

Abstract
Trends in increased tuberculosis infection and a fatality rate of ∼23% have necessitated the search for alternative biomarkers using newly developed postgenomic approaches. Here we provide a systematic analysis of Mycobacterium tuberculosis (Mtb) by directly profiling its gene products. This analysis combines high-throughput proteomics and computational approaches to elucidate the globally expressed complements of the three subcellular compartments (the cell wall, membrane, and cytosol) of Mtb. We report the identifications of 1044 proteins and their corresponding localizations in these compartments. Genome-based computational and metabolic pathways analyses were performed and integrated with proteomics data to reconstruct response networks. From the reconstructed response networks for fatty acid degradation and lipid biosynthesis pathways in Mtb, we identified proteins whose involvements in these pathways were not previously suspected. Furthermore, the subcellular localizations of these expressed proteins provide interesting insights into the compartmentalization of these pathways, which appear to traverse from cell wall to cytoplasm. Results of this large-scale subcellular proteome profile of Mtb have confirmed and validated the computational network hypothesis that functionally related proteins work together in larger organizational structures.
INTRODUCTION
Tuberculosis (TB) is an airborne infection caused by the bacterium Mycobacterium tuberculosis (Mtb). Its global incidence is growing at ∼3 million cases per annum, and various drug-resistant strains of Mtb are now emerging as new threats (WHO, 2003). The genome of the virulent strain, Mtb H37Rv, has been completely sequenced (Cole et al., 1998) and this provides an important resource for our understanding of the pathogenicity of this mycobacterium at the integrated systems level. So far proteomic studies of Mtb and other organisms have mainly involved two-dimensional PAGE (2DGE) to resolve proteins followed by mass spectrometry (MS) to identify them (Kaji et al., 2000; Taoka et al., 2000; Mattow et al., 2001a, 2001b;). However, because of the limitations of 2DGE-based separation methods, only 341 proteins have so far been identified in the Mtb proteome (Mollenkopf et al., 1999; Mattow et al., 2001a, 2001b) thus far, compared with the 3924 protein coding sequences (CDS) predicted from the genome (Camus et al., 2002). Only recently has SDS-PAGE been used to separate membrane proteins of Mtb for further liquid chromatography (LC)-MS/MS analyses (Gu et al., 2003a). Automated two-dimensional, capillary high-performance LC coupled with MS (2DLC/MS) has proven to be very efficient in the analysis of complex protein/peptide mixtures (Isobe et al., 1991). The unbiased nature of this technique has allowed the identification of different protein classes, including both hydrophobic and membrane proteins. It has been previously used for the sequence analysis of protein complexes and the proteomic profiling of other organisms (Takahashi et al., 1985; Link et al., 1999; Washburn et al., 2001; Kaji et al., 2003; Mawuenyega et al., 2003). Using 2DLC/MS we provide a comprehensive subcellular analysis of the protein complements isolated from the cell wall, membrane and cytosol of the Mtb H37Rv virulent strain. We further integrated this data with a comprehensive bioinformatic approach in a systematic investigation of possible functional networks/pathways critical for Mtb pathogenicity. These networks were initially drawn and predicted from previously known genetic and biochemical/metabolic information using a computational method called protein functional networks (Marcotte et al., 1999). On the basis of the proteins identified in the corresponding Mtb subcellular locations, we have redefined the functional context of these networks/pathways. Thus, our proteomic study reveals both the existence and localization of >25% of the Mtb proteome and validates these genome-based network predictions. The availability of the subcellular distributions of the global protein expression profile also allowed us to reconstruct certain functional networks responsible for lipid synthesis and degradation, a characteristic feature of this organism. This systematic analysis may lead to the identification of new targets (individual proteins or functional links) or virulence factors responsible for Mtb persistence and pathogenicity, which can be used both in the diagnosis of TB and for proteomics based vaccine development.
MATERIALS AND METHODS
Mtb Cell Culture and Sample Preparation
Mtb strain H37Rv was cultured in 2 L of glycerol alanine salts medium (Takayama et al., 1975) in roller bottles for 14 d at 37°C with gentle agitation, washed with phosphate-buffered saline (PBS) pH 7.4, and inactivated by gamma-irradiation. The culture supernatant and cells were first separated by filtration through a 0.22-mm membrane before the cells were disrupted in a French press. The disrupted cells were separated into cell wall, membrane, and cytosolic fractions by differential-centrifugation as previously described (Hirschfield et al., 1990; Lee et al., 1992) and as illustrated in Figure 1. To remove protein components that may be simply sticking together during the purification procedure, the cell wall and membrane pellets were washed with ice-cold PBS, then Tween 80, and then PBS again. Approximately 190 mg of cell wall protein, 13 mg of cell membrane protein, and 100 mg of cytosolic protein were recovered. The proteins in each of the fractions were deglycosylated with PNGase F as described by the manufacturer (New England Biolabs, Beverly, MA), treated with acetone to remove lipids, and digested with trypsin as previously described (Mawuenyega et al., 2003). The tryptic digests were dried in a speedvac and reconstituted in water twice and then in 5% acetonitrile, 0.1% formic acid solution, pH 3.0. The precipitates thus formed were removed by centrifugation and the various samples were subjected immediately to 2DLC-MS/MS analyses.
Figure 1.

Subcellular fractionation of Mtb cell lysate. Differential centrifugation was utilized to obtain three subcellular compartments: the cell wall, a membrane fraction, and a cytosolic fraction. The cell wall, a membrane fractions were extensively washed to remove loosely attached and potential contaminant proteins. The separation method is illustrated in the figure.
Automated 2DLC-MS/MS Analysis of Peptides
Each of the fractionated samples was separated on the 2DLC and subsequently analyzed using MS. The automated 2DLC separation of tryptic digests was carried out on a capillary 2DLC system per manufacturer's instructions (Micro-Tech Scientific, Sunnyvale, CA). In brief, the chromatography was carried out using strong cation exchange (SCX) in the first dimension, followed by reversed phase (RP) LC. The first SCX-LC was performed on a polysulfoethyl A, 320 μm id × 100 mm L, 5-μm particles column (PolyLC, Ellicott City, MD), and the second RP-LC was on Biobasic C18, 150 μm id × 100 mm L, 300 μm particles column (Thermo Hypersil-Keystone, Bellefonte, PA). The entire LC-MS system was controlled through Thermo Finnigan's Xcalibur software with a MicroTech Scientific's driver for the LC control. Approximately 20 μg of each fraction was loaded onto the columns and analyzed as described (Mawuenyega et al., 2003). The eluted peptides were sprayed into the LCQDECA mass spectrometer, equipped with a microflow electrospray interface. The total analysis time for a complete run on a sample was ∼17 h, after which bioinformatic searches and data processing were carried out to identify the proteins.
Protein Identification from Tandem Mass Spectrometry and Data Analyses
Electrospray ionization mass spectrometry was carried out using a Finnigan LCQDECA ion-trap mass spectrometer (Thermo Finnigan, San Jose, CA) that was operated by the method files in the sequence setup window of Xcalibur software. The temperature of the heated capillary was maintained at 200°C for 3 microscans at an injection time of 30 msec, in the TunePlus window. The instrument was operated in a data-dependent mode, with dynamic exclusion analysis set for a repeat count of 2 and a repeat duration of 0.5 min, in an exclusion list size of 25, with a 1.5-min exclusion duration window. A full MS scan was acquired between 300 and 1800 m/z followed by an MS/MS scan of the four most intense ions with collision energy of 35%. Amino acid sequences of peptides were inferred from the MS/MS spectra and the Bioworks/TurboSequest software (Thermo Finnigan) automated this process. A peptide was considered to be a match by utilizing the criteria described (Link et al., 1999; Washburn et al., 2001). Also, the database searches were repeated with a reversed Mtb H37Rv genome sequence database to validate the identified proteins and eliminate false positives. Often multiple peptides from the same protein were detected, which permitted completely unambiguous protein identification with a minimal examination of spectra.
Network/Pathway Construction and Analysis
A cellular network is described by a connected graph (i.e., labeled or color coded), with nodes (vertices) coding for genes and proteins that are connected by edges. These edges refer to different types of interactions, such as a physical contact between two proteins, or an interaction through chemical reactions. Small chemicals connected by edges in typical biochemical textbook pathways are nodes in the cellular networks. Conversely, chemicals that are nodes in the biochemical reaction network are edge-labels in the cellular network. A method was developed to identify the so-called “response networks” in cellular interaction networks, which are subgraphs of a larger network spanned by preselected “root nodes.” This method (Forst, 2004) (Mawuenyega, Forst, Dobos, Belisle, J. Chen, E. M. Bradbury, A.R.M. Bradbury, X. Chen, unpublished results) reconstructs cellular networks from genome-context data and external sources.
In detail, we represented the cellular network with proteins as nodes and interactions as edges. We labeled the interactions according to specific types (protein-protein interactions, or the name of chemical that is used in a subsequent reaction between the two proteins that are connected by this particular edge). To construct a cellular network from genomic data, we used the following approach. We first identified all genes that code for proteins. For this purpose we used the 2002 revised annotation provided by Cole et al. (1998). In a first iteration of the construction of a cellular network we intentionally included as many proteins as possible, consisting of annotated genes with known functions as well as conserved unknown proteins and hypothetical genes. In successive iterations we refined our criteria and restricted our set of proteins to those proteins with known interactions with other proteins. We used essentially two distinct computational methods to identify functional links between proteins from genomic data, the first being the Rosetta approach and the second a metabolic reaction method using enzyme annotations.
With respect to protein-protein interaction we used the Rosetta approach developed by Marcotte et al. (1999). Their hypothesis is based on the observation that individual genes in one organism that are fused in another organism into a single chain, the Rosetta sequence, probably have a functional relationship and may interact physically with each other. Huynen et al. (2000) performed a statistical analysis on Mycoplasma genitalium to study the significance of functional links identified among genes through the Rosetta method. They concluded that the identification of genes in one organism that match a fused gene in a second organism provide an 80% statistically significant evidence of a similar function of the gene pairs, a presence in a protein complex by 70%, and a physical interaction by 60%. By restricting predicted protein-protein interactions to interactions with absolute Z-scores of three or lower, Enright et al. (1999) utilized nonoverlapping sequence-domains and showed that no false positives exist in these cases. A Z-score is a statistical measure, indicating the distance and direction of a particular measurement from the mean, in units of standard deviations. Thus our prediction of true protein-protein interactions from fusion genes is statistically significant and close to 100% reliability. Second, we also combined the metabolic reaction data of Mtb from the BioCyc (Krieger et al., 2004) and KEGG databases (Ogata et al., 1999) with the protein interaction network of Mtb. These two metabolic network databases provide information on the annotated Mtb genome, enzyme, and reaction data as well as biochemical pathway and network information. Although the reaction and network information bases on almost a century long of experimental knowledge of biochemical pathways, the specific metabolic network information for Mtb found in these databases were constructed by computational means and thus are used with caution. Nevertheless, we are using this network information in this article and have verified the expression of the genes by our proteomic profile outlined in the results section. The full construction process yielded a network of ∼1000 unique nodes (proteins) and 70,000 unique interactions between these nodes. To construct a network with metabolic reactions relevant to fatty acid/lipid metabolism we applied a two-step process. First, we only considered metabolic reactions involving unambiguous substrates. Thus, ambiguous substrates, such as water, ATP, NADH, and ammonia (top 20 listed in Supplementary Figure 2), that function in many different reactions and, so, have a far higher degree of connectivity (>10) than specific substrates, were ignored. Consistent with this, we ignored reactions involving acyl-CoA, even though this substance possesses a relevant role in fatty acid/lipid metabolism. The acyl-CoA substance class is used in ∼270 reactions in our network and its inclusion obscures prominent visual features of the identified subnetworks without adding further information (the acyl-CoA network is available as Supplementary Figure 1). Protein interactions were labeled with assigned Z-scores that refer to statistical measures of similarity for each pair of interacting proteins (Marcotte, 2000; Enright and Ouzounis, 2001). Second, we pruned the large, computationally obtained network data by utilizing the proteomics profile information. For this purpose, we developed a method to identify experimentally relevant subnetworks (so-called response networks) in large cellular networks (Forst, 2004). The principal idea of this method is the superimposition of a large computed cellular network on a biological data, such as a catalogue of identified proteins through proteomic techniques. This method in itself is capable of scoring subnetworks in cellular networks, by using a variety of experimental data, e.g., expression profiles. However, we did not take this approach, but explored the method's capability to identify pathways and subnetworks by “tagging” specific, experimentally identified proteins and by finding pathways in the network between these tagged proteins. In detail, we chose a set of identified proteins, 33 and 8, respectively, from our proteomics profile, shown to be involved in fatty acid degradation and lipid biosynthesis pathways, and tagged the corresponding nodes in the network. We referred to these tagged nodes as “root nodes and are differently colored in Figure 6. By a graph-theoretical approach, we then identified pathways between all possible pairs of root nodes. Specifically we used the method of finding “k-shortest paths” between a pair of nodes using the Recursive Enumeration Algorithm (REA) by Jiménez and Marzal (Jiménez, 1999). For the purpose of finding k-shortest paths, without further experimental data such as expression values (see above), we considered all edges in the network as of equal importance, e.g., length = 1. The REA algorithm essentially provides us with the shortest and alternative longer paths between two root-node pairs. To control the complexity of the obtained k-shortest paths, we introduced a pathway length restriction. Whenever the length of a computed pathway exceeds a particular, predefined length l, this particular path is disregarded and not included in the subnetwork. The collective set of all identified pathways between these root nodes then defines a subnetwork in the larger cellular network.
Figure 6.

Response networks and subnetworks created by computational methods. The overview pathways were created for (A) the fatty acid degradation and (B) lipid biosynthesis. In both networks, rectangular nodes represent proteins classified as belonging to the fatty acid degradation pathway, oval nodes as belonging to the lipid biosynthesis (The Wellcome Trust Sanger Institute), and hexagonal nodes as belonging to neither. Red node proteins were identified in the cytoplasm, blue in the cell wall, and green in the membrane. Proteins found in both the cell membrane and cell wall were colored pale blue, and proteins found in both membrane and cytoplasm were colored orange. No common proteins were found in the cell wall and cytoplasm, which validates the fractionation procedure and reflects a true compartmentalization of the proteins. White node proteins were not identified. Black connections represent those identified biochemically (Marcotte et al., 1999), whereas green connections represent Rosetta Stone predictions (Marcotte, 2000; Enright and Ouzounis, 2001). The pathway involving accA3, accD3, fabD, kasA, Rv2182c, kasB, and fabD was found in both networks in A and B.
In summary, the algorithm described in the sections “Response Network Analysis” and “Network Scoring” is used to perform the following procedure: i) construct a large biological network from genomic information and interaction data, ii) compile a list of root nodes, iii) compute all possible pairs of root nodes from above list, iv) calculate shortest and k-shortest paths between each pair of root nodes using Dijkstra and REA algorithms with a restricted maximal path-length l, v) record all nodes and edges on identified paths with desired and provided maximal path-length, vi) filter, i.e., delete or hide all other nodes and edges that are not on the selected paths, and vii) reiterate the procedure for refinement. Figure 2 shows a simple flow diagram of the algorithm with references to the individual steps.
Figure 2.

Flow diagram of the Dijkstra and REA algorithms used to identify response networks. Small cap roman numbers at each subprocess refer to specific tasks discussed in the text.
By mathematical means, the above procedure yields a subnetwork within the large biological network that is spanned by root nodes and the pathways between each pair of root nodes that hold the provided properties of k-shortest paths with maximal, overall path length l. Because of the comprehensive proteomic analysis, most of the proteins along a particular pathway between tagged proteins have been identified and thus added value to the constructed network. However, we did find proteins in the subnetwork predictions that were not identified in our proteomic profile (proteins not colored in Figure 6), but are true predictions of the computationally based network construction method.
Response Network Analysis
The following definitions are used.
Definition 1. A “typical” graph Γ = (V, E) = (V(Γ), E(Γ)) consists of a vertex set V with vertices (or nodes) v ∈ V and an edge set E ⊆ V with edges ε ∈ E.
Populating a graph Γ with biological information defines a biological network N with following definition:
Definition 2. Let N = N(V, E, π()) be a network with vertices (nodes) v ∈ V and edges ε ∈ E as well as a function π: X → P that maps vertices and edges, onto their respective properties, p ∈ P, X ∈{V, E}.
In the case of biological networks, depending on the particular network representation, node properties include gene, protein or chemical names, edge properties may refer to specific interactions, such as binding or catalysis.
The mapping π: X → P is at least subjective, because for all p ∈ P, there exists an x ∈ X with π(x) = p.
Network Scoring
Network scoring uses experimental values (such as expression values or uniform weights as in our case) as metrics for weighted edges in the network. Depending on the coding of the biological network as graph, either genes, which expression are measured, are coded as edged in the graph, or genes are represented as nodes and an edge is weighted by the mean expression value of the two genes that are connected by the particular edge. In the case that the biological network is represented as edge graph, edges representing genes, proteins and other cellular components and nodes referring to interactions, in a node graph, genes, proteins and other cellular components are coded as nodes in the biological network, connected by edges. Because the subnetwork filter algorithm assumes weights on edges for scoring, such edge-weights have to be calculated from node scores.
To calculate a total score of the subnetwork N we then sum the weighted edges zm over all m given the constraint of the k-shortest paths with maximal path-length l between each two root nodes in N. As mentioned above in our case of a protein network without specific expression values all zm are set to 1:
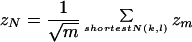 |
Given a particular set of root nodes, the shortest path approach already guarantees a best scored subnetwork.
RESULTS
Comparative Proteomics View of the Mycobacterium Subcellullar Compartments
Each fraction was analyzed twice under the same conditions and we report those nonredundant proteins that were identified on both occasions. We used a fully integrated and automatic analytical platform for the gel-free, large-scale identification of 1044 nonredundant proteins that gave 703 (67%) more proteins than those found by 2DGE approaches (Supplementary Table). For example, the most acidic protein (PE_PGRS, Rv3512) has an isoelectric point (pI) of 3.89, whereas the most basic (rps2, a 30S ribosomal protein) has a pI of 12.18. We also identified low molecular mass (Mr) proteins and those at the extremes of high Mr range; e.g., the 230,621-Da polyketide synthase (ppsC). Based on these findings and others (unpublished data), the 2DLC-MS/MS approach should ideally be able to detect more than 99% of Mtb gene products, on the basis of predicted Mr and pI (Mawuenyega et al., 2003). Our dataset represents a first step toward an exhaustive analysis of the mycobacterial proteins at the extremes of pI and Mr. Although 2DGE should be able to detect proteins with an Mr between 8 and 200 kDa and a pI between 4 and 10, previous 2DGE studies of the Mtb proteome have identified only ∼30 proteins larger than 50 kDa and an even smaller number of proteins with pI values higher than 9 (Jungblut et al., 1999). In a recent study of the Mtb membrane fraction using the SDS-PAGE LC-MS/MS approach, the number of membrane proteins identified, 739 (Gu et al., 2003a), compares very well with the 705 proteins identified in the membrane fraction in this study. This is evident in the wide range of proteins identified at the extremes of pI (∼3.5-12 in both cases) and molecular mass (∼6 to 300 kDa in both cases). The number of proteins identified in the three fractions is summarized in Figure 3. Interestingly, the 705 proteins in the membrane fraction represented more than half of the total number of proteins identified, and shared ∼20% of its proteins with other fractions. Of the 705 membrane proteins, 464 were unique. In comparison, 306 and 356 proteins were identified in the cell wall and cytosol fractions, respectively. As shown in Figure 3, the cell wall fraction had 131 unique proteins, whereas the cytosolic fraction had 173. Only 47 proteins, representing ∼4.5% of the total proteins identified, were found in all three fractions showing a very low probability of cross-contamination. In summary, 1044 nonredundant Mtb H37Rv proteins, representing 26% of the coding sequences (CDSs) predicted from the genome, were profiled from these three subcellular compartments. We did not include a very large number of proteins identified with one peptide, as their validity could not be verified. Also, hundreds of MS/MS spectra remain unassigned, due to their failure to meet the TurboSequest results acceptance criteria. We hope to eliminate these shortcomings with on-going projects to “mass-tag” the Mtb proteome and these peptides by metabolic incorporation of specific stable isotope amino acids. Therefore, more proteins may be amenable to identification (Gu et al., 2002, 2003b).
Figure 3.

A Venn diagram indicating the distribution of proteins identified. Peptides identified from the analysis of each fraction were matched to 1044 nonredundant proteins. The number of proteins detected in the cytosolic fraction was 356 (173 unique), cell wall 306 (131 unique), and the membrane 705 (464 unique).
Functional Characterization of the Proteins Expressed in Individual Cellular Compartments
All the identified proteins (Supplementary Table) were arranged according to their cellular localizations and sorted based on the hierarchical list of predicted protein-coding genes organized by functional category and summarized in Table 1 (The Wellcome Trust Sanger Institute). The H37Rv strain has a total of 3924 protein CDSs (Cole et al., 1998) of which ∼40% have predicted biological roles and another 44% have similarity to proteins in other species. The remaining 16% are unique to mycobacteria. For each functional category in Table 1, the number of proteins identified in the cytosol, membrane, and cell wall fractions were compared with the predicted products of the CDSs in the Mtb genome. In most cases, the percentages of identified functional proteins mirrored those of the functional groups predicted for the genome as a whole (Table 1 and Figure 4). However, gene products characterized as “unknown,” “hypothetical conserved” or as belonging to the PE/PPE family were significantly under-represented (11 vs. 15%, 19 vs. 23%, and 1 vs. 4%, respectively), whereas those identified as being involved in energy metabolism, synthesis and modification of macromolecules, and degradation were all overrepresented (10 vs. 7%, 11 vs. 5%, and 7 vs. 4%, respectively). For those predicted CDSs, which were undetected by MS, their low abundances might be beyond the detection limit of our current methods, or they may not be expressed under the experimental conditions used. Furthermore, under-representation may reflect erroneous prediction.
Table 1.
Functional profiles of expressed MTB proteins
| Cellular roles | Cell wall | Membrane | Cytosol | Proteome (%) | Genome (%) |
|---|---|---|---|---|---|
| V Hypothetical conserved | 54 | 131 | 51 | 195 (19) | 915 (23) |
| V.I Unknown | 34 | 65 | 40 | 111 (11) | 606 (15) |
| II.C Cell envelope | 23 | 55 | 27 | 83 (8) | 360 (9) |
| I.B Energy Metabolism | 31 | 71 | 45 | 109 (10) | 292 (7) |
| II.A Synthesis and modification of macromolecules | 51 | 77 | 48 | 117 (11) | 215 (5) |
| I.J Broad regulatory functions | 11 | 34 | 18 | 52 (5) | 187 (5) |
| IV.C PE and PPE families | 2 | 8 | 2 | 10 (1) | 167 (4) |
| I.A Degradation | 12 | 50 | 20 | 68 (7) | 163 (4) |
| IV.B IS elements, Repeated sequences, and Phage | 10 | 18 | 11 | 28 (3) | 135 (3) |
| III.A Transport/binding proteins | 4 | 24 | 8 | 27 (3) | 123 (3) |
| I.G Biosynthesis of cofactors, prosthetic groups and carriers | 5 | 22 | 9 | 32 (3) | 117 (3) |
| I.D Amino acid biosynthesis | 11 | 26 | 12 | 33 (3) | 95 (2) |
| II.B Degradation of macromolecules | 5 | 16 | 9 | 25 (2) | 87 (2) |
| I.H Lipid Biosynthesis | 9 | 12 | 6 | 18 (2) | 66 (2) |
| IV.H Miscellaneous transferases | 3 | 14 | 3 | 15 (1) | 61 (2) |
| I.F Purines, pyrimidines, nucleosides and nucleotides | 4 | 13 | 7 | 18 (2) | 60 (2) |
| I.C Central intermediary metabolism | 4 | 9 | 5 | 16 (2) | 45 (1) |
| I.I Polyketide and non-ribosomal peptide synthesis | 9 | 18 | 10 | 19 (2) | 41 (1) |
| IV.A Virulence | 1 | 8 | 2 | 10 (1) | 38 (1) |
| III.F Detoxification | 1 | 5 | 5 | 8 (1) | 22 (1) |
| IV.F Cytochrome P450 enzymes | 1 | 2 | 1 | 4 (<1) | 22 (1) |
| III.C Cell division | 5 | 6 | 4 | 11 (1) | 19 (<1) |
| IV.I Miscellaneous phosphatases, lyases, and hydrolases | 2 | 2 | 2 | 6 (1) | 18 (<1) |
| III.B Chaperones/Heat shock | 6 | 9 | 4 | 10 (1) | 16 (<1) |
| III.D Protein and peptide secretion | 2 | 4 | 3 | 6 (1) | 14 (<1) |
| IV.D Antibiotic production and resistance | 0 | 1 | 0 | 1 (<1) | 14 (<1) |
| III.E Adaptations and atypical conditions | 3 | 4 | 0 | 6 (1) | 12 (<1) |
| IV.J Cyclases | 1 | 1 | 2 | 2 (<1) | 6 (<1) |
| IV.E Bacteriocin-like proteins | 0 | 0 | 2 | 2 (<1) | 3 (<1) |
| IV.G Coenzyme F420-dependent enzymes | 0 | 0 | 0 | 0 | 3 (<1) |
| IV.K Chelatases | 1 | 0 | 0 | 1 (<1) | 2 (<1) |
| I.E Polyamine synthesis | 1 | 0 | 0 | 1 (<1) | 1 (<1) |
| 306 | 705 | 356 | 1044 (100) | 3925 (100) |
The identified proteins, arranged to reflect their cellular localizations in the Supplementary Table, were sorted into various functional classes based on the hierarchical list of predicted protein-coding genes arranged by functional category at the The Wellcome Trust Sanger Institute, UK. The number of nonredundant proteins identified in the three compartments were summed up into the proteome, expressed as a percentage and compared with that of the genome. A graphical illustration is shown in Figure 4.
Figure 4.

The plot of variations in functional distribution of genome-encoded proteins compared with those of the proteome. The numerical labels on the pie charts correspond to the functional classes shown on the right-hand side. The sizes of the various pies of the chart show varying percentages of proteins predicted in the genome or identified in our proteome profile. Functional classes listed in Table 1 that did not vary in both the genome and proteome were grouped together and listed under “Others,” which include (I.J) Broad regulatory functions, (IV.B) IS elements, Repeated sequences, and Phage, (III.A) Transport/binding proteins, (I.G) Biosynthesis of cofactors, prosthetic groups and carriers, (II.B) Degradation of macromolecules, (I.H) Lipid Biosynthesis, (I.F) Purines, pyrimidines, nucleosides and nucleotides, (IV.A) Virulence, (III.F) Detoxification, (IV.D) Antibiotic production and resistance, (IV.J) Cyclases, (IV.E) Bacteriocin-like proteins, (IV.G) Coenzyme F420-dependent enzymes, (IV.K) Chelatases, and (I.E) Polyamine synthesis.
Our procedure for subcellular fractionation described in Figure 1 has proved to be effective and reproducible in separating the cell wall and membrane proteins from those of the cytosol, with known protein markers being found in their appropriate compartments. An attractive feature of the subcellular fractionation is that it identified the cellular localization of proteins and protein complexes, regardless of their solubility. However, it was found that the isolated membrane fraction also contained membrane proteins from the cytoplasm and the outer lipid layer. Similarly, the cytosolic fraction contained soluble material released from the cell wall during disruption of the bacilli (Takayama et al., 1975; Hirschfield et al., 1990; Lee et al., 1992). With the exception of these documented incidences, great care was taken to maintain the cellular localization of the proteins in their subcellular compartments. The presence of proteins at unexpected cellular localizations may have biological significance, e.g., cross-trafficking, rather than be a result of cross-contamination during fractionation. Cross-contamination was shown by the presence of 47 proteins (4.5%) in all compartments. Most of these were housekeeping proteins involved in macromolecule metabolism pathways, such as macromolecule synthesis and modifications (12 proteins in functional class II.A), polyketide and nonribosomal peptide synthesis (5 proteins in the class I.I), hypothetical conserved proteins (5 proteins in the class V), and others. The polyketide synthases mentioned above were probably released into the cytosolic and membrane fractions because of their solubility in the protein extraction buffer. These are highly expressed proteins that produce phthiocerol dimycocerosate, a very abundant small molecule in the cell wall. As stated above, these proteins account for <5% of all proteins identified in all three fractions and, therefore, are not a significant number. In the following sections we describe representative proteins identified from each subcellular fraction, inferred to be involved in the persistence/pathogenicity of Mtb.
Cytosol
The cytosolic fraction contains mainly cytoplasmic soluble proteins together with those released from the membrane and cell wall during the disruption of the bacilli. The unique proteins were largely involved in energy metabolism (17%). This finding is reasonable as most of the proteins involved in energy metabolism are located in the cytosol. In the cytoplasm's profile, these consist of NADH dehydrogenases and other miscellaneous oxidoreductases and oxygenases. A few transmembrane (TM) proteins were detected in all three fractions, e.g., (MmpL11 [TM 12] and mmpL10 [TM 11]), and some such as the cation-transporting P-type ATPases ctpB (TM 7) and ctpH (TM 5), were found in both the membrane and cytosol. Of note were those TM proteins found only in the cytosol, which also appeared to have intact signaling peptides, e.g., mmpL5 (TM 12), mmpL12 (TM 11), and mmpL8 (TM 12), required to locate those proteins to their target functional site in either the cell membrane or the cell wall. These TM proteins, as well as an additional 16 cell-envelope proteins detected in the cytosol may be recently synthesized products on route to localization from the cytosol.
Membrane
Membrane proteins comprise two broad classes; integral and peripheral, based on the nature of the membrane-protein interactions. Integral membrane proteins include transmembrane proteins and lipid-anchored proteins. In the Mtb membrane, lipids form a gradient in membrane fluidity so that the region of highest fluidity lies at the outside surface of the pathogen (Liu et al., 1995). Thus the most fluid side of the membrane communicates with the outside world through the temporal association of peripheral membrane macromolecular complexes. Therefore, it is of no surprise that in our study, more than half of the proteins we identified were found in the membrane profile. Of these, the largest class of proteins identified is involved in small-molecule metabolism, in which fatty acid degradation is prominent. Another unique class of membrane proteins was involved in cell processes such as the binding and transportation of amino acids, ions, carbohydrates, and organic acids across the lipid bilayer. Three drug-efflux proteins (Rv1145, Rv1250, and Rv1819c) were also found in the membrane.
Applying TMHMM, a transmembrane protein prediction program based on a hidden Markov model (Krogh et al., 2001) to the 705 membrane proteins in our profile, 80 proteins were predicted as having two or more TM segments. For the entire Mtb proteome dataset, this number rose to 104 proteins. Consideration of proteins with at least one TM-helix increases the number of TM proteins to 144. In addition, 26 others appear to have signal peptides and, therefore, are secreted or targeted to specific cellular localizations. When the membrane proteins analyzed here were compared with those identified by the integrated SDS-PAGE-LC-MS/MS method (Gu et al., 2003a), there were 328 common proteins out of which 30 transmembrane proteins were found by both methods. The separation of proteins instead of peptides by SDS-PAGE method has enabled a slightly higher number of proteins to be identified in the membrane fraction and a better sequence coverage for individual proteins.
An analysis of the distribution of these different predicted transmembrane proteins in the different subcellular compartments reveals that, as expected, most are found in the membrane compartment, although a significant number are also found in the cell wall and cytoplasm. A comparison of the percentages of predicted membrane (and secreted) proteins in the genome with those actually found in the proteome (Figure 5), reveals that secreted proteins are equally represented. Proteins with fewer than 7 TM-helices tend to be under-represented, whereas those with 10, 11 or 12 TM-helices (with a peak at 11 TM-helices) are somewhat over-represented. Membrane proteins with 12 transmembrane helices tend to be involved in the transport of ions or amino acids, and as such may be more abundant, and so more easily identified than transmembrane proteins with fewer transmembrane segments. Coupled with the identification of a unique class of membrane proteins involved in the binding and transportation of small molecules across the lipid bilayer discussed above, and the prominence of proteins responsible for fatty acid metabolism, we hypothesize that the degradation of fatty acids may be occurring between the cytosol and membrane and the end products transported across the lipid bilayer for lipid biosynthesis in the cell wall.
Figure 5.

Number of TM-helices in membrane proteins identified in the different cellular compartments as they compare to predicted domains in the genome. The different profiles were color-coded as shown above. The percentage of occurrence of membrane proteins domains with a given number of TM-helices were expressed as a percentage of the respective number of proteins found in each category. The relative number of TM proteins identified in the proteome corresponds with that of Mtb H37Rv genome, which possesses a large number of proteins with a smaller number of TM-helices, i.e., <7, and the number of proteins with >7 TM-helices decreases drastically. Also, proteins with TM-helices >10 predominate in the proteomics profile, which has the highest number of TM proteins in the membrane compartment, as expected, followed by cytosolic membrane proteins and then TM proteins found in the cell wall. SP, secreted proteins.
The Cell Wall
In our proteomic profile of the cell wall fraction, three abundant components involved in lipid biosynthesis were identified; antigen 85A mycolyltransferase (fbpA) and beta-ketoacyl-acyl-carrier-protein synthases 1 and 2 (kasA and kasB; Supplementary Table). The lipids and covalently associated mycolic acids of the cell wall produced by these highly abundant proteins form the primary hydrophobic barrier by packing into a tight, closely packed lipid layer (Liu et al., 1995). This finding is consistent with the fact that the resistance of the pathogen to chemical injury, dehydration, and certain antibiotics is directly related to the low permeability of the unique wall to small hydrophilic molecules (Jarlier and Nikaido, 1994; Trias and Benz, 1994; Liu et al., 1995; Barry, 2001). Importantly, large numbers of the enzymes responsible for fatty acid metabolism such as acyl-CoA synthases (fadD) and enoyl-CoA hydratase/isomerase (echA) were also profiled in both the cell wall and the membrane fractions (Supplementary Table and Figure 6). Further, the presence of large numbers of unique lipoproteins such as beta-hexosaminidase A, conserved large membrane proteins, surface epimerases such as UDP-glucose-4-epimerase, and antigenic proteins such as antigen 84 (aka wag31) add to the unique complexity of the Mtb cell wall. This complex nature of the cell wall accounts for many of the spore-like intrinsic properties of the genus (Brennan and Nikaido, 1995; Barry et al., 1998; Daffe and Draper, 1998). The systematic identification and prominence of these proteins provides a solid basis for reconstructing the response networks involved in fatty acid degradation and lipid biosynthesis pathways that may reveal the mechanisms of Mtb persistence in its hosts (see Figure 6). Other unique proteins found in the cell wall may play a role in more subtle biological processes including those responsible for the synthesis and modification of macromolecules, as well as those involved in protein and peptide secretion (such as the protein-export membrane protein SecF), adaptations and atypical conditions (such as spore coat protein SA [otsA] and cold shock protein A [cspA]). Because of their locations in the cell wall, these unique proteins and their complexes may interact directly with components of the host cells.
Fatty Acid Degradation and Lipid Biosynthesis Response Networks
The comprehensive subcellular protein profile provided a solid basis to construct and refine cellular networks that control the survival of the mycobacterium or its persistence against host defense mechanisms. By employing the Rosetta Stone approach (Marcotte, 2000; Enright and Ouzounis, 2001) we identified 113 possible interacting gene products derived from the Mtb genome and used further for the construction of a functional linkage subnetwork (green connections in Figure 6). In Figure 6, black connections denote interactions through biochemical reactions with restrictions on ambiguous chemicals or substrates (see Supplementary Figure 2) outlined in the methods section, whereas the gene products are represented as nodes of different shapes. After validation of proteins, removal of the ambiguous chemicals and their links, the constructed cellular network of the whole Mtb genome consisted of 671 nodes (proteins) and 4153 edges (protein functional links). After determining 33 and 8 experimentally identified proteins involved in fatty acid degradation and lipid biosynthesis pathways, respectively, and tagging these proteins as root nodes in the network, we identified a connected subgraph of 47 and 12 nodes and 83 and 23 edges, respectively, within the Mtb cellular networks represented in Figure 6. Thus, the root nodes make up 70 and 67%, respectively, of the proteins involved in the subnetworks in Figure 6. Exceptions and thus computational predictions of functional proteins in the network are the proteins shown in Table 2. Twelve proteins were predicted, out of which 5 were not previously annotated in the 2004 revision of the original publication by Cole et al. (1998). The annotation shown in Table 2 was obtained from the KEGG database and has been verified by a BLAST search against the NCBI nr database with a conservative E-value of 10-50 or smaller. Examples of such proteins are Rv1136 and Rv2182c (Figure 6 and Table 2). Specifically we have high confidence in our identifications with predicted protein-protein links (green links in Figure 6) compared with other experimentally identified proteins. In addition to the Rosetta method, which only uses sequence information without relying on error-prone annotation, we also used predicted protein-protein linkages with assigned Z-scores, and only when the corresponding Z-score of these predictions lied between specific values. Enright et al. (1999) have shown that with an absolute cutoff Z-score of three or less, as we used in our network, the Rosetta method results in virtually no false positive prediction. Thus the confidence in the predicted protein-protein interactions is approximately equal to 100%. On the other hand, proteins not experimentally identified and otherwise predicted, but with well-established annotation are already in known and established metabolic reactions (black links).
Table 2.
Computationally “predicted” Mtb Proteins shown in networks of Figure 6
| Gene | Synonym (ORF number) | Protein |
|---|---|---|
| fadD4 | Rv0214 | Long-chain fatty acid CoA ligase |
| fadD30 | Rv0404 | Putative fatty acid CoA ligase |
| fabH | Rv0533c | 3-oxoacyl-[acyl-carrier-protein] synthase III |
| Rv0892 | Probable monooxygenase | |
| accD2 | Rv0974c | Propionyl-CoA carboxylase beta chain |
| Rv1136 | Hypothetical protein (probable acyl-CoA hydratase) | |
| fadD21 | Rv1185c | Putative fatty acid CoA ligase |
| fadD24 | Rv1529 | acyl-CoA synthase |
| Rv2182c | Putative 1-acyl-sn-glycerol-3-phosphate O-acyltransferase | |
| fabD | Rv2243 | malonyl CoA-acyl carrier protein transacylase |
| Rv3534c | Putative 4-hydroxy-2-oxovalerate aldolase | |
| Rv3536c | Putative 2-keto-4-pentenoate hydratase |
These proteins have not been identified in our proteomics profiles, but were predicted from computationally derived interactions with identified proteins or with other predicted proteins (the only example for this latter case assembles fabH in Figure 6A) in the network. Gene names are only shown if the annotation of The Wellcome Trust Sanger Institute, UK could be verified after a BLAST search.
DISCUSSION
The subcellular fractionation procedure described has been evaluated for its capacity to generate discreet fractions on several occasions (notably, the work conducted by Takayama et al., 1975; Hirschfield et al., 1990; and Lee et al., 1992, in addition to continued evaluation by members of the Belisle and Brennan laboratories at CSU). The identification of proteins in membrane and cell wall fractions usually presents major challenges for separation and detection methods. This is partially overcome by the 2DLC-MS/MS analyses of peptides derived from hydrophobic proteins. In this study, many hydrophobic proteins were identified that are involved in important membrane processes. It became clear from the results that fatty acid degradation is prominent in the membrane (colored green in Figure 6A), but occurs also in the cell wall and cytoplasm (colored blue and red, respectively). Coupled with the identification of a unique class of membrane proteins involved in the binding and transportation of small molecules across the lipid bilayer discussed above, and the prominence of proteins responsible for fatty acid metabolism in the membrane, we hypothesize that the degradation of fatty acids may be occurring between the cytosol and membrane and the end products transported across the lipid bilayer to the cell wall, where lipid biosynthesis occurs. As predicted and shown in Figure 6, we have identified those protein factors in a network governing these reactions. Our comprehensive analysis of protein expression showed that most of the proteins found in the cytosol are involved in energy metabolism and thus, the Mtb can respond to a wide variety of growth conditions and changing metabolic needs. In one predicted mechanism, the chromosomal clustering and coexpression of genes govern the cosynthesis of a large number of proteins required to form a complex. This has been demonstrated in the network of 52 proteins that were involved in fatty acid metabolism and lipid biosynthesis (Figure 6). Within this subnetwork, two features are prominent and both involve the interactions of highly connected proteins. Rv2182c, with 7 predicted connections in the lipid biosynthetic network, and 17 in the fatty acid degradation network, has been annotated as a hypothetical protein (Cole et al., 1998), although it has sequence homologies with acyltransferases. It would appear from an analysis of the networks shown in Figure 6 that this protein plays an important role in both fatty acid degradation and lipid synthesis, possessing functional links with FadD proteins in the fatty acid degradation pathways, as well as with the four kasA/B and plsB1/2 proteins in lipid biosynthesis, forming the center of this latter subnetwork.
The second feature is the cluster formed around fadB2 and fadB3 that exhibit strong functional linkages with the echA proteins in the fatty acid degradation pathways. FadB2 and fadB3 are both 3-hydroxyacyl-CoA dehydrogenases. These global identifications of proteins in the Mtb proteome further validate the computational network hypothesis that functionally related proteins work together in larger organizational structures. Interestingly, some proteins previously identified as belonging to the lipid biosynthetic pathway (e.g., kasA and kasB; The Wellcome Trust Sanger Institute) are nevertheless well connected within the fatty acid degradation pathway, due to their role in the initial steps of lipid biosynthesis utilizing the fatty acid degradation product acetyl-CoA in the form of acetyl-Acyl carrier protein. It is striking that the identified proteins involved in fatty acid degradation (Figure 6A) were distributed between the different cellular compartments in an almost exclusive manner. For example, in the subnetwork centered on fadB2 and fadB3; echA16, echA9, echA3, fadB, fadB2, and echA10 are found in the cytoplasm, echA7, echA12, echA17, echA20, echA21, fadB2, and echA10 in the membrane, and echA4 and echA6 in the cell wall, and fadB2, and echA10 being found in two compartments (membrane and cytoplasm). A similar, but slightly less striking, exclusive distribution is found in the subnetwork centered around Rv2182c, with 16 of 21 proteins found in only one cellular compartment, and five found in two (fadD9, kasA, fadD31, fadD11, fadD32). With one exception (fadD9—membrane and cytoplasm), proteins found in two compartments were found in cell wall and membrane. This probably represents a true compartmentalization of the pathways themselves, with fatty acid degradation, probably occurring mainly in the cytoplasm and membrane, and lipid biosynthesis occurring in cell wall and membrane, with proteins found in two compartments representing functional links between those compartments. Also this functional compartmentalization may account for the differential lipid fluidity in the membrane and cell wall.
In conclusion, this exhaustive analysis of the Mtb proteins did not only elucidate the compartmentalization of functional networks, but also enabled us to find unique proteins in the cytosol, membrane, and cell wall. The resistance of the microbial pathogen to chemical injury, dehydration, and certain antibiotics is directly related to the low permeability of the unique wall to small hydrophilic molecules, mostly lipids. Interestingly, we have identified 3 genes (fadB2 fadB3, and Rv2182c), around which fatty acid degradation and lipid biosynthesis were centered. This, when validated will provide basis for arresting this microbial pathogen at an early stage of development. There is still a long way to go before the complete implications are understood, but this hypothesis of protein-protein interactions shown in the responder networks will invigorate research for the development of new drugs, diagnostic probes or targets for vaccines.
Supplementary Material
Acknowledgments
We acknowledge Lisa Wolfe for her technical assistance during preparation of mycobacterial subcellular fractions. This work is supported by Los Alamos National Laboratory Directed Research Development (LDRD) grants 20020048 DR, 20030508ER, and 20040184ER; DOE Human Genome Instrumentation Grant ERW9840; and DOE grant KP1103010 (to X.C.) and National Institutes of Health grant N01-AI-75320 awarded to Colorado State University. X.C. is a recipient of Presidential Early Career Award for Scientists and Engineers (PECASE).
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E04-04-0329. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-04-0329.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Barry, C. (2001). Interpreting cell wall `virulence factors' of Mycobacterium tuberculosis. Trends Microbiol. 9, 237-241. [DOI] [PubMed] [Google Scholar]
- Barry, C., Lee, R., Mdluli, K., Sampson, A., Schroeder, B., Slayden, R., and Yuan, Y. (1998). Mycolic acids: structure, biosynthesis and physiological functions. Prog. Lipid Res. 37, 143-179. [DOI] [PubMed] [Google Scholar]
- Brennan, P., and Nikaido, H. (1995). The envelope of mycobacteria. Annu. Rev. Biochem. 64, 29-63. [DOI] [PubMed] [Google Scholar]
- Camus, J. C., Pryor, M. J., Medigue, C., and Cole, S. T. (2002). Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology 148, 2967-2973. [DOI] [PubMed] [Google Scholar]
- Cole, S. et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete. Nature 393, 537-544. [DOI] [PubMed] [Google Scholar]
- Daffe, M., and Draper, P. (1998). The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39, 131-203. [DOI] [PubMed] [Google Scholar]
- Enright, A., Iliopoulos, I., Kyrpides, N., and Ouzounis, C. (1999). Protein interaction maps for complete genomes based on gene fusion events. Nature 402, 86-90. [DOI] [PubMed] [Google Scholar]
- Enright, A., and Ouzounis, C. (2001). Functional associations of proteins in entire genomes by means of exhaustive detection of fusion genes. Genome Biology. Available at: http://genomebiology.com/2001/2/9/research/0034. Accessed December 3, 2004. [DOI] [PMC free article] [PubMed]
- Gu, S., Chen, J., Dobos, K. M., Bradbury, E. M., Belisle, J. T., and Chen, X. (2003a). Comprehensive proteomic profiling of the membrane constituents of a Mycobacterium tuberculosis strain. Mol. Cell. Proteomics 2, 1284-1296. [DOI] [PubMed] [Google Scholar]
- Gu, S., Pan, S., Bradbury, E., and Chen, X. (2002). Use of deuterium-labeled lysine for efficient protein identification and. Anal. Chem. 74, 5774-5785. [DOI] [PubMed] [Google Scholar]
- Gu, S., Pan, S., Bradbury, E., and Chen, X. (2003b). Precise peptide sequencing and protein quantification in the human. J. Am. Soc. Mass Spectrom. 14, 1-7. [DOI] [PubMed] [Google Scholar]
- Hirschfield, G., McNeil, M., and Brennan, P. (1990). Peptidoglycan-associated polypeptides of Mycobacterium tuberculosis. J. Bacteriol. 172, 1005-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynen, M., Snel, B., Lathe, W., 3rd, and Bork, P. (2000). Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res. 10, 1204-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe, T., Uchida, K., Taoka, M., Shinkai, F., Manabe, T., and Okuyama, T. (1991). Automated two-dimensional liquid chromatographic system for mapping. J. Chromatog. 588, 115-123. [DOI] [PubMed] [Google Scholar]
- Jarlier, V., and Nikaido, H. (1994). Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol. Lett. 123, 11-18. [DOI] [PubMed] [Google Scholar]
- Jiménez, V. and Marzal, A. (1999). Proceedings of the 3rd International Workshop on Algorithm Engineering, WAE'99, Lecture Notes in Computer Science. New York: Springer-Verlag.
- Jungblut, P., Schaible, U., Mollenkopf, H., Zimny Arndt, U., Raupach, B., Mattow, J., Halada, P., Lamer, S., Hagens, K., and Kaufmann, S. (1999). Comparative proteome analysis of Mycobacterium tuberculosis and Mycobacterium bovis BCG strains: towards functional genomics of microbial pathogens. Mol. Microbiol. 33, 1103-1117. [DOI] [PubMed] [Google Scholar]
- Kaji, H., Saito, H., Yamauchi, Y., Shinkawa, T., Taoka, M., Hirabayashi, J., Kasai, K., Takahashi, N., and Isobe, T. (2003). Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat. Biotechnol. 21, 667-672. [DOI] [PubMed] [Google Scholar]
- Kaji, H., Tsuji, T., Mawuenyega, K., Wakamiya, A., Taoka, M., and Isobe, T. (2000). Profiling of Caenorhabditis elegans proteins using two-dimensional gel. Electrophoresis 21, 1755-1765. [DOI] [PubMed] [Google Scholar]
- Krieger, C. J., Zhang, P., Mueller, L. A., Wang, A., Paley, S., Arnaud, M., Pick, J., Rhee, S. Y., and Karp, P. D. (2004). MetaCyc: a multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res. 32(Database issue), D438-D442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. (2001). Predicting transmembrane protein topology with a hidden Markov model. J. Mol. Biol. 305, 567-580. [DOI] [PubMed] [Google Scholar]
- Lee, B., Hefta, S., and Brennan, P. (1992). Characterization of the major membrane protein of virulent Mycobacterium. Infect. Immun. 60, 2066-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link, A., Eng, J., Schieltz, D., Carmack, E., Mize, G., Morris, D., Garvik, B., and Yates, J. (1999). Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 17, 676-682. [DOI] [PubMed] [Google Scholar]
- Liu, J., Rosenberg, E., and Nikaido, H. (1995). Fluidity of the lipid domain of cell wall from Mycobacterium chelonae. Proc. Natl. Acad. Sci. USA 92, 11254-11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte, E. (2000). Computational genetics: finding protein function by nonhomology methods. Curr. Opin. Struct. Biol. 10, 359-365. [DOI] [PubMed] [Google Scholar]
- Marcotte, E., Pellegrini, M., Thompson, M., Yeates, T., and Eisenberg, D. (1999). A combined algorithm for genome-wide prediction of protein function. Nature 402, 83-86. [DOI] [PubMed] [Google Scholar]
- Mattow, J., Jungblut, P., Muller, E., and Kaufmann, S. (2001a). Identification of acidic, low molecular mass proteins of Mycobacterium. Proteomics 1, 494-507. [DOI] [PubMed] [Google Scholar]
- Mattow, J., Jungblut, P., Schaible, U., Mollenkopf, H., Lamer, S., Zimny Arndt, U., Hagens, K., Muller, E., and Kaufmann, S. (2001b). Identification of proteins from Mycobacterium tuberculosis missing in attenuated Mycobacterium bovis BCG strains. Electrophoresis 22, 2936-2946. [DOI] [PubMed] [Google Scholar]
- Mawuenyega, K., Kaji, H., Yamauchi, Y., Shinkawa, T., Saito, H., Taoka, M., Takahashi, N., and Isobe, T. (2003). Large-scale identification of Caenorhabditis elegans proteins by multidimensional liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2, 23-35. [DOI] [PubMed] [Google Scholar]
- Mollenkopf, H., Jungblut, P., Raupach, B., Mattow, J., Lamer, S., Zimny Arndt, U., Schaible, U., and Kaufmann, S. (1999). A dynamic two-dimensional polyacrylamide gel electrophoresis database: the mycobacterial proteome via internet. Electrophoresis 20, 2172-2180. [DOI] [PubMed] [Google Scholar]
- Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., and Kanehisa, M. (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, N., Ishioka, N., Takahashi, Y., and Putnam, F. W. (1985). Automated tandem high-performance liquid chromatographic system for separation of extremely complex peptide mixtures. J. Chromatogr. 326, 407-418. [DOI] [PubMed] [Google Scholar]
- Takayama, K., Schnoes, H., Armstrong, E., and Boyle, R. (1975). Site of inhibitory action of isoniazid in the synthesis of mycolic acids. J. Lipid Res. 16, 308-317. [PubMed] [Google Scholar]
- Taoka, M., Wakamiya, A., Nakayama, H., and Isobe, T. (2000). Protein profiling of rat cerebella during development. Electrophoresis 21, 1872-1879. [DOI] [PubMed] [Google Scholar]
- The Wellcome Trust Sanger Institute, U.K. http://www.sanger.ac.uk/Projects/M_tuberculosis/Gene_list. [DOI] [PubMed]
- Trias, J., and Benz, R. (1994). Permeability of the cell wall of Mycobacterium smegmatis. Mol. Microbiol. 14, 283-290. [DOI] [PubMed] [Google Scholar]
- WHO. (2003). Global Tuberculosis Control. Surveilance, Planning, Financing. http://www.who.int/gtb/publications/globrep/index.html.
- Washburn, M., Wolters, D., and Yates, J. (2001). Large-scale analysis of the yeast proteome by multidimensional protein. Nat. Biotechnol. 19, 242-247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.