

Abstract
Grazoprevir is a potent pan-genotype and macrocyclic inhibitor of hepatitis C virus (HCV) NS3/4A protease and was developed for treating chronic HCV infection. In HCV genotype (GT) 1a, grazoprevir maintains potent activity against a majority of NS3 resistance-associated amino acid substitutions, including the highly prevalent and naturally occurring Q80K polymorphism that impacts simeprevir, another NS3/4A protease inhibitor. The basis for an unexpected difference in the clinical impact of some NS3 substitutions was investigated. Phenotypic analysis of resistance-associated substitutions identified in NS3 from GT1a-infected patients who failed therapy with grazoprevir (in combination with elbasvir, an inhibitor of HCV NS5A protein) showed that positions 56, 156, and 168 in NS3 were most impactful because they diminished protein-inhibitor interactions. Although an amino acid substitution from aspartic acid to alanine at position 168 (D168A) reduced the potency of grazoprevir, its combination with R155K unexpectedly nullified this effect. Molecular dynamics and free-energy surface studies indicated that Asp-168 is important in anchoring Arg-155 for ligand binding but is not critical for Lys-155 because of the inherent flexibility of its side chain. Moreover, modeling studies supported a strong direct cation-heterocycle interaction between the Lys-155 side chain of the double substitution, R155K/D168A, and the lone pair on the quinoxaline in grazoprevir. This unique interaction provides a structural basis for grazoprevir's higher potency than simeprevir, an inhibitor to which the double substitution confers a significant reduction in potency. Our findings are consistent with the detection of R155K/D168A in NS3 from virologic failures treated with simeprevir but not grazoprevir.
Keywords: crystal structure, drug resistance, hepatitis C virus (HCV), molecular dynamics, protease inhibitor
Introduction
Chronic hepatitis C virus (HCV)2 infection afflicts ∼170 million people worldwide. Although the infection can remain asymptomatic for 20–30 years, if untreated it can lead to liver cirrhosis and hepatocellular carcinoma (1, 2). In 2011, the introduction of protease inhibitors used in combination with pegylated interferon-α and ribavirin significantly improved sustained virologic response (SVR) rates compared with pegylated interferon/ribavirin alone (3). Further improvements in treatment responses have been achieved through the development of all-oral, interferon-free, direct-acting antiviral (DAA) regimens. This advance has further improved responses and also shortened treatment duration as well as eliminated the adverse effects associated with interferon-based treatment (4). Although cure rates of >90% are now achievable for the majority of HCV-infected patients, unmet medical needs still exist for some HCV patients, including those infected with genotype 3 and may have liver cirrhosis or who may also suffer from co-morbidities such as chronic kidney disease or inherited blood disorders. It is anticipated that a combination of DAAs targeting multiple pathways, including nonstructural (NS) 3/4A protease inhibitors, will be required for these hard-to-treat patients.
The HCV NS3 protein and the NS4A cofactor assemble as a catalytically active viral serine protease. Following translation of the viral RNA, host enzymes together with the NS3/4A protease process the polyprotein precursor into functional proteins (5). NS3/4A catalyzes four specific cleavages in the viral polyprotein precursor to release the nonstructural NS3, NS4A, NS4B, NS5A, and NS5B proteins. Inhibition of this activity prevents maturation of the polyprotein into its functional components thereby blocking viral replication. This crucial activity makes the protease an attractive target for intervention in viral replication. Significant efforts in drug discovery to block the activity of the protease have resulted in two major NS3/4A protease inhibitor classes, linear and macrocyclic (6–11). The first approved linear ketoamide inhibitors were slowly reversible inhibitors that covalently targeted the active site serine residue (Ser-139). These inhibitors, exemplified by boceprevir (6) and telaprevir (7), were largely restricted to GT1 infections. Subsequently, more potent noncovalent macrocyclic inhibitors have been developed; these newer NS3/4A protease inhibitors exemplified by simeprevir (8), paritaprevir (9), and grazoprevir (10, 11) show improved potency across many genotypes.
Grazoprevir is a novel quinoxaline macrocyclic NS3/4A protease inhibitor that contains a P2 to P4 macrocyclic constraint (Fig. 1). It is broadly active against HCV genotypes in vitro and also maintains potent activity against many clinically relevant resistance-associated substitutions, including those selected by the linear inhibitors. The medicinal chemistry efforts that led to its discovery and optimization of its activity have been reported (10, 11). The potency of grazoprevir derives, in part, from critical lipophilic interactions at the P2 position in addition to contributions from the P2-P4 constraint among others (10). The clinical activity of grazoprevir in combination with elbasvir, an HCV NS5A replicase complex inhibitor, has been studied in HCV-infected patients. The combination is highly effective resulting in ≥94% SVR in treatment-naive GT1- and GT4-infected patients following a 12-week course of the regimen (12). Although the presence of viruses bearing NS3 resistance-associated substitutions in patients prior to receiving therapy (i.e. those at baseline) did not impact treatment response (13), additional treatment-emergent NS3 substitutions were observed in viruses isolated from patients who failed to achieve SVR. In an effort to obtain a deeper understanding of the impact of NS3 polymorphisms on inhibitor potency, these substitutions were further studied by engineering them into replicon cells. The impact of single amino acid substitutions at most positions could be readily rationalized from a structural standpoint. However, in a limited number of cases, a detailed analysis of the enzyme-inhibitor interactions was needed to address the basis of the resistance or lack thereof. In this report, we discuss the impact of clinically relevant NS3 resistance-associated amino acid substitutions in NS3/4A and the structural basis for their effects on grazoprevir potency.
Figure 1.

Chemical structures of NS3/4A protease macrocyclic inhibitors grazoprevir (A) and simeprevir (B), and linear inhibitor boceprevir (C).
Results
Grazoprevir is a potent pan-genotype NS3/4A protease inhibitor
Grazoprevir is a quinoxaline macrocyclic compound that inhibits HCV NS3/4A protease activity with IC50 values in the picomolar to subnanomolar range in enzyme assays across HCV genotypes (Table 1). Although grazoprevir maintained potent activity across all genotypes, the least susceptible was the purified enzyme from GT3, whereas those from GT1 were the most susceptible. To assess the antiviral activity of grazoprevir in cell culture, the inhibitor was evaluated against replicons representing HCV clinical isolates. Grazoprevir demonstrated an EC50 in the subnanomolar to single-digit nanomolar range against HCV GT1–6 replicons (Table 2). As observed in the enzyme assay, it was less potent against the homologous full-length GT3a replicon compared with the other replicons. Interestingly, in a chimeric replicon bearing only the GT3a NS3 catalytic domain in the background of GT2a_JFH-1, the potency of grazoprevir was improved by greater than 10-fold compared with the full-length GT3_S52 replicon (Table 2). Thus, sequences outside of the catalytic domain influence inhibitor binding interactions. The data demonstrated that the in vitro enzyme activity of grazoprevir translates well to the cell-based replicon assay. More importantly, the data indicated that grazoprevir would have activity against a broad set of clinical isolates.
Table 1.
Potency of grazoprevir against NS3/4A proteases isolated from HCV genotypes
| Enzymea | IC50 ± S.D.b |
|---|---|
| nm | |
| GT1a (H77; NC_004102) | 0.007 ± 0.001 |
| GT1b (BK; M58335) | 0.004 ± 0.001 |
| GT2a (JFH1; AB047639) | 0.067 ± 0.024 |
| GT2b (J8; D10988) | 0.135 ± 0.051 |
| GT3a (NZL1; D17763) | 0.690 ± 0.194 |
| GT4a (ED43; GU814265) | 0.062 ± 0.031 |
| GT5a (1209A014c) | 0.067 ± 0.022 |
| GT6a (6a77; DQ480512) | 0.034 ± 0.007 |
a The strain and GenBankTM accession numbers for the HCV reference sequences are indicated in parentheses.
b The standard deviation is n ≥ 3.
c This is the patient sequence identifier.
Table 2.
Potency of grazoprevir against a panel of replicons bearing NS3/4A from HCV genotypes 1–6 reference sequences
| Replicon | EC50 ± S.D.a |
|---|---|
| nm | |
| GT1a (H77; NC_004102) | 0.4 ± 0.3 |
| GT1b (Con1; AJ238799) | 0.5 ± 0.3 |
| GT2a (JFH-1; AB047639) | 2.3 ± 1.2 |
| GT2b (MD6; AY232740) | 3.7 ± 1.1 |
| GT2b (MD2; AY232732) | 2.9 ± 0.4 |
| GT3a (S52; GU814263)b | 2.1 ± 1.0 |
| GT3a (S52; GU814263) | 35.0 ± 15 |
| GT4a (ED43; GU814265) | 0.3 ± 0.2 |
| GT5a (SA1; KJ925150 | 0.4 ± 0.1 |
| GT6 (GZ52557; DQ278892) | 0.2 ± 0.04 |
a The standard deviation is n ≥ 3.
b The chimeric replicon bearing the catalytic domain of GT3a NS3 is in the GT2a_JFH-1 background.
Grazoprevir maintains activity against critical GT1a NS3 amino acid substitutions
NS3/4A protease inhibitors (PI) are effective in patients infected with GT1; however, a number of amino acid substitutions have been observed particularly among HCV GT1a patients who failed to achieve SVR following treatment with NS3/4A PI-containing regimens (14). Thus, we tested grazoprevir against several of these GT1a NS3 substitutions to evaluate its activity. We selected a number of frequently detected amino acid substitutions in virologic failures as test cases. The substitutions span key positions in NS3/4A that have been associated with resistance to the PI class. In addition, the examples chosen have been broadly identified in GT1a patients who failed to achieve SVR when treated with NS3/4A protease inhibitors. Grazoprevir maintained broad activity against substitutions at a majority of these key positions in GT1a NS3/4A (Table 3). Among the substitutions that did not impact grazoprevir activity were V36M, T54S, V55I, V107I, and S122R. Another group of replicons conferred only a modest reduction (<5-fold) in inhibitory activity. These amino acid substitutions included R155K, A156G, D168N, and I170T. The least susceptible group of substitutions for grazoprevir were observed at positions 56 and 168 (Table 3). Generally, substitutions primarily selected by linear α-ketoamide inhibitors such as boceprevir (for example those located at positions 36, 54, and 55) did not confer a reduction in potency to grazoprevir. Substitutions that were more impactful to grazoprevir also conferred resistance to simeprevir, another Food & Drug Administration-approved macrocyclic inhibitor that was tested as another member of the NS3/4A macrocyclic class of protease inhibitors.
Table 3.
Potency of grazoprevir and NS3/4A protease inhibitors in replicons bearing HCV GT1a NS3 amino acid substitutions
| GT1a replicon | Grazoprevir |
Boceprevir |
Simeprevir |
|||
|---|---|---|---|---|---|---|
| EC50± S.D.a | -Fold shift | EC50± S.D.a | -Fold shift | EC50± S.D.a | -Fold shift | |
| nm | nm | nm | ||||
| WT | 0.4 ± 0.2 | 275 ± 138 | 3.2 ± 2.6 | |||
| V36M | 0.3 ± 0.2 | 0.8 | 919 ± 451 | 3.3 | 11.2 ± 8.2 | 3.5 |
| T54S | 0.3 ± 0.2 | 0.8 | 1559 ± 1002 | 5.7 | 5.1 ± 2.3 | 1.6 |
| V55I | 0.2 ± 0.1 | 0.5 | 207 ± 89 | 0.8 | 4.9 ± 4.9 | 1.5 |
| Y56H | 5.7 ± 4.3 | 14 | 432 ± 179 | 1.6 | 40 ± 22 | 13 |
| Q80K | 0.3 ± 0.3 | 0.8 | 182 ± 88 | 0.7 | 17 ± 7 | 5.3 |
| V107I | 0.3 ± 0.1 | 0.8 | 340 ± 80 | 1.2 | 7.1 ± 2.9 | 2.2 |
| S122R | 0.4 ± 0.1 | 1.0 | 110 ± 44 | 0.4 | 114 ± 53 | 36 |
| R155K | 1.3 ± 0.7 | 3.3 | 980 ± 676 | 3.6 | 19 ± 9 | 5.9 |
| A156G | 1.7 ± 0.1 | 4.3 | 1240 ± 116 | 4.5 | 106 ± 10 | 33 |
| D168A | 29 ± 13 | 73 | 154 ± 126 | 0.6 | 66 ± 52 | 21 |
| D168E | 4.9 ± 4.2 | 12 | 215 ± 93 | 0.8 | 29 ± 22 | 9.1 |
| D168G | 9.6 ± 6.9 | 24 | 285 ± 19 | 1.0 | 20 ± 11 | 6.3 |
| D168N | 0.9 ± 0.4 | 2.3 | 230 ± 135 | 0.8 | 21 ± 20 | 6.6 |
| D168V | 10.5 ± 5.0 | 26 | 834 ± 124 | 3.0 | >2000 | >625 |
| D168Y | 6.9 ± 5.2 | 17 | 246 ± 131 | 0.9 | 1269 ± 735 | 397 |
| I170T | 0.7 ± 0.3 | 1.8 | 440 ± 175 | 1.6 | 22 ± 18 | 6.9 |
a The standard deviation is n ≥ 3.
Among the GT1a substitutions, the impact of Q80K was of particular interest as this naturally occurring polymorphism is observed in 40–50% of NS3 sequences in GT1a-infected patients and has been reported to confer resistance to simeprevir (15, 16). The Q80K substitution remained susceptible to grazoprevir. The crystal structures of grazoprevir and simeprevir bound to NS3/4A protease from GT1a provided a structural rationale for the lack of an effect of Q80K on grazoprevir's activity. As shown in Fig. 2, Gln-80 is located at the periphery of the S2 subsite. The backbone C=O of Gln-80 makes a hydrogen bond with the guanidinium group of Arg-155 to stabilize the salt bridge between Arg-155 and Asp-168. The stability of Arg-155 could also benefit from a second hydrogen bond with the side chain of Gln-80. The Q80K substitution would lose this hydrogen bond because the Lys-80 side chain will move away from Arg-155 due to electrostatic repulsion between the two positively charged residues and therefore affect the stability of Arg-155 slightly. As grazoprevir only has limited interactions with Arg-155 through the P2-P4 linker, it is not surprising that this substitution has little impact on the binding of grazoprevir. In contrast, simeprevir makes extended van der Waals contacts with the guanidinium group of Arg-155, and the Q80K substitution would have been expected to have a greater impact on simeprevir as seen.
Figure 2.
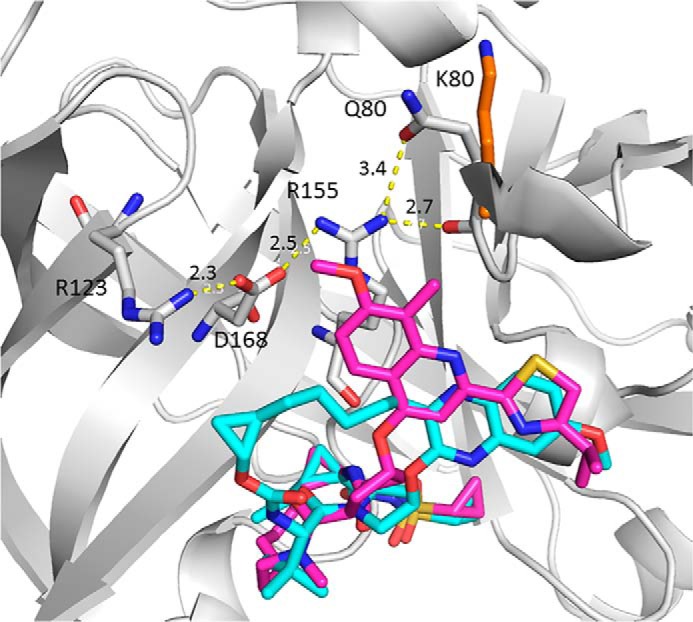
Superposition of the crystal structures of grazoprevir (cyan) and simeprevir (magenta) bound to HCV NS3/4A protease (PDB codes 3SUD and 3KEE). The protein is represented as ribbon, and only residues Arg-123, Asp-168, Arg-155, and Gln-80/Lys-80 that form the salt bridge/hydrogen bonding network are shown (gray, carbon; red, oxygen; blue, nitrogen). The Lys-80 side chain is also highlighted (orange, carbon). The dashed yellow lines represent the distance in angstroms as labeled.
Treatment-emergent substitutions in patients treated with a grazoprevir-containing regimen
Next, we evaluated the clinical significance of the in vitro findings by reviewing the prevalence of NS3/4A substitutions in patients who failed to achieve SVR in clinical studies with a grazoprevir-containing regimen. NS3/4A substitutions detected in a pooled analysis of treatment-naive mono-infected (12) and treatment-naive HIV co-infected HCV GT1a patients (17) were studied. The majority of substitutions among the 37 GT1a virologic failures occurred at amino acid position 168 in NS3 followed by changes at positions 156 and 56 (Fig. 3). All the substitutions that were detected in virologic failures were engineered into GT1a replicon cells and characterized phenotypically. As reported in Table 3, the Y56H and A156G substitutions conferred a 14- and 4.3-fold reduction in grazoprevir activity, respectively. The other substitutions at position 156 (A156T/A156V) resulted in replicons that were unfit and failed to grow in cell culture; they were not characterized any further. The D168A/D168G/D168N/D168V/D168Y substitutions resulted in fold shift reductions in potency for grazoprevir ranging from 2.3- to 73-fold. The NS3 substitutions also conferred potency reductions to simeprevir, another member of the macrocyclic class of inhibitors, but not to the α-ketoamide linear inhibitor boceprevir (Table 3).
Figure 3.

Prevalence of treatment-emergent (TE) NS3 amino acid substitutions as a percentage of all TE NS3 substitutions observed in GT1a virologic failures (n = 37) treated with a grazoprevir-containing regimen in phase 2 and 3 clinical trials. The type of substitution and number of virologic failures detected with that TE substitution is indicated. Note that the number of patients in whom substitutions were detected is indicated in parentheses: V36L (1); V36M (3); Y56F (1); Y56H (5); V107I (2); R155I (1); R155K (1); A156G (2); A156T (11); A156V (1); V158I (2); D168A (13); D168G (1); D168N (1); D168V (2); and D168Y (1).
As multiple resistance-associated mutations can occur on the same viral genome, a more comprehensive view of the impact of substitutions involving combinations of amino acids that have been observed in patients who failed therapy with NS3/4A PIs were also characterized. Some of the double substitutions were engineered in transient replicons if a low replicon fitness was evident in the stable replicon. The summary of the impact of the combination of amino acid substitutions on grazoprevir's antiviral activity in the assay is presented in Table 4. The impact of almost all of the combination of substitutions (except R155K and D168A) on grazoprevir activity could be rationalized based on data from the individual substitutions and prior structural analysis of the interaction between grazoprevir and NS3/4A (11, 18). The impact of V36M and R155K on protease inhibitors has been described previously (14, 19). For the Y56H/D168A double substitution, both amino acid changes together conferred a further potency loss compared with each individual substitution. In the crystal structure of the grazoprevir-NS3/4A complex (Fig. 4), the methoxyquinoxaline group of grazoprevir is in hydrophobic contact with one edge of the Tyr-56 phenyl ring. Mutation of Tyr-56 to a smaller histidine will disrupt this interaction and cause potency loss. This is also true for simeprevir where the isopropylthiazole group makes similar contact with Tyr-56. The potency loss is correlated with the extent of this hydrophobic interaction; grazoprevir and simeprevir both make more contact with Tyr-56 and hence have larger potency shifts. Because the Tyr-56 and Asp-168 residues are separated spatially with a distance of >14 Å, the additive effect of this double substitution on inhibitor potency can be rationalized by the independent or noninteracting nature of the two residues. A lack of an impact on the potency of boceprevir by the combination of changes at positions 56 and 168 was not unexpected based on the effects of the individual substitutions (Table 3).
Table 4.
Potency of grazoprevir and simeprevir against a panel of HCV GT1a NS3 substitutions in replicon assay
| NS3 substitution | Grazoprevir |
Boceprevir |
Simeprevir |
|||
|---|---|---|---|---|---|---|
| EC50 ± S.D. | -Fold shift | EC50 ± S.D. | -Fold shift | EC50 ± S.D. | -Fold shift | |
| nm | nm | nm | ||||
| GT1a_WTa | 0.65 ± 0.32 | 1.0 | 1214 ± 374 | 1.0 | 12.2 ± 6.3 | 1.0 |
| GT1a_R155Ka | 2.9 ± 1.3 | 4.5 | 2582 ± 113 | 2.1 | 484 ± 140 | 40 |
| GT1a_D168Aa | 86 ± 17 | 132 | 962 ± 107 | 0.8 | 4850 ± 76 | 398 |
| GT1a_R155K/D168Aa | 2.7 ± 1.3 | 4.1 | 3266 ± 320 | 2.7 | 5072 | 416 |
| GT1a_WTb | 0.4 ± 0.2 | 1.0 | 275 ± 138 | 1.0 | 3.2 ± 2.6 | 1.0 |
| V36M/R155Kb | 3.5 ± 2.4 | 8.8 | 4785 ± 837 | 17 | 745 ± 233 | 233 |
| Y56H/D168Ab | 655 ± 437 | 1638 | 213 ± 53 | 0.8 | 517 ± 93 | 162 |
a Data are from the transient replicon.
b Data are from the stable replicon.
Figure 4.
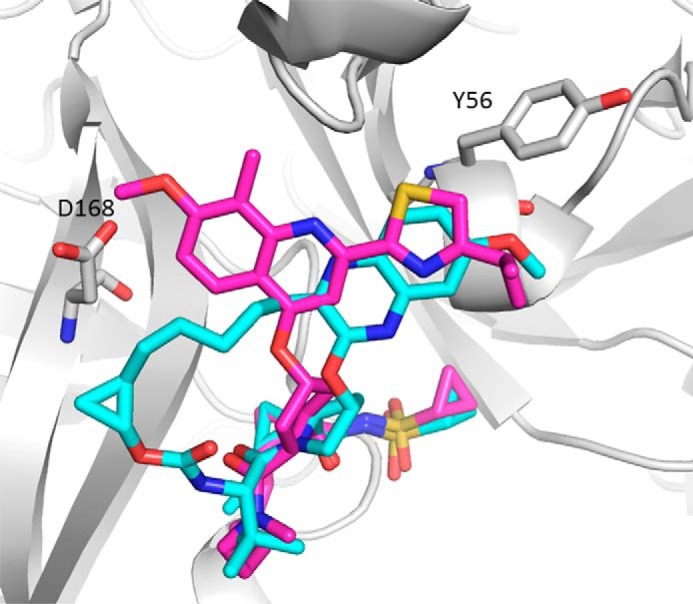
Superposition of grazoprevir (cyan) and simeprevir (magenta) bound to the NS3/4A protease using the crystal structure of PDB code 3SUD as template from GT1a. The protein is represented as ribbon, and Tyr-56 and Asp-168 are shown in stick representation.
R155K/D168A, the nullifying effect of Asp-168 on grazoprevir
Unlike Y56H/D168A, where the double amino acid substitutions conferred an additional loss in inhibitor potency to grazoprevir compared with each single substitution, the impact of the R155K/D168A substitution was unanticipated. Although the independent R155K and D168A substitutions conferred a 4.5- and 132-fold potency reduction, respectively, for grazoprevir, the combination of R155K and D168A had no enhanced impact on inhibitor potency and remained somewhat equivalent to that of R155K alone; in effect, the contribution from D168A was nullified. This observation was not expected, and an obvious explanation for this nullifying effect was not evident. An in-depth investigation was therefore conducted using different computational techniques to obtain insights into the structural basis for the observation.
MD simulation to explore interactions, wild-type (WT) NS3/4A protease
To understand the role of Asp-168 in the stabilization of Arg-155, we carried out all-atom explicit solvent MD simulations of the wild-type (WT) protein and monitored the distance between Asp-168 and Arg-155. The protein structure of NS3/4A bound to grazoprevir (PDB code 3SUD) was used as the starting coordinates for the simulation. In the crystal structure, Arg-155 and Asp-168 form a salt bridge between the cationic guanidinium of arginine and the carboxylate of the aspartic acid residues (Fig. 5a). This noncovalent interaction is maintained during the 100-ns simulation. As shown in Fig. 5b, the distance between Arg-155(NH1) and the Asp-168(OD1) fluctuates around 3.2 Å. Although there were occasional attempts for the two residues to break away from each other, having a distance as large as 7 Å, they quickly re-established the salt bridge, as illustrated in the inset of Fig. 5b.
Figure 5.

a, crystal structure of grazoprevir (green carbon, stick) bound to HCV NS3/4A protease (PDB code 3SUD). The salt-bridge network between Arg-123, Asp-168, and Arg-155 is indicated. b, distance between Arg-155(NH1) and Asp-168(OD1) during the 100-ns MD simulation of the WT protein. The inset on the left is the distribution of the distance, and those on the right are two snapshots highlighting the breaking and regaining of salt bridge between Arg-155 and Asp-168.
MD simulation to explore interactions, R155K NS3/4A protease
Similarly, all-atom explicit solvent MD simulations of the R155K-substituted enzyme were performed using the protein structure of R155K (PDB code 3SUE) as the starting coordinates. In the crystal structure, there is no salt bridge between Lys-155 and Asp-168. Instead, the ammonium of the Lys-155 side chain forms hydrogen bonds with the backbone carbonyl oxygen of Asp-79 and the quinoxaline nitrogen of the inhibitor via a water bridge (Fig. 6a). In our 100-ns MD simulations, despite attempts to form such interactions at the beginning 10 ns, no salt bridge between Lys-155 and Asp-168 was maintained; the distance between the Lys-155(NZ) and Asp-168(OD1) fluctuates around 7 Å, which is greater than the distance of <4 Å considered for the salt bridge (Fig. 6b). Furthermore, the relatively large fluctuation in the distance between Lys-155 and Asp-168 indicates that the Lys-155 side chain is far more mobile than Arg-155, consistent with its inherent flexibility.
Figure 6.

a, crystal structure of grazoprevir (green carbon, stick) bound to the R155K mutant of the HCV NS3/4A protease (PDB code 3SUE). The salt bridge between Arg-123 and Asp-168, and the hydrogen bonds of Lys-155 via a water bridge are indicated. b, distance between Arg-155(NH1) and Asp-168(OD1) during a 100-ns MD simulation of the mutant protein. The inset is the distribution of the distance.
Metadynamics to characterize the free energy landscape
The above MD simulations offered a glimpse of the dynamic behavior of the interactions between Asp-168 and Arg-155 or Lys-155 and highlighted the important role of Asp-168 in the stabilization of Arg-155 through a salt bridge and the lack of such a role for Lys-155. Because these MD simulations have limited sampling of the configuration space, and obtaining meaningful statistics through standard MD would require an impractical amount of computational time, we decided to carry out meta-dynamics simulations to explore the relevant free energy landscape, specifically, to investigate how the D168A substitution affects the conformational free energy of residue 155 of the WT and mutant proteins.
Meta-dynamics simulation is a powerful computational technique to explore complex free energy landscape. Unlike standard MD, the sampling in meta-dynamics simulations is enhanced by systematically introducing biasing potentials to lower the barrier between the free energy wells. The free energy surface (FES), represented as a function of a few chosen degrees of freedom, or collective variables (CVs), is then constructed. The CVs in our simulations are the torsional angles of residue Arg-155 for the WT and D168A mutant and of residue Lys-155 for R155K and R155K/D168A mutants. These torsion angles describe the different conformations of residue 155 of the protein in the native state. Four meta-dynamics simulations were performed, one for each of the proteins. Because no solvent was included in the simulation, these analyses consider protein intra-molecular interactions only.
Fig. 7 shows the free energy surface for the WT and mutant proteins projected onto the Cα-Cβ and Cδ-Cγ torsional space. Each surface features multiple free energy basins separated by barriers. For the WT protein, it is known from the crystal structures that Arg-155 can adopt two distinct conformations, depending on the inhibitor class; boceprevir and telaprevir (linear α-ketoamide inhibitors) bind to the same Arg-155 conformation as the substrate or the unbound protein, and the macrocyclic inhibitors such as grazoprevir and simeprevir bind to an extended Arg-155 conformation induced by the large P2 groups of these inhibitors. These two conformations are mapped onto the torsional free energy surface, as indicated in Fig. 7a. Clearly, the unbound or substrate-bound Arg-155 conformation is the lowest in free energy, characterized by a large basin. A second basin corresponds to the macrocycle-bound Arg-155 conformation. Although the second Arg-155 conformation is higher in free energy, it is nonetheless a stable conformation. With the D168A substitution, the second basin is replaced by a high free energy region, as shown in Fig. 7b. In other words, the D168A substitution changed the macrocycle-bound Arg-155 conformation from a stable low free energy state to an unstable high free energy state. Interestingly, Fig. 7b also indicates that the Arg-155 conformation corresponding to linear ketoamide inhibitor binding is not affected by this amino acid substitution because the corresponding basin remains the same.
Figure 7.

Torsion free energy surface of residue Arg-155 in the WT (a) and D168A (b) and Lys-155 in R155K (c) and R155K/D168A (d) proteins. The x axis is the torsion angle between the Cα and Cβ carbons, and the y axis is the torsion angle between the Cδ and Cγ carbons. Blue indicates low energy and red high energy. The units are in kilocalories/mol. The torsional space of Arg-155 corresponding to the macrocyclic bound and unbound in a and b.
Fig. 7, c and d, shows how the D168A substitution affects the torsional free energy surface of Lys-155 in the R155K and R155K/D168A proteins. The two plots have very similar profiles, as the D168A mutation does not change the overall characteristics of the free energy surface of Lys-155 in these mutants. This is in direct contrast to the significant difference in the free energy surface of Arg-155 in the WT and the D168A protein just discussed.
Cation-pyridine lone pair interaction between Lys-155 and grazoprevir
The above analyses from the MD and meta-dynamics simulations focused on the protein and how the D168A substitution affects the stability of the WT and the mutant proteins differently. Because protein-inhibitor binding involves both binding partners and their interactions, we further investigated the protein-inhibitor inter-molecular interactions. As shown in Fig. 8, modeling indicates that in the R155K/D168A double mutant, Lys-155(NZ) could interact favorably with the quinoxaline lone pair of grazoprevir, contributing to the inhibitor potency. In addition, modeling indicates that this interaction is absent in the case of simeprevir.
Figure 8.
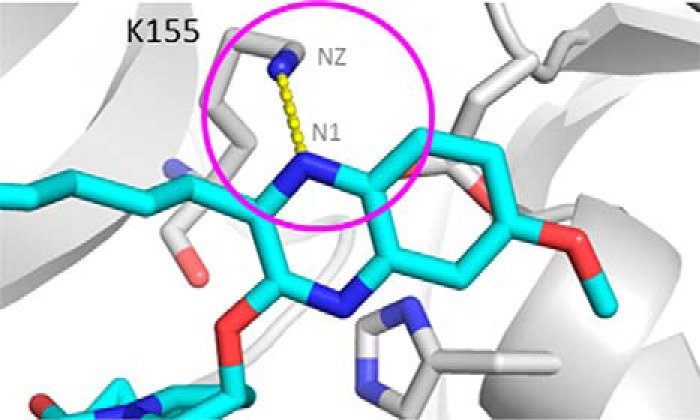
Model of cation-lone pair interaction between Lys-155(NZ) of R155K_D168A (gray carbon, stick) and the nitrogen of grazoprevir (cyan carbon, stick).
Discussion
As add-ons to the standard-of-care, early NS3/4A protease inhibitors, boceprevir and telaprevir, improved response rates for patients infected with HCV GT1; however, the selection of virus-resistant variants, in part, limited their efficacy. Critical GT1a amino acid changes included substitutions at V36M, T54S, and R155K for these earlier linear α-ketoamide inhibitors. Subsequently, the naturally occurring Q80K polymorphism in NS3/4A has been shown to impact response rates for simeprevir, a newer macrocyclic NS3/4A protease inhibitor. Hence, it was of interest to profile grazoprevir against clinical isolates and also in GT1a replicons bearing NS3/4A resistance-associated substitutions. Our data show grazoprevir is a potent pan-genotype NS3/4A inhibitor across HCV genotypes. The activity of grazoprevir in enzyme assays largely mirrored its activity in HCV replicons. The inhibitor also retained activity against a majority of the common resistance-associated substitutions observed in patients who fail therapy with the NS3/4A-containing regimen, including the highly prevalent GT1a Q80K variant. The data indicated that nanomolar concentrations of grazoprevir may result in inhibition of viral replication across a broad range of genotypes in infected hepatocytes.
Although a survey of frequently identified clinically relevant resistance-associated substitutions showed potent activity of grazoprevir in a number of the replicons as described above and reported in Table 3, the basis of virological failure among patients treated with the inhibitor required a direct assessment of selected amino acid changes. It has been demonstrated in a number of clinical trials that the presence of baseline NS3/4A polymorphisms (those present before administration of study drug) in GT1 patients do not impact the efficacy of the combination of grazoprevir and elbasvir (the components of ZepatierTM) (12, 17). However, NS3/4A amino acid substitutions are detected in patients who fail therapy with a grazoprevir-containing regimen. Hence, we reviewed the prevalence of NS3/4A substitutions in patients who failed to achieve SVR in clinical studies with a grazoprevir-containing regimen. An investigation revealed that substitutions at amino acid positions 168 and 156 in NS3 were most prevalent. Re-engineering of the detected substitutions in replicons showed that those located at position 156 tend to confer low replicative fitness to replicons. Amino acid substitutions at position 168 were fit and conferred potency losses in the range of 2.3–73-fold to grazoprevir. This spurred us to investigate other changes, including those involving double substitutions observed with other NS3/4A inhibitors that involved position 168. Although the double substitution at positions 56 and 168 (Y56H/D168A) resulted in a further decrease in susceptibility, the R155K/D168A double substitution did not confer a reduction in potency beyond that observed for R155K; this result was unexpected.
Many of the resistance-associated substitutions in HCV NS3/4A protease have been characterized and understood by means of X-ray crystallography and modeling, such as the Y56H and Y56H/D168A variants discussed in this work. However, the nullifying effect of the D168A substitution on the binding of grazoprevir to the R155K/D168A protein could not be explained readily. A series of computational studies were therefore conducted to investigate the role of D168A on the binding of grazoprevir to the WT and the R155K proteins in an attempt to understand the structural basis for the observed potency change. MD simulations revealed the important role of Asp-168 in anchoring Arg-155 through a salt bridge, facilitating the binding of grazoprevir to the WT protein; the D168A amino acid change will lose this salt bridge capability. However, the lack of such interactions between Lys-155 and Asp-168 suggests that Asp-168 is not a partner for Lys-155 as it is for Arg-155. Therefore, the D168A substitution will have a greater effect on Arg-155 in the WT and the D168A protein than on Lys-155 in the R155K and R155K/D168A proteins. This provides a plausible explanation for the relatively large potency shift from WT to D168A and virtually no shift from R155K to R155K/D168A.
The impact of the amino acid substitutions can also be understood from a thermodynamic standpoint. The FES analysis from meta-dynamics simulations depicts the torsional free energy surface of residue 155 (Arg or Lys) in the WT and the mutant proteins. The FES analysis successfully captured the two distinct conformations of Arg-155 observed in the crystal structure of the WT protein and quantified the relative stability of the two conformations of Arg-155; the largest basin corresponds to the substrate and linear ketoamide inhibitor-bound form, whereas the second basin corresponds to the macrocyclic inhibitor-bound form (Fig. 7a). In the D168A protein, the largest basin remains unchanged, which means substrate and ketoamide inhibitor binding is not affected. However, the second basin disappeared and converted to a high free energy region instead (Fig. 7b). This would increase the free energy cost for the binding of macrocyclic inhibitors and is a key component contributing to the potency loss of grazoprevir. This is consistent with the observation from the MD simulations (data not shown), where the D168A mutation destabilizes the Arg-155 conformation due to the loss of the salt bridge, and it provides a thermodynamic link from the free energy of the conformational states to the structural changes. It is noteworthy that our meta-dynamics simulations were performed in a vacuum, and the exceptionally high energy barrier (up to 17 kcal/mol) from the FES analysis will be reduced in water due to the solvent-screening effect. In terms of protein-ligand interactions, the predicted favorable direct cation-quinoxaline lone pair interaction unique to grazoprevir binding to the R155K/D168A mutant should inspire experimental investigations such as X-ray crystallography study.
Molecular modeling and simulations have become an essential component for the investigation of biomolecular systems and protein-ligand interactions. Understanding the molecular mechanisms underlying drug resistance helps the design of experimental studies and the development of more effective therapeutics targeting mutant proteins. With the increase in computer power and advances in computational algorithms and workflow development, computational methods are being widely used in drug discovery such as inhibitor optimization, mutation studies, and enzyme design. The combination of these techniques together with molecular biology and biochemical analyses enabled a deeper understanding of the structural features that influence the potency of grazoprevir against GT1a resistance-associated substitutions in HCV NS3/4A protease. These findings are consistent with detection of the R155K/D168A double substitution in some patients who failed to achieve SVR with simeprevir but not observed in those treated with grazoprevir. Grazoprevir (in combination with elbasvir, a HCV NS5A inhibitor) has now been approved for the treatment of patients chronically infected with HCV genotypes 1, 3, or 4 depending on the jurisdiction.
Experimental procedures
Protein purification
The full-length HCV NS3/4A proteases from genotypes 1–6 and substituted amino acids were expressed and purified from Escherichia coli as His-tagged constructs by immobilized metal-ion affinity chromatography. Briefly, cell lysates were incubated with TALON metal affinity resin (Clontech), washed with 5 times the bed volume, and eluted (with 350 mm imidazole) also with 5 times the bed volume in 1-ml fractions as per the suggested protocol from the manufacturer (Clontech). Collected fractions were analyzed by denaturing PAGE, and activity was determined for the pooled protein fractions (Clontech user manual for TALON metal affinity resin). Amino acid substitutions were engineered into GT1a H77 and GT1b BK constructs using standard molecular biology techniques (20).
Enzyme kinetics
The catalytic activity of NS3/4A protease was monitored continuously by following the cleavage of a modified peptide substrate by time-resolved fluorescence in a buffer containing 50 mm HEPES, 150 mm NaCl, 0.1% PEG-8000, 15% glycerol, 0.15% Triton X-100, 1 mm DTT at a pH of 7.6 (21). The modified peptide, Ac-C(Eu)DDMEEAbu(COO)ASK (QSY7)-amide, was used as substrate to assay for enzyme activity. NS3/4A protease-catalyzed cleavage of the ester bond (aminobutyric acid(COO)) separates the fluorescent europium cryptate from the quencher (QSY7) and results in an increase in fluorescence (excitation wavelength = 340 nm, emission wavelength = 615 nm). The long-lived fluorescence of europium allows for use of time-resolved measurements that reduce background fluorescence and increases the sensitivity of the assay. Thus, NS3/4A protease concentrations as low as 15 pm were used to enable quantitation of a high level of inhibition by very potent compounds.
Enzyme inhibition
Potencies were determined for NS3/4A protease inhibitors using the assay described above (under enzyme kinetics) with modifications (as indicated below). End-point reactions were completed over 60 min at room temperature following a 30-min pre-incubation of enzyme and inhibitor. For IC50 determinations, varying compound concentrations were prepared via a 3-fold serial dilution with typically 1 mm as the highest concentration in DMSO. NS3/4A protease (at a final concentration of 15 pm) was pre-incubated with the appropriate concentration of each compound for 30 min at room temperature in a 384-well plate. The reaction was initiated by the addition of substrate (at a final concentration of 25 nm) to each well. Following assay termination, the plate was sealed and incubated for 2.5 h (away from light) at room temperature before reading. The product fluorescence was detected using PheraStar Plus plate reader (excitation, 340 nm; emission, 620 nm). IC50 values were calculated by fitting the data to a four-parameter dose-response equation with the aid of the PRISM software application (GraphPad Software, La Jolla, CA).
Cell culture
Human hepatoma cell line Huh7 or Huh7.5 (22) was cultured in Dulbecco's minimal essential medium (DMEM) supplemented with 2 mm glutamine, nonessential amino acids, 0.075% sodium bicarbonate, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS) in 10 mm HEPES at a pH of 7.5 (all reagents were obtained from Bio Whittaker (Lancaster, MA). Stable replicons generated in Huh7 or Huh7.5 cells were cultured in 0.5 mg/ml G418 (Cellgro, Manassas, VA). The generation and establishment of HCV replicons in Huh cell lines were previously described (22, 23).
Construction of replicons bearing resistance-associated amino acid substitutions in NS3
The GT1a replicon sequence used in this study was described by Yi and Lemon (24). Amino acid substitutions were introduced into GT1a replicon constructs using either a QuikChange mutagenesis kit (Stratagene, La Jolla, CA) or by chemically synthesizing the gene bearing the mutation and the appropriate restriction sites (GeneWiz, Piscataway, NJ). The QuikChange template for GT1a was a pCR-XL-TOPO vector (Invitrogen) containing the GT1a NS3 gene. The KpnI-NsiI fragment, which contained a portion of EMCV IRES and the entire NS3 gene, except for the last eight C-terminal amino acids, was sequenced to confirm the presence of the mutations and used to replace the same region in the full-length GT1a subgenomic replicon sequence. The methodology for the generation of replicons has been described previously (22, 23).
Inhibition studies in stable replicon cells
To measure cell-based inhibitory activity of grazoprevir, Huh7 (or Huh7.5) replicon-containing cells were seeded at 1000 (or 2000) cells/well in 384-well collagen I-coated Biocoat plates (BD Biosciences). Twenty four hours post-seeding, the relevant concentration of grazoprevir was added to the cells in a final DMSO concentration of 0.5% (v/v) in 5% FBS with no addition of G418. The cells were treated with inhibitor for 72 h at which point they were washed with PBS and lysed (Cell lysis buffer, Ambion, Carlsbad, CA). The amount of replicon RNA was measured by real time quantitative PCR (TaqMan assay). The PCR primers for GT1a replicons were located in HCV internal ribosome entry site (IRES) and were as follows: IRES_F, 5′TGCGGAACCGGTGAGTACA3′; IRES_R, 5′GCGGGTTTATCCAAGAAAGGA3′; the probe sequence was 6-carboxyfluorescein-labeled 5′CGGAATTGCCAGGACGACCGG3′. The real time RT-PCRs were run on an ABI PRISM 7900HT sequence detection system using the following program: 48 °C for 30 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s, 60 °C for 1 min. The CT values were plotted against log inhibitor concentration and fitted to a four-parameter sigmoid dose-response model. The EC50 was the concentration necessary to achieve ΔCT = 1 over the projected baseline threshold, whereas the EC90 was the concentration necessary to achieve ΔCT = 3.2 over the baseline threshold.
All TaqMan reagents were from Life Technologies, Inc.
Inhibition studies in transient replicons
The GT1a H77S.3 HCV replicon containing the Gaussia luciferase gene (G-luc) integrated in-frame with the viral polypeptide was obtained from Dr. Stanley M. Lemon (25). This construct was used to analyze various polymorphisms. The substitutions of interest were produced by cloning a cassette of the modified target gene (made by gene synthesis into the GT1a H77S.3 background). Transfection of this replicon RNA, transcribed from cDNA using T7 Megascript (Ambion, Carlsbad, CA) into Huh7.5 cells, results in replication of the HCV RNA and expression of the G-luc protein. The levels of expressed G-luc protein directly correlate with HCV RNA copy number and viral protein translation. Briefly, 5 × 106 Huh7.5 cells were transfected by electroporation with 5 μg of replicon RNA on a Bio-Rad Gene Pulser Xcell using the exponential protocol at 270 volts, 950 microfarad capacitance, and resistance of 100 Ω. Cells were transferred to 75-ml flasks with 1/100th transferred to a well of a 24-well plate.
To measure fitness, culture supernatant was collected from cells growing in the 24-well plate after 6 h and each day thereafter. All collected supernatants were stored at 4 °C until luciferase measurement. After 7 days, all lysate samples were assayed for Gaussia luciferase activity using the BioLux Gaussia Luciferase Assay kit (New England Biolabs, Ipswich, MA) as per the manufacturer's protocol. The fitness was determined by comparing the relative increase in luciferase produced by a variant to that produced by the wild-type genome.
To measure the effect of genome mutations on compound potency, 3 days after transfection cells were transferred into 96-well plates and allowed to adhere overnight. The following day, serial 2-fold dilutions of compounds in 0.5% DMSO were added. After 72 h, the supernatants were assayed for luciferase activity using the reagent described above. Luciferase activity was measured on an Envision plate reader (PerkinElmer Life Sciences). Concentrations of compounds reducing the luciferase measurements by 50 or 90% compared with the DMSO-only control were taken as EC50 and EC90, respectively, using Prism analysis of a sigmoidal dose-response curve.
Molecular dynamics simulations
All the molecular dynamics (MD) simulations were carried out with AMBER14 suite (26) and the ff14SB force field. Starting with the crystal structure of the protein, the system was prepared using MOE molecular modeling package (MOE-2015.10, Chemical Computing Group, Montreal, Canada). The C and N termini were capped with N-methyl and acetyl groups, respectively. The protonation state of the amino acid residues aspartic acid, glutamic acid, and histidine was determined at pH 7.4 using Protonate 3D; alanine 139 was mutated back to serine (note this catalytic residue was mutated to alanine in the crystal structure). The catalytic histidine 57 was protonated. The prepared system was neutralized by adding Cl− ions and solvated in a water box extending at least 11 Å in each direction from the protein. First, the system was energy-minimized using 250 steps of steepest descent and 750 steps of conjugate-gradient restraint minimizations to remove steric clashes. The system was then heated to 300 K during a 50-ps simulation using a time step of 2 fs and periodic boundary conditions in the NVT ensemble, followed by a 100-ns production simulation. Nonbonded interactions were truncated with a cutoff of 9 Å and the long-range Coulomb interactions were treated by the particle-mesh Ewald method.
Metadynamics
The metadynamics simulations were performed using the Desmond package (Maestro release 2015-1, Schrödinger, LLC, New York). The OPLS-2005 force field (27) was used to model the interactions of the system under study. The Protein Preparation workflow (28) in Maestro was used to prepare the system such as adding hydrogens, assigning the proper protonation states to ionizable residues, incorporating the N-terminal acetyl and C-terminal amide caps, etc. For the metadynamics simulations, the torsion angles Cα-Cβ-Cγ and Cβ-Cγ-Cδ-XE were defined as the collective variables, where X is nitrogen for Arg-155 and carbon for Lys-155. These torsional angles describe the distinct conformational states of residue 155 for the wild-type and mutant proteins. The history-dependent Gaussian bias potential was defined by the following parameters: height = 0.03, width = 5°, update interval = 0.09 ps. For each protein, metadynamics simulations in vacuum were performed for 10 ns using the NPT ensemble at temperature 300 K and pressure 1.01325 bar. The particle-mesh Ewald method (27) and periodic boundary conditions were used. To maintain structural integrity during the simulation, a harmonic restraint of force constant 1.0 KJ/mol was applied to the protein except residue Arg-155/Lys-155 and the surrounding residues Arg-123, Asp-168/Ala-168, Ala-156, and Gln-80. The Desmond workflow and parameter settings for the MD simulation were used (27).
Author contributions
Z. G. and Y. H. performed the modeling and molecular dynamics experiments and interpreted the data. S. B. and S. C. performed NS3/4A protease inhibitor studies in stable replicons; R. C. and P. I. performed inhibition studies in transient replicons and conducted cytotoxicity assays; P. M. performed molecular biology and generated the cell lines bearing the amino acid substitutions; E. A. A. conceived and coordinated the study and served as primary author. All authors reviewed the results, contributed to writing the paper, and approved the final version of the manuscript.
Acknowledgments
We thank Fred Lahser for discussions during the study and preparation of the manuscript. We also thank Brad Sherborne for valuable input on the computational experiments.
All authors are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ.
- HCV
- hepatitis C virus
- DAA
- direct-acting antiviral agent
- G-luc
- Gaussian luciferase
- MD
- molecular dynamics
- SVR
- sustained virologic response
- GT
- genotype
- PDB
- Protein Data Bank
- FES
- free energy surface
- CV
- collective variable
- NS
- nonstructural
- IRES
- internal ribosome entry site
- TE
- treatment-emergent
- PI
- protease inhibitor.
References
- 1. Lavanchy D. (2009) The global burden of hepatitis C. Liver Int. 29, 74–81 [DOI] [PubMed] [Google Scholar]
- 2. Messina J. P., Humphreys I., Flaxman A., Brown A., Cooke G. S., Pybus O. G., and Barnes E. (2015) Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 61, 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ghany M. G., Nelson D. R., Strader D. B., Thomas D. L., Seeff L. B., and American Association for Study of Liver Disease (2011) An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54, 1433–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization. (2016) Guidelines for the screening, care and treatment of persons with chronic hepatitis C infection updated version. April 2016, WHO Document Production Services, Geneva, Switzerland: [PubMed] [Google Scholar]
- 5. Kolykhalov A. A., Mihalik K., Feinstone S. M., and Rice C. M. (2000) Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′-nontranslated region are essential for virus replication in vivo. J. Virol. 74, 2046–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Malcolm B. A., Liu R., Lahser F., Agrawal S., Belanger B., Butkiewicz N., Chase R., Gheyas F., Hart A., Hesk D., Ingravallo P., Jiang C., Kong R., Lu J., Pichardo J., et al. (2006) SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of α-interferon in replicon cells. Antimicrob. Agents Chemother. 50, 1013–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perni R. B., Almquist S. J., Byrn R. A., Chandorkar G., Chaturvedi P. R., Courtney L. F., Decker C. J., Dinehart K., Gates C. A., Harbeson S. L., Heiser A., Kalkeri G., Kolaczkowski E., Lin K., Luong Y. P., et al. (2006) Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3–4A serine protease. Antimicrob. Agents Chemother. 50, 899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosenquist Å., Samuelsson B., Johansson P. O., Cummings M. D., Lenz O., Raboisson P., Simmen K., Vendeville S., de Kock H., Nilsson M., Horvath A., Kalmeijer R., de la Rosa G., and Beumont-Mauviel M. (2014) Discovery and development of simeprevir (TMC435), a HCV NS3/4A protease inhibitor. J. Med. Chem. 57, 1673–1693 [DOI] [PubMed] [Google Scholar]
- 9. Pilot-Matias T., Tripathi R., Cohen D., Gaultier I., Dekhtyar T., Lu L., Reisch T., Irvin M., Hopkins T., Pithawalla R., Middleton T., Ng T., McDaniel K., Or Y. S., Menon R., et al. (2015) In vitro and in vivo antiviral activity and resistance profile of the hepatitis C virus NS3/4A protease inhibitor ABT-450. Antimicrob. Agents Chemother. 59, 988–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liverton N. J., Holloway M. K., McCauley J. A., Rudd M. T., Butcher J. W., Carroll S. S., DiMuzio J., Fandozzi C., Gilbert K. F., Mao S.-S., McIntyre C. J., Nguyen K. T., Romano J. J., Stahlhut M., Wan B.-L., et al. (2008) Molecular modeling based approach to potent P2-P4 macrocyclic inhibitors of hepatitis C NS3/4A protease. J. Am. Chem. Soc. 130, 4607–4609 [DOI] [PubMed] [Google Scholar]
- 11. Harper S., McCauley J. A., Rudd M. T., Ferrara M., DiFilippo M., Crescenzi B., Koch U., Petrocchi A., Holloway M. K., Butcher J. W., Romano J. J., Bush K. J., Gilbert K. F., McIntyre C. J., Nguyen K. T., et al. (2012) Discovery of MK-5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor. ACS Med. Chem. Lett. 3, 332–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lawitz E., Gane E., Pearlman B., Tam E., Ghesquiere W., Guyader D., Alric L., Bronowicki J. P., Lester L., Sievert W., Ghalib R., Balart L., Sund F., Lagging M., Dutko F., et al. (2015) Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial. Lancet 385, 1075–1086 [DOI] [PubMed] [Google Scholar]
- 13. Black S., Pak I., Ingravallo P., McMonagle P., Chase R., Shaughnessy M., Hwang P., Haber B., Harrigan P. R., Brumme C., and Howe A. Y. (2015) J. Hepatol. 62, S677–S678 [Google Scholar]
- 14. Lontok E., Harrington P., Howe A., Kieffer T., Lennerstrand J., Lenz O., McPhee F., Mo H., Parkin N., Pilot-Matias T., and Miller V. (2015) Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology 62, 1623–1632 [DOI] [PubMed] [Google Scholar]
- 15. Jacobson I. M., Dore G. J., Foster G. R., Fried M. W., Radu M., Rafalsky V. V., Moroz L., Craxi A., Peeters M., Lenz O., Ouwerkerk-Mahadevan S., De La Rosa G., Kalmeijer R., Scott J., Sinha R., and Beumont-Mauviel M. (2014) Simeprevir with pegylated interferon α 2a plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-1): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 384, 403–413 [DOI] [PubMed] [Google Scholar]
- 16. Lenz O., Verbinnen T., Fevery B., Tambuyzer L., Vijgen L., Peeters M., Buelens A., Ceulemans H., Beumont M., Picchio G., and De Meyer S. (2015) Virology analyses of HCV isolates from genotype 1-infected patients treated with simeprevir plus peginterferon/ribavirin in Phase IIb/III studies. J. Hepatol. 62, 1008–1014 [DOI] [PubMed] [Google Scholar]
- 17. Rockstroh J. K., Nelson M., Katlama C., Lalezari J., Mallolas J., Bloch M., Matthews G. V., Saag M. S., Zamor P. J., Orkin C., Gress J., Klopfer S., Shaughnessy M., Wahl J., Nguyen B.-Y., et al. (2015) Efficacy and safety of grazoprevir (MK-5172) and elbasvir (MK-8742) in patients with hepatitis C virus and HIV co-infection (C-EDGE CO-INFECTION): a non-randomised, open-label trial. Lancet HIV 2, e319-e327 [DOI] [PubMed] [Google Scholar]
- 18. Romano K. P., Ali A., Aydin C., Soumana D., Ozen A., Deveau L. M., Silver C., Cao H., Newton A., Petropoulos C. J., Huang W., and Schiffer C. A. (2012) The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog. 8, e1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bae A., Sun S. C., Qi X., Chen X., Ku K., Worth A., Wong K. A., Harris J., Miller M. D., and Mo H. (2010) Susceptibility of treatment-naive hepatitis C virus (HCV) clinical isolates to HCV protease inhibitors. Antimicrob. Agents Chemother. 54, 5288–5297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Green M. R., and Sambrook J. (2012) Molecular Cloning: A Laboratory Manual, 4th Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 21. Mao S. S., DiMuzio J., McHale C., Burlein C., Olsen D., and Carroll S. S. (2008) A time-resolved, internally quenched fluorescence assay to characterize inhibition of hepatitis C virus nonstructural protein 3-4A protease at low enzyme concentrations. Anal. Biochem. 373, 1–8 [DOI] [PubMed] [Google Scholar]
- 22. Blight K. J., Kolykhalov A. A., and Rice C. M. (2000) Efficient initiation of HCV RNA replication in cell culture. Science 290, 1972–1974 [DOI] [PubMed] [Google Scholar]
- 23. Lohmann V., Körner F., Koch J., Herian U., Theilmann L., and Bartenschlager R. (1999) Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113 [DOI] [PubMed] [Google Scholar]
- 24. Yi M., and Lemon S. M. (2004) Adaptive mutations producing efficient replication of genotype 1a hepatitis C virus RNA in normal Huh7 cells. J. Virol. 78, 7904–7915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shimakami T., Welsch C., Yamane D., McGivern D. R., Yi M., Zeuzem S., and Lemon S. M. (2011) Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 140, 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Case D. A., Botello-Smith W., Cerutti D. S., Cheatham T. E. 3rd., Darden T. A., Duke R. E., Giese T. J., Gohlke H., Goetz A. W., Homeyer N., Izadi S., Janowski P., Kaus J., Kovalenko A., Lee T. S., et al. (2016) AMBER 2016, Version 16 University of California, San Francisco [Google Scholar]
- 27. Kaminski G. A., Friesner R. A., Tirado-Rives J., and Jorgensen W. L. (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 105, 6474–6487 [Google Scholar]
- 28. Jakalian A., Jack D. B., and Bayly C. I. (2002) Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641 [DOI] [PubMed] [Google Scholar]