

Abstract
Gain-of-function (GOF) mutations in the STAT1 gene are critical for the onset of chronic mucocutaneous candidiasis (CMC) disease. However, the molecular basis for the gain of STAT1 function remains largely unclear. Here, we investigated the structural features of STAT1 GOF residues to better understand the impact of these pathogenic mutations. We constructed STAT1 alanine mutants of the α3 helix residues of the coiled-coil domain, which are frequently found in CMC pathogenic mutations, and measured their transcriptional activities. Most of the identified GOF residues were located inside the coiled-coil domain stem structure or at the protein surface of the anti-parallel dimer interface. Unlike those, Arg-274 was adjacent to the DNA-binding domain. In addition, Arg-274 was found to functionally interact with Gln-441 in the DNA-binding domain. Because Gln-441 is located at the anti-parallel dimer contact site, Gln-441 reorientation by Arg-274 mutation probably impedes formation of the dimer. Further, the statistical analysis of RNA-seq data with STAT1-deficient epithelial cells and primary T cells from a CMC patient revealed that the R274Q mutation affected gene expression levels of 66 and 76 non-overlapping RefSeq genes, respectively. Because their transcription levels were only slightly modulated by wild-type STAT1, we concluded that the R274Q mutation increased transcriptional activity but did not change dramatically the repertoire of STAT1 targets. Hence, we provide a novel mechanism of STAT1 GOF triggered by a CMC pathogenic mutation.
Keywords: genetics, Janus kinase (JAK), pathogenesis, signal transducers and activators of transcription 1 (STAT1), transcription regulation
Introduction
Signal transducer and activator of transcription 1 (STAT1) is an essential mediator of interferon (IFN)-α/β- and IFN-γ-induced signaling that activates the immune response against various infectious pathogens (1, 2). IFN-γ activation of receptor-bound Janus kinase 1/2 (JAK1/2) leads to tyrosine phosphorylation of the IFN-γ receptor, resulting in the creation of docking sites for cytosolic STAT1. On the receptor, STAT1 becomes phosphorylated by JAKs, dimerizes via phosphorylation-dependent interaction, and finally translocates to the nucleus. On a target gene promoter, the STAT1 dimer recognizes interferon-γ-activated sites and drives downstream gene transcription that encodes proteins for anti-viral and pro-inflammatory functions. After an immune response, dephosphorylation of STAT1 by tyrosine phosphatases reverses JAK-mediated phosphorylation to attenuate IFN-γ signaling. Precise regulation of both the magnitude and duration of STAT1 activation is critical for the orchestration of a wide variety of immunological processes.
Recent progress in the methodologies of genome sequencing has revealed a wide variety of single nucleotide polymorphisms in the STAT1 gene locus (3, 4). These mutations frequently change the resultant STAT1 amino acid sequence but rarely generate critical damage to its immunoregulatory functions. In the latest version of the Human Gene Mutation Database (HGMD), 51 missense mutations in the STAT1 gene have been registered as disease-causing mutations. These include mutations with a phenotype of autosomal recessive STAT1 deficiency, autosomal dominant STAT1 deficiency, and autosomal dominant gain of STAT1 activity (Fig. 1A). The loss-of-function (LOF)2 mutations have been associated with the pathogenesis of Mendelian susceptibility to mycobacterial disease, which is a rare syndrome characterized by infections with weakly virulent mycobacteria (5, 6). On the other hand, its gain-of-function (GOF) mutations exhibit different immunological defects. The patients suffer from persistent or recurrent infection of the skin, nails, and mucous membranes with Candida albicans, referred to as chronic mucocutaneous candidiasis (CMC) (7–9). Assigning structural changes caused by these pathogenic amino acid substitutions has shed light on the molecular mechanisms of STAT1-mediated immunodeficiencies as well as the fundamental roles of the original residue in normal STAT1 functions.
Figure 1.

Effects of amino acid substitutions on the transcriptional activity in the α3 helix region of the STAT1 CCD. A, arrowheads indicate positions of the STAT1 residues targeted by pathogenic missense mutations. The GOF mutations are red. Mutations coupled with STAT1 LOF and STAT1 deficiency are blue and gray, respectively. Amino acid number is indicated above. Lollipops indicate positions of the known phosphorylation sites. B, location of pathogenic GOF mutation in the crystal structure of the DNA-bound STAT1 dimer. One subunit of the dimer is illustrated as a ribbon diagram, and another is shown in a surface model. The four CCD helices are indicated as α1–α4. The locations of the CMC-hot-spot GOF mutations are colored red, and those of other known GOF mutations are colored orange. The domains are shown in gray (ND), green (CCD), cyan (DBD and LK), and dark blue (SH2). C, IFN-γ-stimulated transcriptional activities of the α3 mutants were measured by a Dual-Luciferase reporter system with the reporter driven by 5× IFN-γ-response elements of the IRF1 promoter. RLU of the mutants were normalized to wild-type STAT1 activity. The experiments shown in the lanes were performed in triplicate. Error bars, S.D. of each condition. Asterisks, significant differences of the mean values of the mutant RLU compared with the mean values of the STAT1 RLU. p values were determined with Student's test: *, p < 0.05; **, p < 0.01. D, protein levels of the α3 mutants were analyzed by Western blotting with anti-STAT1 antibody (top). The amount of the loaded protein (10 mg) was visualized by Coomassie Brilliant Blue (CBB) staining of the immunoblotted membrane (bottom).
STAT1 consists of six domains common to STAT family proteins (10). From the N to the C terminus of the protein, they are designated as the N-terminal domain (ND), the coiled-coil domain (CCD), the DNA-binding domain (DBD), a structural linker domain (LK), the Src-homology 2 domain (SH2), and a residual C-terminal domain (CTD). There is no apparent preferential localization of LOF residues in the domain structure (Fig. 1A). However, the critical residues for DNA binding and phosphorylation have been identified as Gln-463 and Tyr-701, respectively (5, 6). In contrast, GOF mutations clearly accumulate in the CCD and the DBD (Fig. 1A). Because the CCD protrudes outward from the bound DNA (11) (Fig. 1B), it was initially postulated that the CCD acts as a docking platform for transcription factors to cooperatively support STAT1-mediated transcription and that GOF mutations in the CCD increased their interactions (12). Recently, another mechanism of STAT1 GOF was proposed (7, 9). Dephosphorylation of Tyr-701 proceeds via anti-parallel conformation of the STAT1 dimer through phospho-Tyr-701-independent interaction between the CCD and DBD (13). Some of the pathogenic GOF residues accumulate at this anti-parallel dimer interface. Thus, it has been hypothesized that stabilization of an anti-parallel dimer is critical for normal function of STAT1. However, it remains largely unclear which residues innately present a risk of disease-causing missense mutation.
In this report, we describe our investigation of the molecular and structural basis for gain of STAT1 function caused by CMC pathogenic mutations. Because these mutations frequently converged on the α3 helix of the CCD, we generated a series of Ala mutants of this region and measured the mutant activities using our optimized luciferase reporter assay. We especially focused on one particularly frequent GOF residue, Arg-274, because it was known as a CMC hot spot (7, 9) and possessed structural features distinct from the other GOF residues. Most of the GOF residues are toward the inside of four helical bundles of the CCD or occupied on the protein surface near the interface of anti-parallel dimer formation. In contrast, according to the crystallographic structure of STAT1, Arg-274 is located at the bottom of a deep cleft between the CCD and the DBD, implying a role for this residue in the configuration of these two domains. In fact, Arg-274 is sufficiently close to interact with Gln-441 of the DBD. Disruption of this mutual association by these combinatorial mutations leads to aberrant increases in STAT1 transcription activities. Because Gln-441 is located near the interface of the anti-parallel STAT1 dimer, a pathogenic Arg-274 mutation would probably impede and destabilize Gln-441-mediated dimer stabilization. Hence, we provide a novel mechanism for STAT1 GOF mutation associated with the onset of CMC. Extending previous reports, these findings may help to predict the impacts of uncharacterized missense mutations in the STAT1 gene and construct a predictive risk model for association of these mutations with CMC onset and development.
Results
Implication of three distinct structural disorders of STAT1 caused by pathogenic GOF mutations of the CCD
The STAT1 CCD is composed of four α-helices (11). CMC hot spots were reported to converge on the residues around the third helix (3), implying that the local structure of this helix might be indispensable for the regulation of STAT1 activity (Fig. 1B). In support of this idea, the amino acid sequence of the third helix is conserved among various organisms as well as other members of the STAT family of human proteins (supplemental Fig. S1). To investigate the influence of amino acid changes in this region, we optimized a luciferase reporter assay to measure a series of mutants. Because it seemed critical to select a highly sensitive IFN-γ-response element to detect differences between mutants, luciferase reporter driven by enhancer elements from three representative STAT1-regulated genes (i.e. GBP, IRF1, and LY6E) was tested (14) (supplemental Fig. S2). These three reporters were individually transfected along with a wild-type STAT1 expression vector into STAT1-deficient cells (15). After exposure to IFN-γ, reporter activities increased ∼92.3-, 254.1-, and 5.3-fold when we examined the GBP, IRF1, and LY6E elements, respectively. Hence, we adopted the IFN-γ-response element of IRF1 for measurement of mutant activities. Next, we assessed transcriptional activities of a series of Ala mutants, of which 31 residues of the third helix, except for original Ala residues, were substituted (Fig. 1C).
Using our optimized luciferase reporter assay, a total of 11 Ala mutants exhibited 2–4-fold increases in their transcription activities relative to that of wild type (Fig. 1C). Only two Ala mutants (i.e. E268A and Q272A) showed repressive activities. These mutants were equally expressed in cells at the protein level (Fig. 1D). Thus, up-regulation of the activity of each mutant was presumably due to its enhanced transcriptional activities. According to the crystallographic structure (11), among the GOF mutants, residues Leu-259, Ser-269, Leu-283, Glu-284, Tyr-287, and Thr-288 were toward the center of the four-helix bundles (supplemental Fig. S3, A and B). In contrast, other GOF residues (i.e. Gln-275, Lys-278, Glu-282, and Lys-286) appeared to form the protein surface. As shown in previous reports, these results supported the hypothesis that two structurally distinct mechanisms were involved in the gain of STAT1 function that was associated with CMC pathogenesis (7). However, in a structural aspect, Arg-274 appeared unique because it possessed both of these structural properties. Whereas Arg-274 was toward the outside of the CCD (supplemental Fig. S3C), the occupied surface area was almost covered with the projecting DBD of the same molecule (supplemental Fig. S3A). We then focused on the mechanism underlying the aberrant increase of activity caused by the Arg-274 mutation.
Influence of R274Q mutations on the regulation of Tyr-701 phosphorylation
Amino acid changes of Arg-274 to Gln, Trp, and Gly have been found to be pathogenic missense mutations (3), suggesting that the integrity of the Arg residue was indispensable for normal STAT1 regulation. To evaluate the tolerance for its substitution, we assessed the transcriptional activities of a series of mutants in which the original Arg-274 was replaced by 19 other amino acids. Based upon the results of the luciferase assay as described above, the activities of all of the mutants were increased relative to wild type (Fig. 2A). Because these mutants were equally expressed in cells at the protein level (Fig. 2B), up-regulation of each mutant activity was due to their enhanced transcriptional activities, not their increased protein stability. Among the amino acid variations, the R274K mutation had the smallest effect on the activity, indicating that these two amino acids are “interchangeable,” because both have a positive charge. In contrast, proline residues are known to destabilize the α-helix structure by steric hindrance, and the proline substitution had the greatest impact. Hence, these data suggested that maintenance of the local CCD structure surrounded by Arg-274 was needed for normal STAT1 regulation.
Figure 2.
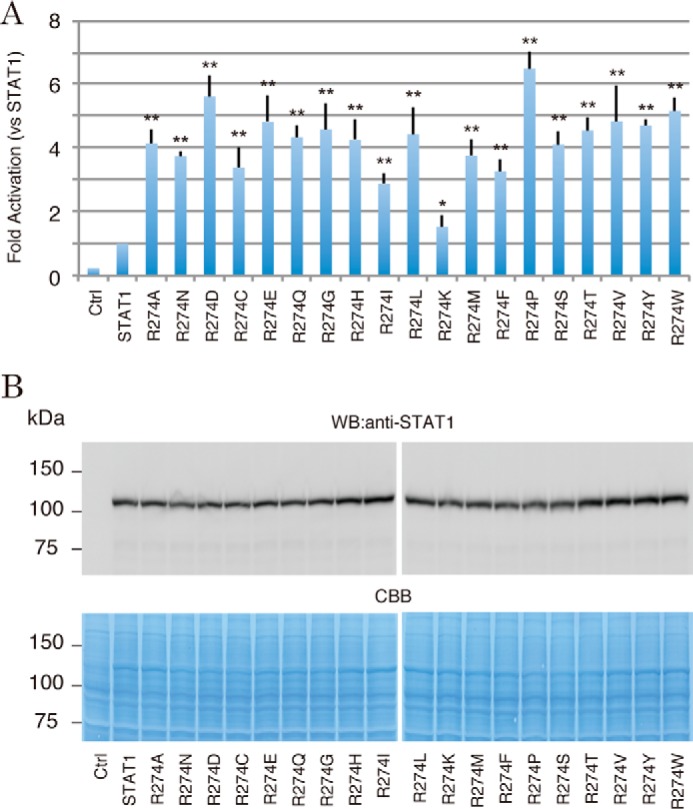
The transcriptional activities of the Arg-274 mutants with other amino acid residues. A, IFN-γ-stimulated transcriptional activities of a series of Arg-274 mutants were measured by the Dual-Luciferase reporter system. The RLU of the mutants were normalized to the value of wild-type STAT1 activity. The experiments shown in the lanes were performed in triplicate. Error bars, S.D. of each condition. Asterisks, significant differences of the mean values of the mutant RLU compared with the mean values of the STAT1 RLU. p values were determined with Student's test: *, p < 0.05; **, p < 0.01. B, protein levels of Arg-274 mutants were analyzed by Western blotting with anti-STAT1 antibody (top). The amount of the loaded protein (10 μg) was visualized by Coomassie Brilliant Blue (CBB) staining of the immunoblotted membrane (bottom).
It has been generally assumed that CMC-causing GOF mutations of STAT1 were coupled with a gain of Tyr-701 phosphorylation (7). We next asked whether the R274Q mutation influenced Tyr-701 phosphorylation in three cellular states that mimicked sequential cellular responses after IFN-γ stimulation (Fig. 3A) (i.e. in the absence of IFN-γ stimulation (basal), 2 h after IFN-γ treatment (stimulated), and 2 h after treatment of IFN-γ-prestimulated cells with staurosporine, an inhibitor of JAK (attenuated)). We assessed R274Q, wild-type STAT1, and an established Tyr-701 dephosphorylation mutant, F76A/L77A/F172W (13, 16–18). These proteins were expressed in HEK293 cells, and their phosphorylation levels were monitored by Western blotting. In the “basal” state, no phosphorylation was observed with R274Q, wild type, or F76A/L77A/F172W (Fig. 3B, lanes 2, 5, and 8). In the presence of IFN-γ (“stimulated” state), slightly greater phosphorylation of R274Q and F76A/L77A/F172W was detected than that of wild type (Fig. 3B, lanes 3, 6, 9). After the addition of staurosporine to the culture medium (“attenuated” state), three STAT1 constructions showed significantly different levels of phosphorylation from one another. Whereas phosphorylation of wild type was reduced back to the basal level (Fig. 3B, lanes 4), that of F76A/L77A/F172W was almost equal to the stimulated level (Fig. 3B, lanes 10). The dephosphorylation of R274Q was abrogated like F76A/L77A/F172W, but the extent was milder than F76A/L77A/F172W (Fig. 3B, lanes 7). Similarly, increased phosphorylation of R274Q was confirmed by detecting its increased dimer formation (Fig. 3C). Together, these data supported the idea that mutation of Arg-274 influenced a regulatory mechanism of Tyr-701 phosphorylation (7).
Figure 3.

Impacts of R274Q mutations on the IFN-γ-induced regulatory mechanisms of STAT1 phosphorylation and dimerization. A, time line and workflow of cell preparations for the experiments in B and C. γ, IFN-γ; Sp, staurosporine. B and C, Tyr-701 phosphorylation levels (B) and dimerization efficiencies (C) of STAT1 mutants. Phospho-Tyr-701 level (top) and the amount of loaded protein (bottom) in the cell lysates expressed with the indicated STAT1 constructs were visualized by Western blotting. The immunoprecipitated FLAG-tagged proteins and the bound Halo-tagged dimeric counterpart were visualized by STAT1 antibody (top). Expression levels of the FLAG-tagged proteins were visualized with anti-FLAG antibody (bottom). *, nonspecific band. HT, Halo-tagged STAT1; FLAG, FLAG-tagged STAT1; Endo, endogenous STAT1. D and E, time course of IFN-γ-induced phosphorylation (D) and staurosporine-induced dephosphorylation (E) of the R274Q mutant. The cells were recovered at the indicated time points and subjected to Western blotting with the indicated antibodies. F, transcriptional activities of the indicated mutants were measured by Dual-Luciferase reporter assays with the reporter driven by the IFN-γ-response element of IRF1. The RLU of the mutants were normalized against wild-type STAT1 activity. The experiments shown in the lanes were performed in triplicate. Error bars, S.D. of each condition. Asterisks, significant differences of the mean values of the mutant RLU compared with the mean values of the STAT1 RLU. p values were determined with Student's test: n.s., not significant; **, p < 0.01. HT, Halo-tagged STAT1; Endo and FLAG, endogenous and FLAG-tagged STAT1. *, nonspecific band.
Next, we inquired whether JAK and the Tyr-701 phosphatase were involved in this mechanism. First, we assessed the R274Q mutant for the kinetics of addition and removal of phospho-Tyr-701. Halo-tagged STAT1 and Halo-tagged R274Q were expressed in HEK293 cells, and they were recovered from the cells 15, 30, 60, and 120 min after treatment with IFN-γ or recovered from the IFN-γ-prestimulated cells 15, 30, 60, and 120 min after exposure to staurosporine. At every time point, phosphorylation of R274Q was greater than that of wild type (Fig. 3, D and E). Next, in a reporter assay, an additional R274Q mutation of the F76A/L77A/F172W dephosphorylation mutant failed to increase the transcriptional activity of F76A/L77A/F172W (Fig. 3F). Together, these results implied that R274Q-induced GOF was caused by reduced dephosphorylation, similar to that in an F76A/L77A/F172W dephosphorylation mutant, rather than by acceleration of JAK-mediated phosphorylation.
Molecular and structural bases of the aberrant increase in STAT1 activity by the R274Q mutation
It was reported that dephosphorylation of STAT1 proceeded through reorientation of parallel STAT1 dimer subunits to an anti-parallel form (13, 16–18). To evaluate whether the residues stabilizing this intermediate might contribute to the efficiency and kinetics of dephosphorylation, we next investigated the locations of Arg-274 and the other GOF residues in the structure of the anti-parallel STAT1 dimer. To visualize a dimeric assembly at the amino acid level, we made a map to illustrate the distances between two residues of each dimeric component (17) (Fig. 4A). This map demonstrated that there were seven major contact sites of anti-parallel subunits of the STAT1 dimer (i.e. 3 for CCD α1-DBD, 2 for CCD α3-DBD, and 2 for DBD-DBD interactions). Because most of the surface of GOF residues (like Gln-275, Lys-278, Glu-282, and Lys-286 (Fig. 1C)) was sufficiently close to contacting the DBD of the dimeric counterpart (Fig. 4B), their mutations would induce destabilization of the anti-parallel dimer. On the other hand, although the contact map showed Arg-274 similarly positioned near the dimer interface (Fig. 4A), it was not likely that Arg-274 participated in direct interaction with the dimeric counterpart, because the DBD from the same STAT1 molecule interfered (Fig. 4B). According to the intramolecule distance map of STAT1 (Fig. 4C), Arg-274 was covered by the residues on the loop between the β9 and β10 strands of the DBD (residues 437–445). More specifically, in an enlarged view of the crystal structure (Fig. 4D), Arg-274 was found close to the side chain of Gln-441 (7.56 Å) and main chains of Pro-442 (5.01 Å) and Gly-443 (4.90 Å). Because this loop was located on the interaction surface of the anti-parallel STAT1 dimer (red circle in Fig. 4A), an amino acid change of Arg-274 for the others might alter dimer stability through this DBD-DBD interaction. To investigate this idea, we asked whether a series of Ala mutations in the loop between the β9 and β10 strands of the DBD influenced STAT1 activity (Fig. 5A). As expected, most mutations showed GOF effects (Fig. 5B). The degrees of GOF were probably correlated with the distances between the mutated residues and the dimeric contact site (Fig. 4C), and they peaked with the Gln-441 residue. To confirm that the increased activity of the Arg-274 mutant was actually derived from impairment of the interaction with this loop structure, we measured transcriptional activities of mutants of Arg-274 and Gln-441, and their combinations. Although one point mutation of Q441R showed an increase in transcriptional activity (Fig. 5C), the increase of activity by the Q441R mutation, but not the Q441D mutation, was cancelled by an additional mutation, R274Q (Fig. 5D). This offset effect was not observed by swapping R274P and Q441R (Fig. 5E). Thus, the results suggested that the interaction between arginine and glutamine could be important in the regulation of STAT1 activity. Taken together, the data suggested that intramolecular bridging between Arg-274 of the CCD and Gln-441 of the DBD was required for maintaining the appropriate configuration of STAT1 and also suggested that the R274Q-induced transformation of this local structure probably impaired anti-parallel dimer formation and subsequent dephosphorylation of Tyr-701.
Figure 4.

Structural features of Arg-274 in an anti-parallel STAT1 dimer structure. A, interfaces of the anti-parallel conformation of the STAT1 dimer. The heat map depicts the distances from a residue of one component of the anti-parallel STAT1 dimer to that of the other. Possible contact sites of the dimer are marked with a red square or circle. All distances were calculated based on barycentric coordinates of the residues in an anti-parallel dimer structure (Protein Data Bank entry 1YVL). Scale is shown at the bottom. Amino acid number of the residues (right and bottom) and domain and secondary structure of the protein (left and top) are illustrated beside the heat map. Red arrowheads, position of CMC-related GOF mutations. B, locations of the GOF residues characterized in Fig. 1C are illustrated in an anti-parallel STAT1 structure. C, heat map illustrates the distance between two residues of the same STAT1 molecule. All distances were calculated as described in A by using the coordinates of a parallel dimer structure (Protein Data Bank entry 1BF5). A possible attachment site of CCD with DBD is circled in red. D, intramolecular interaction between Arg-274 and Gln-441. The structure surrounded by Arg-274 indicated by a dashed circle in B is enlarged. Each subunit of the dimer is shown as a ribbon diagram and protein surface, respectively. The domains are colored in gray (ND), green (CCD), cyan (DBD and LK), and dark blue (SH2). Distances between the indicated atoms were calculated by the measurement tool packaged in PyMOL.
Figure 5.

Interaction of Arg-274 with Gln-441 is indispensable for regulated STAT1 activity. A, sequence homology of the loop between β9 and β10 of STAT family proteins. Secondary structure elements surrounding the loop are illustrated above the alignment. The sequences were aligned with ClustalW. B–E, transcriptional activities of the indicated mutants were assessed by the Dual-Luciferase reporter system. The RLU of the mutants were normalized against the value of wild-type STAT1 activity (B and C), R274Q (D), or R274P (E). The experiments shown in the lanes were performed in triplicate. The error bars represent S.D. of each condition. Asterisks, significant differences of the mean values of the mutant RLU compared with the mean values of the STAT1 RLU. p values were determined with Student's test: n.s., not significant; **, p < 0.01.
Impact of an Arg-274 mutation on the genome-wide expression profile
In a previous report, a DNA-binding transcription factor, interferon regulatory factor 9 (IRF9), was found to interact with the CCD of STAT1, enabling it to modify the original function of STAT1 (12). In this scenario, pathogenic CCD mutations might not only influence innate STAT1 activity but also reconstitute the repertoire of STAT1 target genes by altering assembly of the transcription factor complexes (19). To examine this hypothesis, we performed RNA-seq analysis of R274Q-modulated transcription. First, either STAT1 or R274Q was expressed in the STAT1-deficient cells, and the cells were treated with IFN-γ. Then a series of RNA-seq libraries were prepared from the mRNA of the IFN-γ-stimulated cells, and they were subjected to massive parallel sequencing. We obtained an average of 23.2 million raw reads with an average quality score of 37.6, and an average of 90.3% of reads aligned with the human genome (supplemental Table S1A). Most reads were mapped in the CDS regions encoding registered RefSeq genes, whereas others were in intron or intergenic regions. The expression levels of individual RefSeq genes were calculated from the sequencing reads as the value of fragments per kilobase of exon per million reads (FPKM). As presented in a scatter plot depicting the log-transformed FPKM values of Ctrl, STAT1, and R274Q samples, each biological duplicate's transcription profile was well reproduced (Fig. 6A).
Figure 6.

Effect of the R274Q mutation on the IFN-γ-induced genome-wide gene expression profile. A and B, scatter plots depict different genome-wide expression profiles of experimental duplicates (A) or of the IFN-γ-stimulated U3C cells expressing the indicated STAT1 constructs (B). Each dot represents log-transformed FPKM values of RefSeq gene transcripts. The red dots indicate those transcripts with statistically significant differences between the x axis and the y axis libraries. The statistical relationship between the duplicates is indicated by Spearman's rank correlation coefficient ρ. The experiments were performed in duplicate. C, a Venn diagram demonstrates the number of RefSeq genes with statistically significant differences in comparison with the transcripts of the indicated libraries. The gene symbols are listed in supplemental Table S2. D, heat map presents expression profile of 76 of the R274Q-sensitive genes. Gene symbols are indicated (right).
Next, to investigate STAT1-modulated and R274Q-modulated genes, the FPKM values of each RefSeq gene assessed in the STAT1 sample or the R274Q sample were statistically compared with those in the Ctrl sample. The Cuffdiff2 tool calculated the change in expression of each RefSeq gene along with the statistical scores for these changes and then determined the RefSeq genes with “significant” changes based on p values less than the default false discovery rate (p < 0.05) after Benjamini-Hochberg correction for multiple testing (20). In a comparison of Ctrl and STAT1 samples, significant changes were found with 179 of the RefSeq genes in duplicate experiment 1 and 162 of the RefSeq genes in duplicate experiment 2. Among these, 134 genes were common in biological duplicates (Fig. 6B (top) and supplemental Table S2A). Similarly, comparing the FPKM values of Ctrl and R274Q samples, significant changes in expression were found with 266 and 259 of the RefSeq genes in duplicate 1 and 2 experiments, respectively, and 206 of these genes overlapped (Fig. 6B (middle) and supplemental Table S2B). Of note, a Venn diagram showed that among 206 of the significant R274Q genes, 76 did not overlap with 134 of the significant STAT1 genes (Fig. 6C). Although no statistically significant difference between Ctrl and STAT1 samples was detected with these 76 specific genes, STAT1 appeared to slightly change most of their expression levels (Fig. 6D). This implied that the R274Q mutation might simply promote expressions of STAT1 target genes. In fact, the genome-wide transcription profile of STAT1 and R274Q samples demonstrated a very similar trend (Fig. 6B, bottom).
Direct targets of R274Q
To investigate the behavior of wild-type STAT1 and R274Q mutants on endogenous STAT1 target genes, we attempted to obtain a list of STAT1 binding sites on the genome from ChIP-seq data registered in the gene expression omnibus (21) (GSE15353). As previously reported, 13,060 and 43,692 STAT1 peaks were found with the IFN-γ-unstimulated and IFN-γ-stimulated samples, respectively. Among 134 STAT1-modulated genes (Fig. 6C), 82 genes harbored IFN-γ-dependent STAT1 peaks within 0.5 kb of the transcription start sites (Fig. 7, A and B). This suggested that half of the modulated genes were direct targets and the others were secondary targets. Similarly, STAT1 accumulations were observed on R274Q-modulated gene promoters with approximately the same ratio (Fig. 7A, bottom). Furthermore, 76 R274Q-specific genes were slightly modified by STAT1 (Fig. 6D), and STAT1 occupied the promoters of these genes in an IFN-γ-dependent manner (Fig. 7B, bottom). Hence, these results supported the idea that 76 R274Q-specific genes might be potential targets of STAT1.
Figure 7.

Effect of the R274Q mutation on IFN-γ-dependent regulation of the STAT1 target gene. A, averaged ChIP signal of STAT1 was plotted for the promoters of the STAT1- or R274Q-regulated RefSeq genes detected in Fig. 6. A pie chart shows the number of RefSeq genes harboring STAT1 peaks within 1 kb of the transcription start site. TSS, transcription start site. B, heat map demonstrates the intensity of the STAT1 occupancy of the STAT1-regulated (top) or R274Q-specific (bottom) genes shown in Fig. 6C in the presence (middle) or the absence (left) of IFN-γ. -Fold changes of FPKM values of STAT1 relative to Ctrl are displayed on the right of the panel. C and D, enrichment of the reads of ChIP-seq for STAT1 (top) and RNA-seq of R274Q-dependent transcription (bottom) on the GBP2 gene (C) and IL6 gene (D). Genome position and exon-intron structure are indicated at the top and bottom, respectively. E, validations of IFN-γ-induced activation of IL6 (left) and GBP2 (right) by RT-qPCR. The Ct value in each condition is normalized against the expression levels of GAPDH. Error bars, S.D. of each condition.
To extend the genome-wide analysis, we further examined IFN-γ-dependent transcriptional regulation of GBP2 and IL6. We focused on these two genes because GBP2 and IL6 harbor STAT1 peaks (Fig. 7, C and D, top, respectively). Further, the Cuffdiff2 tool indicated that STAT1 activated the GBP2 gene by 4.89-fold (Fig. 7C, bottom) but had almost no effect on the IL6 gene (1.22-fold; Fig. 7D, bottom). In turn, R274Q activated the GBP2 and IL6 genes by 6.74- and 2.02-fold, respectively. To verify RNA-seq data by reverse transcription and quantitative real-time PCR, U3C cells were transfected with vectors encoding either STAT1 or the R274Q mutant, and a series of mRNAs were recovered from the cells 2 and 24 h after IFN-γ treatment. As expected, the GBP2 gene was induced by STAT1 and R274Q in a time-dependent fashion, and the -fold change under each condition was greater in R274Q-expressing cells. In turn, the IL6 gene was unexpectedly repressed 2 h after IFN-γ treatment regardless of STAT1 expression. However, in a similar RNA-seq result, R274Q induced a 2–3-fold increase in expression relative to STAT1 after exposure to IFN-γ. The TRANSFAC motif tool found putative IFN-γ-response elements at positions +369 (TTCCCACAA) and +132 (TTCCTCCAA) near the transcription start site of the GBP2 gene, but not in the IL6 gene promoter, implying that other DNA-binding factors could transiently regulate the IL6 gene before STAT1 recruitment. Together, these data reinforce the model that R274Q magnified expression of the large part of the STAT1 target genes but exhibited behavior on the promoter similar to that of wild type.
To further examine these conclusions in a physiological context, we performed RNA-seq analysis with T lymphocytes that were collected from a healthy individual and a CMC patient who carried the heterozygous R274Q mutation (Fig. 8A). A series of mRNAs was prepared from wild-type and R274Q CD4+ T cells after additional culture with or without IFN-γ treatment. We obtained an average of 8.22 million raw reads with an average quality score of 38.5. An average of 89.8% of the reads aligned with the human genome (supplemental Table S1B). In the patient's T cells, 48% of the STAT1 mRNA appeared to be expressed from the R274Q allele (Fig. 8B). The FPKM values of each RefSeq gene were calculated from the resultant sequencing reads. By differential analysis with the Cuffdiff2 tool, significant IFN-γ-dependent changes in transcription were detected on 103 RefSeq genes with wild-type T cells, whereas significant changes were detected in 147 RefSeq genes with R274Q T cells (supplemental Table S2C). As shown in a scatter plot depicting the log-transformed FPKM values of wild-type and R274Q T cells, expression of the most significant RefSeq genes was increased in an IFN-γ-dependent manner (Fig. 8C). Among 147 significant genes detected in R274Q T cells, 81 genes overlapped with the genes detected in wild-type T cells, and the residual 66 genes were detected only in R274Q T cells (Fig. 8D). Importantly, these 66 genes did not overlap with the 76 genes detected by the analysis with the U3C cells. This result suggested that the cell-specific promoter context (like nucleosome positioning and occupancy by other transcription factors) might influence the IFN-γ response of the genes. To assess IFN-γ-induced transcription of these R274Q-significant genes in wild-type T cells, their log-transformed FPKM values were highlighted in the scatter plot and histogram (Fig. 8E). Consistent with the results obtained with U3C cells (Fig. 6D), as well as the 81 overlapped genes (red), the 66 specific genes (pink) were slightly increased in response to IFN-γ stimulation (Fig. 8E, top). Taken together, these data indicated that the R274Q mutation increased transcriptional activity but would not dramatically change the repertoire of the STAT1 target genes.
Figure 8.

The IFN-γ-dependent gene expression profile in T cells. A, schematic diagram of RNA and NGS library preparation from T cells. B, visualization of expression from R274Q allele. The BAM file of the WT sample (top) and the R274Q sample (bottom) were loaded onto the Integrated Genome Viewer. C, scatter plots depict different genome-wide expression profiles of the WT sample (top) and the R274Q sample (bottom). Each dot represents log-transformed FPKM values of RefSeq gene transcripts. The red dots indicate those transcripts with statistically significant differences between the x axis and y axis libraries. D, a Venn diagram demonstrates the number of IFN-γ-dependent genes with statistically significant differences. The gene symbols are listed. E, expression profiles of the R274Q-regulated genes in the WT sample. In the scatter plot (top), each dot represents log-transformed FPKM values of RefSeq gene transcripts. The statistical significances of the colored dots are indicated in the panel. The histogram shows the distribution of the RefSeq genes with the indicated log-transformed FPKM values of the wild-type T cells stimulated with IFN-γ (bottom). Blue, density within 25,069 of the total registered RefSeq genes; red, density within 147 of the R274Q significant genes.
Discussion
The ready availability of next-generation sequencing technology has increased the use of genetic testing in the diagnosis of CMC disease. If the identified mutation were registered as a STAT1 GOF mutation in the disease-related variation databases (Fig. 1A and supplemental Table S3), the carrier patient could be easily diagnosed with CMC. However, the available databases have not covered all of the potential STAT1 GOF mutations. In this study, to help anticipate the pathogenic risks of an unregistered STAT1 missense mutation, we investigated the molecular and structural bases of STAT1 GOF by use of amino acid substitution. We focused on the mechanism of GOF mutation caused by alterations at the Arg-274 CMC-hot-spot residue (7). Substitution of the Arg-274 residue significantly increased STAT1 transcriptional activity (Fig. 2) and impeded dephosphorylation at phospho-Tyr-701 (Fig. 3). We further demonstrated that Arg-274 physically interacted with Gln-441 of the DBD of the same molecule (Fig. 4) and that this association could be disrupted by the pathogenic R274Q mutation (Fig. 5). Because Gln-441 constituted one of the seven contact sites of anti-parallel STAT1 subunit dimers (Fig. 4A), the R274Q mutation probably destabilizes and decreases the formation of this type of dimer.
Recent proposals have attempted to describe the molecular impact of removing phosphate from Tyr-701 in which SH2 interaction occurs (13). The two reciprocal Tyr(P)–SH2 bonds of phosphorylated STAT1 structurally support stable dimerization in parallel conformation, whereas the phosphorylation-independent ND-ND or CCD-DBD interaction constitutes an anti-parallel conformation. The anti-parallel form was evident in both in vivo and in vitro assessments. To permit exposure of phospho-Tyr-701 on the protein surface for access to phosphatase activity, reorientation of the STAT1 dimer from parallel to anti-parallel conformation has been proposed as a rate-limiting step for phosphatase activity. To support this possible dephosphorylation mechanism, many of the residues that were targeted by the reported CMC-pathogenic GOF mutations, but not the pathogenic LOF mutations, were also observed at the interaction surfaces of the anti-parallel dimer (7). Moreover, these pathogenic GOF mutations led to persistent Tyr-701 phosphorylation in vivo as well as partially decreased dephosphorylation by STAT1 phosphatase in vitro. Of note, the side chain of Arg-274 was not oriented to the interaction surface but instead toward the loop between β9 and β10 strands of the DBD in the same molecule. In this study, Ala mutations of most residues constituting this loop led to increases in STAT1 transcriptional activity. As for the crystallographic structure, the loop located on the protein surface was sufficiently close to the dimeric counterpart to stabilize the anti-parallel conformation. Thus, the increase in STAT1 transcription activity caused by the R274Q mutation evoked an alternative mechanism of STAT1 GOF. Unlike the GOF mutations of the residues exposed at the protein surface, R274Q substitution could not directly destabilize dimer formation. Rather, the substitution would alter the orientation of the loop involved in the anti-parallel dimer formation and indirectly modify the subsequent dephosphorylation process. Thus, our finding provides an additional destabilization mechanism of STAT1 GOF caused by CMC-associated mutation.
Based on the crystallographic structure of STAT1, the GOF residues of the α3 helix (Fig. 1C) were classified into at least three distinct groups (supplemental Fig. S3) (i.e. GOF residues on the protein surface, residues toward the inside of the CCD, and residues facing the DBD). The GOF residues in the α3 helix, such as Gln-275, Lys-278, Glu-282, and Lys-286 (Fig. 1C), appeared to belong to the first group, and these residues were located on the protein surface. Among the residues of the CCD targeted by CMC-related mutations, Asp-165, Asp-168, Asp-171, Phe-172, Asn-179, Lys-278, Gln-285, and Lys-286 probably corresponded to this category (supplemental Table S3). In the structure of the anti-parallel STAT1 dimer (supplemental Fig. S4A), the CMC-pathogenic GOF residues categorized in this group were clearly localized to the contact site of the DBD of its counterpart. This suggested that the wild-type residues would contribute to formation and stabilization of the anti-parallel dimer and help to facilitate the subsequent Tyr-701 dephosphorylation process. In fact, in addition to the GOF residues of the CCD, the DBD residues facing the α3 helix of the dimeric counterpart, such as Gly-384, Thr-385, and Lys-388, were also frequently identified as CMC pathogenic mutations (3, 4). Hence, missense mutations substituting Gln-275 or Glu-282, which were newly identified in this report as GOF residues and that surrounded the interface of the anti-parallel dimer, could give a high risk for onset and development of CMC disease.
Next, the side chains of the second GOF group were located toward the inside of the α-helical bundle core of the CCD. It is likely that these residues fasten the four helices and maintain a rigid coiled-coil stem. From the results of the reporter analysis (Fig. 1C), the identified GOF residues (Leu-259, Ser-269, Leu-283, Glu-284, Tyr-287, and Thr-288) possessed this type of structural feature (supplemental Fig. S3, B and C). Similarly, some residues registered in the CMC-related mutation database, like Ile-156, Leu-163, Gln-167, Tyr-170, Cys-174, Met-202, Arg-210, Glu-235 Val-266, Ala-267, Tyr-287, Thr-288 and Ile-294, share the structural property of this group (supplemental Fig. S4B). Therefore, the DNA sequences of the STAT1 gene encoding Leu-259, Ser-269, Leu-283, and Glu-284 would be potential sites yielding a CMC pathogenic mutation. Although the CMC-related Val-266 residues of the α3-helix appeared to belong to this category, our assessment failed to detect risks of substitutions of these residues (Fig. 1C). Because of the similarity of the chemical properties between Ala and a partially hydrophobic residue, like Cys, Tyr, Met, and Val, it was presumably difficult to detect increases in their transcriptional activities. To circumvent this technical limitation of our method, an approach using the 19 saturated mutants will be needed to fully cover the predictions of all of the potential GOF residues related to CMC.
Finally, the third class of GOF residues of the α3 helix was positioned at the bottom of a deep groove surrounded by the CCD and the DBD. As demonstrated in this study, it was assumed that these GOF residues, like Arg-274, were involved in maintenance of the STAT1 configuration via intramolecular association of the CCD and the DBD. Among the residues targeted by the CMC pathogenic mutations (supplemental Table S3), Gln-271 possessed this type of structural feature as well as Arg-274 (supplemental Fig. S4C). The side chain of Gln-271 of the CCD was sufficiently close to Glu-353 of the β3 sheet of the DBD (3.57 Å) for hydrophilic interaction (supplemental Fig. S5). Corresponding to the location of the Gln-441 residue, Glu-353 also constituted a contact interface of the other anti-parallel dimer subunit (red circle in Fig. 4A). This suggested that the interaction between Gln-271 and Glu-351 would be involved in stabilization of dimer formation and the regulation of dephosphorylation of phospho-Tyr-701. Of note, the Q271P mutation was registered as one of the CMC-causing mutations (7), implying that this pathogenic mutation of CCD might increase STAT1 activity by impairment of the intramolecular association with the DBD. However, the reporter analysis in Fig. 1C showed that the activity of the Q271A mutant was equivalent to wild-type activity. Based upon this this unexpected result, we speculated that the requirement at this site was not simply side chain size or hydrophobicity. For a more detailed examination of the effect of Gln-271 amino acid substitution, we constructed a series of Gln-271 mutants in which the original Gln residue was exchanged for the other 19 residues. These were expressed in cells, and the activities were measured (supplemental Fig. S6, A and B). Half of the Gln-271 mutants showed 1.5–3.0-fold increases in their transcriptional activities relative to that of wild type. Substitutions of the Gln-271 residue for negatively charged residues (Asp and Glu) induced significant STAT1 GOF. These results indicated that replacement of an electrostatically neutral Gln-271 residue by a negatively charged one generated electrostatic repulsion against Glu-353 and negated the interaction between the CCD and the DBD. Also, STAT1 GOF was observed when Gln-271 was replaced by a hydrophobic and bulky residue (Trp, Tyr, Phe, Leu, or Ile) or a residue that probably disrupted the α3-helix of CCD (Pro or Gly). Although the effects of the Gln-271 substitutions were less than those of the related Arg-274 substitutions (Fig. 2), it was assumed that the degree to which a mutant contributed to STAT1 GOF was significantly influenced by the chemical properties of the original residue. In summary, the Gln-271 residue was involved in the mechanism of interaction between the CCD and the DBD, and the defect in this Gln-271-mediated interaction was probably associated with the onset of CMC disease.
Generally, the in silico approach has been applied for the prediction of the functional effects of uncharacterized missense mutations. Many popular prediction tools, like SIFT, Polyphen, and PROVEN, utilize algorithms to score the position of the protein sequence based on its conservation throughout evolution and its similarity to homologous protein sequences. In this aspect, the prediction tools tend to give a higher score for a more conserved residue irrespective of its structural importance. In fact, SIFT gave a high score to the substitutions of the Arg-274 residue, which was highly conserved among both orthologous (STAT1s) and paralogous (STAT families) proteins (supplemental Figs. S1B and S6C). Although the roles of Gln-271 and Arg-274 in the structure formation and function of STAT1 were basically similar to one another, the scores for the substitutions of the less conserved Gln-271 residue gave relatively low estimates. Therefore, in comparison with the activities measured by a reporter assay, the predictive errors based on the calculated scores appeared to occur most often with substitutions of Gln-271, such as its substitution by a hydrophilic residue or a hydrophobic and bulky residue (supplemental Fig. S6C). These results showed that to accurately predict the risk of an uncharacterized STAT1 missense mutation, the structural and functional importance of the given residues should also be considered as well as their amino acid conservation in the STAT1 sequence. Taken together, we believe that the data in this study provide a partial framework to construct a predictive risk model for uncharacterized mutations involved in the onset of CMC disease.
Experimental procedures
Plasmid and site-directed mutagenesis
A vector for expressing halo-tagged STAT1 was obtained from the Kazusa cDNA/ORF clone collection (FHC013013). Multiple site-directed mutations were introduced into the plasmid by a PCR-based method with a series of relevant oligonucleotide DNAs. Five tandem elements that were derived from IRF1 (TTCCCCGAA) were inserted into multiple cloning sites of the pGL4.24 vector (Promega, Inc.). A TK/pRL internal control vector for the Dual-Luciferase system was purchased from Promega, Inc.
Cell culture and reagents
HEK293 (Riken BioResource Center) and U3C cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS). Cells were treated with recombinant human IFN-γ (PeproTech, Inc.) and staurosporine (Wako Chemicals, Inc.) at final concentrations of 10 ng/ml and 500 nm, respectively. The antibodies against STAT1 (C-24, Santa Cruz Biotechnology, Inc.) and phosphorylated STAT1 at Tyr-701 (M135, Abcam, Inc.) were used for Western blotting analysis at 1:500 and 1:1000 dilutions, respectively.
T-cell preparation and stimulation
Peripheral blood mononuclear cells from healthy individuals and a patient carrying a heterozygous GOF mutation in STAT1, R274Q, were separated by density gradient separation using Lymphoprep (Axis-Shield). The CD4+ T cells were then separated by IMagTM (557767, BD Biosciences) according to the manufacturer's protocol. The separated CD4+ T cells were suspended at a density of 106 cells/ml in RPMI1640 with 10% FBS. They were then cultured in the presence of IFN-γ (1,000 IU/ml) for 8 h. After cultivation, RNA was prepared from the stimulated cells and subjected to RNA-sequencing analysis.
Luciferase reporter assays
The vectors expressing a series of the STAT1 mutants were transfected along with a test reporter and TK-driven internal control reporter into exponentially growing U3C cells. After transfection (24 h), cells were treated with IFN-γ and further incubated for 24 h. Dual-Luciferase assays were performed according to the manufacturer's instructions (Promega). All results shown in the figures are displayed as the -fold change in relative luciferase units (RLU) of the mutants compared with wild-type STAT1. Each assay was performed in triplicate, in a single experiment.
Contact map preparation
The distance between two amino acids depicted in the contact map was calculated from centroid coordinates of the residues. Atomic coordinates for each atom of a residue were based on the values of 1BF5 (phosphorylated STAT1) and 1YVL (unphosphorylated STAT1) in the Protein Data Bank.
RNA-seq
Exponentially growing U3C cells were transfected with the STAT1 or R274Q expression vector. Following transfection (48 h), the cells were further cultured in the presence or the absence of IFN-γ for 24 h. RNA was obtained from the cells by using TRIzol reagent (Life Technologies, Inc.). A series of RNA-seq libraries was prepared from 3 μg of total RNA with a SureSelect Strand-Specific RNA Library Prep kit (Agilent Technologies, Inc.) according to the manufacturer's instructions. Each library was subject to Illumina Hiseq-2000, and ∼20,000,000 single reads of 50 bp were obtained. Each assay was performed in duplicate, in a single experiment.
Processing of RNA-seq and ChIP-seq data
For RNA-seq experiments, differential gene expression between the samples was analyzed with TopHat and Cufflinks. In brief, RNA-seq reads were quality-checked with ShortRead (version 1.23.12), aligned to the hg19 human genome with TopHat (version 2.0.12), and assembled into transcripts with Cufflinks (version 2.0.2). For identification of statistically significant differences between experimental conditions, the assembled data sets were subject to Cuffdiff (version 2.0.2) and visualized by plotting with CummeRbund (version 2.7.2).
To determine genome-wide STAT1 binding sites, ChIP-seq data were obtained from a GEO database (GSE15353) and processed with Bowtie and MACS. Briefly, ChIP-seq reads were mapped with Bowtie (version 0.12.9) by using alignment option “-n 3” and “-e 200”, and reporting option “-m 1” and “–best”. For peak calling, mapped reads were subject to MACS (version 1.4.1) in the default setting. The peaks were assigned to the genes by using the CreateRegulatoryDomains tool of GREAT (genomic regions enrichment of annotations tool). For this process, a range to survey the associated genes was set with “5 kb” upstream and “1 kb” downstream from the transcription start site and up to “10 kb” extension as distal regulatory regions. The average signal of ChIP enrichment surrounding the transcription start sites of the gene of interest was extracted by Sitepro (version 0.6.6) in the CEAS package (version 1.0.2). By using Cluster version 3.0, these ChIP signals surrounding the transcription start sites were organized by hierarchical clustering with a centroid linkage method.
Author contributions
R. F. performed all of the experimental work, analyzed data, and wrote the manuscript. A. H. and T. S. computed the effect of STAT1 mutations. M. K. contributed to patient care and T-cell preparation. O. O. and S. O. participated in detailed discussion of the study and designed and supervised the project.
Supplementary Material
Acknowledgments
We especially thank Dr. Jean-Laurent Casanova for providing U3C cells. We are also grateful to Masaki Takazawa for data processing, to Kazuhiro Sato for constructing a series of expression vectors of STAT1 mutants, and to Miyuki Tsumura and Shiho Nishimura for supporting RNA preparation from the patient's T cells.
This work was supported in part by research grants from the Platform for Drug Design, Informatics, and Structural Lifescience (PDIS) and by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development (AMED). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1–S3 and Figs. S1–S6.
- LOF
- loss-of-function
- GOF
- gain-of-function
- CMC
- chronic mucocutaneous candidiasis
- ND
- N-terminal domain
- CCD
- coiled-coil domain
- LK
- linker domain
- CTD
- C-terminal domain
- FPKM
- fragments per kilobase of exon per million reads
- RLU
- relative luciferase units
- Ctrl
- control
- RNA-seq and ChIP-seq
- RNA- and ChIP-sequencing, respectively.
References
- 1. Shuai K., and Liu B. (2003) Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 3, 900–911 [DOI] [PubMed] [Google Scholar]
- 2. Stark G. R., and Darnell J. E. Jr. (2012) The JAK-STAT pathway at twenty. Immunity 36, 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boisson-Dupuis S., Kong X. F., Okada S., Cypowyj S., Puel A., Abel L., and Casanova J. L. (2012) Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 24, 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Casanova J. L., Holland S. M., and Notarangelo L. D. (2012) Inborn errors of human JAKs and STATs. Immunity 36, 515–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chapgier A., Boisson-Dupuis S., Jouanguy E., Vogt G., Feinberg J., Prochnicka-Chalufour A., Casrouge A., Yang K., Soudais C., Fieschi C., Santos O. F., Bustamante J., Picard C., de Beaucoudrey L., Emile J. F., et al. (2006) Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dupuis S., Dargemont C., Fieschi C., Thomassin N., Rosenzweig S., Harris J., Holland S. M., Schreiber R. D., and Casanova J. L. (2001) Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 293, 300–303 [DOI] [PubMed] [Google Scholar]
- 7. Liu L., Okada S., Kong X. F., Kreins A. Y., Cypowyj S., Abhyankar A., Toubiana J., Itan Y., Audry M., Nitschke P., Masson C., Toth B., Flatot J., Migaud M., Chrabieh M., et al. (2011) Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 208, 1635–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Toubiana J., Okada S., Hiller J., Oleastro M., Lagos Gomez M., Aldave Becerra J. C., Ouachée-Chardin M., Fouyssac F., Girisha K. M., Etzioni A., Van Montfrans J., Camcioglu Y., Kerns L. A., Belohradsky B., et al. (2016) Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 127, 3154–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van de Veerdonk F. L., Plantinga T. S., Hoischen A., Smeekens S. P., Joosten L. A., Gilissen C., Arts P., Rosentul D. C., Carmichael A. J., Smits-van der Graaf C. A., Kullberg B. J., van der Meer J. W., Lilic D., Veltman J. A., and Netea M. G. (2011) STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 365, 54–61 [DOI] [PubMed] [Google Scholar]
- 10. Hoey T., and Schindler U. (1998) STAT structure and function in signaling. Curr. Opin. Genet. Dev. 8, 582–587 [DOI] [PubMed] [Google Scholar]
- 11. Chen X., Vinkemeier U., Zhao Y., Jeruzalmi D., Darnell J. E. Jr., and Kuriyan J. (1998) Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell 93, 827–839 [DOI] [PubMed] [Google Scholar]
- 12. Horvath C. M., Stark G. R., Kerr I. M., and Darnell J. E. Jr. (1996) Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex. Mol. Cell. Biol. 16, 6957–6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mertens C., Zhong M., Krishnaraj R., Zou W., Chen X., and Darnell J. E. Jr. (2006) Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev. 20, 3372–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Darnell J. E. Jr., Kerr I. M., and Stark G. R. (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421 [DOI] [PubMed] [Google Scholar]
- 15. Bonjardim C. A. (1998) JAK/STAT-deficient cell lines. Braz. J. Med. Biol. Res. 31, 1389–1395 [DOI] [PubMed] [Google Scholar]
- 16. Chen X., Bhandari R., Vinkemeier U., Van Den Akker F., Darnell J. E. Jr, and Kuriyan J. (2003) A reinterpretation of the dimerization interface of the N-terminal domains of STATs. Protein Sci. 12, 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao X., and Chen X. (2005) Crystallization and X-ray crystallographic analysis of human STAT1. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 666–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhong M., Henriksen M. A., Takeuchi K., Schaefer O., Liu B., ten Hoeve J., Ren Z., Mao X., Chen X., Shuai K., and Darnell J. E. Jr. (2005) Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc. Natl. Acad. Sci. U.S.A. 102, 3966–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hartman S. E., Bertone P., Nath A. K., Royce T. E., Gerstein M., Weissman S., and Snyder M. (2005) Global changes in STAT target selection and transcription regulation upon interferon treatments. Genes Dev. 19, 2953–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trapnell C., Hendrickson D. G., Sauvageau M., Goff L., Rinn J. L., and Pachter L. (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robertson G., Hirst M., Bainbridge M., Bilenky M., Zhao Y., Zeng T., Euskirchen G., Bernier B., Varhol R., Delaney A., Thiessen N., Griffith O. L., He A., Marra M., Snyder M., and Jones S. (2007) Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4, 651–657 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.