support a critical role for synaptic dysfunction in the etiology of a variety of cognitive disorders (Zoghbi and Bear, 2012; Cooke and Bear, 2012; Morrison and Baxter, 2012; Nithianantharajah and Hannan, 2013; Buffington et al., 2014; Balu and Coyle, 2015). Synaptic proteins including receptors, adhesion molecules, and scaffolding proteins have been shown to be involved in regulating synaptic transmission and plasticity (Fields and Itoh, 1996; Yang et al., 2004; Derkach et al., 2007; Dityatev et al., 2008; Xu, 2011; Verpelli et al., 2012; Ebert and Greenberg, 2013; Fan et al., 2014). Postsynaptic NMDARs are essential for synaptic development and plasticity and their dysfunction has been implicated in neurological and psychiatric diseases.
Recently, it has become possible to evaluate genetic variation in the healthy human population through extensive exome sequencing. At present, 425 missense variants have been identified in GRIN2B (gnomAD browser β, genome Aggregation Database evaluated on 12-12-2016), which is fewer than would be expected from overall rate of genetic variation (see Fig. 1E in Ogden et al., 2017). This reduced degree of genetic variation compared with the exome places GRIN2B in the top 1 percentile in terms of intolerance (Petrovski et al., 2013). Furthermore, there are regions of extreme intolerance to variation within the protein, including the transmembrane helices, which show virtually no variants in the healthy population (Swanger et al., 2016; Ogden et al., 2017). Indeed, loss-of-function variants are likely deleterious given that GluN2B is essential for viability in mice.
Whole-exome sequencing has also produced an enormous amount of data linking genetic composition to disease phenotype. Missense mutations located both in extracellular domains and transmembrane elements have been identified in patient cohorts with developmental disability or delays, epileptic encephalopathy, autism spectrum disorders (ASDs), and Alzheimer's disease (Endele et al., 2010; Tarabeux et al., 2011; de Ligt et al., 2012; Lemke et al., 2013; Lesca et al., 2013; Andreoli et al., 2014; Lemke et al., 2014; Fromer et al., 2014; Zhu et al., 2015; Swanger et al., 2016; for review, see Yuan et al., 2015 and Hu et al., 2016). Although there are a growing number of variants being identified in patients with neurological disease, functional data exist for <10% of the rare variants and de novo mutations reported for the GRIN family of genes. To understand whether the pathophysiology results from changes in mutant receptor response properties, functional evaluation of the mutant receptors is necessary.
The C terminus shows substantial genetic variation and largely appears tolerant to genetic changes (Swanger et al., 2016). However, numerous sites for posttranslational modification and binding to intracellular scaffolding proteins exist in this region, suggesting that some variants could influence receptor function and thus human disease. Despite the description of numerous variants in the C terminus (Yuan et al., 2015; Hu et al., 2016), there has yet to be any systematic exploration of the effect on NMDAR localization or function for any of the variants located in the C terminus. We have examined several missense variants in the GluN2B C-terminal domain to evaluate potential phenotypes in trafficking, synaptic protein binding, and function. Our results show clear changes in surface expression and subcellular localization that could contribute to patient phenotypes.
We targeted rare missense variants located in the C terminus of GluN2B, none of which had been studied using either in vitro or in vivo functional assays. Our rationale was twofold. First, we hoped to learn about the molecular consequences of these variants that could provide insight into the underlying pathophysiology of disease. Second, because the C termini of NMDAR are large and hard to evaluate systematically regarding their role in trafficking and functional modulation, we hypothesized that targeting critical residues revealed from patient sequencing would provide a powerful clue to understanding molecular determinants that might influence NMDAR synaptic expression and/or functional properties.
We focused on specific residues within the GluN2B C terminus that might affect trafficking and synaptic function and first evaluated surface expression and MAGUK binding in a preliminary screen, which allowed us to narrow the study to three specific variants. Among these three rare variants, we found one that had clear deficits in plasma membrane expression, as well as a decrease in MAGUK binding. Our findings are consistent with human GluN2B S1415, L1424, and S1452 affecting MAGUK binding to the GluN2B C terminus. It may seem unexpected that all three of the variants evaluated had defects in MAGUK binding because none was within the PDZ ligand. However, there are residues upstream of the PDZ ligand that regulate non-PDZ binding to SAP102 (Chen et al., 2011) as well as PSD-95 (Cousins and Stephenson, 2012). The variants in the cluster between S1415 and S1452 may be involved in structural or posttranslational regulation of the primary MAGUK-binding region.
Only one mutant evaluated, GluN2B S1415L, showed defects in both surface expression and spine density. It is interesting that this variant was identified in an autism patient because spine changes are often implicated in autism spectrum disorders (ASDs). In many cases, there is an overabundance of spines, especially in cortical pyramidal neurons in autism patients and animal models (Comery et al., 1997; Irwin et al., 2000; Hutsler and Zhang, 2010; Penzes et al., 2011; Santini et al., 2013; Tang et al., 2014), but deficits are also reported (Zoghbi and Bear, 2012; Belichenko et al., 2009). For example, spine density is largely increased in tuberous sclerosis neurons, whereas it is reduced in Angelman syndrome and Down syndrome neurons (Tang et al., 2014; Hethorn et al., 2015; Belichenko et al., 2004). In one of the most popular ASD animal models, Shank3 KO mice have decreased spine density in striatal medium spiny neurons (Peça et al., 2011). Similarly, loss-of-function Shank3 plays a dominant-negative role in regulating spine number (Durand et al., 2012). Interestingly, increased spine density was observed in Purkinje cells of TSC1-null mice (Tsai et al., 2012), whereas reduction of spine density was reported in hippocampal and cortical neurons lacking TSC1 (Tavazoie et al., 2005; Meikle et al., 2008). However, the same group reported no change in spine density or morphology of acute loss of Tsc1 neuron in slices and suggested that this was due to different hippocampal activity levels in vivo and in slice cultures (Bateup et al., 2011). In addition, a mutation in neuroligin 4X identified in an autistic patient is associated with changes in the number of functional spines (Bemben et al., 2015). Therefore, there are a variety of examples of aberrant spine density in animal models of ASDs and our findings support a model in which GluN2B dysfunction contributes to a dendritic spine deficit in ASDs.
In addition to changes in spine density, there are also reports of changes in spine morphology or dendritic arborizations. For example, in both Fragile X and Rett syndrome, there is no change in spine number, but rather a change in spine morphology with more filopodia-like and thin spines (Phillips and Pozzo-Miller, 2015). In addition, the Shank3 KO mouse model displays increases in dendritic arborizations. Indeed, there is evidence that both NMDAR function and MAGUK binding are important determinants in regulating dendritic spine morphology (Chen et al., 2011). Our finding that GluN2B S1413L affects spine density is consistent with the overall literature of spine phenotypes with ASD. More precise studies on potential effects of this variant on dendritic complexity or changes in spine morphology are important topics for further studies.
We observed a variety of abnormal phenotypes for GluN2B S1413L, including deficits in NMDAR trafficking, synaptic NMDAR currents, and spine density. What might be the molecular basis for these deficits? This residue is not part of any known motif or previously described binding domain. Although serines are often targets for phosphorylation, we did not find S1413 to be a phosphorylated residue in any database or in the literature (for review, see Sanz-Clemente et al., 2013a). To test this, we performed immunoprecipitation of native GluN2B from mouse brain synaptosomal fractions and evaluated using mass spectrometry, but also did not find S1413 to be phosphorylated in vivo under basal conditions (S.L., unpublished results). However, we certainly cannot exclude the possibility that synaptic activity during development might trigger phosphorylation of this residue by an unidentified kinase.
In summary, we have used information from whole-exome sequencing or Sanger sequencing of NMDAR genes of patients to investigate mechanisms underlying NMDAR dysfunction. Specifically, we found that three GRIN2B missense variants generated GluN2B subunits that exhibited deficient binding to MAGUKs. In addition, surface expression of one variant, GluN2B S1413L, was significantly reduced. We also found that S1413L affected spine density. Furthermore, we found that GluN2B S1413L resulted in a deficit in the rescue of NMDAR synaptic currents in mice, but was without effect on normal NMDAR function. This result is consistent with a deficit in the number of synaptic NMDARs. Together, these data support the idea that this variant has multiple effects on protein binding and synaptic function that could be relevant to spine function.
Autism is a disorder thought to involve dysfunction at the level of the spine. In an autistic spectrum disorder cohort study, human GluN2B S1415L was found in one of 428 ASD patients and was inherited from one parent (validated by Sanger sequencing). This variant was absent in 283 Caucasian control individuals (Endele et al., 2010; Tarabeux et al., 2011) and was neither annotated in dbSNP nor reported in ClinVar. It is not observed in 1000 Genomes and National Heart, Lung, and Blood Institute Exome Sequencing Project. Two individuals of South Asian descent reported with this variant in the gnomAD browser were identified with low confidence based on the quality of the data. Although we lack the clinical information to determine pathogenicity of human S1415L, our functional assays on rodent S1413L reveal a possible mechanism whereby a single amino acid substitution in the GluN2B C terminus could alter synaptic and spine function and thus potentially contribute to diseases with complex genetics such as ASD. Further studies are needed to fully understand how this single variant might affect neural function.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke–National Institutes of Health (NINDS Intramural Program Grant to S.L., M.V., L.Z., A.S.-C., J.D.B., W.L. and K.W.R. and Grants NS036654 and NS092989 to S.F.T.), the Eunice Kennedy Shriver National Institute of Child Health and Human Development–NIH (Grant R01-HD082373 to H.Y.), the National Center for Advancing Translational Sciences–NIH (Grant UL1-TR000454 to H.Y.), the National Institute on Aging–NIH (Career Transition Award R00 AG041225 to A.S.-C.), and Portuguese Foundation for Science and Technology (FCT-Fundacao para a Ciencia e a Tecnologia, Grant SFRH/BI/106010/2015 to M.V.). We thank Jing Zhang and Phuong Le for technical assistance and the NINDS light imaging facility for their expertise.
S.F.T. is a consultant of Janssen Pharmaceuticals, Inc., Pfizer Inc, Boehringer Ingelheim Pharma GmbH & Co. KG, and co-founder of NeurOp Inc. The other authors declare no competing financial interest.
References
- Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, Tanzi RE, Bertram L (2008) Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet 40:827–834. 10.1038/ng.171 [DOI] [PubMed] [Google Scholar]
- Andreoli V, De Marco EV, Trecroci F, Cittadella R, Di Palma G, Gambardella A (2014) Potential involvement of GRIN2B encoding the NMDA receptor subunit NR2B in the spectrum of Alzheimer's disease. J Neural Transm (Vienna) 121:533–542. 10.1007/s00702-013-1125-7 [DOI] [PubMed] [Google Scholar]
- Arning L, Saft C, Wieczorek S, Andrich J, Kraus PH, Epplen JT (2007) NR2A and NR2B receptor gene variations modify age at onset in Huntington disease in a sex-specific manner. Hum Genet 122:175–182. 10.1007/s00439-007-0393-4 [DOI] [PubMed] [Google Scholar]
- Balu DT, Coyle JT (2015) The NMDA receptor 'glycine modulatory site' in schizophrenia: D-serine, glycine, and beyond. Curr Opin Pharmacol 20:109–115. 10.1016/j.coph.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL (2011) Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J Neurosci 31:8862–8869. 10.1523/JNEUROSCI.1617-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC (2004) Synaptic structural abnormalities in the Ts65Dn mouse model of Down syndrome. J Comp Neurol 480:281–298. 10.1002/cne.20337 [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Wright EE, Belichenko NP, Masliah E, Li HH, Mobley WC, Francke U (2009) Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol 514:240–258. 10.1002/cne.22009 [DOI] [PubMed] [Google Scholar]
- Bemben MA, Nguyen QA, Wang T, Li Y, Nicoll RA, Roche KW (2015) Autism-associated mutation inhibits protein kinase C-mediated neuroligin-4X enhancement of excitatory synapses. Proc Natl Acad Sci U S A 112:2551–2556. 10.1073/pnas.1500501112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, Huang W, Costa-Mattioli M (2014) Translational control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci 37:17–38. 10.1146/annurev-neuro-071013-014100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Holderith NB, Nusser Z, DiGregorio DA, Cull-Candy SG (2005) Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat Neurosci 8:1310–1318. 10.1038/nn1534 [DOI] [PubMed] [Google Scholar]
- Chen BS, Thomas EV, Sanz-Clemente A, Roche KW (2011) NMDA receptor-dependent regulation of dendritic spine morphology by SAP102 splice variants. J Neurosci 31:89–96. 10.1523/JNEUROSCI.1034-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Gray JA, Sanz-Clemente A, Wei Z, Thomas EV, Nicoll RA, Roche KW (2012) SAP102 mediates synaptic clearance of NMDA receptors. Cell Rep 2:1120–1128. 10.1016/j.celrep.2012.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Huang YH, Lau LF, Huganir RL (2004) Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci 24:10248–10259. 10.1523/JNEUROSCI.0546-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT (1997) Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A 94:5401–5404. 10.1073/pnas.94.10.5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SF, Bear MF (2012) Stimulus-selective response plasticity in the visual cortex: an assay for the assessment of pathophysiology and treatment of cognitive impairment associated with psychiatric disorders. Biol Psychiatry 71:487–495. 10.1016/j.biopsych.2011.09.006 [DOI] [PubMed] [Google Scholar]
- Cousins SL, Stephenson FA (2012) Identification of N-methyl-D-aspartic acid (NMDA) receptor subtype-specific binding sites that mediate direct interactions with scaffold protein PSD-95. J Biol Chem 287:13465–13476. 10.1074/jbc.M111.292862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367:1921–1929. 10.1056/NEJMoa1206524 [DOI] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR (2007) Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci 8:101–113. 10.1038/nrn2055 [DOI] [PubMed] [Google Scholar]
- Dityatev A, Bukalo O, Schachner M (2008) Modulation of synaptic transmission and plasticity by cell adhesion and repulsion molecules. Neuron Glia Biol 4:197–209. 10.1017/S1740925X09990111 [DOI] [PubMed] [Google Scholar]
- Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, Montcouquiol M, Sans N (2012) SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry 17:71–84. 10.1038/mp.2011.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME (2013) Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493:327–337. 10.1038/nature11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endele S, et al. (2010) Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 42:1021–1026. 10.1038/ng.677 [DOI] [PubMed] [Google Scholar]
- Epi KC, et al. (2013) De novo mutations in epileptic encephalopathies. Nature 501:217–221. 10.1038/nature12439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Jin WY, Wang YT (2014) The NMDA receptor complex: a multifunctional machine at the glutamatergic synapse. Front Cell Neurosci 8:160. 10.3389/fncel.2014.00160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Itoh K (1996) Neural cell adhesion molecules in activity-dependent development and synaptic plasticity. Trends Neurosci 19:473–480. 10.1016/S0166-2236(96)30013-1 [DOI] [PubMed] [Google Scholar]
- Freunscht I, Popp B, Blank R, Endele S, Moog U, Petri H, Prott EC, Reis A, Rübo J, Zabel B, Zenker M, Hebebrand J, Wieczorek D (2013) Behavioral phenotype in five individuals with de novo mutations within the GRIN2B gene. Behav Brain Funct 9:20. 10.1186/1744-9081-9-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, et al. (2014) De novo mutations in schizophrenia implicate synaptic networks. Nature 506:179–184. 10.1038/nature12929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA (2011) Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71:1085–1101. 10.1016/j.neuron.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, Patry L, Massicotte C, Ambalavanan A, Spiegelman D, Diallo O, Henrion E, Dionne-Laporte A, Fougerat A, Pshezhetsky AV, Venkateswaran S, Rouleau GA, Michaud JL (2014) De novo mutations in moderate or severe intellectual disability. PLoS Genet 10:e1004772. 10.1371/journal.pgen.1004772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Tajima N, Risgaard R, Perszyk RE, Jørgensen L, Vance KM, Ogden KK, Clausen RP, Furukawa H, Traynelis SF (2013) Structural determinants of agonist efficacy at the glutamate binding site of N-methyl-D-aspartate receptors. Mol Pharmacol 84:114–127. 10.1124/mol.113.085803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hethorn WR, Ciarlone SL, Filonova I, Rogers JT, Aguirre D, Ramirez RA, Grieco JC, Peters MM, Gulick D, Anderson AE, L Banko J, Lussier AL, Weeber EJ (2015) Reelin supplementation recovers synaptic plasticity and cognitive deficits in a mouse model for Angelman syndrome. Eur J Neurosci 41:1372–1380. 10.1111/ejn.12893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Chen W, Myers SJ, Yuan H, Traynelis SF (2016) Human GRIN2B variants in neurodevelopmental disorders. J Pharmacol Sci 132:115–121. 10.1016/j.jphs.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H (2010) Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res 1309:83–94. 10.1016/j.brainres.2009.09.120 [DOI] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT (2000) Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10:1038–1044. 10.1093/cercor/10.10.1038 [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M (2004) PDZ domain proteins of synapses. Nat Rev Neurosci 5:771–781. 10.1038/nrn1517 [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M (1996) Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 16:333–344. 10.1016/S0896-6273(00)80051-3 [DOI] [PubMed] [Google Scholar]
- Lemke JR, et al. (2013) Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet 45:1067–1072. 10.1038/ng.2728 [DOI] [PubMed] [Google Scholar]
- Lemke JR, Hendrickx R, Geider K, Laube B, Schwake M, Harvey RJ, James VM, Pepler A, Steiner I, Hörtnagel K, Neidhardt J, Ruf S, Wolff M, Bartholdi D, Caraballo R, Platzer K, Suls A, De Jonghe P, Biskup S, Weckhuysen S (2014) GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann Neurol 75:147–154. 10.1002/ana.24073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesca G, et al. (2013) GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet 45:1061–1066. 10.1038/ng.2726 [DOI] [PubMed] [Google Scholar]
- Lester RA, Clements JD, Westbrook GL, Jahr CE (1990) Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature 346:565–567. 10.1038/346565a0 [DOI] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ (2008) Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 28:5422–5432. 10.1523/JNEUROSCI.0955-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Baxter MG (2012) The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci 13:240–250. 10.1038/nrn3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RA, Casals F, Gauthier J, Hamdan FF, Keebler J, Boyko AR, Bustamante CD, Piton AM, Spiegelman D, Henrion E, Zilversmit M, Hussin J, Quinlan J, Yang Y, Lafrenière RG, Griffing AR, Stone EA, Rouleau GA, Awadalla P (2011) A population genetic approach to mapping neurological disorder genes using deep resequencing. PLoS Genet 7:e1001318. 10.1371/journal.pgen.1001318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithianantharajah J, Hannan AJ (2013) Dysregulation of synaptic proteins, dendritic spine abnormalities and pathological plasticity of synapses as experience-dependent mediators of cognitive and psychiatric symptoms in Huntington's disease. Neuroscience 251:66–74. 10.1016/j.neuroscience.2012.05.043 [DOI] [PubMed] [Google Scholar]
- Ogden KK, Chen W, Swanger SA, McDaniel MJ, Fan LZ, Hu C, Tankovic A, Kusumoto H, Kosobucki GJ, Schulien AJ, Su Z, Pecha J, Bhattacharya S, Petrovski S, Cohen AE, Aizenman E, Traynelis SF, Yuan H (2017) Molecular mechanism of disease-associated mutations in the Pre-M1 Helix of NMDA receptors and potential rescue pharmacology. PLoS Genet 13:e1006536. 10.1371/journal.pgen.1006536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki T, Sakurai K, Dou H, Toru M, Yamakawa-Kobayashi K, Arinami T (2001) Mutation analysis of the NMDAR2B (GRIN2B) gene in schizophrenia. Mol Psychiatry 6:211–216. 10.1038/sj.mp.4000808 [DOI] [PubMed] [Google Scholar]
- Parsons DW, et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. 10.1126/science.1164382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G (2011) Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472:437–442. 10.1038/nature09965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14:285–293. 10.1038/nn.2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB (2013) Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9:e1003709. 10.1371/journal.pgen.1003709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M, Pozzo-Miller L (2015) Dendritic spine dysgenesis in autism related disorders. Neurosci Lett 601:30–40. 10.1016/j.neulet.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Huganir RL (1995) Synaptic expression of the high-affinity kainate receptor subunit KA2 in hippocampal cultures. Neuroscience 69:383–393. 10.1016/0306-4522(95)00253-F [DOI] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E (2013) Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493:411–415. 10.1038/nature11782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Matta JA, Isaac JT, Roche KW (2010) Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 67:984–996. 10.1016/j.neuron.2010.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Nicoll RA, Roche KW (2013a) Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19:62–75. 10.1177/1073858411435129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Gray JA, Ogilvie KA, Nicoll RA, Roche KW (2013b) Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep 3:607–614. 10.1016/j.celrep.2013.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Chen W, Wells G, Burger PB, Tankovic A, Bhattacharya S, Strong KL, Hu C, Kusumoto H, Zhang J, Adams DR, Millichap JJ, Petrovski S, Traynelis SF, Yuan H (2016) Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. Am J Hum Genet 99:1261–1280. 10.1016/j.ajhg.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83:1131–1143. 10.1016/j.neuron.2014.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarabeux J, et al. (2011) Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry 1:e55. 10.1038/tp.2011.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL (2005) Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8:1727–1734. 10.1038/nn1566 [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M (2012) Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488:647–651. 10.1038/nature11310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpelli C, Schmeisser MJ, Sala C, Boeckers TM (2012) Scaffold proteins at the postsynaptic density. Adv Exp Med Biol 970:29–61. 10.1007/978-3-7091-0932-8_2 [DOI] [PubMed] [Google Scholar]
- Wills TA, Klug JR, Silberman Y, Baucum AJ, Weitlauf C, Colbran RJ, Delpire E, Winder DG (2012) GluN2B subunit deletion reveals key role in acute and chronic ethanol sensitivity of glutamate synapses in bed nucleus of the stria terminalis. Proc Natl Acad Sci U S A 109:E278–287. 10.1073/pnas.1113820109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W. (2011) PSD-95-like membrane associated guanylate kinases (PSD-MAGUKs) and synaptic plasticity. Curr Opin Neurobiol 21:306–312. 10.1016/j.conb.2011.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XL, Xiong WC, Mei L (2004) Lipid rafts in neuregulin signaling at synapses. Life Sci 75:2495–2504. 10.1016/j.lfs.2004.04.036 [DOI] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF (2009) Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci 29:12045–12058. 10.1523/jneurosci.1365-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF (2015) Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol Pharmacol 88:203–217. 10.1124/mol.115.097998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, et al. (2015) Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med 17:774–781. 10.1038/gim.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF (2012) Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4: pii: a009886. 10.1101/cshperspect.a009886 [DOI] [PMC free article] [PubMed] [Google Scholar]