

Abstract
Many allosteric proteins form homo-oligomeric complexes to regulate a biological function. In homo-oligomers, subunits establish communication pathways that are modulated by external stimuli like ligand binding. A challenge for dissecting the communication mechanisms in homo-oligomers is identifying intermediate liganded states, which are typically transiently populated. However, their identities provide the most mechanistic information on how ligand-induced signals propagate from bound to empty subunits. Here, we dissected the directionality and magnitude of subunit communication in a reengineered single-chain version of the homodimeric transcription factor cAMP receptor protein. By combining wild-type and mutant subunits in various asymmetric configurations, we revealed a linear relationship between the magnitude of cooperative effects and the number of mutant subunits. We found that a single mutation is sufficient to change the global allosteric behavior of the dimer even when one subunit was wild type. Dimers harboring two mutations with opposite cooperative effects had different allosteric properties depending on the arrangement of the mutations. When the two mutations were placed in the same subunit, the resulting cooperativity was neutral. In contrast, when placed in different subunits, the observed cooperativity was dominated by the mutation with strongest effects over cAMP affinity relative to wild type. These results highlight the distinct roles of intrasubunit interactions and intersubunit communication in allostery. Finally, dimers bound to either one or two cAMP molecules had similar DNA affinities, indicating that both asymmetric and symmetric liganded states activate DNA interactions. These studies have revealed the multiple communication pathways that homo-oligomers employ to transduce signals.
Keywords: allosteric regulation, conformational change, cooperativity, cyclic AMP (cAMP), transcription factor, single-chain dimer, subunit communication, cAMP receptor protein (CRP)
Introduction
Allosteric proteins are the basic building blocks in the transmission of biological signals, allowing communication between and within cells and from the extracellular environment to the cytosol (1). Because many allosteric proteins form homo-oligomeric complexes to modulate a ligand-induced biological response (2, 3), intersubunit communication must play a crucial role in the transduction of an allosteric signal (4, 5). Identifying the molecular mechanisms of intersubunit communication has important implications, from understanding how biological systems detect and transduce signals (6, 7) to reengineering of signaling proteins (8–10) and to developing allosteric therapeutic modulators with enhanced affinities and specificities (11, 12). Despite its importance, dissecting the mechanisms of transduction of allosteric signals from one protein subunit to another has proven difficult because it requires monitoring intermediate liganded states that are poorly populated, especially if the protein displays positive ligand binding cooperativity. Therefore, one of the remaining unresolved issues in allostery is dissecting the directionality of pathways of signal transmissions across protein subunits.
A widely used strategy to perturb and examine the mechanisms of intersubunit communication in homo-oligomeric systems is to evaluate a mutation or combinations of mutations on binding or catalytic activities. However, in homo-oligomers mutations are present in all subunits, making it difficult to quantitatively dissect mutational effects from one subunit to another. To overcome this obstacle, in this study we reengineered the homodimeric transcription factor cAMP receptor protein (CRP)3 into a single chain by covalently linking the two identical CRP subunits through an unstructured polypeptide linker (Fig. 1A). The CRP single-chain dimer (CRPSC) allowed us to construct asymmetric CRP dimers harboring either a wild-type and a mutant subunit or subunits with different mutation types. Specifically, the mutations S62F and D53H have been shown to reduce and enhance cAMP binding cooperativity, respectively. By placing these two mutations in various asymmetric configurations, we dissected the directionality and magnitude of mutational perturbations from one subunit to another, thereby providing a unique opportunity to examine communication pathways within and across CRP subunits.
Figure 1.

Design and construction of a CRP single-chain dimer (CRPSC). A, model of a CRPSC that connects the two CRP subunits (cyan and orange) through a flexible polypeptide linker (red). The model was rendered in PyMOL based on the CRP structure bound to cyclic nucleotide (PDB 1G6N). B, structure of CRP in the unliganded (left, PDB 2WC2) and cAMP-bound conformations (right, PDB 1G6N). cAMP is shown in magenta.
Each CRP subunit has two functionally and structurally distinct domains, a cAMP-binding domain in the N terminus that is also responsible for dimer formation and a C-terminal DNA-binding domain (Fig. 1B). CRP binds two cAMP molecules and undergoes a conformational change in the DNA-binding domains that enables the protein to interact with high affinity and specificity with DNA promoter sequences (13–17). The molecular architecture of CRP is ideal for quantitative studies on the mechanisms of transduction of allosteric signals because: 1) cAMP binding reports on intersubunit communication and cooperative interactions, and 2) DNA binding reports on intrasubunit interactions and cAMP-induced conformational changes of the protein (13, 18).
Our results show that CRPSC is indistinguishable from the wild-type CRP homodimer based on solution structure, thermodynamic stability, and cAMP binding affinities and cooperativity. As seen in the wild-type CRP (13), the DNA binding activity of CRPSC is allosterically controlled by cAMP. We find that combinations of wild-type and mutant subunits in CRPSC result in cAMP binding affinities and cooperativities that are different from those of the parental proteins (i.e. wild-type CRP or variants with the same mutation in both subunits). Furthermore, we show that mutations with opposite effects on cAMP binding affinity have dramatically different consequences on cooperative interactions whether the mutations are in the same subunit or in different ones. Finally, we find that asymmetric mutants bound to one cAMP molecule have indistinguishable DNA binding affinity constants compared with the doubly liganded CRPSC, which suggests that a single cAMP molecule bound to CRP is sufficient to allosterically drive the conformational changes required for robust interactions with DNA, and it underscores the role of asymmetric liganded states in the regulation of gene expression (19).
Results
Biophysical and functional characterization of a CRP single-chain dimer
We engineered a CRP single-chain dimer (CRPSC) that connects the C terminus of the first CRP subunit to the N terminus of the second one through a flexible polypeptide linker of the repeat sequence (SGGGG)7 (Fig. 1A). Based on the CRP structures in the unliganded and cAMP-bound states (14–16), the distance between the C terminus of one subunit and the N terminus of the other is ∼75 Å. The designed linker spans a larger distance (∼130 Å) to accommodate the protein's conformational changes induced by cAMP binding. CRPSC migrated as a 48-kDa protein in an SDS denaturing gel, twice the molecular mass of one CRP subunit (Fig. 2A). During the purification process, gel filtration chromatograms of CRPSC had the same elution volume as of wild-type CRP corroborating that in native conditions CRPSC eluted as a pseudodimer (Fig. 2B). During consecutive size exclusion runs, no peaks were observed before CRPSC elution (data not shown), which otherwise would have indicated the formation of a dimer of dimers or other high-order oligomeric states. Moreover, CRPSC and wild-type CRP had identical CD spectra (Fig. 2C), indicating that the global fold and secondary structure content of the two proteins are the same. We evaluated the effect of the polypeptide linker on the thermodynamic stability of CRPSC by monitoring changes in tryptophan fluorescence (Fig. 2D) and circular dichroism (data not shown) as a function of GdnHCl concentration. At the experimental conditions, CRPSC and wild-type CRP displayed a single unfolding transition and had similar fitted thermodynamic parameters (ΔG0 and m values) that are in agreement with previously published data (supplemental Table S1) (20).
Figure 2.
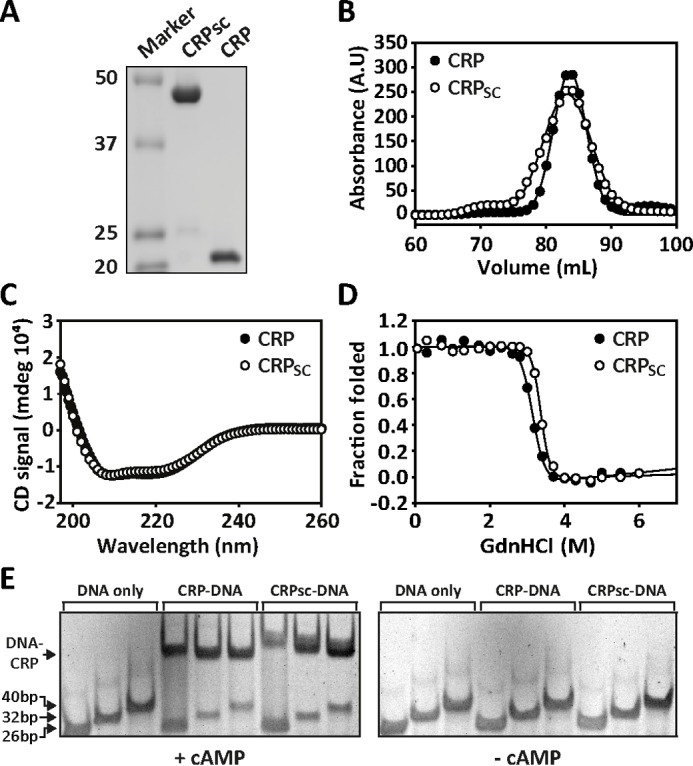
Biophysical and functional characterization of CRPSC. A, SDS-PAGE showing the molecular mass (in kDa) of CRPSC versus wild-type CRP (labeled CRP in all figure panels). B, size-exclusion chromatogram of CRPSC and wild-type CRP. C, CD spectra of CRPSC and wild-type CRP. D, chemical denaturation of CRPSC and wild-type CRP monitored by changes in tryptophan fluorescence. The line corresponds to the fit of a two-state unfolding model as described under “Materials and Methods.” E, CRP-DNA interactions monitored by electrophoretic mobility shift assay using increasing lengths of the lac promoter in the absence and presence of 200 μm cAMP for CRPSC and wild-type CRP.
Having established that the solution structure and the thermodynamic stability between CRPSC and wild-type CRP are very similar, we investigated whether the polypeptide linker affected the basic function of CRP, namely the allosteric activation of DNA binding by cAMP. We used electrophoretic mobility shift assay (EMSA) to monitor the interaction of CRPSC and wild-type CRP to three different lengths of the lac promoter made by 26, 32, and 40 bp (Fig. 2E). The different promoter lengths were used to test whether the polypeptide linker in the CRPSC construct contributed to nonspecific binding to DNA flanking sequences. In the presence of saturating amounts of cAMP, we observed robust DNA binding for both CRPSC and wild-type CRP. Moreover, for all promoter lengths we observed a single band, indicating that the polypeptide linker does not contribute to nonspecific DNA binding. In the absence of cAMP, no DNA binding was observed for both proteins. These results indicate that the polypeptide linker in CRPSC does not affect the cAMP-dependent allosteric activation of the protein.
Quantification of cAMP binding and DNA interactions in CRPSC
To quantitatively determine whether CRPSC had cAMP-binding association constants similar to those reported for wild-type CRP, we used a published method based on changes in 8-anilino-1-naphthalenesulfonic acid (ANS) fluorescence to measure cAMP binding (13, 21). Fig. 3A shows that the cAMP titration curves for CRPSC and wild-type CRP almost completely overlapped, resulting in indistinguishable microscopic association constants using a two binding-site model (supplemental Table S1). We further validated the fluorescence data using isothermal titration calorimetry (ITC). The ITC data revealed similar microscopic cAMP-binding association constants between CRPSC and wild-type CRP (supplemental Fig. S1 and supplemental Table S2). As with wild-type CRP, the sequential binding of two cAMP molecules to CRPSC showed an initial exothermic phase followed by an endothermic phase. This biphasic behavior agrees with previous ITC studies (13, 22) and corroborates that the polypeptide linker does not affect the affinities nor the thermodynamic signatures associated with cAMP binding.
Figure 3.
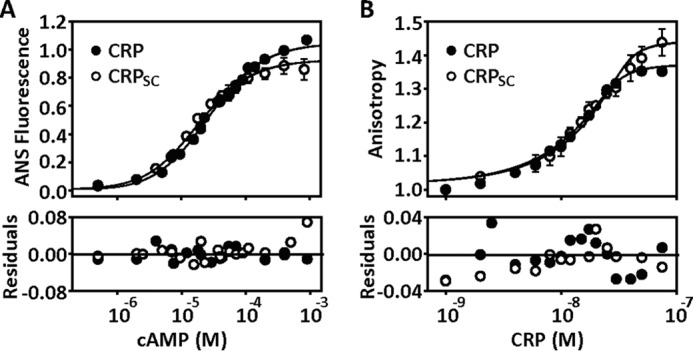
Quantification of the functional behavior of CRPSC. A, cAMP binding to CRPSC and wild-type CRP (labeled CRP in all figure panels) monitored by changes in ANS fluorescence. The solid lines represent the fit using a two-site binding model as described under “Materials and Methods.” B, binding of CRPSC or wild-type CRP to a 32-bp fluorescein-labeled lac promoter monitored by changes in fluorescence anisotropy. The solid lines represent the fit as described in Ref. 32. Residuals of the fit for both experiments are shown below the titrations.
DNA binding constants were obtained by monitoring anisotropy changes during the formation of the complex CRP-DNA using a 32-bp fluorescein-labeled lac promoter (Fig. 3B). The data showed that both CRPSC and wild-type CRP have similar binding affinities for the lac promoter fragment in the presence of 200 μm cAMP, a concentration wherein both binding sites are occupied. Noteworthy, the maximal anisotropy levels were slightly higher with the CRPSC, suggesting a more rigid protein-DNA complex. Control experiments in the absence of cAMP showed negligible DNA binding (supplemental Table S1). Altogether, the results from these quantitative studies demonstrate that the presence of the polypeptide linker that connects the two CRP subunits does not perturb cAMP binding or DNA binding activities.
Asymmetric CRP dimers composed of wild-type and mutant subunits
Two previously well characterized mutations, S62F and D53H (13, 21, 23), were used in our studies to perturb cAMP binding affinity and cooperativity in the CRPSC construct. In wild-type CRP, the mutation S62F decreases the affinity to cAMP and generates negative cooperativity between the two cAMP-binding domains. In contrast, the mutation D53H has small effects on cAMP binding affinity to the first site, but it significantly increases the affinity for the second one, thereby generating positive binding cooperativity.
We first placed these mutations in both subunits of the CRPSC (also referred as symmetric mutants) to corroborate the same mutational effects reported for wild-type CRP. The symmetric CRPSC mutant harboring S62F (CRPSCS/S) had an association constant for the first cAMP-binding site, k1, 10 times lower than that for the wild-type CRPSC (CRPSCWT/WT), and it displayed negative cooperativity (cS/S = k2/k1 = 0.12). In contrast, the symmetric CRPSC mutant harboring D53H (CRPSCD/D) had similar k1 values compared with CRPSCWT/WT, and it showed positive cooperativity (cD/D = 8.8) (Fig. 4A and Table 1). These results are quantitatively very similar to what has been reported by others (13, 21, 23) and thus show that these two mutations retain their effect in CRPSC as described for the wild-type CRP.
Figure 4.

Effect of symmetric and asymmetric mutations on cAMP binding affinity. A, cAMP titrations to CRPSCWT/WT (green circles) and the symmetric mutants CRPSCD/D (red circles) and CRPSCS/S (dark purple circles). B, cAMP titrations to the single asymmetric mutants CRPSCD/WT (light pink squares) and CRPSCS/WT (light purple squares). C, cAMP titrations to the double asymmetric mutants CRPSCS+D/WT (dark brown diamonds) and CRPSCS/D (beige diamonds). For comparison, the dashed lines corresponding to the fits of the parent symmetric proteins obtained from A were included in B and C. The solid lines in all three panels represent the fit using a two-site binding model as described under “Materials and Methods.” Error bars correspond to the standard deviation of at least three repeats.
Table 1.
cAMP and DNA binding affinity constants to CRPSC
Error corresponds to standard deviation from fitted parameters using a two-site binding model as described in Equation 1 under “Materials and Methods.” The units of k1 and k2 are 104 m−1 and c = k2/k1. The units of kDNA(empty) and kDNA(cAMP-2) are 107 m−1.
| cAMP binding affinity and cooperativity (c) |
DNA binding affinity |
||||
|---|---|---|---|---|---|
| k1 | k2 | c | kDNA(empty) | kDNA(cAMP-2) | |
| CRPSCWT/WT | 3.4 ± 0.6 | 6.1 ± 2.3 | 1.82 | 0.6 ± 0.1 | 6.8 ± 0.7 |
| CRPSCS/S | 0.34 ± 0.06 | 0.04 ± 0.03 | 0.12 | 1.2 ± 0.1 | 6.3 ± 0.1 |
| CRPSCD/D | 4.4 ± 1.9 | 38.9 ± 8.9 | 8.79 | 1.0 ± 0.2 | 20.2 ± 1.7 |
| CRPSCS/WT | 2.3 ± 0.3 | 0.47 ± 0.06 | 0.20 | 0.5 ± 0.1 | 23.0 ± 4.0 |
| CRPSCD/WT | 4.2 ± 0.8 | 17.1 ± 3.4 | 4.08 | 0.3 ± 0.1 | 19.8 ± 1.0 |
| CRPSCS/D | 2.8 ± 0.7 | 0.7 ± 0.3 | 0.25 | 0.7 ± 0.1 | 8.9 ± 2.3 |
| CRPSCS+D/WT | 1.5 ± 0.2 | 1.7 ± 0.2 | 1.12 | 1.1 ± 0.2 | 20.0 ± 7.4 |
We then constructed asymmetric CRPSC mutants harboring either S62F or D53H in one subunit while keeping the neighboring subunit wild type (referred as single asymmetric mutants). We evaluated the directionality and magnitude of the mutational effect on cAMP binding affinity by comparing the binding affinities of the first and second sites (k1 and k2) of the single asymmetric CRPSC mutants with the parental symmetric proteins, namely CRPSCWT/WT, CRPSCS/S, and CRPSCD/D. Fig. 4B shows cAMP titrations for the single asymmetric mutants S62F (CRPSCS/WT) and D53H (CRPSCD/WT), respectively. Interestingly, for both asymmetric CRPSC mutants, k1 and k2 did not correspond to a simple linear combination of affinities of the wild-type or symmetric mutant proteins (Table 1). For example, k1 for CRPSCS/WT was 7 times higher than for CRPSCS/S and 1.5 times lower than for CRPSCWT/WT, whereas k2 was 10 times higher than for CRPSCS/S and 13 times lower than for CRPSCWT/WT. For CRPSCD/WT, k1 was similar to CRPSCWT/WT and CRPSCD/D, but k2 was 3 times higher than for CRPSCWT/WT and 2 times lower than for CRPSCD/D. Altogether, these results clearly show bidirectional effects between mutant and wild-type CRP subunits and underscore that a single mutation in a single subunit is sufficient to alter the global allosteric behavior of the dimer even when the other subunit remained wild type.
Asymmetric CRP dimers harboring mutant subunits with opposite cooperative effects
The effects of asymmetric mutants on cAMP binding affinities resulted in intermediate values of cAMP binding cooperativity compared with symmetric CRPSC mutants. The cooperativities for CRPSCS/WT and CRPSCD/WT were cS/WT = 0.2 and cD/WT = 4.1, whereas for CRPSCS/S and CRPSCD/D, the cooperativities were cS/S = 0.12 and cD/D = 8.8, respectively. Interestingly, although the mutations S62F and D53H have opposite cooperative effects, the magnitude of their cooperative effect over the affinity of the second cAMP-binding site was similar. For example, CRPSCS/WT and CRPSCD/WT reduced and increased the affinity for the second cAMP-binding site 5- and 4-fold, respectively, whereas CRPSCS/S and CRPSCD/D reduced and increased the affinity 8- and 9-fold, respectively (Table 1).
To investigate the roles of intersubunit communication and intrasubunit interactions in cAMP binding cooperativity, we placed the S62F and D53H mutations in CRPSC in two different configurations (referred as double asymmetric mutants). To study intersubunit communication, we constructed a CRPSC harboring the S62F mutation in one subunit and the D53H in the neighboring one (CRPSCS/D). To study intrasubunit interactions, we constructed a CRPSC harboring both mutations in the same subunit, whereas the other one remained wild type (CRPSCS+D/WT). Given the similar magnitudes in their cooperative effects, the two CRPSC mutant configurations allowed us to determine to what extent intersubunit communication or intrasubunit interactions neutralize the opposite cooperative effects of S62F and D53H.
The cAMP titrations of CRPSCS/D and CRPSCS+D/WT revealed important differences in cAMP binding affinity and cooperativity (Fig. 4C). CRPSCS/D, with the two mutations in opposing subunits, displayed negative cooperativity (cS/D = 0.25) as seen in CRPSCS/S and CRPSCS/WT. However, the cooperativity in CRPSCS/D was not as low as in CRPSCS/S and CRPSCS/WT, indicating that the positive cooperative effects exerted by mutation D53H were transduced to the neighboring subunit harboring the mutation S62F. Surprisingly, CRPSCS+D/WT, with the two mutations in the same subunit, showed neutral cooperativity (cS+D/WT = 1.1), albeit k1 and k2 were 50% lower than those of the wild-type protein. Thus, despite the fact that the two mutations in CRPSCS+D/WT significantly reduced the cAMP binding affinity in both binding sites, any cooperative effects transduced from the double mutant subunit to the neighboring wild-type subunit were negligible. Thus, these results show that opposing cooperative effects are better counterbalanced through intrasubunit interactions than via intersubunit communication.
One could speculate that the functional differences between CRPSCS/D and CRPSCS+D/WT were due to destabilization of the secondary structures introduced by the mutations. However, chemical denaturation experiments monitored by CD and tryptophan fluorescence showed little differences in ΔG0 and m values between these two proteins (supplemental Fig. S2 and supplemental Table S3), which argues that the mutations do not alter the global fold of the protein, but instead their effects over cooperative interactions may arise from changes in the motions of the protein. In fact, previous studies have shown a correlation between protein motions and cAMP binding cooperativity in CRP (23, 29). Therefore, our results show that CRPSC is an ideal construct to examine how changes in protein motions in a single subunit modulate the behavior of the neighboring one through the dimer interface, i.e. intersubunit communication mediated by quaternary interactions.
DNA interactions with asymmetric CRP configurations
It is well established that CRP bound to two cAMP molecules interacts with high affinity and specificity to DNA promoter sequences (13). Less understood, however, is the role of the singly cAMP-bound conformation in transcription regulation and DNA interactions. Because both CRPSCWT/WT and the wild-type CRP displayed positive cooperativity between the two cAMP-binding domains (Table 1 and supplemental Table 1), the singly cAMP-bound intermediate state is poorly populated and thus difficult to isolate and characterize. However, the asymmetric mutants CRPSCS/D, CRPSCS/WT, and CRPSCS/S displayed negative cAMP binding cooperativity, enabling us to populate the singly cAMP-bound conformation and examine its interaction and binding affinity for the lac promoter.
Based on the cAMP binding data for CRPSCS/D and CRPSCS/WT, the highest ratio of singly to doubly cAMP-bound populations was obtained when [cAMP] = 30 μm. At this cAMP concentration, the distribution of populations of unliganded, singly cAMP-bound, and doubly bound states is ∼34, 60, and 6%, respectively (supplemental Fig. S3). Because of the negligible interaction between the unliganded CRP and the lac promoter, the total change in anisotropy at 30 μm cAMP, which reflects the formation of the CRP-DNA complex, corresponds to ∼91% to the singly cAMP-bound conformation and 9% to the doubly bound state. Fig. 5A shows the DNA binding data of CRPSCS/D using [cAMP] = 0, 30, and 1000 μm. Surprisingly, the titrations at 30 and 1000 μm revealed that both the singly cAMP-bound and doubly cAMP-bound conformations had very similar DNA binding affinities constants: kDNA(cAMP) = 8.0·107 m−1 and kDNA(cAMP-2) = 8.9·107 m−1, respectively. We estimated the DNA binding affinity of the unliganded conformation, kDNA(empty), assuming a similar total change in anisotropy as seen for the singly and doubly cAMP-bound conformations. This assumption provided an upper limit on the affinity of the unliganded conformation (∼7.0·106 m−1), which was at least 10 times lower than the affinity of the singly and doubly cAMP-bound conformations (Fig. 5B). Similar results were obtained for CRPSCS/WT, namely the singly and doubly cAMP-bound conformations interacted with the lac promoter with high affinity, 8.0·107 and 2.2·108 m−1, respectively, whereas the affinity of the unliganded conformation was more than 20 times lower, 3.0·106 m−1 (Fig. 5B).
Figure 5.
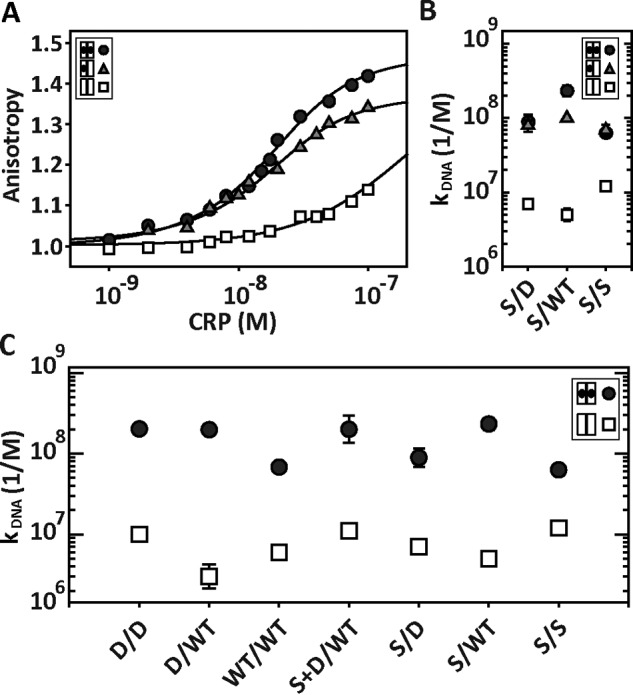
DNA-CRPSC interactions using saturating and non-saturating cAMP concentrations. A, interaction of CRPSCS/D with the 32-bp lac promoter using 0, 30, and 1000 μm cAMP, which correspond to unbound (open squares), singly (gray triangles), and doubly (black circles) cAMP-bound states. The solid lines represent the fit as described in Ref. 32. B, DNA binding affinity constants of CRPSCS/D, CRPSCS/WT, and CRPSCS/S when the proteins are in the unbound (open squares), singly (gray triangles), and doubly (black circles) cAMP-bound states. C, DNA binding affinity constants obtained from fluorescence anisotropy experiments for symmetric and asymmetric CRPSC in the absence (open squares) and presence of saturating cAMP concentrations (black circles). For CRPSCWT/WT, CRPSCD/D, and CRPSCD/WT, [cAMP] = 200 μm. For CRPSCS/S, [cAMP] = 2000 μm. For CRPSCS/D, CRPSCS/WT, and CRPSCS+D/WT, [cAMP] = 1000 μm.
For CRPSCS/S, the highest ratio between singly and doubly cAMP-bound populations was obtained with [cAMP] = 200 μm (supplemental Fig. S3). At this concentration, the population of doubly cAMP-bound is ∼2%, whereas the unliganded and singly cAMP-bound populations were ∼40 and ∼58%, respectively. In agreement with the results from CRPSCS/D and CRPSCS/WT, the singly and doubly cAMP-bound conformations for CRPSCS/S also have comparable affinities, 6.9·107 and 6.3·107 m−1, respectively (Fig. 5B and supplemental Methods).
Altogether, our data suggest that binding of a single cAMP molecule to CRP is sufficient to allosterically drive the conformational changes required for robust interactions with DNA promoter sequences. Importantly, all the CRPSC mutants studied here displayed significantly higher DNA binding affinity constants in saturating cAMP concentrations than in the absence of cAMP. Thus, the basic allosteric activation mechanism in all CRPSC constructs remained unperturbed (Fig. 5C).
Discussion
Covalent linkage of an allosteric protein complex
In this study we used several quantitative approaches to demonstrate that the wild-type CRP can be successfully reengineered and expressed as CRPSC without compromising the fold, stability, and function of the protein. The strategy of linking individual subunits in a homo-oligomeric protein offers important advantages to study hybrid functional states (i.e. combinations of mutant and wild-type subunits) (24). First, it eliminates the statistical degeneracy that occurs when mixing unlinked chains of mutant and wild-type subunits (25, 26), which can be further complicated if the mutant and wild-type subunit have different oligomerization association constants. Second, because of the high local concentration of protein subunits in the linked oligomer, subunit exchange throughout the course of an experiment is largely minimized. Finally, hybrid functional states allow for a detailed quantitative examination of the mechanism of intersubunit communication and coordination in homo-oligomeric proteins (24). Here, we compared symmetric and asymmetric CRPSC mutant configurations to investigate the mechanisms of communication within and across CRP subunits, and we determined how different communication pathways play unique roles in cooperative interactions.
Intersubunit communication independent of ligand binding
An important question in the field of allostery is whether coupling interactions between protein subunits emerge exclusively from ligand binding or, instead, the native state ensemble already manifests coupling interactions whose magnitude is amplified by the presence of the ligand (5, 27, 28). A comparison of the cAMP binding data between symmetric and asymmetric CRPSC configurations allowed us to address this question.
We found that for the asymmetric mutants CRPSCS/WT and CRPSCS/D, the affinity for the first cAMP-binding site, k1, was reduced by ∼35% compared with the parental symmetric proteins CRPSCWT/WT and CRPSCD/D. Given that k1 for CRPSCWT/WT and CRPSCD/D was ∼10 times higher than for CRPSCS/S, it is more likely that the wild-type or the D53H subunit in CRPSCS/WT and CRPSCS/D binds cAMP before the S62F subunit does. Therefore, the ∼35% reduction in k1 cannot be mediated by cAMP because the S62F subunit was unliganded but still able to transduce a perturbing effect to the neighboring subunit.
Similarly, CRPSCS+D/WT revealed cAMP-independent perturbations between the double mutant subunit and the wild-type subunit. Based on the affinities of the parental symmetric proteins, we expected k1 for CRPSCS+D/WT to be between 2.4·104 and 3.4·104 m−1. The former value corresponds to the average affinity of CRPSCD/D and CRPSCS/S, whereas the latter value corresponds to the affinity of the wild-type subunit in CRPSCWT/WT. However, the results for CRPSCS+D/WT revealed k1 ∼1.5·104 m−1, an affinity much lower compared with a simple interpolation from the average affinities of the parental proteins. This indicates that the double mutant subunit in CRPSCS+D/WT significantly affected the wild-type subunit or else k1 would have been similar or close to the value seen in CRPSCWT/WT. Altogether, the results from the asymmetric mutants favor a model in which the native state ensemble of CRP manifests coupling interactions between its subunits that are likely amplified by cAMP binding.
Arrangement of asymmetric mutations plays a crucial role in cAMP binding cooperativity
Interestingly, our results show that a single mutation in one CRP subunit was sufficient to drive the same cooperative behavior seen in the parental proteins but with an intermediate magnitude (Table 1), indicating that the effects of mutations over cooperative interactions between the two cAMP-binding domains scale linearly proportional with the number of mutant subunits.
Furthermore, our results show that the arrangement of the mutations has dramatic consequences depending on whether intrasubunit interactions or intersubunit communications are at play. In fact, by just placing the mutations S62F and D53H in different configurations, we engineered a family of CRPSC variants with fine-tuned binding energies covering a broad range of positive-to-negative cooperativities (Fig. 6). The results obtained with the CRPSC construct therefore illustrate how the different proteins' communication pathways can be exploited to modulate binding and enzymatic activities without directly altering active binding pockets or interaction surfaces.
Figure 6.
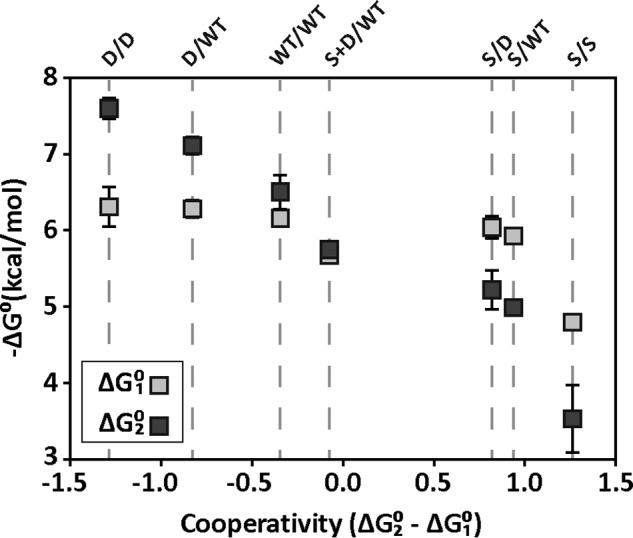
cAMP-binding energies and cooperativities of CRPSC variants. Light and dark squares correspond to the cAMP-binding energy for the first (ΔG01) and second (ΔG02) sites, respectively. The order of CRPSC mutants is sorted from most positive cooperativity (ΔG02 − ΔG01 < 0) to most negative (ΔG02 − ΔG01 > 0) as indicated by the dashed lines.
Role of asymmetric conformations in the mechanism of CRP activation and interaction with DNA
The main structural transition of CRP that accompanies cAMP binding involves an ∼60° rotation of the DNA-binding domains relative to the cAMP-binding domains (14–16). Such structural transition enables tight interactions with the major groove of the DNA and provides a structural basis for the activation and affinity enhancement of CRP for DNA promoter sequences. Although high resolution structures offer detailed information about the unliganded and doubly cAMP-bound conformations, the allosteric activation pathway of CRP must involve, in addition, a singly cAMP-bound intermediate. This intermediate is not only important for a mechanistic understanding of the activation pathway of CRP, but it may also represent an additional conformer in the regulation of gene expression as proposed by others (19). In this study, we took advantage of the negative cAMP binding cooperativity of CRPSCS/D and CRPSCS/WT to interrogate the DNA binding properties of CRP bound to one cAMP molecule.
We found that in the singly cAMP-bound state, CRPSCS/D, CRPSCS/WT, and CRPSCS/S have a similarly high affinity for the lac promoter compared to the doubly cAMP-bound conformation (Fig. 5B). These results suggest that a single cAMP-binding event triggers a conformational change in CRP that allows tight interactions with the DNA. NMR studies with the CRP homodimer showed that cAMP binding to either wild-type or D53H subunits elicits an active conformation of the DNA-binding domains (30). Thus, one possible model to explain our results is that after the first cAMP-binding event, the cAMP-bound subunit elicits a reorientation of the DNA-binding domain of the neighboring unliganded subunit that is compatible with DNA binding (Fig. 7).
Figure 7.
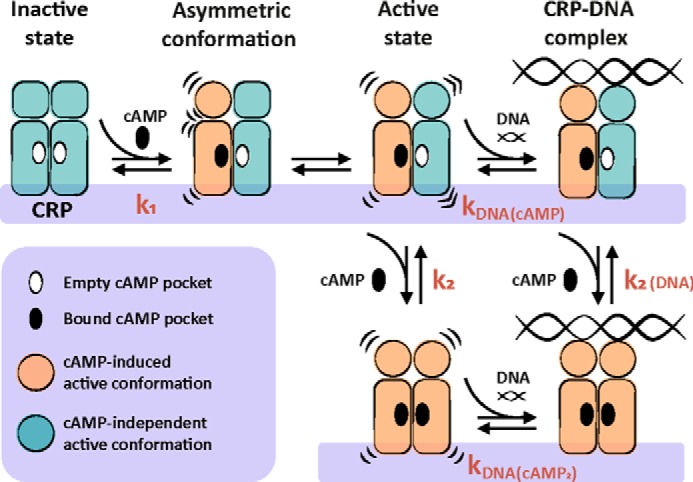
Mechanism of CRP activation and interaction with DNA. In the absence of cAMP, CRP is in an inactive state. Binding of cAMP to one CRP subunit triggers a conformational change in the DNA-binding domain of the bound subunit, generating an asymmetric conformation within the dimer. The cAMP-bound subunit induces a re-orientation in the unliganded neighboring subunit that is compatible with strong DNA interactions. Binding of a second cAMP molecule can occur to either the singly cAMP-bound CRP or to the ternary complex DNA-CRP-cAMP.
An alternative model is that DNA interactions occur with a CRP dimer in a hybrid conformation, namely the cAMP-bound subunit is in the active conformation, whereas the unliganded one remains in the inactive state. A DNA-CRP complex formed with only one active DNA-binding domain would establish approximately half of the interactions and thus result in half of the binding energy compared with a CRP homodimer with both DNA-binding domains active. The observed DNA binding energies (ΔG0 = −RT·ln(kDNA)) for the singly and the doubly cAMP-bound states were essentially the same, around −10.7 kcal/mol. Therefore, we favor a model in which binding of cAMP to one CRP subunit elicits a conformational change in the neighboring one. Such a conformational change in the unliganded subunit does not need to be the exact same as in the cAMP-bound subunit but needs to be compatible with strong DNA interactions. This model also provides a plausible explanation for the transduction of positive cooperative effects from one subunit to another seen in asymmetric CRPSC mutants, especially for CRPSCS/D where the D53H subunit bound to cAMP increases the affinity of the S62F subunit by 2-fold.
The proposed model in Fig. 7 provides a framework to further dissect the thermodynamic cycle describing the linkage between cAMP and DNA interactions. Specifically, we interrogated the effect of the CRP-DNA interaction over cAMP binding cooperativity, which in Fig. 7 corresponds to the binding affinity constant for the second cAMP molecule when CRP is bound to DNA, k2(DNA), and it can be calculated from the relationship k2(DNA) = k2·kDNA(cAMP-2)/kDNA(cAMP). Using the results from this study, we obtained values of k2(DNA) that are slightly larger than k2, indicating that DNA interactions with a CRP molecule in intermediate liganded states also modulate the protein's response to cAMP concentration. This result sheds lights into the bidirectional interplay between DNA interactions and cAMP binding cooperativity during transcription regulation. Future studies using asymmetric CRPSC mutants that simultaneously perturb cAMP binding and DNA interactions will be used to further dissect the communication pathways between the cAMP- and DNA-binding domains within and across protein subunits.
Materials and methods
Cloning, expression, and purification of CRPSC
The DNA sequence of wild-type CRP from Escherichia coli was used to synthesize (GenScript) the CRP single-chain dimer (CRPSC) with a sequence encoding (SGGGG)7 as linker connecting the two CRP subunits. The protein purification protocol is described in detail in the supplemental Methods and supplemental Fig. S4. The wild-type CRP was kindly provided by Dr. James C. Lee from the University of Texas Medical Branch.
Circular dichroism (CD)
Measurements were performed in an Aviv Model 202-01 spectrometer with 10 μm protein in PBS buffer, pH 7.4, over the range of 195–260 nm. For each sample, two repetitive scans were performed, averaged, and baseline-corrected.
Chemical denaturation with GdnHCl
Protein unfolding was monitored by changes in tryptophan fluorescence (λex = 295 nm and λem = 340 nm) and circular dichroism absorption (at 222 nm) using 10 μm protein in 20 mm Tris, 50 mm NaCl, 1 mm EDTA, pH 7.8. At least two independent titrations were performed for each protein and corrected for buffer absorption. Data were fitted to a two-state unfolding model according to the linear extrapolation method (31).
cAMP binding monitored by ITC
Experiments were performed in 20 mm Tris, 50 mm NaCl, 1 mm EDTA, 0.2 mm tris(2-carboxyethyl)phosphine, pH 7.8, at 25 °C in a VP-ITC microcalorimeter (1.4-ml chamber volume). All solutions were filtered and degassed prior use. The protein and cAMP concentrations were 16 μm and 0.57 mm, respectively. The experiment consisted of a first 5-μl injection, followed by 17 injections of 18 μl each. A reference titration of cAMP into buffer was subtracted from the cAMP titration to the protein. The data were analyzed using a sequential two-site binding model (MicroCal ITC-Origin).
EMSA
Reaction mixtures contained 40 nm DNA, 1.25 μm wild-type CRP or CRPSC, 10 mm DTT, and 200 μm cAMP in 20 mm Tris, 50 mm NaCl, 1 mm EDTA, pH 7.8. After a 45-min equilibration at room temperature, samples were loaded onto an 8.5% polyacrylamide gel in 0.5× TBE buffer. Gels were run at 10 V/cm for 55 min in 0.5× TBE buffer with 1 mm DTT and 200 μm cAMP.
cAMP binding monitored by ANS fluorescence
Measurements were collected with a PTI spectrometer. All experiments were conducted in 20 mm Tris, 50 mm NaCl, 1 mm EDTA, pH 7.8, at 25 °C. The reaction mixture contained 47.7 μm ANS and 3.6 μm protein. cAMP binding to wild-type CRP or CRPSC was measured by the quenching of the fluorescent signal from the CRP-ANS complex (λex = 350 and λem = 480 nm). Intensity count as a function of cAMP concentration was fitted to a two-site binding model as in Equation 1,
| (Eq. 1) |
where F480 nm is the observed signal; F0, F1, and F2 correspond to the fluorescent signal of the free, singly, and doubly cAMP-bound states; k1 and k2 correspond to the microscopic cAMP affinity constant for the first and second binding events, and x to the concentration of cAMP.
DNA binding monitored by anisotropy
Measurements were collected with a PTI spectrometer using a 32-bp lac promoter (5′-GCAATTAATGTGAGTTAGCTCACTCATTAGGC-3′) covalently linked to a fluorescein molecule (λex = 480 nm and λem = 518 nm). The reaction mixture contained 5–10 nm of fluorescein-labeled DNA and various concentrations of cAMP (see Fig. 5 legend). Data were analyzed as described previously by Heyduk and Lee (32).
Author contributions
M. F. L. designed, conducted, and analyzed research and wrote the manuscript. F. G. conducted and analyzed research and wrote the manuscript. A. J. E. conducted research. R. A. M. designed and analyzed research and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Aleksandra Gmyrek for support on ITC experiments and Courtney Hodges and Steven Metallo for comments on the manuscript.
This work was supported with startup funds from Georgetown University. The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains supplemental Methods, Figs. S1–S4, and Tables S1–S3.
- CRP
- cAMP receptor protein
- CRPsc
- CRP single-chain dimer
- ITC
- isothermal titration calorimetry
- GdnHCl
- guanidine hydrochloride
- ANS
- 8-anilinonaphthalene-1-sulfonic acid
- c
- cooperativity
- PDB
- Protein Data Bank.
References
- 1. Smock R. G., and Gierasch L. M. (2009) Sending signals dynamically. Science 324, 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dayhoff J. E., Shoemaker B. A., Bryant S. H., and Panchenko A. R. (2010) Evolution of protein binding modes in homooligomers. J. Mol. Biol. 395, 860–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goodsell D. S., and Olson A. J. (2000) Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 29, 105–153 [DOI] [PubMed] [Google Scholar]
- 4. Changeux J.-P., and Edelstein S. J. (2005) Allosteric mechanisms of signal transduction. Science 308, 1424–1428 [DOI] [PubMed] [Google Scholar]
- 5. Motlagh H. N., Wrabl J. O., Li J., and Hilser V. J. (2014) The ensemble nature of allostery. Nature 508, 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Motlagh H. N., and Hilser V. J. (2012) Agonism/antagonism switching in allosteric ensembles. Proc. Natl. Acad. Sci. U.S.A. 109, 4134–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kornev A. P., Taylor S. S., and Ten Eyck L. F. (2008) A generalized allosteric mechanism for cis-regulated cyclic nucleotide binding domains. PLoS Comput. Biol. 4, e1000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi J. H., Laurent A. H., Hilser V. J., and Ostermeier M. (2015) Design of protein switches based on an ensemble model of allostery. Nat. Commun. 6, 6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ha J. H., and Loh S. N. (2012) Protein conformational switches: from nature to design. Chemistry 18, 7984–7999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Makhlynets O. V., Raymond E. A., and Korendovych I. V. (2015) Design of allosterically regulated protein catalysts. Biochemistry 54, 1444–1456 [DOI] [PubMed] [Google Scholar]
- 11. Kar G., Keskin O., Gursoy A., and Nussinov R. (2010) Allostery and population shift in drug discovery. Curr. Opin. Pharmacol. 10, 715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wenthur C. J., Gentry P. R., Mathews T. P., and Lindsley C. W. (2014) Drugs for allosteric sites on receptors. Annu. Rev. Pharmacol. Toxicol. 54, 165–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin S. H., and Lee J. C. (2002) Communications between the high-affinity cyclic nucleotide binding sites in E. coli cyclic AMP receptor protein: effect of single site mutations. Biochemistry 41, 11857–11867 [DOI] [PubMed] [Google Scholar]
- 14. McKay D. B., and Steitz T. A. (1981) Structure of catabolite gene activator protein at 2.9 resolution suggests binding to left-handed B-DNA. Nature 290, 744–749 [DOI] [PubMed] [Google Scholar]
- 15. Popovych N., Tzeng S.-R., Tonelli M., Ebright R. H., and Kalodimos C. G. (2009) Structural basis for cAMP-mediated allosteric control of the catabolite activator protein. Proc. Natl. Acad. Sci. U.S.A. 106, 6927–6932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharma H., Yu S., Kong J., Wang J., and Steitz T. A. (2009) Structure of apo-CAP reveals that large conformational changes are necessary for DNA binding. Proc. Natl. Acad. Sci. U.S.A. 106, 16604–16609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reznikoff W. S. (1992) Catabolite gene activator protein activation of lac transcription. J. Bacteriol. 174, 655–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin S. H., Kovac L., Chin A. J., Chin C. C., and Lee J. C. (2002) Ability of E. coli Cyclic AMP receptor protein to differentiate cyclic nucleotides: effects of single site mutations. Biochemistry 41, 2946–2955 [DOI] [PubMed] [Google Scholar]
- 19. Lin S.-H., and Lee J. C. (2002) Linkage of multiequilibria in DNA recognition by the D53H Escherichia coli cAMP receptor protein. Biochemistry 41, 14935–14943 [DOI] [PubMed] [Google Scholar]
- 20. Li J., and Lee J. C. (2011) Modulation of allosteric behavior through adjustment of the differential stability of the two interacting domains in E. coli cAMP receptor protein. Biophys. Chem. 159, 210–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dai J., Lin S.-H., Kemmis C., Chin A. J., and Lee J. C. (2004) Interplay between site-specific mutations and cyclic nucleotides in modulating DNA recognition by Escherichia coli cyclic AMP receptor protein. Biochemistry 43, 8901–8910 [DOI] [PubMed] [Google Scholar]
- 22. Gorshkova I., Moore J. L., McKenney K. H., and Schwarz F. P. (1995) Thermodynamics of cyclic nucleotide binding to the cAMP receptor protein and its T127L mutant. J. Biol. Chem. 270, 21679–21683 [DOI] [PubMed] [Google Scholar]
- 23. Gekko K., Obu N., Li J., and Lee J. C. (2004) A linear correlation between the energetics of allosteric communication and protein flexibility in the Escherichia coli cyclic AMP receptor protein revealed by mutation-induced changes in compressibility and amide hydrogen-deuterium exchange. Biochemistry 43, 3844–3852 [DOI] [PubMed] [Google Scholar]
- 24. Martin A., Baker T. A., and Sauer R. T. (2005) Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature 437, 1115–1120 [DOI] [PubMed] [Google Scholar]
- 25. Moreau M. J., McGeoch A. T., Lowe A. R., Itzhaki L. S., and Bell S. D. (2007) ATPase site architecture and helicase mechanism of an archaeal MCM. Mol. Cell 28, 304–314 [DOI] [PubMed] [Google Scholar]
- 26. Li P. T., Scott D. J., and Gollnick P. (2002) Creating hetero-11-mers composed of wild-type and mutant subunits to study RNA binding to TRAP. J. Biol. Chem. 277, 11838–11844 [DOI] [PubMed] [Google Scholar]
- 27. Tsai C.-J., del Sol A., and Nussinov R. (2008) Allostery: absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 378, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boehr D. D., Nussinov R., and Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu S., Maillard R. A., Gribenko A. V., and Lee J. C. (2012) The N-terminal capping propensities of the D-helix modulate the allosteric activation of the Escherichia coli cAMP receptor protein. J. Biol. Chem. 287, 39402–39411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tzeng S.-R., and Kalodimos C. G. (2012) Protein activity regulation by conformational entropy. Nature 488, 236–240 [DOI] [PubMed] [Google Scholar]
- 31. Santoro M. M., and Bolen D. W. (1988) Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27, 8063–8068 [DOI] [PubMed] [Google Scholar]
- 32. Heyduk T., and Lee J. C. (1990) Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction DNA-protein interaction. Proc. Natl. Acad. Sci. U.S.A. 87, 1744–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.