

Abstract
The hyperpolarization-activated and cyclic nucleotide-modulated ion channel (HCN) drives the pacemaker activity in the heart, and its malfunction can result in heart disorders. One such disorder, familial sinus bradycardia, is caused by the S672R mutation in HCN, whose electrophysiological phenotypes include a negative shift in the channel activation voltage and an accelerated HCN deactivation. The outcomes of these changes are abnormally low resting heart rates. However, the molecular mechanism underlying these electrophysiological changes is currently not fully understood. Crystallographic investigations indicate that the S672R mutation causes limited changes in the structure of the HCN intracellular gating tetramer, but its effects on protein dynamics are unknown. Here, we utilize comparative S672R versus WT NMR analyses to show that the S672R mutation results in extensive perturbations of the dynamics in both apo- and holo-forms of the HCN4 isoform, reflecting how S672R remodels the free energy landscape for the modulation of HCN4 by cAMP, i.e. the primary cyclic nucleotide modulator of HCN channels. We show that the S672R mutation results in a constitutive shift of the dynamic auto-inhibitory equilibrium toward inactive states of HCN4 and broadens the free-energy well of the apo-form, enhancing the millisecond to microsecond dynamics of the holo-form at sites critical for gating cAMP binding. These S672R-induced variations in dynamics provide a molecular basis for the electrophysiological phenotypes of this mutation and demonstrate that the pathogenic effects of the S672R mutation can be rationalized primarily in terms of modulations of protein dynamics.
Keywords: allosteric regulation, cell signaling, cyclic AMP (cAMP), nuclear magnetic resonance (NMR), protein dynamic, free energy landscape, HCN
Introduction
The cardiac pacemaker current is regulated by hyperpolarization-activated cyclic nucleotide-modulated (HCN)2 ion channels and, in particular, by the isoform HCN4, which is the most abundant in the sinoatrial node (1, 2). These channels open during the diastolic phase resulting in an ion influx that cause a depolarizing shift in the membrane's resting potential toward its activation threshold. The channel opening is predominantly regulated by a voltage sensor in its trans-membrane domain (TMD) that responds to membrane hyperpolarization. However, the intracellular region (IR) provides an additional means of modulation via cyclic nucleotide-regulated auto-inhibition (3–7) The IR includes the following two domains critical for regulating channel gating: the C-linker and the cAMP-binding domain (CBD) (Fig. 1A). The C-linker includes six helices (A′–F′) that facilitate cAMP-dependent activation by promoting tetramerization through intermolecular interactions between the A′ and B′ helices of one protomer and the C′ and D′ helices of another (Fig. 1A) (4, 6–8). The tetramerization competency of the C-linker is influenced by conformational changes within the CBD that occur upon cAMP binding (9, 10).
Figure 1.

Structural architecture of the HCN4 intracellular region. A, topology of tetrameric HCN4 channels. The TMD is connected to the CBD through an α-helical domain known as the C-linker, which also serves as the main point of contact between protomers within the gating tetramer. Ser-672 and cAMP are shown as gray and black spheres, respectively. B, overlay of the apo (red; PDB code 2MNG) and cAMP-bound (gray; PDB code 3OTF) CBDs of WT HCN4. Select regions (i.e. N3A, BBR, and Lid) that undergo cAMP-dependent conformational changes are indicated with dashed boxes. cAMP and Ser-672 are depicted as in A. C, structural overlay of holo-WT (black, PDB code 3OTF) and holo-S672R (blue, PDB code 4HBN) HCN4 CBDs. D, changes in key contacts mediated by Ser-672 (black ribbon, yellow sticks) and S672R (blue ribbon, cyan sticks). Polar contacts for Ser-672 and S672R are depicted with green and red dashed lines, respectively. Selected residues that form interactions with S672(R) or that experience side-chain reorientations are shown as sticks. cAMP is shown as white spheres. E, four-state thermodynamic cycle to model allostery for the HCN CBD, in which the auto-inhibitory equilibrium is coupled to the cAMP-binding equilibrium through the active versus inactive state selectivity of cAMP. A hypothetical mechanism for the bradycardia-induced S672R mutant posits that the auto-inhibitory CBD equilibrium is shifted by the mutation further toward the inactive state (red lines). F, simplified free energy diagram illustrating that the hypothesized mutation-induced shift of the auto-inhibitory equilibrium toward the inactive state may arise either from a stabilization of the inactive state and/or from a destabilization of the active state. This scheme assumes that the state-specific association constants are not significantly affected by the mutation.
The CBD consists of a contiguous β-subdomain and a non-contiguous α-subdomain (Fig. 1B). The β-subdomain spans the primary cAMP-binding site, which includes a variable β-hairpin motif referred to as the base-binding region (BBR) (Fig. 1B) and a highly conserved anchoring site, known as the phosphate-binding cassette (PBC) (Fig. 1B and supplemental Fig. S1). In addition, cAMP binding is gated by a C-terminal helix (or C-helix in short) and a segment following the C-helix (or C-terminal Lid, CTL). Together the C-helix and the CTL define the CBD lid (Fig. 1B), which is indispensable for cAMP signaling via HCN channels (9, 11). Deletion of the lid, even partially, is sufficient to render HCN channel gating insensitive to cAMP (9, 11). This is because the cAMP-driven lid closure is coupled to conformational changes in the N-terminal α-subdomain (N3A) (Fig. 1B), which controls the IR tetramerization by removing steric clashes between the C-linker and the rigid β-subdomain of the apo-CBD (8, 9, 12, 13). In the absence of the steric clashes arising from the apo-CBD, the C-linker forms a stable IR tetramer, and the tonic HCN auto-inhibition is removed (9).
The coupling between cAMP binding, CBD activation, and channel gating is of central importance to the physiological function of HCN (9, 14). Mutations that perturb or disrupt this coupling cause pathological conditions for the heart and pacemaker current (11, 15–19). For example, alteration of the highly conserved Ser-672 residue in the HCN4 CBD by the heterozygous S672R mutation results in bradycardia, i.e. abnormally low resting heart rates that are reduced by ∼30% (17, 18). Electrophysiological profiling using integral HCN4 channels revealed that the S672R HCN4 variant is activated at voltages that are ∼10% more negative than wild type in either the absence or the presence of endogenous cAMP (18). The negative S672R versus WT voltage shift was independently confirmed using chimeric constructs in which the HCN2 trans-membrane channel was combined with the HCN4 IR (20), indicating that S672R represents a loss-of-function mutation with enhanced resistance to channel opening, i.e. S672R causes a reduced inward current flow in the diastolic phase, explaining the slower than normal heart rate observed in patients affected by the mutation (17, 18). Furthermore, another S672R-included change consistently revealed by electrophysiology is a reduction on the deactivation time constant, i.e. the S672R mutation causes a significant acceleration of HCN channel deactivation (18, 20).
As a first step toward elucidating the molecular mechanism for the observed electrophysiological phenotypes of the bradycardia S672R mutation, the structure of the S672R HCN4 IR mutant was solved in the presence of cAMP (Fig. 1C) (20). However, the crystal structure of the cAMP-bound S672R HCN4 IR tetramer did not reveal any major change relative to wild type (Fig. 1C, r.m.s.d. 0.41 Å), with the exception of the BBR (Fig. 1C), which exhibited increased local disorder for four loop residues, as indicated by missing electron density and side chain re-packing of selected surrounding residues (Fig. 1D) (5, 8, 20). No appreciable variations in subunit assembly as a result of the mutation were detected (5, 8, 20). Although these initial structural results provide an essential framework for further investigations of the S672R variant, several questions about the molecular mechanism underlying the S672R bradycardia-inducing mutation still remain open.
It is still not clear how the S672R mutation perturbs the apo-CBD, and in general the CBD dynamics, which has been recognized as a key determinant of the auto-inhibitory CBD function in HCN gating and in other cAMP-dependent systems (9, 13, 21–29). One of the aspects of CBD dynamics that is most relevant for auto-inhibition is the equilibrium between inactive and active conformations (Fig. 1, B and E) (30–38). The coupling of this two-state dynamic conformational equilibrium with the cAMP-binding equilibrium results in a four-state thermodynamic cycle (Fig. 1E), which provides the simplest allosteric model to rationalize the cAMP-dependent modulation of HCN channels, i.e. a shift to less negative activation voltages, as well as to rationalize the effect of disease-related mutations. Considering that the S672R mutant leads to a shift to more negative activation voltages, i.e. an effect opposite to cAMP, while still preserving similar K½ values for integral HCN4 channels (17, 18), we hypothesize that the S672R mutation compromises the cAMP-dependent activation either by giving rise to a partial reversal of the two-state auto-inhibitory equilibrium toward the inactive conformation (Fig. 1, E and F) and/or by partially stabilizing additional states, distinct from the fully active or fully inactive states, in which the activation function of the CBD is compromised. To test these hypotheses, here we comparatively analyze the S672R and WT HCN4 CBDs in both apo- and holo-forms using NMR spectroscopy, which reports on dynamics at residue resolution and over multiple time scales (30, 39). Furthermore, we examined the S672R versus WT relaxation and chemical shift changes, which are exquisitely sensitive atomic reporters of subtle but functionally relevant allosteric perturbations that are often elusive to other structural techniques (27, 30).
Results
S672R mutation leads to pervasive perturbations of the HCN4 CBD in both Apo- and Holo-forms
As a first step toward understanding how the bradycardia-linked S672R mutation affects the apo- and holo-HCN4 CBD, we assigned the 1H-15N HSQC spectra of the S672R HCN4(563–724) construct (supplemental Fig. S2) in the absence of cAMP (Fig. 2A) and in the presence of saturating amounts of cAMP (Fig. 2B). The comparison with the corresponding spectra of WT HCN4(563–724) (9) reveals that the S672R mutation results in pervasive changes for both apo- and holo-forms (Fig. 2, A–C), well beyond that previously anticipated based on X-ray structure comparisons (Fig. 1C) (20). In both apo- and holo-forms, the most pronounced chemical shift changes are observed not only at the site of the mutation but extend to loci located within a 15-Å radius from Ser-672 (Fig. 2, C and D), including the β2–3 loop, the PBC, the BBR, and, in the case of the holo sample, the C-terminal lid as well (Fig. 2C). In addition, several residues throughout the β-subdomain and the C-terminal helix in both apo- and holo-forms exhibits notable ppm changes (> 0.1 ppm; Fig. 2, C, E, and F).
Figure 2.

WT versus S672R chemical shift perturbations in the apo- and holo-HCN4 CBD. Overlay of NH-HSQC spectra for WT (black) and S672R (blue) HCN4(563–724) in the apo (A) and cAMP-bound (B) samples. C, CCS differences between the WT and S672R variant for the apo (red) and cAMP-bound (blue) samples. The black circles along the bottom of the plot depict residues within 15 Å of the mutation site. The protein's secondary structure is depicted along the top as white (α-helices) and gray (β-strands) rectangles, and the BBR and several other motifs are highlighted with gray background. D, residue-specific local WT versus S672R r.m.s.d. values (vertical bars) computed in MOLMOL (56) by aligning three adjacent residues from the WT and S672R cAMP bound X-ray structures (PDB codes 3OTF (8) and 4HBN (20), respectively), as was described previously (27, 41). The solid and dashed black horizontal lines depict the average and average + S.D. r.m.s.d. values, and the horizontal red line represents a 15-Å distance from S672R. The blue and red lines represent the backbone amide nitrogen distances from residue S672(R) for the cAMP bound structure (4HBN) and the apo NMR ensemble (2MNG), respectively. The mutation site (S672R) is highlighted with a red asterisk in both C and D. E and F, residues with CCS changes greater than the dashed line in C were mapped onto the crystal structure as spheres and surface for both the apo (E) and the cAMP-bound (F) samples.
Some of the largest S672R versus WT ppm variations arise either from losses of key interactions mediated by the Ser-672 hydroxyl, as in the case of Glu-625 and adjacent residues in the β2–3 loop (Fig. 1D), or from local structural changes, as in the case of the BBR that exhibits prominent S672R versus WT r.m.s.d. values based on the available crystal structures (Fig. 2D). However, most residues outside of the BBR display only relatively subtle S672R versus WT structural changes, as confirmed by marginal r.m.s.d. values (Fig. 2D) (8, 20), and by the lack of significant S672R versus WT differences in the secondary structure profiles computed based on the secondary chemical shifts (supplemental Fig. S3). These observations suggest that the S672R versus WT ppm variations of Fig. 2C may also reflect mutation-dependent modulations in dynamics. In fact, chemical shifts of CBDs are known to report on the position of the dynamic auto-inhibitory equilibrium of CBDs (Fig. 1E), i.e. they encode for the relatively inactive versus active state populations. Specifically, NMR peak positions reflect population-weighted averages of the respective ppm values for the pure inactive and active CBD states, which exchange rapidly on the chemical shift NMR time scale (9, 40). A simple but effective means to extract state populations from average chemical shifts is the projection analysis (9, 21, 28, 29, 30, 41), in which changes in HSQC cross-peak positions arising from the mutation are evaluated relative to a reference vector defined by the WT apo and WT holo samples. For example, Fig. 3, A–C, shows how the residue-specific S672R holo versus WT holo vectors are analyzed in terms of the angles defined with the reference vectors (θ)and of the normalized projections onto the reference vectors (X). The cos(θ) values (Fig. 3, A and B) report on the direction of the mutation-induced shifts in the inhibitory equilibrium (i.e. cos(θ) <0 for inactivation) and on deviations from the linearity expected based on pure two-state equilibria (i.e. |cos(θ)| <1), whereas the X values (Fig. 3C) quantify the degree of mutation-induced fractional inhibition or activation (9, 21, 28–30, 41).
Figure 3.

Chemical shift projection analysis (CHESPA) of the HCN4 S672R mutation. A, cos(θ) values computed as in Equation 3, as explained in the text. Selected residues reported in E–H are labeled. B, distribution of cosθ values from A, revealing a net predominance of negative cos(θ) values. C, fractional activation (X) values for the perturbation vector relative to the activation vector, computed as per Equation 4. The red box highlights a subset of lid residues with markedly negative X values, pointing to a significant shift toward the inactive state. Selected residues reported in E–H are labeled. D, 3D map of CHESPA outliers, defined as residues with either |cosθ| <0.9 or |X| >1.1. Such outliers reveal deviations from the two-state equilibrium, such as those arising from nearest neighbor effects or population of a third state. E, representative NH-HSQC cross-peaks for a residue from the C-terminal lid region with a holo-S672R X value close to −0.4 (C, red box). F, similar to E, but for a representative residue with a holo-S672R X value close to −0.1. G, representative residue (Val-642) from the S672R holo spectra that exhibited a non-linear chemical shift change relative to the WT apo- and holo-states. H, representative residues from the S672R apo spectra compared with WT apo- and holo-states that displayed non-linear chemical shift changes. This non-linearity is also evident from the CHESPA analysis of the apo-S672R HCN4 CBD, shown in I and J, revealing a nearly uniform cos(θ) distribution (I) between the −1 and 1 extremes. Selected residues reported in E–H are labeled in I. K, X values for apo-S672R residues. L, similar to D but for the apo-S672R CHESPA outliers.
S672R mutation shifts the dynamic inhibitory equilibrium of the HCN4 CBD toward an inactive but partially De-correlated state
The chemical shift projection analysis (CHESPA) of holo-S672R shows that negative cos(θ) values are observed for the majority of the residues (Fig. 3A), with a cos(θ) distribution highly skewed toward −1 (Fig. 3B), reflecting a clear prevalence of mutation-induced shifts in cross-peak positions from the WT holo toward the WT apo, i.e. the auto-inhibitory state. However, the extents of these shifts, i.e. the magnitudes of the X values, are highly residue-dependent (Fig. 3B). Residue-specific variations in X values are expected for sites proximal to either the mutation and/or cAMP, such as the BBR and the β2–3 loop (Fig. 3A), because these loci are affected by nearest neighbor effects (NNEs). Nevertheless, in our case residue-specific variations in X values are also observed for sites more removed from the mutation and cAMP, such as the N-terminal helices and the C-terminal region (Fig. 3C). For example, the X values of CTL residues 714–724 range from −0.2 to ∼−1 with an average X value of −0.4 (Fig. 3, C and E), whereas in the N-terminal N3A significantly lower X values close to −0.1 are measured (Fig. 3, C and F). The variations in residue-specific X values represent a clear deviation from the pattern of nearly residue-independent X values expected for these regions based on the two-state model (Fig. 1E), indicating that the response of the holo-CBD to the S672R mutation cannot be fully recapitulated by a simple two-state auto-inhibitory equilibrium (Fig. 1E). Deviations from the two-state model are also confirmed by the appearance of |cos(θ)| values <1 for several residues, including those not affected by NNE (Fig. 3, A and D) (27), pointing to the presence of non-linear patterns (Fig. 3G). Overall, the CHESPA analysis of holo-S672R reveals that S672R causes a partial reversal of the inhibitory equilibrium toward a less activated state (Fig. 3B), but such reversion is not as correlated as expected based on a purely two-state model because it is significantly more pronounced in the C-terminal lid than in other regions largely insensitive to NNEs (Fig. 3, C, E, and F).
To investigate whether the mutation-induced de-correlation is unique to the holo sample or applies also to the apo-form, we extended the CHESPA analysis to the apo-S672R sample (Fig. 3, I–L). Although the S672R holo-CHESPA analysis clearly shows a shift toward inactivation, this shift is not expected to affect the auto-inhibitory S672R apo-CHESPA. The S672R-induced auto-inhibitory shift arises from a partial stabilization of the inactive state and/or from partial destabilization of the active state (Fig. 1F). However, in either case, the WT apo inhibitory equilibria of our construct are known to be already almost quantitatively skewed toward the inactive state (Fig. 1E) (9, 40). Hence, the Ser-672-induced auto-inhibitory shift is expected to be negligible under our experimental conditions, suggesting that the S672R apo-CHESPA analysis should reflect primarily de-correlative effects of the mutation, i.e. deviations from the two-state model. This prediction is confirmed by the cos(θ) values measured for S672R apo (Fig. 3I), exhibiting a broad scattered distribution spanning the full −1 to 1 range (Fig. 3J), which is in marked contrast to the inactivation-skewed pattern observed for S672R holo (Fig. 3B). Furthermore, the abundance of residues with |cos(θ)| <1 (Fig. 3, H and J) and the scatter detected in the residue-specific X values (Fig. 3K) confirm that the perturbations caused by S672R cannot be modeled exclusively based on a two-state model (Fig. 1E). These deviations from the two-state transition point to mutation-induced de-correlations and possibly to enhanced dynamics. The latter were gauged through S672R versus WT comparative 15N-relaxation analyses (Fig. 4; supplemental Figs. S4–S7; supplemental Tables S1–S3). Internal dynamics were surveyed using spin relaxation experiments (15N-T1, 15N-T2, and 1H15N NOEs) analyzed in terms of N-H S2 order parameters (Fig. 4, A and B) and reduced spectral densities (supplemental Figs. S4–S6; Fig. 4, C–J). Significant changes in picosecond to nanosecond and microsecond to millisecond dynamics, identified as explained under “Experimental procedures,” were mapped on the crystal structure of S672R HCN4 (Fig. 4, K–N). The S2 distributions shown in Fig. 4, A and B, provide a global picture of the mutant-induced variations in fast (picosecond to nanosecond) dynamics, whereas Fig. 4, C–J and K and L, offer a region and residue-resolution dissection of dynamics, respectively.
Figure 4.

Comparative S672R versus WT analysis of HCN4 CBD dynamics. Order parameter (S2) distribution for WT (black) and S672R (red) in the absence (A) or presence of saturating amounts of cAMP (B). Data were fit with either one (solid line) or two (dashed line) Gaussian distributions and as summarized in supplemental Table S1. C–J, two-dimensional reduced spectral density plots for the apo-WT (black) and S672R (red) HCN4 CBD. The following four regions were probed: the β2-β3 loop (aa 621–636), BBR (aa 639–652), extended PBC (aa 659–676), and Lid region (aa 699–724), for both apo (top row) and cAMP-bound (bottom row) samples. The Lid is further dissected into the Lid and CTL in supplemental Fig. S6. The solid black line represents the expected spectral densities for an isotropic rigid rotor, and the black and red ellipsoids are centered on the average spectral density values with axes defined by ±2 S.D. for the WT and mutant, respectively. K, map of apo residues with S672R versus WT enhanced picosecond to nanosecond (red spheres and surface) and microsecond to millisecond (orange spheres and surface) dynamics. L, map of apo residues with S672R versus WT quenched picosecond to nanosecond (blue sphere and surface) and microsecond to millisecond (cyan sphere and surface) dynamics. M and N, as in K and L except for the cAMP-bound sample.
Comparative S672R versus WT analysis of internal dynamics for the Apo-CBD reveals that the S672R mutation results in a significant overall enhancement in the amplitude of the picosecond to nanosecond internal motions with local increases in millisecond to microsecond dynamics near cAMP phosphate recognition sites
The overall S2 distributions observed in the absence of cAMP for the WT and S672R CBD reveal that the mutation results in a marked net shift toward lower S2 values (Fig. 4A). This result points to increased amplitudes of picosecond to nanosecond motions in S672R versus WT apo-CBD. Furthermore, considering that order parameters for picosecond to nanosecond motions are a proxy for conformational entropy (30), the S672R-induced shift toward reduced S2 values suggests a widening of the well in the free-energy landscape minimum representing the apo-CBD (Fig. 5A). The S672R versus WT dynamics enhancements of the apo-CBD are not limited to the picosecond to nanosecond time scale, but they include also microsecond to millisecond motions (Fig. 4K). In particular, the β2-β3 loop proximal to the mutated site (Fig. 1D) displays some of the greatest residue-specific enhancements of dynamics, as observed for both low and high frequency spectral density functions (supplemental Fig. S4; Fig. 4, C and K). Specifically, residue Glu-625, which forms a hydrogen bond with Ser-672 (Fig. 1D), exhibits a remarkable ∼3-fold increase in J(0) (supplemental Fig. S4A; Fig. 4C), consistent with a large increase in microsecond to millisecond dynamics. In addition, several residues adjacent to Glu-625 display enhanced picosecond to nanosecond dynamics (Fig. 4K; supplemental Fig. S4C). These data suggest that by disrupting the interaction between Ser-672 and Glu-625, the S672R mutation destabilizes the entire β2-β3 loop. Because the β2-β3 loop lies adjacent to the PBC, we checked whether this destabilization could extend to this cAMP-binding motif. Although the dynamic characterization of the apo-PBC is sparse due to line broadening, several residues at the edges of the PBC do indeed exhibit enhanced microsecond to millisecond dynamics (Fig. 4G; supplemental Fig. S4A), confirming that the β2-β3 enhancement of dynamics does at least partially extend to the PBC, which is a primary cAMP recognition site. So next we checked whether the S672R causes significant changes in dynamics at the other main cAMP contact sites, i.e. the BBR and the lid.
Figure 5.

S672R versus WT cAMP-binding kinetics from protein- and ligand-based NMR experiments. A, schematic diagram depicting the effects of the reduced S672R versus WT order parameters (Fig. 4A) and higher conformational entropy on the free energy landscape of binding. B, schematic illustration of the rationale for measuring apo-holo exchange rates through HSQC spectra at sub-stoichiometric ligand concentrations. Further details are available in the text. C and E, representative cross-peak from the NH-HSQC spectra of the WT (C) and S672R HCN4 CBD (E). The red and blue peaks represent the apo and fully bound states, respectively, whereas the green peaks are from a sample containing 100 μm HCN4 CBD and 25 μm cAMP. For both WT and mutant CBDs, the ΔνA-H is similar (80 versus 81 Hz) but the exchange regime changes from slow for the WT (C) to fast for S672R (E). D and F, sorted plots of ΔνA-H versus residue label arranged from largest to smallest for WT (D) and S672R (F). Indicated within the plots with colors are the chemical exchange regimes: blue, slow; yellow, intermediate; and red, fast. Insets show representative residues close to the boundaries between slow-intermediate and fast-intermediate exchange regimes. Gly-715 spectra are shown in supplemental Fig. S8. The dashed lines highlight the ΔνA-H range corresponding to the intermediate exchange regime. G, ROESY spectra of 50 μm WT HCN4 CBD in the presence of excess cAMP (500 μm). The diagonal peaks indicated in the spectra correspond to the H8 proton of cAMP in its bound (H8b) and free (H8f) states. The cross-peak for the chemical exchange from bound to free H8 is also indicated as H8b → f. Black contours indicate positive peaks; red contours indicate negative peaks. The H8b → f preserves the same sign as the diagonal, as expected for chemical exchange cross-peaks. H, plot of H8b → f versus H8b intensity ratios versus ROESY mixing times. The initial slopes of the buildups based on the first order approximation are reported in the figure. For the S672R mutant, the bound ligand diagonal peak was detectable only up to a ROESY mixing time of 30 ms due to rapid decay. In contrast, the WT bound H8 signal was detectable up to a ROESY mixing time of 50 ms. The solid black line depicts the slope of the two WT points shown in the plot, and the dashed line depicts the slope that includes two additional points at mixing times of 40 and 50 ms.
As to the BBR, whereas the 645–650 region in the WT apo-CBD displays higher J(ωN+ωH) values than adjacent residues, pointing to flexibility in the picosecond to nanosecond time scale, no further significant enhancements of dynamics could be detected for S672R apo under our experimental conditions (supplemental Fig. S4C; Fig. 4E). Although BBR residues Lys-645 and Lys-648 did exhibit slight decreases in J(0) (Fig. 4E; supplemental Fig. S4A), Lys-648 displayed quenched dynamics at the ωN frequency (supplemental Fig. S4B), and for either residue no significant changes at the J(ωN+ωH) spectral density were detected. In contrast, the apo C-helix exhibited quenching of microsecond to millisecond dynamics as seen by decreased J(0) values for Thr-705 and Ala-707 (Fig. 4L; supplemental Figs. S4A and S6A). Meanwhile, our probes for the BBR and lid do not support a major net S672R versus WT enhancement in picosecond to nanosecond dynamics in these regions (Fig. 4, E and I). However, it should be considered that these regions are already flexible in the picosecond to nanosecond time scale in the WT apo-CBD, as shown by a significantly increased J(ωN+ωH) value relative to those expected for overall tumbling (supplemental Fig. S4C). Beyond the cAMP-binding sites, significant dynamic changes were found at the interface between α- and β-subdomains and even in parts of the N-terminal C-linker (Fig. 4, K and L; supplemental Fig. S4). The net effect of these pervasive variations in dynamics in the apo-CBD is that the S672R mutation results in an enhancement in the amplitude of fast (picosecond to nanosecond) motions (Fig. 4A) and in the associated entropy of the conformational ensemble sampled by the apo-CBD (Fig. 5A). To evaluate the effect of the S672R mutation on the holo-CBD dynamics, the comparative S672R versus WT 15N-relaxation analyses were extended to the data acquired in the presence of excess cAMP (Fig. 4, B, D–J, M, and N; supplemental Figs. S5 and S6).
In the Holo-CBD, the S672R mutation results in localized enhancements in the dynamics of key cAMP recognition and gating regions (PBC and Lid)
Unlike the apo-CBD (Fig. 4A), in the case of the holo-CBD no net S672R versus WT shift to larger amplitude picosecond to nanosecond motions is observed in the overall distribution of S2 order parameters (Fig. 4B). However, several localized S672R versus WT enhancements of dynamics were detected also for the holo-CBD (Fig. 4, H, J, M, and N; supplemental Figs. S5 and S6). The most dramatic increase in dynamics was found in two loci within the cAMP-binding pocket, i.e. PBC and lid, but significant enhancements of dynamics were also identified in selected sites within the N3A and C-linker (Fig. 4, H, J and M; supplemental Fig. S5). The PBC, which is nearly completely assigned in the cAMP-bound state and better sampled than in the apo-state, experiences large enhancements of microsecond to millisecond dynamics, as indicated by several significant increases in the J(0) (Fig. 4H; supplemental Fig. S5A). Unlike the apo-CBD, the mutation induced increase in dynamics for the PBC did not extent to the adjacent β2-β3 loop (Fig. 4D). Similarly to the apo-CBD, for the BBR, i.e. the other cAMP contact region in the β-subdomain, only a few significant changes in dynamics were recorded, and these exhibited a mixed pattern, including both enhancements and quenches (Fig. 4F; supplemental Fig. S5).
Besides the PBC, the other region of the holo-CBD experiencing major dynamic enhancements is the C-helix and the adjacent C-terminal lid segment (Fig. 4, J, M, and N; supplemental Figs. S5 and S6). The S672R mutation caused a significant enhancement of microsecond to millisecond dynamics in the C-helix, as seen by increased J(0) values for Glu-704, Thr-705, Ala-707, and Leu-708 (supplemental Fig. S6B). In addition, Ile-714, which forms the capping interactions with the adenine base of cAMP, displayed a remarkable 2-fold increase in the corresponding J(0) value (supplemental Figs. S5A and S6D; Fig. 4, J and M). Furthermore, several residues in the CTL exhibited significant enhancements in nanosecond dynamics as seen by J(ωN) (supplemental Fig. S5B) and picosecond to nanosecond dynamics as revealed by increased J(ωN+ωH) values (Fig. 4M; supplemental Fig. S5C).
Interestingly, for the S672R holo sample the picosecond to nanosecond dynamics in the lid region, as probed by the average J(ωN+ωH) spectral densities for the C-helix and CTL, lie in-between those of the WT apo and WT holo samples (supplemental Tables S2 and S3). This result is in agreement with the fractional activations observed by CHESPA for this region, which also shows a partial reversion toward the apo-inactive state (Fig. 3C). Together, the 15N-relaxation data and the CHESPA comparative analyses for the holo-CBD consistently point to a partial mutation-induced disengagement of the CTL, which is part of a key gating element for cAMP. Furthermore, the mutation also enhances the dynamics of key cAMP recognition elements, such as the PBC (Fig. 4, G and H), suggesting that S672R may lead also to altered on-off cAMP exchange kinetics. This hypothesis is also supported by the overall S672R versus WT net shift toward lower order parameters, i.e. higher conformational entropy, observed for the apo-CBD (Fig. 4A). The wider free energy well of the mutant is in agreement with reduced free energy barriers for the apo-holo exchange (Fig. 5A), corroborating the hypothesized S672R versus WT acceleration of cAMP-binding kinetics. To further test this hypothesis, we analyzed the cAMP-binding dynamics utilizing protein-based and ligand-based NMR experiments.
S672R mutation accelerates the on/off rates of the HCN4 CBD-cAMP complex
The cAMP-binding kinetics were probed by comparative analyses of chemical exchange regimes in HSQC spectra (Fig. 5, B–F) and ROESY buildup curves (Fig. 5, G and H). The HSQC analysis involved monitoring residue-specific transitions from slow to intermediate and from intermediate to fast for apo-holo exchange regimes in HSQC spectra acquired at sub-stoichiometric ligand/protein ratios, as illustrated in Fig. 5B. At a given protein/ligand stoichiometric ratio, the effective apo-holo exchange constant, kex, is defined by Equation 1,
| (Eq. 1) |
A particular value of kex results in different exchange regimes for different residues, depending on the respective differences in Hz between the apo and holo resonance frequencies (ΔνA-H). If kex ≪ ΔνA-H for a selected residue, then that amino acid will appear in the slow exchange regime, which is characterized by two peaks appearing at the positions of the apo- and holo-states with intensities proportional to the populations of ligand-bound and -unbound protein (Fig. 5B, blue peaks). However, under the same experimental conditions, and therefore with the same kex, for other residues that experience lower ΔνA-H values, it is possible that kex ≈ ΔνA-H, i.e. the residues fall within the intermediate exchange regime in which peaks broaden significantly, often beyond detection (Fig. 5B, black peak). For amino acids with lower ΔνA-H so that kex ≫ ΔνA-H, the fast exchange regime applies, and only a single peak is observed whose chemical shift is a population weighted average of the apo and holo chemical shifts (Fig. 5B, red peak). The difference in residue-specific exchange regimes provides an opportunity for a simple estimation of kex by assessing the range of ΔνA-H values that result in intermediate chemical exchange. For this purpose, the ΔνA-H values were sorted in decreasing order, and the chemical exchange regime was determined for each residue to define the boundaries between slow, intermediate, and fast exchange (Fig. 5, D and F). The kex was then assessed based on the range of ΔνA-H values that resulted in intermediate chemical exchange (Fig. 5, D and F). Overall, Fig. 5, D and F, illustrate how the ranked ΔνA-H frequency differences define a “built-in” scale for gauging kex. This approach is not as quantitative as other methods (e.g. Nz-exchange spectroscopy (26, 42)), but it is applicable to both HCN4 WT and mutant, unlike Nz-exchange experiments, as explained in the supplemental Materials and Methods.
The HSQC-based kex assessment was performed for both the WT and S672R HCN4 CBD in the presence of sub-stoichiometric amounts of cAMP (Fig. 5, C–F). Evidence that kex increases in going from S672R to WT is already provided by residues that experience similar apo versus holo frequency differences but fall in the slow exchange regime in WT and in the fast exchange regime in S672R. This is the case, for example, for Ala-597 as shown in Fig. 5, C and E. In WT HCN4, two separate peaks are detected for Ala-597 (Fig. 5C), consistent with slow apo-holo exchange, although in S672R a single peak is observed (Fig. 5E), consistent with fast apo-holo exchange. Considering that the ΔνA-H values of WT and S672R are comparable (Fig. 5, C and E), the slow versus fast exchange regimes observed for Ala-597 in WT versus S672R, suggests that the mutation results in an increased kex value. The two-peak versus one-peak patterns shown in Fig. 5, C and E, are the basis for determining whether a given residue experiences slow or fast apo-holo exchange (see supplemental Materials and Methods and supplemental Fig. S8 for further details) so that the plots of the sorted ΔνA-H values are partitioned into the three different kinetic regimes (Fig. 5, D and F). The comparison of Fig. 5, D and F, reveals marked WT versus S672R differences in the positions of the apo-holo exchange regimes within the scales defined by the sorted ΔνA-H values (Fig. 5, D and F). For WT, the slow to fast transition occurs in the 30–40 Hz range (Fig. 5D), and for S672R, the intermediate regimes fall in the 150–260 Hz window (Fig. 5F), confirming that the mutation leads to a significantly enhanced kex value.
The S672R-induced kex enhancement revealed by the HSQC analyses (Fig. 5, B–F) reflects a primarily kinetic effect, as no significant S672R versus WT differences were observed for the thermodynamic affinities of the HCN4 CBD, with Kd values in the 2–10 μm range for both mutant and WT (Fig. 6). Specifically, based on supplemental Equation S3, the assessed koff values for WT and S672R are expected to fall in the 23–31 and 113–202 s−1 ranges, respectively, pointing to a major S672R-induced acceleration in the cAMP on/off dynamics. This result was independently confirmed through ligand-based NMR experiments, i.e. ROESY spectra (Fig. 5, G and H), which complement the protein-based NMR data, i.e. the semi-quantitative HSQC analysis outlined above. The ROESY data were acquired in the presence of excess ligand, and slow exchange was observed between the free and bound ligand peaks, as confirmed by ROESY cross-peaks with the same sign as the diagonal (Fig. 5G). The initial slopes of the mixing time buildup measured for the bound-to-free exchange cross-peak fall in the 18–32 and 110–134 s−1 ranges for WT and S672R, respectively (Fig. 5H). These values are consistent with the koff ranges independently estimated based on the HSQC data and confirm that the S672R mutation leads to an at least ∼3-fold enhancement in the on-off exchange rates of cAMP.
Figure 6.
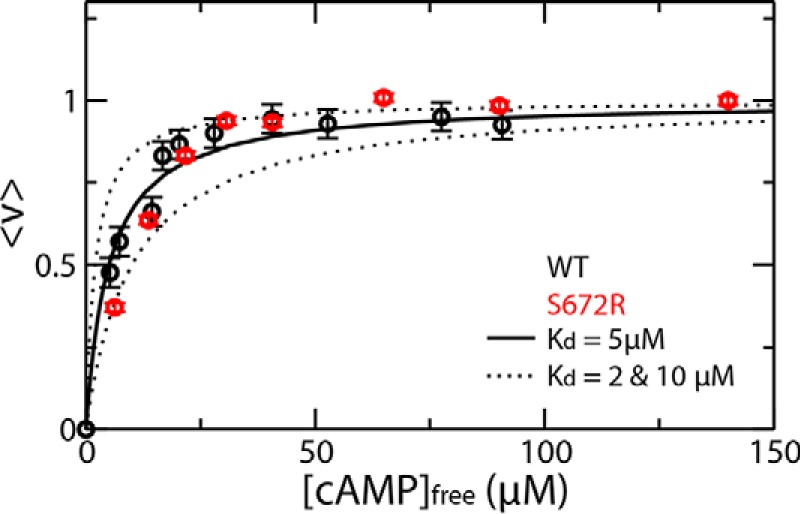
Binding isotherms of S672R (red) human HCN4(563–724) with cAMP monitored by STD NMR. Data were fitted with a rectangular hyperbola for a 1:1 binding stoichiometry providing a Kd in the 2–10 μm range. The fractional saturation was measured by normalizing the STD amplification factor (9). The corresponding data for WT human HCN4(563–724) (9) is shown (black dots) for the convenience of comparison. The low micromolar affinity reported here for the S672R variant is in agreement with the range of Kd values of 0.8 and 11.2 μm measured by fluorescence anisotropy and isothermal titration calorimetry by Xu et al. (20), respectively. In addition, considering that the apo-WT state is already primarily sampling the inactive state, based on the scheme in Fig. 1F, a mutation-induced shift toward inactivation is not expected to result in major variations in the observed affinity for cAMP.
Discussion
Bradycardia-related S672R mutation remodels the free energy landscape and the dynamic profiles critical for the cAMP-dependent modulation of HCN4 channels
The comparative NMR analyses presented here (Figs. 2–6; supplemental Figs. S3–S8) have revealed how the S672R bradycardia-inducing mutation remodels the free energy landscape (FEL) for the cAMP-dependent regulation of the HCN4 channels, as illustrated in Fig. 7A. The FEL remodeling caused by S672R (Fig. 7A) is recapitulated by two main perturbations. First, the mutation results in a shift of the auto-inhibitory CBD equilibrium toward inactivation, as shown by the CHESPA data (Fig. 3), consistent with the stabilization of inactive conformations and/or the destabilization of the active state (Fig. 7A, vertical red arrows). Second, the S672R variant leads to a widening of the apo-inactive free energy basin (Fig. 7A, horizontal red arrows), as indicated by a redistribution of the apo-CBD order parameters for fast (picosecond to nanosecond) motions toward lower values (Fig. 4A). The broadening of the apo-inactive conformational sub-set is also confirmed by the CHESPA data (Fig. 3), which indicate that the mutation-induced shift to inactive states cannot be fully captured by a simple two-state exchange model, i.e. rather than a single cooperative transition, S672R causes a partial de-correlation, which accounts for the observed residue dependence of the fractional activation values (X) and for the |cos(θ)| <1 value observed at multiple sites (Fig. 3). Considering that the apo-CBD samples primarily inactive conformations, the broadening of the free energy well caused by S672R (Fig. 7A) is expected to lower the free energy barrier for apo-holo exchange (Fig. 5A). This prediction was confirmed by the observation that S672R accelerates the cAMP-binding kinetics by a factor of at least 3-fold, as measured by both protein- and ligand-based NMR methods (Fig. 5, C–H).
Figure 7.
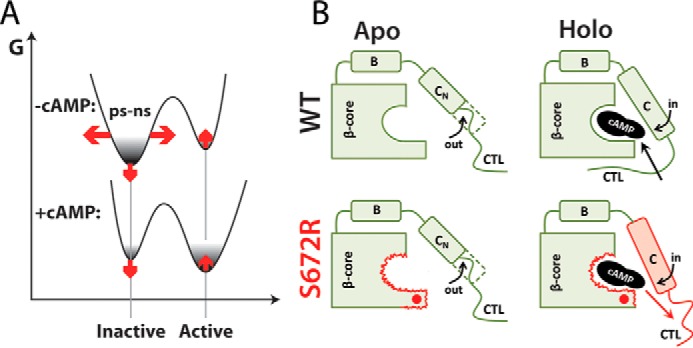
Proposed mechanism of S672R-induced bradycardia based on free energy landscape re-modeling. A, schematic free energy landscape for the allosteric cAMP-dependent modulation of the HCN4 CBD and its perturbation by the S672R mutation (red arrows). The horizontal red arrows denote the S672R versus WT enhanced conformational entropy of the inactive state as indicated by the S672R versus WT decreased S2 parameters of the apo samples (Fig. 4A). The vertical red arrows illustrate that the S672R mutation results in a CBD shift toward inactivation either by destabilizing the active state and/or by stabilizing the inactive state. B, summary of critical S672R versus WT changes in dynamics. Only the β-core and the C-terminal α-subdomain (B and C helices and the adjacent CTL) are shown for simplicity. Red wavy lines indicate S672R versus WT enhanced dynamics in the HCN4 CBD. According to the proposed model, the enhanced dynamics at sites lining the cAMP-binding pocket as well as the disengagement of the CTL in the cAMP-bound sample lead to an acceleration of the kinetics of cAMP binding and unbinding. The red dot indicates the site of the S672R mutation. The red outline and red shading denote areas with enhanced S672R versus WT dynamics, leading to an increased koff rate for cAMP (red arrow).
The mutation-induced enhancement of dynamics is not limited only to the fast (picosecond to nanosecond) time scale, but it extends also to the microsecond to millisecond dynamic window (Fig. 4, C–N). The S672R variant exhibits enhanced millisecond to microsecond dynamics at sites critical for cAMP recognition and binding, such as the PBC (Fig. 7B). The PBC enhancement of millisecond to microsecond dynamics was detected in both apo- and holo-forms and extends to the adjacent β2–3 loop in the apo-CBD, whereas for the holo-CBD it affects the C-terminal lid, referred to as CTL (Fig. 7B). The CTL is part of the lid region critical for the cAMP-dependent HCN channel activation (14) and for gating cAMP binding in WT (Fig. 1, B and C). However, in the holo-S672R, the CTL becomes partially disengaged, as consistently indicated by both the negative CHESPA fractional activation values (Fig. 3, B, C, and E) and the concurrent mutation-induced enhancement of both fast and slow dynamics (Fig. 4; supplemental Fig. S6D and Table S3). The enhanced lid dynamics in S672R versus WT is also in full agreement with the faster binding kinetics observed for the mutated variant. Overall, the picture emerging from the NMR investigation presented here suggests that the S672R-induced perturbations extend well beyond what was previously anticipated based on X-ray crystallography of the cAMP-bound intracellular region (20). The crystal structure of S672R and WT holo-HCN4 IR tetramers differs primarily at the BBR, whereas NMR chemical shift changes show that for both apo- and holo-forms S672R affects multiple sites within an ∼15-Å radius, spanning not only the BBR but also several other loci in the β-subdomain and lid regions (Fig. 2, D–F).
S672R-induced FEL remodeling provides an initial molecular explanation for the electrophysiological changes underlying familial bradycardia
The S672R-induced remodeling of the free energy landscape for the cAMP-dependent modulation of HCN (Fig. 7) provides a molecular framework to rationalize the two main electrophysiological signatures of this bradycardia-related mutation, i.e. the negative shift in the activation voltage and the faster deactivation. The former phenotype may arise as a result of a reduction in cAMP affinity (20) and/or of a constitutive S672R-induced variation in the position of the auto-inhibitory CBD equilibrium toward inactivation (17, 18). Our data support the notion that the second mechanism significantly contributes to the negative S672R-induced shift in the activation voltage observed by electrophysiology. Considering that activation voltages of full-length HCN4 have been shown to correlate with the position of the auto-inhibitory CBD equilibrium as probed by CHESPA (9), the partial inactivation observed for the S672R variant (vertical red arrows in Fig. 7A) is consistent with the negative S672R versus WT change of the activation voltage observed by electrophysiology for the full-length channel (18, 20). Furthermore, the position of the auto-inhibitory CBD equilibrium in reconstituted apo full-length channels may be affected by the presence of dimers known to be populated in the absence of cAMP (43, 44). If dimerization causes a partial shift of the WT apo-CBD toward activation, the S672R mutation may result in an appreciably increased sampling of inactive states also in the absence of cAMP, explaining the constitutive negative shifts in activation voltages observed for apo-channels (18).
The other putative mechanism, i.e. a reduction of cAMP affinity, does not appear to be sufficient alone to explain the observed negative S672R-induced shift in the activation voltage without any constitutive mutation-induced partial inactivation. This conclusion is supported by several independent observations. First, S672R is known to cause a negative shift in activation voltages even in the absence of cAMP (18). Second, we did not observe any major S672R versus WT increases in the cAMP Kd values measured under our experimental conditions by STD for the monomeric HCN4 CBD, as both Kd values fall in the 2–10 μm range (Fig. 6). Third, no significant S672R-induced enhancements in the EC50 values were reported for the full-length HCN4 channels either (18). Fourth, although S672R versus WT K½ increase was reported for either HCN2 or hybrid HCN2/4 channels (20), a significant mutation-induced reduction in integral HCN4 affinity would decrease the sensitivity to physiological levels of cAMP and result in chronotropic incompetence, which, however, was not observed in patients affected by the S672R mutation (17, 18). These observations consistently suggest that the negative S672R-induced shift in the activation voltage cannot be fully accounted for by a reduction of cAMP affinity alone without any constitutive S672R-induced variation in the position of the auto-inhibitory CBD equilibrium.
The other electrophysiological signature of the bradycardia-related mutation, i.e. the faster deactivation, is explained at least in part by the S672R-induced acceleration of the cAMP off kinetics, as seen by our combined analyses of protein- and ligand-based NMR experiments (Fig. 5). This result is in agreement with the S594R versus WT koff enhancement reported for the unbinding of a fluorescent cAMP analog from full-length mHCN2 channels and is rationalized by both the S672R-induced widening of the apo free energy well (Fig. 5A) and the enhancement of dynamics observed in the holo-CBD for the PBC and the lid, two critical elements of the cAMP-binding sites (Fig. 7). Unlike the PBC and the lid, the remaining cAMP recognition region, i.e. the BBR, does not appear to be subject to significant mutation-induced enhancements of dynamics (Fig. 4).
In conclusion, we have shown how the bradycardia-related S672R mutation remodels the FEL for the cAMP-dependent modulation of HCN4 channel gating. The S672R-induced FEL remodeling is recapitulated by two main effects. First, S672R results in the constitutive stabilization of inactive versus active states, contributing to the negative S672R versus WT shift in the activation voltage observed by electrophysiology for HCN4 channels. Second, S672R leads to enhanced dynamics at sites pivotal for cAMP binding, such as the PBC and the lid, which in turn leads to an acceleration of the cAMP-unbinding kinetics, explaining at least in part the faster deactivation reported for S672R versus WT HCN4 channels. Hence, our results provide a mechanistic explanation of the main electrophysiological determinants of the familial bradycardia diagnosed in patients carrying the S672R mutation. In addition, the NMR approaches outlined here will be useful to map how other disease-related mutations remodel functionally relevant free energy landscapes in other systems as well (45–48).
Experimental procedures
HCN4 sample preparation
The expression and purification of HCN4(563–724), referred to here as HCN4 CBD for simplicity, was adapted from a previously established protocol (9). A rare codon optimized SUMO-HCN4 fusion construct was subcloned into a pET302NT-His vector (Invitrogen) and expressed in Escherichia coli BL-21(DE3) cells in M9 minimal media isotopically enriched with [15N]ammonium chloride (Sigma). Expression was induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside (Thermo Fisher Scientific) at an A600 of 0.6 and incubated for a further 16–18 h at 20 °C. Cells were lysed with a cell disruptor, and the cell lysate was centrifuged at 20,000 × g for 1 h. The cell lysate was then filtered with 0.8/0.2-μm syringe filters (VWR Scientific) and purified by Ni-NTA affinity chromatography following the same protocol as in Akimoto et al. (9). Following proteolytic cleavage of the His6-SUMO tag with His6-tagged TEV protease, the sample was purified again with Ni-NTA affinity chromatography to remove both the SUMO tag and the TEV protease. The cleaved protein was then diluted 3-fold with Sp Wash buffer (10 mm MES, 10 mm KCl, and 10 mm 2-mercaptoethanol, pH 6.0) and further purified by cation exchange chromatography with a 5-ml HiTrap Sp column (GE Healthcare). Following injection, the protein was eluted using a 100% gradient over 100 min with Sp elution buffer (10 mm MES, 1 m KCl, and 10 mm 2-mercaptoethanol, pH 6.0). The purified protein was then dialyzed overnight in NMR buffer (20 mm MES, 75 mm KCl, 2 mm EDTA, 2 mm EGTA, 1 mm DTT, and 0.02% sodium azide, pH 6.5) and stored at 4 °C. HCN4(563–724) S672R was created by site-directed mutagenesis using a variation of the QuikChange protocol. The reaction used the KOD Hot Start master mix (Novagen) with primers synthesized in-house. The reaction protocol followed the manufacturer's specifications for the KOD Hot Start kit. Following the reaction, parental DNA was degraded with DpnI for 1 h at 37 °C, and the reaction product was transformed in E. coli top10 competent cells. Plasmids were later isolated from single colonies grown in liquid LB media using a GeneJET Plasmid Miniprep kit (Thermo Fisher Scientific) and sequenced to confirm the mutation. HCN4(563–724) S672R was expressed and purified similarly to the WT with the exception that the cation exchange gradient was changed to 100% Sp elution buffer over 25 min.
NMR spectroscopy
NMR experiments were acquired on a Bruker Avance 700 MHz spectrometer equipped with a 5-mm TCI cryo-probe, unless otherwise specified. All experiments were acquired at 300 K in the NMR buffer defined above with 5% D2O unless otherwise specified. Spectra were processed either with NMRpipe (49) or directly in Bruker's Topspin and analyzed in NMRFAM-SPARKY (50). Chemical shift assignments of S672R were obtained through spectral comparison with WT spectra (9), if no ambiguities were present, and through triple resonance experiments (i.e. HNCO, HNCA, and HN(CO)CACB) otherwise. The Talos+ server was used to predict the protein's secondary structures using HN, Cα, Cβ, and C′ chemical shifts and to cross-check assignments based on acceptable backbone torsional angles (51).
Chemical shift analyses
Compounded chemical shift differences (ΔCCS) were computed as described previously (9, 27, 29, 40, 41), using Equation 2,
| (Eq. 2) |
where the δ symbols represent the proton (H) and nitrogen (N) chemical shifts (in ppm) for states 1 and 2. The CHESPA protocol has been described elsewhere (9, 23, 29). Briefly, the WT apo and WT holo cross-peaks for a given residue define a reference vector A, whereas the S672R (either apo or holo) and its corresponding WT state define a perturbation vector B. The cosθ and fractional activation (X) values are then computed for residues with mutant versus WT ΔCCS >0.05 ppm, using Equations 3 and 4.
| (Eq. 3) |
| (Eq. 4) |
NMR spin relaxation measurements and reduced spectral density mapping
The longitudinal and transverse relaxation rates, as well as the {1H-15N} steady state NOEs were measured for 150 μm WT and S672R HCN4(563–724) samples in their apo-form and cAMP-bound form (2 mm cAMP). Pseudo-3D versions of the inversion recovery and CPMG pulse sequences modified to include a water flip-back pulse and sensitivity enhancement were used to measure T1 and T2 rates, respectively (52, 53). Both experiments included 1024 complex points with a spectral width of 14.28 ppm in the t2 dimension and 256 complex points with a spectral width of 31.82 ppm in the t1 dimension. The T1 experiment was acquired with 16 scans, 128 dummy scans, a recycle delay of 2.5 s, and the following relaxation delays: 100 ms (×2); 200, 300, and 400 ms (×2); and 500, 600, 800, and 1000 ms (×2) (where ×2 indicates duplicate spectra). The T2 experiment was acquired with 32 scans, 128 dummy scans, a recycle delay of 1.8 s, and the following CPMG relaxation delays: 8.42 and 16.96 ms (×2); 25.44, 33.92, and 42.40 ms (×2); and 50.88, 59.36, 76.32, and 93.28 ms (×2). The HN NOE data for WT had been previously acquired (9), and for the S672R mutant the HN NOE were measured similarly to WT. In brief, the HN-NOE experiment used a 10-s recycle delay with a 5-s proton saturation period. NOE experiments were acquired in 10–12 sets of saturated and unsaturated spectra, which were co-added and processed similarly to the HSQC spectra noted above. The HN-NOEs were then computed from the intensity ratios of saturated/non-saturated peaks.
Error bars were determined from replicate measurements. T1 and T2 experiments were processed in Topspin using cosine squared window functions and residual water deconvolution. Spectra were then transferred to Sparky and fitted to an exponential decay to extract T1 and T2 rates. Reduced spectral densities were computed as described previously (24, 25). Significant changes in dynamics were identified by computing the average spectral density values for all residues along with their standard deviations (S.D.) and by excluding from subsequent averaging calculations any residues outside the average ± 1 S.D. This computation of progressively trimmed averages and standard deviations was completed three times, after which any further iteration resulted only in marginal changes, i.e. ∼1%. At this point, the average spectral density was considered to arise primarily from overall molecular tumbling as opposed to internal motions.
The assessment of spectral densities reflecting overall tumbling served as a reference to identify significant S672R versus WT variations in internal dynamics based on the following criteria. (i) The spectral density of a residue for either WT or S672R has to be outside the average ± 5 S.D. (ii) The S672R versus WT spectral density change for a given residue must exceed five times the S.D. obtained from the overall tumbling assessment or, if greater, the sum of the respective experimental errors. Residues that met such criteria were deemed subject to significant S672R versus WT changes in internal dynamics and mapped onto the crystal structure of S672R HCN4. Residues undergoing S672R versus WT enhancement (suppression) in the picosecond to nanosecond backbone dynamics were identified through increases (decreases) in J(ωH+ωN), whereas residues with S672R versus WT enhancement (suppression) in millisecond to microsecond backbone dynamics were identified through increases (decreases) in J(0), typically with values above those computed for overall tumbling, in the absence of J(ωH+ωN) changes, as well as through 850 versus 700 MHz R2 comparisons. The T2 relaxation rates were also measured on a Bruker Avance 850 MHz spectrometer equipped with a TXI probe to further examine residues with significant contributions from Rex arising from dynamics in the millisecond to microsecond time scale. The pulse sequence employed for this analysis was a pseudo-3D version of the CPMG experiment with sensitivity enhancement and a water flip-back pulse. Spectra were acquired with 2048 complex points and a spectral width of 14 ppm in the direct dimension and 256 complex points and a spectral width of 36 ppm in the indirect dimension. Six CPMG relaxation delays were acquired in duplicate: 16.96, 33.92, 50.88, 67.84, 84.80, and 101.76 ms. The comparison of R2 rates at 850 and 700 MHz are shown in supplemental Fig. S7. The difference in R2 rates at 850 and 700 MHz was then computed, and significant changes in microsecond to millisecond dynamics were identified using an approach similar to the one described above for the reduced spectral density analysis. The S2 order parameters were calculated from J(0) and J(ωN+ωH) as described previously (54, 55). The S2 distributions were modeled through non-linear fitting with both one and two Gaussian curves. Further details are available in supplemental Table S1.
Measurements of binding thermodynamics and kinetics
Binding affinities of cAMP for the WT and S672R HCN4 CBD were determined by measuring binding isotherms through the STD amplification factor, as explained previously (9). For the assessment of the binding kinetics through protein-based experiments, Nz experiments could not be implemented due to weak holo peaks at cAMP concentrations sufficiently low to ensure slow exchange (supplemental Materials and Methods). Hence, a simpler and less quantitative approach was taken based on the analysis of gradient- and sensitivity-enhanced {15N,1H} HSQC spectra, as explained under “Results.” These HSQC datasets were acquired with 1024 complex points and a spectral width of 14.1 ppm in the directly detected 1H dimension and 128 complex points and a spectral width of 31.8 ppm in the indirectly detected 15N dimension. Spectra were processed employing a 60° shifted sine squared bell window function, zero filling, and linear prediction in the indirect dimension. Titrations were acquired with 100 μm total protein concentrations and total cAMP concentrations of 25, 50, 100, 200, 400, 600, 1000, and 1500 μm. The HSQC-based assessment of the cAMP-binding kinetics was complemented by acquiring 2D-ROESY spectra, recorded with mixing times in the 15–50 ms range in the presence of excess ligand. Spectra were acquired with spectral widths of 12 ppm in both dimensions and 2048 and 512 complex t2 and t1 points, respectively. The strength of the continuous wave spin-lock strength was set at 2.5 kHz. The cAMP off kinetics was probed through the initial slope of the plot of the ratio of the bound-to-free (b → f) ROE cross-peak intensity to the bound ligand diagonal peak intensity versus the ROESY mixing times.
Author contributions
S. B., M. A., and G. M. designed the research, S. B., M. A., and S. A. performed the research, S. B. and G. M. analyzed the data and wrote the paper.
Supplementary Material
Acknowledgments
We thank Professors Eric Accili (University of British Columbia), L. Zhou (Virginia Commonwealth University), W. N. Zagotta (University of Washington), Anna Moroni (University of Milan), D. Di Francesco (University of Milan), and Susan S. Taylor (University of California at San Diego) as well as B. VanSchouwen, J. Byun, R. Ahmed, N. Jafari, and M. Gloyd (McMaster) for helpful discussions.
This work was supported by Canadian Institutes of Health Research Grant MOP-68897 (to G. M.) and Natural Sciences and Engineering Research Council of Canada Grant RGPIN-2014-04514 (to G. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This paper is dedicated to the memory of our colleague Alex D. Bain (1948–2016), a brilliant NMR spectroscopist and insightful mentor.
This article was selected as one of our Editors' Picks.
This article contains supplemental Tables S1–S3, Equations S1–S3, Figs. S1–S8, and Materials and Methods.
- HCN
- hyperpolarization-activated cyclic nucleotide-modulated channels
- BBR
- base-binding region
- CBD
- cAMP-binding domain
- CCS
- combined chemical shift (ppm)
- CHESPA
- chemical shift projection analysis
- CTL
- C-terminal lid
- FA
- fluorescence anisotropy
- FEL
- free energy landscape
- HSQC
- heteronuclear single quantum coherence spectroscopy
- IR
- intracellular region
- ITC
- isothermal titration calorimetry
- PBC
- phosphate-binding cassette
- S672R
- HCN4 Ser-672 variant (amino acids 563–724)
- STD
- saturation transfer difference
- TMD
- trans-membrane domain
- aa
- amino acid
- r.m.s.d.
- root mean square deviation
- PDB
- Protein Data Bank
- Ni-NTA
- nickel-nitrilotriacetic acid
- TEV
- tobacco etch virus
- NNE
- nearest neighbor effect
- ROESY
- rotating frame nuclear Overhauser effect spectroscopy.
References
- 1. Benarroch E. E. (2013) HCN channels: function and clinical implications. Neurology 80, 304–310 [DOI] [PubMed] [Google Scholar]
- 2. Craven K. B., and Zagotta W. N. (2006) CNG and HCN channels: two peas, one pod. Annu. Rev. Physiol. 68, 375–401 [DOI] [PubMed] [Google Scholar]
- 3. Wicks N. L., Wong T., Sun J., Madden Z., and Young E. C. (2011) Cytoplasmic cAMP-sensing domain of hyperpolarization-activated cation (HCN) channels uses two structurally distinct mechanisms to regulate voltage gating. Proc. Natl. Acad. Sci. U.S.A. 108, 609–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wainger B. J., DeGennaro M., Santoro B., Siegelbaum S. A., and Tibbs G. R. (2001) Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411, 805–810 [DOI] [PubMed] [Google Scholar]
- 5. Zagotta W. N., Olivier N. B., Black K. D., Young E. C., Olson R., and Gouaux E. (2003) Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 425, 200–205 [DOI] [PubMed] [Google Scholar]
- 6. Ng L. C., Putrenko I., Baronas V., Van Petegem F., and Accili E. A. (2016) Cyclic purine and pyrimidine nucleotides bind to the HCN2 ion channel and variably promote C-terminal domain interactions and opening. Structure 24, 1629–1642 [DOI] [PubMed] [Google Scholar]
- 7. Lolicato M., Nardini M., Gazzarrini S., Möller S., Bertinetti D., Herberg F. W., Bolognesi M., Martin H., Fasolini M., Bertrand J. A., Arrigoni C., Thiel G., and Moroni A. (2011) Tetramerization dynamics of C-terminal domain underlies isoform-specific cAMP gating in hyperpolarization-activated cyclic nucleotide-gated channels. J. Biol. Chem. 286, 44811–44820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu X., Vysotskaya Z. V., Liu Q., and Zhou L. (2010) Structural basis for the cAMP-dependent gating in the human HCN4 channel. J. Biol. Chem. 285, 37082–37091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Akimoto M., Zhang Z., Boulton S., Selvaratnam R., VanSchouwen B., Gloyd M., Accili E. A., Lange O. F., and Melacini G. (2014) A mechanism for the auto-inhibition of hyperpolarization-activated cyclic nucleotide-gated (HCN) channel opening and its relief by cAMP. J. Biol. Chem. 289, 22205–22220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seifert R. (2016) Distinct signaling roles of cIMP, cCMP, and cUMP. Structure 24, 1627–1628 [DOI] [PubMed] [Google Scholar]
- 11. Schweizer P. A., Duhme N., Thomas D., Becker R., Zehelein J., Draguhn A., Bruehl C., Katus H. A., and Koenen M. (2010) cAMP sensitivity of HCN pacemaker channels determines basal heart rate but is not critical for autonomic rate control. Circ. Arrhythm. Electrophysiol. 3, 542–552 [DOI] [PubMed] [Google Scholar]
- 12. Taraska J. W., Puljung M. C., Olivier N. B., Flynn G. E., and Zagotta W. N. (2009) Mapping the structure and conformational movements of proteins with transition metal ion FRET. Nat. Methods 6, 532–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. VanSchouwen B., Akimoto M., Sayadi M., Fogolari F., and Melacini G. (2015) Role of dynamics in the autoinhibition and activation of the hyperpolarization-activated cyclic nucleotide-modulated (HCN) ion channels. J. Biol. Chem. 290, 17642–17654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou L., and Siegelbaum S. A. (2007) Gating of HCN channels by cyclic nucleotides: residue contacts that underlie ligand binding, selectivity, and efficacy. Structure 15, 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verkerk A. O., and Wilders R. (2014) Pacemaker activity of the human sinoatrial node: effects of HCN4 mutations on the hyperpolarization-activated current. Europace 16, 384–395 [DOI] [PubMed] [Google Scholar]
- 16. Baruscotti M., Bucchi A., Milanesi R., Paina M., Barbuti A., Gnecchi-Ruscone T., Bianco E., Vitali-Serdoz L., Cappato R., and DiFrancesco D. (2017) A gain-of-function mutation in the cardiac pacemaker HCN4 channel increasing cAMP sensitivity is associated with familial inappropriate sinus tachycardia. Eur. Heart J. 38, 280–288 [DOI] [PubMed] [Google Scholar]
- 17. DiFrancesco D. (2013) Funny channel gene mutations associated with arrhythmias. J. Physiol. 591, 4117–4124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Milanesi R., Baruscotti M., Gnecchi-Ruscone T., and DiFrancesco D. (2006) Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N. Engl. J. Med. 354, 151–157 [DOI] [PubMed] [Google Scholar]
- 19. Schulze-Bahr E., Neu A., Friederich P., Kaupp U. B., Breithardt G., Pongs O., and Isbrandt D. (2003) Pacemaker channel dysfunction in a patient with sinus node disease. J. Clin. Invest. 111, 1537–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu X., Marni F., Wu S., Su Z., Musayev F., Shrestha S., Xie C., Gao W., Liu Q., and Zhou L. (2012) Local and global interpretations of a disease-causing mutation near the ligand entry path in hyperpolarization-activated cAMP-gated channel. Structure 20, 2116–2123 [DOI] [PubMed] [Google Scholar]
- 21. VanSchouwen B., Selvaratnam R., Giri R., Lorenz R., Herberg F. W., Kim C., and Melacini G. (2015) Mechanism of cAMP partial agonism in protein kinase G (PKG). J. Biol. Chem. 290, 28631–28641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akimoto M., Selvaratnam R., McNicholl E. T., Verma G., Taylor S. S., and Melacini G. (2013) Signaling through dynamic linkers as revealed by PKA. Proc. Nat. Acad. Sci. U.S.A. 110, 14231–14236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boulton S., Akimoto M., VanSchouwen B., Moleschi K., Selvaratnam R., Giri R., and Melacini G. (2014) Tapping the translation potential of cAMP signalling: molecular basis for selectivity in cAMP agonism and antagonism as revealed by NMR. Biochem. Soc. Trans. 42, 302–307 [DOI] [PubMed] [Google Scholar]
- 24. Das R., Chowdhury S., Mazhab-Jafari M. T., Sildas S., Selvaratnam R., and Melacini G. (2009) Dynamically driven ligand selectivity in cyclic nucleotide binding domains. J. Biol. Chem. 284, 23682–23696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Das R., Mazhab-Jafari M. T., Chowdhury S., SilDas S., Selvaratnam R., and Melacini G. (2008) Entropy-driven cAMP-dependent allosteric control of inhibitory interactions in exchange proteins directly activated by cAMP. J. Biol. Chem. 283, 19691–19703 [DOI] [PubMed] [Google Scholar]
- 26. Das R., and Melacini G. (2007) A model for agonism and antagonism in an ancient and ubiquitous cAMP-binding domain. J. Biol. Chem. 282, 581–593 [DOI] [PubMed] [Google Scholar]
- 27. Selvaratnam R., Chowdhury S., VanSchouwen B., and Melacini G. (2011) Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Nat. Acad. Sci. U.S.A. 108, 6133–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Selvaratnam R., Mazhab-Jafari M. T., Das R., and Melacini G. (2012) The auto-inhibitory role of the EPAC hinge helix as mapped by NMR. PLoS One 7, e48707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Selvaratnam R., VanSchouwen B., Fogolari F., Mazhab-Jafari M. T., Das R., and Melacini G. (2012) The projection analysis of NMR chemical shifts reveals extended EPAC autoinhibition determinants. Biophys. J. 102, 630–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boulton S., and Melacini G. (2016) Advances in NMR methods to map allosteric sites: from models to translation. Chem. Rev. 116, 6267–6304 [DOI] [PubMed] [Google Scholar]
- 31. Nussinov R. (2016) Introduction to protein ensembles and allostery. Chem. Rev. 116, 6263–6266 [DOI] [PubMed] [Google Scholar]
- 32. Ma B., and Nussinov R. (2016) Protein dynamics: conformational footprints. Nat. Chem. Biol. 12, 890–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guo J., and Zhou H.-X. (2016) Protein allostery and conformational dynamics. Chem. Rev. 116, 6503–6515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo C., and Zhou H. X. (2016) Unidirectional allostery in the regulatory subunit RIα facilitates efficient deactivation of protein kinase A. Proc. Nat. Acad. Sci. U.S.A. 113, E6776–E6785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo J., and Zhou H.-X. (2015) Dynamically driven protein allostery exhibits disparate responses for fast and slow motions. Biophys. J. 108, 2771–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Greenwood A. I., Rogals M. J., De S., Lu K. P., Kovrigin E. L., and Nicholson L. K. (2011) Complete determination of the Pin1 catalytic domain thermodynamic cycle by NMR lineshape analysis. J. Biomol. NMR 51, 21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cembran A., Kim J., Gao J., and Veglia G. (2014) NMR mapping of protein conformational landscapes using coordinated behavior of chemical shifts upon ligand binding. Phys. Chem. Chem. Phys. 16, 6508–6518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Omidvar R., Xia Y., Porcelli F., Bohlmann H., and Veglia G. (2016) NMR structure and conformational dynamics of AtPDFL2.1, a defensin-like peptide from Arabidopsis thaliana. Biochim. Biophys. Acta 1864, 1739–1747 [DOI] [PubMed] [Google Scholar]
- 39. Kleckner I. R., and Foster M. P. (2011) An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 1814, 942–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moleschi K. J., Akimoto M., and Melacini G. (2015) Measurement of state-specific association constants in allosteric sensors through molecular stapling and NMR. J. Am. Chem. Soc. 137, 10777–10785 [DOI] [PubMed] [Google Scholar]
- 41. Boulton S., Akimoto M., Selvaratnam R., Bashiri A., and Melacini G. (2014) A tool set to map allosteric networks through the NMR chemical shift covariance analysis. Sci. Rep. 4, 7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Farrow N. A., Zhang O., Forman-Kay J. D., and Kay L. E. (1994) A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium. J. Biomol. NMR 4, 727–734 [DOI] [PubMed] [Google Scholar]
- 43. Ulens C., and Siegelbaum S. A. (2003) Regulation of hyperpolarization-activated HCN channels by cAMP through a gating switch in binding domain symmetry. Neuron 40, 959–970 [DOI] [PubMed] [Google Scholar]
- 44. VanSchouwen B., and Melacini G. (2016) Structural basis of tonic inhibition by dimers of dimers in hyperpolarization-activated cyclic-nucleotide-modulated (HCN) ion channels. J. Phys. Chem. 120, 10936–10950 [DOI] [PubMed] [Google Scholar]
- 45. Schuetz A. K., and Kay L. E. (2016) A dynamic molecular basis for malfunction in disease mutants of p97/VCP. eLife 5, e20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sekhar A., Rumfeldt J. A. O., Broom H. R., Doyle C. M., Sobering R. E., Meiering E. M., and Kay L. E. (2016) Probing the free energy landscapes of ALS disease mutants of SOD1 by NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 116, E6939–E6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J., Masterson L. R., Cembran A., Verardi R., Shi L., Gao J., Taylor S. S., and Veglia G. (2015) Dysfunctional conformational dynamics of protein kinase A induced by a lethal mutant of phospholamban hinder phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 112, 3716–3721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nussinov R., and Tsai C.-J. (2013) Allostery in disease and in drug discovery. Cell 153, 293–305 [DOI] [PubMed] [Google Scholar]
- 49. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 50. Goddard T. D., and Kneller D. G. (2008) Sparky3, University of California, San Francisco [Google Scholar]
- 51. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Loh A. P., Guo W., Nicholson L. K., and Oswald R. E. (1999) Backbone dynamics of inactive, active, and effector-bound Cdc42Hs from measurements of 15N relaxation parameters at multiple field strengths. Biochemistry 38, 12547–12557 [DOI] [PubMed] [Google Scholar]
- 53. Wang C., Pawley N. H., and Nicholson L. K. (2001) The role of backbone motions in ligand binding to the c-Src SH3 domain. J. Mol. Biol. 313, 873–887 [DOI] [PubMed] [Google Scholar]
- 54. Bracken C., Carr P. A., Cavanagh J., and Palmer A. G. 3rd (1999) Temperature dependence of intramolecular dynamics of the basic leucine zipper of GCN4: implications for the entropy of association with DNA. J. Mol. Biol. 285, 2133–2146 [DOI] [PubMed] [Google Scholar]
- 55. Popovych N., Sun S., Ebright R. H., and Kalodimos C. G. (2006) Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 13, 831–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koradi R., Billeter M., and Wüthrich K. (1996) MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.