

Abstract
ADP-glucose pyrophosphorylase (AGPase) controls bacterial glycogen and plant starch biosynthetic pathways, the most common carbon storage polysaccharides in nature. AGPase activity is allosterically regulated by a series of metabolites in the energetic flux within the cell. Very recently, we reported the first crystal structures of the paradigmatic AGPase from Escherichia coli (EcAGPase) in complex with its preferred physiological negative and positive allosteric regulators, adenosine 5′-monophosphate (AMP) and fructose 1,6-bisphosphate (FBP), respectively. However, understanding the molecular mechanism by which AMP and FBP allosterically modulates EcAGPase enzymatic activity still remains enigmatic. Here we found that single point mutations of key residues in the AMP-binding site decrease its inhibitory effect but also clearly abolish the overall AMP-mediated stabilization effect in wild-type EcAGPase. Single point mutations of key residues for FBP binding did not revert the AMP-mediated stabilization. Strikingly, an EcAGPase-R130A mutant displayed a dramatic increase in activity when compared with wild-type EcAGPase, and this increase correlated with a significant increment of glycogen content in vivo. The crystal structure of EcAGPase-R130A revealed unprecedented conformational changes in structural elements involved in the allosteric signal transmission. Altogether, we propose a model in which the positive and negative energy reporters regulate AGPase catalytic activity via intra- and interprotomer cross-talk, with a “sensory motif” and two loops, RL1 and RL2, flanking the ATP-binding site playing a significant role. The information reported herein provides exciting possibilities for industrial/biotechnological applications.
Keywords: allosteric regulation, carbohydrate biosynthesis, crystal structure, enzyme mechanism, glycobiology, glycogen, starch
Introduction
Glycogen and starch are the major carbon and energy reserve polysaccharides in nature. Glycogen is a very large branched glucose homopolymer containing ∼90% α-(1→4)-glucoside linkages and 10% α-(1→6)-glucoside linkages (1, 2). Glycogen is found in archaea, heterotrophic bacteria, fungi, and higher eukaryotes, localizing as discrete cytoplasmic granules of <50 nm (3). Eukaryotes utilize UDP-glucose as the activated nucleotide donor for glycogen biosynthesis, whereas archaebacteria and bacteria have selected ADP-Glc, defining two different pathways with distinct regulatory mechanisms and rate-controlling steps (2, 4–7). In bacteria, the basic biosynthetic pathway of glycogen involves the action of three enzymes: ADP-glucose pyrophosphorylase (AGPase),3 glycogen synthase (GS), and branching enzyme (BE; Refs. 6 and 8). AGPase catalyzes the biosynthesis of the nucleotide sugar donor, ADP-Glc (Ref. 9; Fig. 1). The second step is catalyzed by GS, which transfers a glucose residue to the hydroxyl group at position 4 of a glucose residue located at the non-reducing end of glycogen to form an α-(1→4)-glucoside linkage (10–13). It is worth noting that in fungi and higher eukaryotes glycogenin-linked maltooligosaccharides provide the initial glycogen primer for further elongation by GS (14–16). In contrast, bacterial glycogen de novo synthesis seems not to require the presence of α-(1→4)-linked glucans, with GS catalyzing the initiation and elongation reactions (17–19). The third step is catalyzed by BE, which forms the α-(1→6) branch points in glycogen (8). Specifically, BE cleavage α-(1→4)-glucosidic linkage in glycogen, yielding a non-reducing end oligosaccharide chain, with subsequent attachment of the oligosaccharide to the α-(1→6) position of the polymer (20, 21). Glycogen degradation is carried out by glycogen phosphorylase (GP; Ref. 22), which functions as a depolymerizing enzyme, and the debranching enzyme that catalyzes the removal of α-(1→6)-linked ramifications (23).
Figure 1.

Chemical reaction catalyzed by EcAGPase. EcAGPase catalyzes the main regulatory step in bacterial glycogen. EcAGPase catalyzes the reaction between ATP and G1P in the presence of a divalent metal cation, Mg2+, to form ADP-Glc and pyrophosphate (26). The enzymatic reaction is reversible in vitro. However, the hydrolysis of inorganic pyrophosphate by inorganic pyrophosphatase results in an irreversible reaction in vivo in the direction of ADP-Glc biosynthesis (9). ADP-Glc biosynthesis and hydrolysis directions are shown as a full and dotted lines, respectively. The two major positive (FBP) and negative (AMP) allosteric regulators are shown. The biosynthesis of bacterial glycogen in E. coli, essentially involve the action of two other consecutive enzymes, the glycogen synthase and branching enzyme.
The main regulatory step in the bacterial glycogen biosynthetic pathway is carried out by AGPase (9, 24, 25). AGPase catalyzes a condensation reaction between ATP and α-d-glucose-1-phosphate (G1P) to produce ADP-Glc and pyrophosphate (Refs. 9 and 26; Fig. 1). Specifically, the oxygen on the phosphate group of G1P acts as a nucleophile attacking the α-PO4 group of the nucleoside triphosphate, leading to the liberation of pyrophosphate (PPi; 27). The reaction is held in the presence of the divalent metal cation Mg2+, which minimizes the charge repulsion between phosphate groups, inducing/favoring nucleophile activation (28). In addition, two positively charged residues polarize these groups, increasing the nucleophilic nature of the oxygen attacking the phosphor atom (29, 30). AGPase follows a sequential ordered bi-bi mechanism with ATP binding first followed by G1P and ordered release of pyrophosphate and ADP-Glc (31). The hydrolysis of pyrophosphate by the action of inorganic pyrophosphatases results in a global irreversible and energetically expensive reaction in vivo (32, 33). As a consequence, evolution led AGPase to acquire an exquisite allosteric regulation mechanism to control its enzymatic activity by essential metabolites in the energetic flux within the cell (31, 34, 35). AGPase activators are metabolites that reflect signals of high carbon and energy content of a particular bacteria or tissue, whereas inhibitors of the enzyme indicate low metabolic energy levels (9, 36). Based on the specific positive or negative allosteric regulators, AGPases have been classified into nine different classes (9, 34).
Bacterial AGPases are encoded by a single gene, giving rise to a native homotetrameric protein (α4) with a molecular mass of ∼200 kDa (9). In the case of the paradigmatic bacterial AGPase from Escherichia coli (EcAGPase), each protomer is encoded by a single gene (glgC) located inside an operon together with the genes that code for GS (glgA), GP (glgP), BE (glgB), and phosphoglucomutase (pgm; Refs. 37 and 38). To date, two crystal structures of bacterial AGPases have been reported, that of EcAGPase (Fig. 2A; Ref. 26) and Agrobacterium tumefaciens (AtAGPase; Ref. 39). Each protomer comprises two domains, the N-terminal glycosyltransferase-A like domain (GT-A-like), where the active site is located, and the C-terminal left-handed β-helix domain (LβH; Refs. 26 and 39–42). EcAGPase dimerizes by an end-to-end stacking of two β-helix domains, whereas the tetramer assembly is mainly mediated by the interaction of GT-A-like domains. The resulting architecture allows the protomers to communicate with each other, from which cooperativity emerges (26, 39, 43). Very recently, the crystal structures of the paradigmatic EcAGPase were solved in complex with its preferred physiological positive and negative allosteric regulators, fructose 1,6-bisphosphate (FBP) and adenosine 5′-monophosphate (AMP), respectively (Fig. 2A; Ref. 26), defining four common allosteric clefts between the GT-A-like and LβH domains of neighboring protomers in which both allosteric modulators binds to partially overlapping sites. This structural configuration of the EcAGPase regulatory site accounts for the fact that sensitivity to inhibition by AMP is modulated by the concentration of the activator FBP (24, 44). Specifically, each allosteric cleft is communicated with the corresponding active site of the same protomer through a region defined as the sensory motif (SM), a complex structural element constituted by the nucleotide-binding loop NBL, including a G-rich motif involved in ATP binding and a segment rich in short secondary structure elements (26). Altogether we proposed a model in which the binding of the positive and negative energy reporters regulate EcAGPase catalytic activity through the SM and two critical regulatory loops, RL1 and RL2, flanking the active binding site via intra-protomer interactions and inter-protomer cross-talk (26).
Figure 2.

Dissecting the structural determinants of EcAGPase regulation. A, ribbon representation of the overall homotetrameric structure of EcAGPase showing the location of the active and regulatory sites. AMP, FBP, ATP, and G1P molecules are shown in two protomers from different dimers of the EcAGPase homotetramer. Protomers A and C are shown in orange and yellow, respectively. The location of the ATP-binding site in EcAGPase was determined by structural superposition with the crystal structure of N-acetylglucosamine-1-phosphate uridyltransferase in complex with ATP (PDB code 4K6R) and that of AGPase from S. tuberosum in complex with ATP and ADPG (PDB code 1YP3). The G1P-binding site in EcAGPase was determined taking into account the location of the glucose moiety of sucrose in the EcAGPase-AMP-SUC complex (5L6V). B, thermal unfolding for the unliganded form of EcAGPase (green) and the EcAGPase-AMP (blue), EcAGPase-Adenine (black), EcAGPase-5RP (yellow), and EcAGPase-PO4 (red) complexes. C, thermal unfolding for the unliganded form of EcAGPase (green) and the EcAGPase-AMP (blue), EcAGPase-AMP-FBP (gray), and EcAGPase-AMP-F6P complexes.
Here we carefully explore the consequences of single point mutations in the allosteric cleft of EcAGPase in key residues involved in FBP and AMP binding, looking for changes in the allosteric properties, stabilization, and their impact in enzymatic activity. We explore how those mutations impact in the yield of glycogen in the physiological environment of the host. Finally, in combination with the crystal structure of an EcAGPase single point mutant, we provide important mechanistic insights into the regulatory mechanism by which EcAGPase modulates catalysis at the molecular level.
Results and discussion
Dissecting the structural determinants of EcAGPase allosteric regulation
The crystal structures of the EcAGPase-AMP-SUC (PDB code 5L6V) and EcAGPase-FBP (PDB code 5L6S) complexes revealed that the allosteric regulators AMP and FBP bind to a common regulatory cleft defined by the GT-A-like and LβH domains of neighboring protomers (26). AMP is deeply buried into the cleft, defined by (i) the N-terminal β2-β3 hairpin (residues 46–52), α5, and the connecting loop α2-α3 (residues 37–42) and (ii) the C-terminal α15 (residues 419–425) and the connecting loops β28-β29 (residues 384–388) and β25-β26 (residues 367–371; Figs. 2A and 3). In addition, AMP makes strong contacts with neighbor protomers of the homotetramer, leading to the stabilization of the quaternary structure of the enzyme. Specifically, the EcAGPase-AMP complex was ∼4.6 °C more stable than the unliganded form of the enzyme (26). In contrast, FBP is located in a more solvent-exposed environment, mainly comprised by the last C-terminal residues of the enzyme (residues 420–431; Ref. 26). Interestingly, the addition of FBP to the EcAGPase-AMP complex triggered a clear reduction in the apparent melting temperatures (Tm) values, indicating that FBP is able to compete with AMP and to modify the structural arrangement of the EcAGPase-AMP complex, leading to a less stable structure (26, 45). Supporting this notion, (i) the structural comparison of the EcAGPase-AMP-SUC and EcAGPase-FBP crystal structures revealed that the AMP- and FBP-binding sites partially overlap, and (ii) sensitivity to inhibition by AMP is modulated by the concentration of the activator FBP (34).
Figure 3.

Localization of residues in the active and regulatory sites in EcAGPase. A, EcAGPase protomer showing the GT-A-like and LβH domains. B, close view of the AMP regulatory site showing the location of key residues involved in AMP binding and its communication with the active site of EcAGPase. Selected/mutated residues are underlined. C, close view of the FBP regulatory site showing the location of key residues involved in FBP binding and its communication with the active site of EcAGPase. Selected/mutated residues are underlined.
To further advance the understanding of the molecular mechanism of EcAGPase allosteric regulation, we studied the contribution of a series of chemical derivatives of AMP and FBP to the stabilization of the enzyme. To this end, the ellipticity of EcAGPase in the presence of AMP, orthophosphate, d-ribose 5-phosphate, adenine, FBP, and fructose 6-phosphate (F6P) was monitored at 222 nm as a function of temperature. Upon the addition of AMP, the Tm value of EcAGPase increased from 65.6 °C to 73.6 °C (Fig. 2B). In contrast, the presence of orthophosphate, d-ribose 5-phosphate, and adenine did not significantly affect the Tm value of EcAGPase, with 69.0 °C, 69.7 °C, and 69.5 °C values, respectively (Fig. 2B). These experimental observations correlated well with the structural configuration of the AMP allosteric site as visualized in the EcAGPase-AMP-SUC crystal structure (Fig. 3; Ref. 26). Specifically, the α-PO4 of AMP localizes in a pocket composed of Arg-40, His-46, Arg-52, and Arg-386 of the same protomer, with no evident interactions with adjacent protomers of the homotetramer (Fig. 3B). Similarly, the d-ribose moiety of d-ribose 5-phosphate provides two additional interactions with Lys-39 and Thr-79, also located in the same protomer. Interestingly, the EcAGPase-AMP-SUC crystal structure revealed that the adenine heterocycle is stabilized by a strong stacking interaction with Arg-130 (α7) from the GT-A-like domain of a neighboring protomer (Figs. 2A and 3, A and B). However, the nucleobase alone did not provide any measurable stabilization effect.
Different moieties of the AMP chemical scaffold have been studied regarding their modulatory effects on EcAGPase activity. Orthophosphate was characterized as a weak inhibitor of EcAGPase, requiring a much higher concentration to achieve similar inhibitory levels than AMP (24). Most of the AGPase crystal structures reported to date revealed the presence of either orthophosphate or sulfate anions located in the corresponding binding pocket of the α-PO4 moiety of AMP in the regulatory site of EcAGPase (26, 39, 46). Interestingly, structural evidence also revealed the presence of (i) orthophosphate in the active site of EcAGPase (26) and (ii) sulfate, a phosphate mimic, in the active site of Solanum tuberosum AGPase (StAGPase), with the ATP substrate in a non-catalytically active conformation (PDB code 1YP3; Ref. 46). This structural information might suggest a competitive inhibitory effect of these anions with the EcAGPase substrates at high concentrations; however, an intra-protomeric inhibitory effect cannot be discarded. Finally, the presence of d-ribose 5-phosphate or adenine did not alter the activity of EcAGPase (47, 48). Altogether, these experimental observations indicate that the complete AMP scaffold is required for the stabilization of the enzyme, strongly supporting the notion that AMP inhibitory properties are inherently linked to the inter-protomer cross-talk in the negative allosteric mechanism of EcAGPase.
Finally, the addition of FBP to the EcAGPase-AMP complex produced a clear reduction in the Tm value, whereas the presence of F6P did not significantly affect the Tm value of the EcAGPase-AMP complex. Taking into consideration that F6P does not modulate EcAGPase activity (34) together with its inability to alter EcAGPase-AMP stability points toward the PO4 group at position 1 of fructose as a key player in enhancing the enzymatic activity to modulate AMP inhibition and to reverse AMP stabilization effect (Figs. 2B and 3C).
Design of EcAGPase single point mutants in the regulatory cleft and active site
We carefully explored the consequences of single point mutations in the allosteric cleft of EcAGPase in key residues involved in AMP and FBP binding, looking for changes in the allosteric properties, stabilization, and their impact in enzymatic activity (Fig. 3). Residues facing the allosteric cleft, Lys-39, Arg-40, His-46, Arg-130, Arg-386, Arg-419, and Arg-423, were selected and replaced by alanine. Residues Lys-39, Arg-40, His-46, and Arg-130 belong to the N-terminal GT-A-like domain, whereas Arg-386, Arg-419, and Arg-423 are located in the LβH domain (Figs. 2A and 3). Specifically, Lys-39, Arg-40, and His-46 are located in the SM motif, Arg-386 are in the β25-β26 loop of the LβH domain, and residues Arg-419 and Arg-423 are in the C-terminal α15 (26). The crystal structure of the EcAGPase-AMP-SUC revealed that (i) the α-PO4 group of the negative regulator AMP interacts with Arg-40, His-46, and Arg-386, and (ii) the d-ribose moiety is at a van der Waals distance of Lys-39. In addition, the adenine heterocycle is stabilized by a strong stacking interaction with Arg-130 and additional van der Waals interactions with Arg-419 and Arg-386 (Fig. 3B; Ref. 26). Based on the EcAGPase-FBP crystal structure, residues Lys-39, Arg-419, and Arg-423 of the same protomer interact with the PO4 group at position 6 of FBP; meanwhile Arg-130 in the neighbor protomer appears at salt-bridge distance. It is worth noting that Lys-39 proved to be essential for the FBP-mediated enzymatic activation to take place (44).
Single point mutants of the predicted catalytic residues, Lys-42 and Lys-195, were constructed by replacing them by alanine (30, 49, 50). The predicted catalytic residue Lys-42 is located in a key region of the SM motif, at very close distance of several residues participating in both AMP and FBP regulators interactions, in the regulatory cleft. Moreover, the side chain of Lys-42 is in close contact with two aspartic residues, Asp-142 and Asp-276 (26, 39, 51, 52), which were suggested to participate in the interaction of the divalent metal cation Mg2+ during catalysis, as observed in other nucleotidyltransferases (28, 53). Finally, Lys-195 is located in the β12-β13 loop of the sugar-binding pocket, in the active site of the GT-A-like domain, far apart from the allosteric cleft. Lys-195 was proposed to interact with the β-PO4 group of ADP-Glc (26, 46). It is worth noting that no EcAGPase constructs encode additional amino acids when compared with the native enzyme. They were expressed and purified to apparent homogeneity following three main steps including anionic exchange, ammonium sulfate precipitation, and hydrophobic interaction criteria (see “Experimental procedures” for details).
Single point mutants localized in the regulatory cleft impact EcAGPase stabilization
The addition of AMP did not modify the Tm values of the EcAGPase-R40A, EcAGPase-R46A, EcAGPase-R130A, and EcAGPase-R386A mutants (Fig. 4), strongly supporting a role of these residues in AMP binding, as visualized in the crystal structure of the EcAGPase-AMP-SUC complex (Figs. 3 and 4). In contrast, the addition of AMP to the EcAGPase-K39A, EcAGPase-R419A, and EcAGPase-R423A mutants, which are involved in FBP recognition, according to the EcAGPase-FBP crystal structure, triggered a clear increment in the Tm values as observed in the wild-type enzyme (26). As expected, the stabilization of EcAGPase-K42A and EcAGPase-K195A mutants, the predicted catalytic residues, was not affected by the addition of the negative regulator. Interestingly, the addition of FBP to the EcAGPase-K39A and EcAGPase-R419A was unable to revert the stabilization of the enzyme variants mediated by AMP. In contrast, some reversion was observed in EcAGPase-R423A (Fig. 5; Ref. 26). As expected, EcAGPase-K195A displays a similar behavior. However, the Tm value of the EcAGPase-K42A mutant in the presence of AMP or AMP/FBP regulators remained equally stable. Because the side chain of Lys-42 is in close contact with two aspartic residues, Asp-142 and Asp-276 (26, 39, 51, 52), this phenomenon might reflect the communication between the allosteric and active sites.
Figure 4.

Thermal unfolding transitions of EcAGPase and selected EcAGPase variants in complex with AMP. Thermal unfolding transitions were recorded at 222 nm between 20 °C and 90 °C. Tm value for the apo state of EcAGPase and selected EcAGPase variants are shown in green. AMP concentrations are shown in black.
Figure 5.
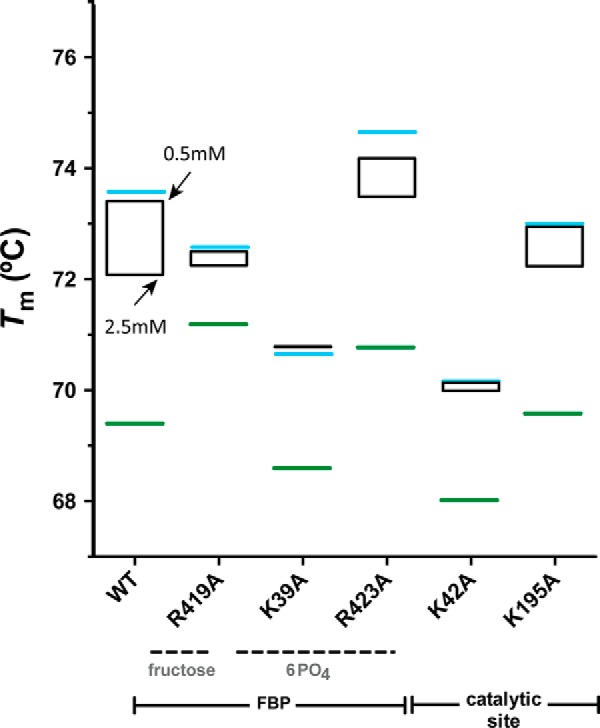
Thermal unfolding transitions of EcAGPase and selected single point mutants in the presence of AMP and FBP. Thermal unfolding transitions were recorded at 222 nm between 20 °C and 90 °C. Tm value for the apo state of EcAGPase and selected EcAGPase variants are shown in green. The Tm values of EcAGPase and selected EcAGPase variants in complex with AMP at 1 mm are shown in blue. FBP concentrations are shown in black.
EcAGPase-R130A deregulates AMP-mediated inhibition of the enzymatic activity, inducing the overproduction of glycogen in vivo
To characterize the impact of the single point mutations on the allosteric properties of EcAGPase, we measured their specific activities by the colorimetric end-point malachite green phosphate assay. EcAGPase-R130A doubled the specific activity compared with the wild-type EcAGPase in the absence of allosteric regulators (Fig. 6). As expected, wild-type EcAGPase is highly activated by FBP, but in contrast, most mutants failed to reach similar levels of activity compared with the EcAGPase-FBP active state (Fig. 6). Interestingly, EcAGPase-R130A was the only mutant activated by FBP, consistent with the requirement of an intra-protomeric signaling for the activation of the enzyme. Nevertheless, its activity did not reach the same levels of the wild type, which could be attributable to the lack of additional mechanism to reach full activation, the interprotomeric cross-talk.
Figure 6.

Activity measurements of EcAGPase and selected EcAGPase variants. The enzymatic activity of EcAGPase and selected EcAGPase variants was measured in the absence of allosteric regulators (A), in the presence of FBP (B), in the presence of AMP (C), and in the presence of both positive and negative regulators FBP and AMP (see “Experimental procedures” for details). The activity values for EcAGPase (green) and EcAGPase-FBP (orange) are shown as lines as a reference.
In the presence of AMP, EcAGPase-R40A, EcAGPase-R40E, and EcAGPase-H46A mutants (the mutated residues interact with the AMP α-PO4) along with the EcAGPase-R130A and EcAGPase-R386A mutants (the mutated residues interact with the adenine heterocycle and d-ribose moieties of AMP, respectively) displayed a similar behavior as that observed for the wild-type enzyme. Specifically, a partial inhibition was shown in all cases, pointing to a compensatory effect of the mutated residues. No inhibition was detected in the EcAGPase-K39A and EcAGPase-R419A mutants (the mutated residues interact with the d-ribose and adenine heterocycle moieties of AMP, respectively). The EcAGPase-R423A displayed behavior similar to the wild-type enzyme, which is consistent with the lack of interactions between this residue and AMP. The effect of the negative regulator AMP was also addressed in the presence of FBP, as sensitivity to inhibition by AMP is modulated by the concentration of the activator FBP (Fig. 6; Refs. 24, 34, and 44). Strikingly, EcAGPase-R130A displayed (i) a similar specific activity than in the presence of FBP and (ii) 7-fold higher than the wild-type enzyme in the presence of both AMP and FBP regulators, revealing a deregulation of the inactivation mediated by AMP for this mutant. Interestingly, the EcAGPase-K39A and EcAGPase-R419A mutants were unable to completely revert the slight activation induced by FBP in the presence of AMP, which is consistent with the results observed in the thermal unfolding transitions in the presence of both modulators (Fig. 5). As expected, EcAGPase-K42A and EcAGPase-K195A mutants, the predicted catalytic residues, were inactive in all conditions tested.
To study the impact of the single point mutations in the in vivo glycogen content of E. coli, the E. coli K-12 ΔglgC pTARA strain was transformed with pET22b-EcAGPase or the corresponding mutants (see “Experimental procedures” for details). The cultures were synchronized, and aliquots were removed every hour before and after the induction for the determination of glycogen content. The growth curves of the differentEcAGPase mutants confirmed that they were in the exponential growth phase in all cases (Fig. 7A). After glycogen extraction, a standard anthrone assay was used for the detection of sugars (Fig. 7B). As a complementary approach, a less sensitive but more specific iodimetric method was also used to measure glycogen content (Fig. 7C). As expected, the EcAGPase-K42A and EcAGPase-K195A mutants carrying out the predicted catalytic residues were unable to accumulate glycogen. Strikingly, EcAGPase-R130A mutant displayed an overproduction of glycogen in vivo, which correlates with the specific activities measured for this variant in vitro, supporting the notion of a hyperactive deregulated enzyme. Interestingly, EcAGPase-R40E mutant displayed a glycogen content similar to that observed with EcAGPase-R130A, although its specific activities did not show clear evidences of being a highly active enzyme. It is worth noting that EcAGPase is mainly activated by FBP; however, it can be also activated by other glycolytic intermediates (34, 54). The change in the charge of EcAGPase-R40E in the regulatory cleft might, therefore, modify the effect of other regulators in vivo, highlighting the complexity of the allosteric mechanism of this key enzyme.
Figure 7.
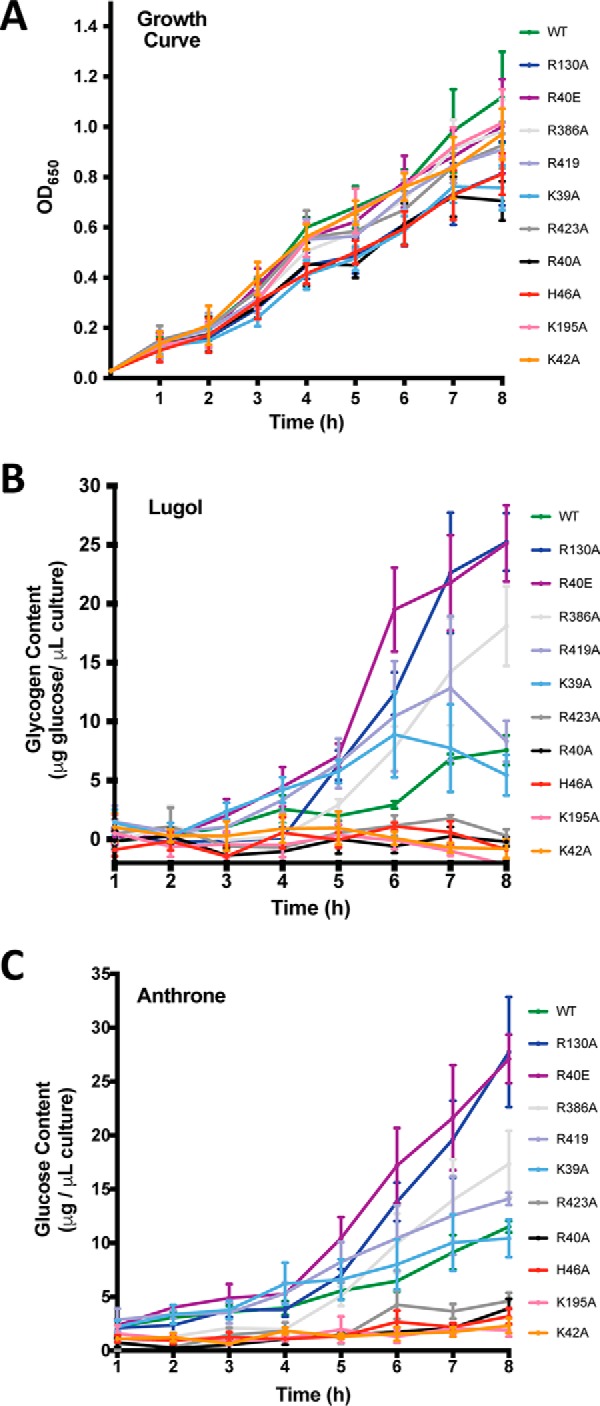
Glycogen content in vivo. A, growth curve. B, glycogen content by using Lugol method (71). C, glycogen content by using anthrone method (72, 82).
The crystal structure of EcAGPase-R130A reveals unprecedented conformational changes in key structural elements involved in EcAGPase allosteric regulation
The crystal structure of EcAGPase-R130A was solved in its unliganded form at 3.09 Å resolution (PDB code 5MNI; supplemental Table S1). EcAGPase-R130A crystallized in space group P 21 with eight molecules in the asymmetric unit, representing two homotetramers of the enzyme. When compared with the EcAGPase-AMP-SUC and EcAGPase-FBP complexes (26), the crystal structure of EcAGPase-R130A revealed significant conformational changes. First, the two dimers of the homotetramer were reoriented, strongly suggesting conformational flexibility of the quaternary structure of EcAGPase (Fig. 8, A and B; supplemental Fig. S1). Moreover, conformational differences were also observed in key elements implicated in the proposed allosteric regulatory mechanism (26). Specifically, residues 26–41 of the SM motif displayed a new extended conformation, partially overlapping the α-PO4- and d-ribose-binding sites of AMP, observed in the EcAGPase-AMP-SUC structure (Fig. 8C; supplemental Fig. S1). The side chain of Arg-419 makes hydrogen-bonding interactions with the main chain carbonyl group of Leu-34 and the side chain of Asn-38. Interestingly, in the other seven protomers this region of the SM was found partially disordered. The EcAGPase-AMP-SUC and EcAGPase-FBP crystal structures revealed that both AMP- and FBP-binding sites partially overlap, pointing to an important role of dynamics and conformational changes of several structural elements located in the regulatory cleft in the signal transduction mechanism. The replacement of Arg-130 by alanine certainly affected the transduction of the negative regulatory signal to the active site, likely due to an interprotomer cross-talk mechanism, as suggested by the EcAGPase-AMP-SUC crystal structure. However, the absence of Arg-130 might also modify the rearrangement/dynamics of other structural elements located in the regulatory site of EcAGPase, most notably the SM motif, contributing to the activation/cooperativity of the enzyme.
Figure 8.
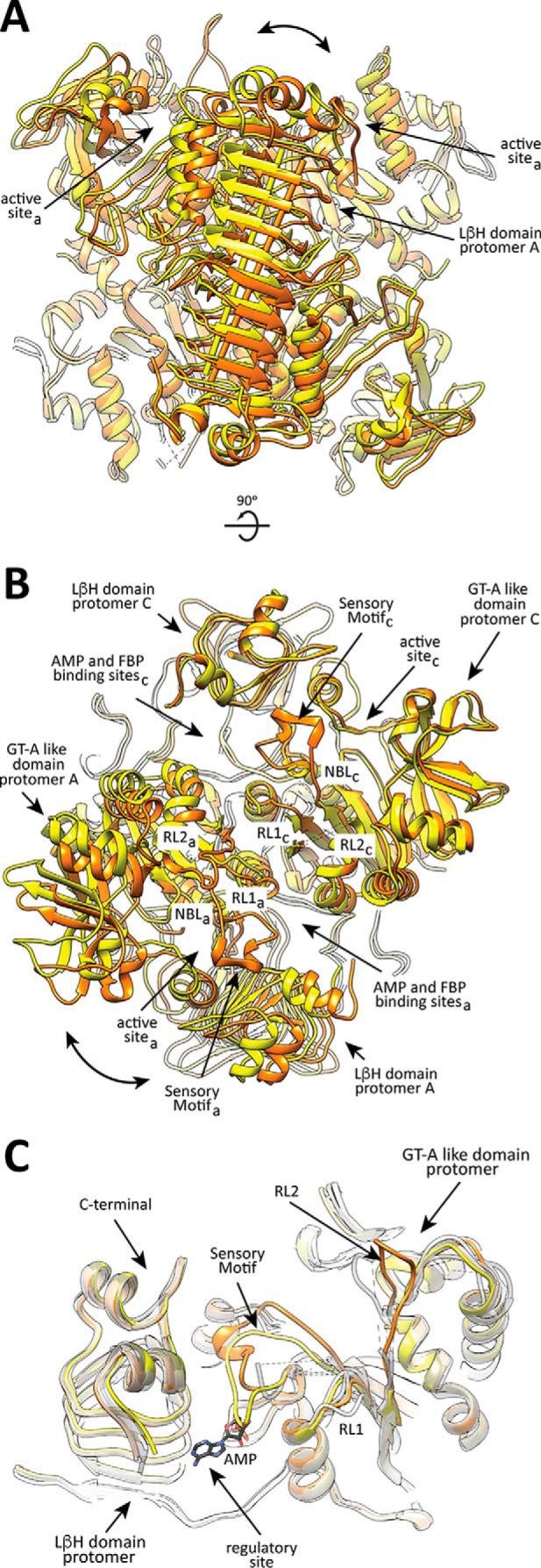
The crystal structure of EcAGPase-R130A. A and B, two views of the structural superposition between the EcAGPase-R130A (yellow) and EcAGPase-FBP (orange) tetramers, based on the alignment of LβH domains pairs (arrow). The relative rotation of both domains is shown (curved arrow). C, EcAGPase-R130A structure (protomer D: yellow) centered at the sensory motif, superimposed with an EcAGPase-AMP-SUC protomer (orange), revealing the prominent conformational changes leading the SM motif to partially occupy the AMP-binding site. The other EcAGPase-R130A protomers of the asymmetric unit appear superposed in gray scale, showing variability in the RL1, RL2 regions and the G-rich loop.
Similarly, the RL1 loop, which is in close contact with the nucleotide-binding loop (NBL) (residues 26–33) including the GGXGXR consensus sequence involved in ATP binding, was also observed to adopt different structural arrangements. Interestingly, the replacement of the highly conserved Tyr-75 per alanine resulted in an inactive enzyme, suggesting a functional role for this residue in the modulation of the enzymatic activity, as reported for the Q74A mutant (45). Finally, RL2 could only be observed/partially modeled in a different conformation with respect to the EcAGPase-AMP-SUC and EcAGPase-FBP structures. Taken together, the new crystal structure of EcAGPase-R130A provides evidence of conformational changes including local flexibility in key elements proposed to be involved in allosteric communication.
EcAGPase shares common sequence signatures for allosteric regulator binding with other enterobacterial AGPases
Multiple primary structure alignment among members of the Enterobacteriaceae family of AGPases revealed Lys-42 and Lys-195, the proposed catalytic residues, along with Arg-40, His-46, Arg-386, and Arg-130, residues involved in AMP binding, highly preserved (Fig. 9). Some experimental data indicated that the C-terminal region of EcAGPase is involved in the recognition of the positive regulator and allosteric activation mechanism of the enzyme (54). Specifically, (i) the 419RXMLRKLXXKQER431 sequence and (ii) the key residue Lys-39, which were observed to interact with the positive regulator FBP in the EcAGPase-FBP complex, are strictly conserved among enterobacterial AGPases known to use FBP as a positive regulator (26, 34, 54). Based on primary structure alignment, enterobacteria families that have not been studied in terms of their allosteric activators and conserve this C-terminal region are predicted to also be regulated by FBP (Shigella flexneri, Shigella boydii, Salmonella choleraesuis, Escherichia fergusonii; Fig. 9). Interestingly, some enterobacterial AGPases not regulated by FBP (34, 54) displayed clear discrepancies in the C-terminal region (Serratia marcescens, Serratia liquefaciens, Hafnia alvei), strongly suggesting that other enterobacteria AGPases lacking this region might have a different positive regulatory mechanism (Yersinia pestis, Pantoea vagans, Erwinia sp., Pectobacterium atrosepticum, Dickeya dadantii, Proteus vulgaris; Figs. 9 and 10).
Figure 9.

Multiple sequence alignment of selected regions among members of the Enterobacteriaceae family of AGPases. Shown are E. coli (UniProt code P0A6V1), Salmonella typhimurium (UniProt code P05415); Enterobacter aerogenes (GenbankTM code A0A0H3FKN1); Enterobacter cloacae (UniProt code G8LJ14); Shigella dysenteriae (UniProt code E7SR47); Klebsiella pneumoniae (UniProt code A6TF49); Citrobacter freundii (GenbankTM code A0A023V613); S. flexneri (UniProt code P0A6V4); S. boydii (UniProt code Q31VJ3); S. choleraesuis (UniProt code Q57IU0); E. fergusonii (UniProt code B7LSE1); S. liquefaciens (GenbankTM code A0A0X8SJ44); S. marcescens (GenbankTM code A0A0P8VY88); H. alvei (GenbankTM code A0A097R790); Cronobacter sakazakii (UniProt code A7MGF4); Y. pestis (UniProt code Q1C1E1); P. vagans (UniProt code E1SGX9); Erwinia sp. (UniProt code E3DJ85); P. atrosepticum (UniProt code Q6CZK2); D. dadantii (UniProt code D2BY77); P. vulgaris (GenbankTM code A0A0G4Q122).
Cell energy metabolism is inherently linked to the evolution of organisms; thus, enterobacterial glycogen synthesis ought to be studied in this context. Based on our observations, AGPase seems to follow the same phylogenetic history of the Enterobacteriaceae family, as described in the Pathosystems Resource Integration Center (55). In this regard, the conserved use of FBP as positive allosteric regulator in EcAGPase seems to be a trait acquired by a subgroup of enterobacterial AGPases sharing the C-terminal 419RXMLRKLXXKQER431 sequence (supplemental Fig. S2).
Conclusion
Allosteric proteins are biological control systems and are considered one of the most elaborate products of molecular evolution (56). A convergence evolution in allosteric molecular mechanisms and symmetry in quaternary architectures have been observed in unrelated proteins. Early studies pointed out homotropic cooperative effect characteristics compatible with Monod-Wyman-Changeaux model of EcAGPase (24). Interestingly, other enzymes of the central energy metabolism display similarities at structural and regulatory levels to EcAGPase, like pyruvate kinase, lactate dehydrogenase (LDH), and glycerol kinase (57, 58, 59). These enzymes are homotetramers with D2 symmetry that binds FBP as allosteric effector in some organisms. As for EcAGPase, in all these enzymes, the allosteric phenomenon was proposed to occur after the Monod-Wyman-Changeaux model (60), where the symmetry of the tetrameric system must be preserved, and the catalytic properties are explained by conformational displacement between the so called “tense” low-activity/affinity state (T-state) and a “relaxed” high-activity/affinity state (R-state) in a concerted fashion among protomers. These structurally distinct conformational states had been structurally observed bound to modulators (61, 62). It is worth noting that although pyruvate kinase and LDH use FBP as positive allosteric modulators, glycerol kinase is inhibited by this metabolite. Altogether, the experimental data suggest that the crystal structure of EcAGPase-AMP-SUC very likely represents the conformational state of the T-state. The allosteric charge cleft is conserved among AGPases providing the place of interaction of different negatively charged modulators. The geometry of the ligands and their specific interaction with the charged side chains might result in differential tensions across the structure, which leads ultimately to local rearrangements and global conformational changes. In this regard, parallelism can be drawn for EcAGPase-R130A compared with LDH, where reduction in positive charge in the tetramer interface deregulates a bacterial LDH and stabilizes its tetrameric structure (63).
Finally, the transformation of plants with E. coli allosteric mutants on the glgC gene significantly increased (i) the rate of starch synthesis in tubers of transgenic potato (64) and (ii) starch content (65, 66). Therefore, the information reported herein provides exciting possibilities for industrial/biotechnological applications.
Experimental procedures
Materials
The pET22b-EcAGPaseK39A, pET22b-EcAGPaseR40A, pET22b-EcAGPaseR40E, pET22b-EcAGPaseK42A, pET22b-EcAGPaseH46A, pET22b-EcAGPaseR130A, pET22b-EcAGPaseK195A, pET22b-EcAGPaseR386A, pET22b-EcAGPaseR419A, and pET22b-EcAGPaseR423A plasmids carrying out the EcAGPase mutants were synthesized/sequenced by ATG:biosynthetics (Merzhausen, Germany) using the pET22b-EcAGPase clone as template (see “Materials” for EcAGPase cloning) and further expressed and purified to apparent homogeneity as described for the recombinant EcAGPase enzyme.
EcAGPase cloning, expression, and purification
The full-length glgC gene from E. coli BL21 was amplified by standard PCR using oligonucleotide primers glgC_NdeI_Fwd (5′-GGGAATTCCATATGGTTAGTT-3′) and glgC_XhoI_Rev (5′-CCGCTCGAGTCATCGCTCCTG-3), Phusion DNA Polymerase (New England BioLabs), and purified genomic DNA as the template. The PCR fragment was digested with NdeI and XhoI and purified by agarose gel electrophoresis. The fragment was ligated to the expression vector pET22b (Novagen) using T4 DNA ligase, generating pET22b-EcAGPase. The recombinant EcAGPase has no additional amino acids when compared with the native enzyme. E. coli BL21(DE3) cells transformed with pET22b-EcAGPase or the corresponding mutants were grown in 2000 ml of LB medium (5 g of yeast extract, 10 g of peptone Tryptone, 10 g of NaCl) supplemented with 100 μg/ml carbenicillin at 37 °C. When the culture reached an A600 of 0.8, EcAGPase expression was induced by the addition of 1 mm isopropyl β-thiogalactopyranoside and further incubated at 18 °C for 20 h. Cells were harvested by centrifugation at 5000 × g and resuspended in 40 ml of 50 mm Hepes, pH 8.0, 5 mm MgCl2, 0.1 mm EDTA, 10% sucrose (w/v; solution A) containing protease inhibitors (Complete EDTA-free; Roche Applied Science) and 10 mg/liter of lysozyme (Sigma). Cells were then disrupted by sonication (24 cycles of 10 s each) and centrifuged for 20 min at 20,000 × g. The supernatant was dialyzed twice against solution A by using a 100,000-Da molecular mass cutoff dialysis membrane. The solution was then applied to a Q-Sepharose packed column (30 ml; GE Healthcare) equilibrated with solution A. Elution was performed with a linear 0–0.5 m NaCl gradient in 100 ml. Enzymatically active fractions were pooled and incubated with 1.2 m ammonium sulfate final concentration (solution B). The resultant suspension was centrifuged for 20 min at 32,000 × g, and the supernatant was applied into a Phenyl Shodex HIC PH-814 column equilibrated in solution B. The enzyme was eluted with a linear gradient of 100% solution B to 100% solution A in 50 ml. The protein was dialyzed against solution A with a 100-kDa molecular mass cutoff overnight and stored at −80 °C. All the mutants were purified following the same protocol.
Thermal unfolding
Thermal unfolding transitions were recorded on a J-815 CD spectropolarimeter (Jasco Corp., Tokio, Japan) equipped with a water-cooled Peltier unit. Measurements were carried out at 222 nm by using Hellma 110-QS quartz cuvettes with a 1-mm optical path length. Samples were 2 μm EcAGPase or EcAGPase mutants in 50 mm Tris-HCl, pH 8.0, 100 mm NaCl. Thermal dependences of the ellipticity were monitored in a range from 30 to 90 °C at 222 nm. Temperature was increased stepwise by 1 °C/min. Ligands effects were assessed in the same conditions for the following concentrations: AMP, 0.25 mm, 0.5 mm and 1 mm; FBP 0.5 mm and 2.5 mm. Transitions were normalized and tentatively fitted according to Equation 1 to obtain the apparent melting temperature (Tm) (67):
| (Eq. 1) |
where y represents the observed CD signal at 222 nm, yf and yu are the y axis intercepts, and mf and mu the slopes of the pre- and post-transition baselines, respectively, T is the temperature in K, Tm is the melting temperature, and ΔHm is the enthalpy change of unfolding at Tm. Curve fitting was performed with KaleidaGraph 4.5.2 (Synergy Software).
EcAGPase enzymatic assay
The enzymatic activity of EcAGPase and EcAGPase mutants was monitored using a microplate colorimetric end-point malachite green phosphate assay (68). EcAGPase catalyzes the reaction of ATP and G1P to produce ADP-Glc and PPi (26). This reaction is coupled with an inorganic pyrophosphatase, which catalyzes the hydrolysis of PPi to orthophosphate (Pi). Pi dosage is determined by the formation of a phosphomolybdate-malachite green complex: 1) enzymatic-coupled reactions,
| (Eq. 2) |
where α-glucose-1P is α-d-glucose-1-phosphate, and
| (Eq. 3) |
2) stop reaction by complexation of Mg2+ with EDTA; 3) molybdate/malachite green reactions,
| (Eq. 4) |
and
| (Eq. 5) |
and 4) the addition of sodium citrate for reaction stabilization. Specifically, samples contained 41.1 μm (8 μg/ml) of EcAGPase and 14.1 μm (1 μg/ml) pyrophosphatase (Sigma), 0.5 mm ATP, 0.5 mm G1P, 50 mm Tris-HCl pH 7.5, 2 mm MgCl2, and 100 mm NaCl with or without the addition of the allosteric regulators AMP, FBP, or AMP/FBP at 0.5 mm in a final volume of 60 μl. Reactions were incubated at 20 °C and stopped at a given time by the addition of 10 μl of 50 mm EDTA, pH 8.0. Afterward, 20 μl of molybdate solution (34 mm ammonium molybdate tetrahydrate in 4n HCl; Fluka) were added to the reaction mixture and incubated at 20 °C for 3 min. Then, 60 μl of malachite green solution (1 mm malachite green oxalate salt in water; Sigma) were added to the mixture and incubated at 25 °C for 5 min. Finally, the solution was stabilized by the addition of 60 μl of sodium citrate (170 mm in water; Sigma) and measured at 620 nm in a Spectra Max M2 plate reader. Measurements were performed in quintuplicate. EcAGPase and EcAGPase mutants were stored in 5.1 μm (1 mg/ml) aliquots in 50 mm Tris-Cl, pH 7.5, and 100 mm NaCl at −80 °C for single use. Specific activities were stable during a period.
Overexpression of EcAGPase mutants in a glgC knock-out E. coli strain
E. coli strain K-12 carrying out a glgC gene deletion (E. coli K-12 ΔglgC; kanamycin-resistant strain; Keio collection; Dharmacon GE) was transformed with pTARA plasmid (chloramphenicol resistant plasmid; Addgene) to provide a controlled expression of T7 RNA polymerase. The resulting strain was subsequently transformed with pET22b-EcAGPase, pET22b-EcAGPaseK39A, pET22b-EcAGPaseR40A, pET22b-EcAGPaseR40E, pET22b-EcAGPaseK42A, pET22b-EcAGPaseH46A, pET22b-EcAGPaseR130A, pET22b-EcAGPaseK195A, pET22b-EcAGPaseR386A, pET22b-EcAGPaseR419A, or pET22b-EcAGPaseR423A plasmid carrying the wild-type EcAGPase or the corresponding mutants. E. coli K-12 ΔglgC cells transformed with pTARA and pET22b-EcAGPase or the corresponding mutants were further synchronized in 10 ml of LB medium supplemented with 34 μg/ml chloramphenicol, 25 μg/ml kanamycin, and 100 μg/ml carbenicillin at 30 °C (69). After 2 passages, bacterial cultures were diluted 1:50. Aliquots of 900 μl were removed every hour and kept on ice. After 4 h, protein expression was induced by the addition of 1 mm isopropyl β-thiogalactopyranoside and 100 μg/ml arabinose. Aliquots of 900 μl were removed every hour and kept on ice for subsequent steps.
Glycogen extraction
Glycogen was extracted as previously described with minor modifications (70). Culture aliquots of 400 μl of the E. coli K-12 ΔglgC strain transformed with pTARA and pET22b-EcAGPase or the corresponding mutants were centrifuged at 1200 × g for 10 min, and the supernatant was discarded. Pellets were resuspended in 100 μl of 8.9 m (50%) KOH and further incubated at 95 °C for 30 min. 300 μl of ice-cold 95% ethanol was added to the sample and centrifuged at 1200 × g for 30 min. Pellets were air-dried and stored at −20 °C.
Glycogen measurement
The production of glycogen in the E. coli K-12 ΔglgC strain transformed with pTARA and pET22b-EcAGPase or the corresponding mutants was determined by two colorimetric methods (71–72). Glycogen pellets were resuspended in 100 μl of water by vigorous shaking for 5 min followed by the addition of 200 μl of 10.3 mm (0.2%) anthrone (Sigma) in 95% H2SO4. Samples were then incubated at 95 °C for 20 min. Aliquots of 200 μl were transferred to a 96-well plate and measured at 650 nm using a Spectra Max M2 plate reader. Glucose standards were treated in a similar manner in parallel. Because anthrone is a general reagent for the detection of sugars, a second less sensitive but specific assay was used. Glycogen pellets were washed with 20 μl of saturated NH4Cl and dry-heated at 95 °C to remove the excess ammonia. 200 μl of freshly prepared iodine reagent, a modified Lugol's iodine staining, obtained by mixing 20 μl of 102.4 mm (2.6%) I2 and 1.6 m (26%) KI in water with 5 ml of saturated CaCl2, were added. Samples were transferred to a 96-well plate and measured at 450 nm using a Spectra Max M2 plate reader. Measurements were performed in 4 and 5 replicates for the iodimetric and anthrone methods, respectively.
EcAGPase crystallization and data collection
Crystallization trials were carried out in sitting-drop 96-well plates by using a mosquito crystal robot (TTP Labtech). Crystals of EcAGPase-R130A were obtained by mixing 0.25 μl of EcAGPase-R130A at 6.3 mg/ml in 50 mm Tris-HCl, pH 7.5, 100 mm NaCl with 0.25 μl of mother liquor containing 14% polyethylene glycol 3.350, 140 mm magnesium formate, 30% ethylene glycol. Crystals grew in 13 days and were directly frozen under liquid nitrogen. A complete dataset of EcAGPase-R130A was collected at I04 beamline (Diamond Light Source (DLS), Didcot, Oxfordshire, UK) with an oscillation angle of 0.15° for a total of 1200 images using a Pilatus 6M-F detector. The EcAGPase-R130 form crystallized in the P 21 space group with 8 molecules in the asymmetric unit and diffracted to a maximum resolution of 3.09 Å (supplemental Table S1).
EcAGPase structure determination and refinement
The crystal structure of EcAGPase-R130A was solved by molecular replacement with the program Phaser (73) using a tetramer from the crystal structure of EcAGPase-AMP-SUC, PDB atomic coordinates 5L6V (26) as the search model. The final structure was obtained using alternate cycles of manual model-building using COOT (74) and Phenix (phenix.refine; Ref. 75) or Refmac5 (76). NCS (non-crystallographic symmetry) restraints were calculated automatically during refinement, and differences between chains were subsequently modeled. During the refinement the structure geometry was validated using Molprobity (77). The root mean square coordinates error (Å) was from a Luzzati plot (78, 79) using SFCHECK (80). Atomic coordinates and structure factors have been deposited with the Protein Data Bank, accession code 5MNI (EcAGPase-R130A). Molecular graphics and structural analyses were performed with the UCSF Chimera package (81).
Enterobacterial AGPases alignment
A representative group of enterobacterial AGPase protein sequences from different species was obtained from UniProt database and sequences aligned using Clustal-Omega. Middle distance BLOSUM62 tree was performed using Jalview.
Author contributions
N. C., J. O. C., M. E. G., conceived the project. N. C., J. O. C., A. M., A. O., and A. E. performed the experiments. N. C., J. O. C., and M. E. G., analyzed the results. N. C., J. O. C., and M. E. G. wrote the manuscript.
Supplementary Material
Acknowledgments
We acknowledge Diamond Light Source (DLS) (beamline I04 and I04–1 under proposals 8302 and 10130) and the French National Synchrotron SOLEIL. Access to structural biology facilities was supported in part by the EU FP7 infrastructure grant BIOSTRUCT-X (contract 283570). We also thank all members of the Structural Glycobiology Group for valuable scientific discussions.
This work was supported by European Commission Contract HEALTH-F3-2011-260872, Spanish Ministry of Economy and Competitiveness Contract BIO2013-49022-C2–2-R, and the Basque Government (to M. E. G.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Table S1 and Figs. S1 and S2.
The atomic coordinates and structure factors (code 5MNI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- AGPase
- ADP-glucose pyrophosphorylase
- EcAGPase
- AGPase from E. coli
- GS
- glycogen synthase
- BE
- branching enzyme
- GP
- glycogen phosphorylase
- G1P
- α-d-glucose-1-phosphate
- GT-A-like
- glycosyltransferase-A like domain
- LβH
- left-handed β-helix domain
- FBP
- fructose 1,6-bisphosphate
- SM
- sensory motif
- F6P
- fructose 6-phosphate
- LDH
- lactate dehydrogenase
- SUC
- sucrose.
References
- 1. Ball S., Guan H. P., James M., Myers A., Keeling P., Mouille G., Buléon A., Colonna P., and Preiss J. (1996) From glycogen to amylopectin: a model for the biogenesis of the plant starch granule. Cell 86, 349–352 [DOI] [PubMed] [Google Scholar]
- 2. Roach P. J., Depaoli-Roach A. A., Hurley T. D., and Tagliabracci V. S. (2012) Glycogen and its metabolism: some new developments and old themes. Biochem. J. 441, 763–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manners D. J. (1991) Recent developments in our understanding of glycogen structure. Carbohydr. Polym. 16, 37–82 [Google Scholar]
- 4. Leloir L. F., and Cardini C. E. (1957) Biosynthesis of glycogen from uridine diphosphate glucose. J. Am. Chem. Soc. 79, 6340–6341 [Google Scholar]
- 5. Recondo E., and Leloir L. F. (1961) Adenosine diphosphate glucose and starch synthesis. Biochem. Biophys. Res. Commun. 6, 85–88 [DOI] [PubMed] [Google Scholar]
- 6. Preiss J. (1984) Bacterial glycogen synthesis and its regulation. Annu. Rev. Microbiol. 38, 419–458 [DOI] [PubMed] [Google Scholar]
- 7. Ball S. G., and Morell M. K. (2003) From bacterial glycogen to starch: understanding the biogenesis of the plant starch granule. Annu. Rev. Plant Biol. 54, 207–233 [DOI] [PubMed] [Google Scholar]
- 8. Preiss J. (2014) Glycogen: biosynthesis and regulation. EcoSal Plus 6, 1–28 [DOI] [PubMed] [Google Scholar]
- 9. Ballicora M. A., Iglesias A. A., and Preiss J. (2003) ADP-glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol. Mol. Biol. Rev. 67, 213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guerin M. E., Buschiazzo A., Ugalde J. E., Ugalde R. A., and Alzari P. M. (2003) Preliminary crystallographic studies of glycogen synthase from Agrobacterium tumefaciens. Acta Crystallogr. D Biol. Crystallogr. 59, 526–528 [DOI] [PubMed] [Google Scholar]
- 11. Buschiazzo A., Ugalde J. E., Guerin M. E., Shepard W., Ugalde R. A., and Alzari P. M. (2004) Crystal structure of glycogen synthase: homologous enzymes catalyze glycogen synthesis and degradation. EMBO J. 23, 3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horcajada C., Guinovart J. J., Fita I., and Ferrer J. C. (2006) Crystal structure of an archaeal glycogen synthase: Insights into oligomerization and substrate binding of eukaryotic glycogen synthases. J. Biol. Chem. 281, 2923–2931 [DOI] [PubMed] [Google Scholar]
- 13. Sheng F., Jia X., Yep A., Preiss J., and Geiger J. H. (2009) The crystal structures of the open and catalytically competent closed conformation of Escherichia coli glycogen synthase. J. Biol. Chem. 284, 17796–17807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gibbons B. J., Roach P. J., and Hurley T. D. (2002) Crystal structure of the autocatalytic initiator of glycogen biosynthesis, glycogenin. J. Mol. Biol. 319, 463–477 [DOI] [PubMed] [Google Scholar]
- 15. Chaikuad A., Froese D. S., Berridge G., von Delft F., Oppermann U., and Yue W. W. (2011) Conformational plasticity of glycogenin and its maltosaccharide substrate during glycogen biogenesis. Proc. Natl. Acad. Sci. 108, 21028–21033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zeqiraj E., Tang X., Hunter R. W., García-Rocha M., Judd A., Deak M., von Wilamowitz-Moellendorff A., Kurinov I., Guinovart J. J., Tyers M., Sakamoto K., and Sicheri F. (2014) Structural basis for the recruitment of glycogen synthase by glycogenin. Proc. Natl. Acad. Sci. U.S.A. 111, E2831–E2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ugalde J. E., Parodi A. J., and Ugalde R. A. (2003) De novo synthesis of bacterial glycogen: Agrobacterium tumefaciens glycogen synthase is involved in glucan initiation and elongation. Proc. Natl. Acad. Sci. U.S.A. 100, 10659–10663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Díaz A., Martínez-Pons C., Fita I., Ferrer J. C., and Guinovart J. J. (2011) Processivity and subcellular localization of glycogen synthase depend on a non-catalytic high affinity glycogen-binding site. J. Biol. Chem. 286, 18505–18514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Díaz A., Díaz-Lobo M., Grados E., Guinovart J. J., Fita I., and Ferrer J. C. (2012) Lyase activity of glycogen synthase: Is an elimination/addition mechanism a possible reaction pathway for retaining glycosyltransferases? IUBMB Life 64, 649–658 [DOI] [PubMed] [Google Scholar]
- 20. Abad M. C., Binderup K., Rios-Steiner J., Arni R. K., Preiss J., and Geiger J. H. (2002) The x-ray crystallographic structure of Escherichia coli branching enzyme. J. Biol. Chem. 277, 42164–42170 [DOI] [PubMed] [Google Scholar]
- 21. Feng L., Fawaz R., Hovde S., Gilbert L., Chiou J., and Geiger J. H. (2015) Crystal structures of Escherichia coli branching enzyme in complex with linear oligosaccharides. Biochemistry 54, 6207–6218 [DOI] [PubMed] [Google Scholar]
- 22. Watson K. A., McCleverty C., Geremia S., Cottaz S., Driguez H., and Johnson L. N. (1999) Phosphorylase recognition and phosphorolysis of its oligosaccharide substrate: answers to a long outstanding question. EMBO J. 18, 4619–4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Møller M. S., Henriksen A., and Svensson B. (2016) Structure and function of α-glucan debranching enzymes. Cell. Mol. Life Sci. 73, 2619–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gentner N., and Preiss J. (1967) Activator-inhibitor interactions in the adenosine diphosphate glucose pyrophosphorylase of Escherichia coli B. Biochem. Biophys. Res. Commun. 27, 417–423 [DOI] [PubMed] [Google Scholar]
- 25. Preiss J., Yung S. G., and Baecker P. A. (1983) Regulation of bacterial glycogen synthesis. Mol. Cell. Biochem. 57, 61–80 [DOI] [PubMed] [Google Scholar]
- 26. Cifuente J. O., Comino N., Madariaga-Marcos J., López-Fernández S., García-Alija M., Agirre J., Albesa-Jové D., and Guerin M. E. (2016) Structural basis of glycogen biosynthesis regulation in bacteria. Structure 24, 1613–1622 [DOI] [PubMed] [Google Scholar]
- 27. Blankenfeldt W., Asuncion M., Lam J. S., and Naismith J. H. (2000) The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA). EMBO J. 19, 6652–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Swift R. V., Ong C. D., and Amaro R. E. (2012) Magnesium-induced nucleophile activation in the guanylyltransferase mRNA capping enzyme. Biochemistry 51, 10236–10243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vithani N., Bais V., and Prakash B. (2014) GlmU (N-acetylglucosamine-1-phosphate uridyltransferase) bound to three magnesium ions and ATP at the active site. Acta Crystallogr. F Struct. Biol. Commun. 70, 703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Führing J., Cramer J. T., Routier F. H., Lamerz A. C., Baruch P., Gerardy-Schahn R., and Fedorov R. (2013) Catalytic mechanism and allosteric regulation of UDP-glucose pyrophosphorylase from Leishmania major. ACS Catal. 3, 2976–2985 [Google Scholar]
- 31. Paule M. R., and Preiss J. (1971) Biosynthesis of Bacterial Glycogen. X. The kinetic mechanism of adenosine diphosphoglucose pyrophosphorylase from Rhodospirillum rubrum. J. Biol. Chem. 246, 4602–4609 [Google Scholar]
- 32. Kornberg A. (1962) On the metabolic significance of phosphorolytic and pyrophosphorolytic reactions. in Horizons in Biochemistry (Kasha H., and Pullman P., eds) pp. 251–264, Academic Press, New York, NY [Google Scholar]
- 33. Lahti R. (1983) Microbial inorganic pyrophosphatases. Microbiol. Rev. 47, 169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Preiss J. (1978) Regulation of adenosine diphosphate glucose pyrophosphorylase. Adv. Enzymol. Relat. Areas Mol. Biol. 46, 317–381 [DOI] [PubMed] [Google Scholar]
- 35. Meyer C. R., Yirsa J., Gott B., and Preiss J. (1998) A kinetic study of site-directed mutants of Escherichia coli ADP-glucose pyrophosphorylase: the role of residue 295 in allosteric regulation. Arch. Biochem. Biophys. 352, 247–254 [DOI] [PubMed] [Google Scholar]
- 36. Ball S., Colleoni C., Cenci U., Raj J. N., and Tirtiaux C. (2011) The evolution of glycogen and starch metabolism in eukaryotes gives molecular clues to understand the establishment of plastid endosymbiosis. J. Exp. Bot. 62, 1775–1801 [DOI] [PubMed] [Google Scholar]
- 37. Ugalde J. E., Lepek V., Uttaro A., Estrella J., Iglesias A., and Ugalde R. A. (1998) Gene organization and transcription analysis of the Agrobacterium tumefaciens glycogen (glg) operon: two transcripts for the single phosphoglucomutase gene. J. Bacteriol. 180, 6557–6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Preiss J., and Romeo T. (1989) Physiology, biochemistry, and genetics of bacterial glycogen synthesis. Adv Microb Physiol 30, 183–238 [DOI] [PubMed] [Google Scholar]
- 39. Cupp-Vickery J. R., Igarashi R. Y., Perez M., Poland M., and Meyer C. R. (2008) Structural analysis of ADP-glucose pyrophosphorylase from the bacterium Agrobacterium tumefaciens. Biochemistry 47, 4439–4451 [DOI] [PubMed] [Google Scholar]
- 40. Albesa-Jové D., and Guerin M. E. (2016) The conformational plasticity of glycosyltransferases. Curr. Opin. Struct. Biol. 40, 23–32 [DOI] [PubMed] [Google Scholar]
- 41. Albesa-Jové D., Giganti D., Jackson M., Alzari P. M., and Guerin M. E. (2014) Structure-function relationships of membrane-associated GT-B glycosyltransferases. Glycobiology 24, 108–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bejar C. M., Ballicora M. A., Gómez-Casati D. F., Iglesias A. A., and Preiss J. (2004) The ADP-glucose pyrophosphorylase from Escherichia coli comprises two tightly bound distinct domains. FEBS Lett. 573, 99–104 [DOI] [PubMed] [Google Scholar]
- 43. Perutz M. F. (1989) Mechanisms of cooperativity and allosteric regulation in proteins. Q. Rev. Biophys. 22, 139–237 [DOI] [PubMed] [Google Scholar]
- 44. Gardiol A., and Preiss J. (1990) Escherichia coli E-39 ADP glucose synthetase has different activation kinetics from the wild-type allosteric enzyme. Arch. Biochem. Biophys. 280, 175–180 [DOI] [PubMed] [Google Scholar]
- 45. Figueroa C. M., Esper M. C., Bertolo A., Demonte A. M., Aleanzi M., Iglesias A. A., and Ballicora M. A. (2011) Understanding the allosteric trigger for the fructose 1,6-bisphosphate regulation of the ADP-glucose pyrophosphorylase from Escherichia coli. Biochimie 93, 1816–1823 [DOI] [PubMed] [Google Scholar]
- 46. Jin X., Ballicora M. A., Preiss J., and Geiger J. H. (2005) Crystal structure of potato tuber ADP-glucose pyrophosphorylase. EMBO J. 24, 694–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Preiss J., Shen L., and Partridge M. (1965) The activation of Escherichia coli ADP-glucose pyrophosphorylase. Biochem. Biophys. Res. Commun. 18, 180–185 [DOI] [PubMed] [Google Scholar]
- 48. Ribéreau-Gayon G., Sabraw A., Lammel C., and Preiss J. (1971) Biosynthesis of bacterial glycogen IX: regulatory properties of the adenosine diphosphate glucose pyrophosphorylases of the Enterobactericeae. Arch. Biochem. Biophys. 142, 675–692 [DOI] [PubMed] [Google Scholar]
- 49. Hill M. A., Kaufmann K., Otero J., and Preiss J. (1991) Biosynthesis of bacterial glycogen: mutagenesis of a catalytic site residue of ADP-glucose pyrophosphorylase from Escherichia coli. J. Biol. Chem. 266, 12455–12460 [PubMed] [Google Scholar]
- 50. Ballicora M. A., Dubay J. R., Devillers C. H., and Preiss J. (2005) Resurrecting the ancestral enzymatic role of a modulatory subunit. J. Biol. Chem. 280, 10189–10195 [DOI] [PubMed] [Google Scholar]
- 51. Frueauf J. B., Ballicora M. A., and Preiss J. (2001) Aspartate residue 142 is important for catalysis by ADP-glucose pyrophosphorylase from Escherichia coli. J. Biol. Chem. 276, 46319–46325 [DOI] [PubMed] [Google Scholar]
- 52. Bejar C. M., Jin X., Ballicora M. A., and Preiss J. (2006) Molecular architecture of the glucose 1-phosphate site in ADP-glucose pyrophosphorylases. J. Biol. Chem. 281, 40473–40484 [DOI] [PubMed] [Google Scholar]
- 53. Steitz T. A., Smerdon S. J., Jäger J., and Joyce C. M. (1994) A unified polymerase mechanism for nonhomologous DNA and RNA polymerases. Science 266, 2022–2025 [DOI] [PubMed] [Google Scholar]
- 54. Ballicora M. A., Sesma J. I., Iglesias A. A., and Preiss J. (2002) Characterization of chimeric ADPglucose pyrophosphorylases of Escherichia coli and Agrobacterium tumefaciens: importance of the C terminus on the selectivity for allosteric regulators. Biochemistry 41, 9431–9437 [DOI] [PubMed] [Google Scholar]
- 55. Wattam A. R., Abraham D., Dalay O., Disz T. L., Driscoll T., Gabbard J. L., Gillespie J. J., Gough R., Hix D., Kenyon R., Machi D., Mao C., Nordberg E. K., Olson R., Overbeek R., Pusch G. D., et al. (2014) PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Monod J., Changeaux J. P., and Jacob F. (1963) Allosteric proteins and cellular control systems. J. Mol. Biol. 6, 306–329 [DOI] [PubMed] [Google Scholar]
- 57. Fushinobu S., Kamata K., Iwata S., Sakai H., Ohta T., and Matsuzawa H. (1996) Allosteric activation of l-lactate dehydrogenase analyzed by hybrid enzymes with effector-sensitive and -insensitive subunits. J. Biol. Chem. 271, 25611–25616 [DOI] [PubMed] [Google Scholar]
- 58. Ormö M., Bystrom C. E., and Remington S. J. (1998) Crystal structure of a complex of Escherichia coli glycerol kinase and an allosteric effector fructose 1,6-bisphosphate. Biochemistry 37, 16565–16572 [DOI] [PubMed] [Google Scholar]
- 59. Valentini G., Chiarelli L., Fortin R., Speranza M. L., Galizzi A., and Mattevi A. (2000) The allosteric regulation of pyruvate kinase. J. Biol. Chem. 275, 18145–18152 [DOI] [PubMed] [Google Scholar]
- 60. Monod J., Wyman J., and Changeux J. P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 [DOI] [PubMed] [Google Scholar]
- 61. Jurica M. S., Mesecar A., Heath P. J., Shi W., Nowak T., and Stoddard B. L. (1998) The allosteric regulation of pyruvate kinase by fructose 1,6-bisphosphate. Structure 6, 195–210 [DOI] [PubMed] [Google Scholar]
- 62. Ikehara Y., Arai K., Furukawa N., Ohno T., Miyake T., Fushinobu S., Nakajima M., Miyanaga A., and Taguchi H. (2014) The core of allosteric motion in Thermus caldophilus l-lactate dehydrogenase. J. Biol. Chem. 289, 31550–31564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Clarke A. R., Wigley D. B., Barstow D. A., Chia W. N., Atkinson T., and Holbrook J. J. (1987) A single amino acid substitution deregulates a bacterial lactate dehydrogenase and stabilizes its tetrameric structure. Biochim. Biophys. Acta 913, 72–80 [DOI] [PubMed] [Google Scholar]
- 64. Sweetlove L. J., Burrell M. M., and ap Rees T. (1996) Starch metabolism in tubers of transgenic potato (Solanum tuberosum) with increased ADPglucose pyrophosphorylase. Biochem. J. 320, 493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Stark D. M., Timmerman K. P., Barry G. F., Preiss J., and Kishore G. M. (1992) Regulation of the amount of starch in plant tissues by ADP glucose pyrophosphorylase. Science 258, 287–292 [DOI] [PubMed] [Google Scholar]
- 66. Tuncel A., and Okita T. W. (2013) Improving starch yield in cereals by overexpression of ADP glucose pyrophosphorylase: expectations and unanticipated outcomes. Plant Sci. 211, 52–60 [DOI] [PubMed] [Google Scholar]
- 67. Sotomayor-Pérez A. C., Subrini O., Hessel A., Ladant D., and Chenal A. (2013) Molecular crowding stabilizes both the intrinsically disordered calcium-free state and the folded calcium-bound state of a repeat in toxin (RTX) protein. J. Am. Chem. Soc. 135, 11929–11934 [DOI] [PubMed] [Google Scholar]
- 68. Fusari C., Demonte A. M., Figueroa C. M., Aleanzi M., and Iglesias A. A. (2006) A colorimetric method for the assay of ADP-glucose pyrophosphorylase. Anal. Biochem. 352, 145–147 [DOI] [PubMed] [Google Scholar]
- 69. Wouter A., Duetz M. C., and Bills G. (2010) Chapter 8: Miniaturization of fermentations, in Manual of Industrial Microbiology and Biotechnology (Lynd L. R., Zhao H., Katz L., Baltz R. H., Bull A. T., Junker B., Masurekar P., Davies J. E., Reeves C. D., and Demain A. L., eds) 3rd Ed., pp. 99–116, American Society for Microbiology, Washington, D. C. [Google Scholar]
- 70. Preiss J., Greenberg E., and Sabraw A. (1975) Biosynthesis of bacterial glycogen. Kinetic studies of a glucose-1-phosphate adenylyltransferase (EC 2.7.7.27) from a glycogen-deficient mutant of Escherichia coli B. J. Biol. Chem. 250, 7631–7638 [PubMed] [Google Scholar]
- 71. Krisman C. R. (1962) A method for the calorimetric estimation of glycogen with iodine. Anal. Biochem. 4, 17–23 [DOI] [PubMed] [Google Scholar]
- 72. Archibald A. R., Fleming I. D., Liddle A. M., Manners D. J., Mercer G. A., and Wright A. (1961) α-1,4-Glucosans. Part XI. The absorption spectra of glycogen- and amylopectin-iodine complexes. J. Chem. Soc. 232, 1183–1190 [Google Scholar]
- 73. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Luzzati V. (1952) Traitement statistique des erreurs dans la determination des structures cristallines. Acta Cryst. 5, 802–810 [Google Scholar]
- 79. Vaguine A. A., Richelle J., and Wodak S. J. (1999) SFCHECK: a unified set of procedure for evaluating the quality of macromolecular structure-factor data and their agreement with atomic model. Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 [DOI] [PubMed] [Google Scholar]
- 80. Laskowski R. A. (2003) Structural quality assurance. Methods Biochem. Anal. 44, 273–303 [PubMed] [Google Scholar]
- 81. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 82. Lodeiro A. R., di Lorenzo O., Petruccelli S., Molina-Ortiz S., and Sorgentini D. (1994) An experiment on glycogen biosynthesis in Escherichia coli. Biochem. Educ. 22, 213–215 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.