

Abstract
Excitation–contraction coupling in cardiac myocytes requires calcium influx through L-type calcium channels in the sarcolemma, which gates calcium release through sarcoplasmic reticulum ryanodine receptors in a process known as calcium-induced calcium release, producing a myoplasmic calcium transient and enabling cardiomyocyte contraction. The spatio-temporal dynamics of calcium release, buffering, and reuptake into the sarcoplasmic reticulum play a central role in excitation–contraction coupling in both normal and diseased cardiac myocytes. However, further quantitative understanding of these cells’ calcium machinery and the study of mechanisms that underlie both normal cardiac function and calcium-dependent etiologies in heart disease requires accurate knowledge of cardiac ultrastructure, protein distribution and subcellular function. As current imaging techniques are limited in spatial resolution, limiting insight into changes in calcium handling, computational models of excitation–contraction coupling have been increasingly employed to probe these structure–function relationships. This review will focus on the development of structural models of cardiac calcium dynamics at the subcellular level, orienting the reader broadly towards the development of models of subcellular calcium handling in cardiomyocytes. Specific focus will be given to progress in recent years in terms of multi-scale modeling employing resolved spatial models of subcellular calcium machinery. A review of the state-of-the-art will be followed by a review of emergent insights into calcium-dependent etiologies in heart disease and, finally, we will offer a perspective on future directions for related computational modeling and simulation efforts.
Keywords: Electrophysiology, mathematical modeling and simulation, calcium dynamics, excitation-contraction coupling, subcellular modeling
Introduction
Excitation–contraction coupling (ECC) in cardiomyocytes requires calcium (Ca) influx through L-type Ca channels (LCCs) in the sarcolemma, which initiates Ca release through ryanodine receptors (RyRs) clustered in the terminal cisternae of the sarcoplasmic reticulum (called junctional SR [jSR]) in a process known as Ca-induced Ca release (CICR). Ca influx via an a intracellular Ca transient (CaT), enabling cardiomyocyte contraction. Ca is removed from the myoplasm, ending the CaT, via the sarco/endoplasmic reticulum Ca-ATPase (SERCA) and by the sarcolemmal Na–Ca exchanger (NCX) as well as the Ca pump (CaP). All RyRs and associated jSR structures that can be activated as a distinct unit are denominated the “calcium release unit” (CRU).
The spatio-temporal dynamics of CICR, buffering, and reuptake into the SR play a central role in ECC in both normal and diseased cardiac myocytes. In cardiac myocytes, it has been proposed that 5–15 LCCs embedded in the sarcolemma appose 50–200 clustered RyRs as distinct structures1 (see Figure 1A, left panel). However, the exact numbers and ratio of LCCs and RyRs in these functional couplons is an area of ongoing research. The dyad is considered to be a single-sided lobe of the jSR apposing the transverse-tubule (t-tubule) membrane, invaginations of the sarcolemma of cardiomyocytes. The dyadic geometry estimated to have a radius of 0.05–0.2 µm, and a height of 10–12 nm, can alter in disease and displays significant interspecies variability.2 Several characteristic properties of ECC, such as high gain and graded Ca release, arise from interactions that occur in and between these local dyadic microdomains. Dyads are clustered along t-tubules. Mammalian ventricular cells typically have a well-developed, regular structure for t-tubules (t-network). Atrial cardiomyocytes from large mammals have been shown to have well-developed t-tubular networks; however, species differences and specifically a lack of defined t-tubular structure in atria myocytes from small mammals has historically led to atrial t-tubules being overlooked. The t-tubular system plays a central role in the synchronization of Ca signaling and ECC in many striated muscle cells; disruption of the t-network contributes to dyssynchronous Ca release and impaired contraction.3–7 CICR in small dyads gives rise to high gain through positive feedback (an all or none event), but the spatial distribution and relative isolation of CRUs allows for sequential recruitment and graded release. The restricted number of molecules in each CRU can mean that approximating dynamics as continuous is inappropriate: processes therein may be better described by stochastic as opposed to deterministic models. Many earlier models of the cardiac action potential did not include descriptions of CICR that accounted for these local mechanisms.8
Figure 1.

CRU organization and t-tubular structure in the normal (left) and failing (right) cardiac myocyte. In healthy ventricular cardiac myocytes, L-type Ca channels directly appose RyRs in each CRU, and t-tubular structure is regular (A and B, left column). However, in the setting of heart failure, disruption of the t-tubular network (B, right column) causes RyR dispersion (A, right column), leading to abnormal calcium transients. Disruption of the t-tubule network (B, right column) and RyR dispersion (A, right colum) leads to abnormal calcium transients. (Adapted from Louch et al.,38 used with permission.)
Ca signaling in the CRU is a fundamentally discrete process;9 short-lived, local increases in intracellular Ca via triggered SR release are known as Ca sparks, which regulate the generation of whole-cell CaT and ECC. For example, spontaneous Ca sparks have durations of 10–100 ms, allowing SR Ca uptake to keep pace with release, a process which cannot happen during a complete triggered release event. Long-lasting sparks with durations of several hundred milliseconds to seconds, so-called “embers,” are also widely observed. Experiments have shown that the transition from normal to long-lasting sparks can occur when RyRs open probability is reduced.10 Dysfunction in Ca handling is central to a number of cardiac pathologies (including heart failure (HF) and atrial fibrillation (AF)) and may lead to mechanical dysfunction as well as arrhythmia. Disruption of dyadic structure is thought to be largely responsible for changes in Ca handling; we will delve further into these aspects in later sections of this review. Further quantitative understanding of cardiomyocytes’ Ca machinery and the study of mechanisms that underlie both normal cardiac function and calcium-dependent etiologies in heart disease thus requires accurate knowledge of cardiac ultrastructure, protein distribution and subcellular function. As current imaging techniques are limited in the spatial resolution to which changes in Ca can be detected, computational models of ECC have thus been increasingly employed to probe these structure–function relationships. Mathematical modeling of ECC in cardiomyocytes is a fundamentally multi-scale problem: it involves gradients on the spatial scale of 100 nm or even less in dyadic clefts and concentration profiles along the 100 µm of the whole cell, as well as the submillisecond timescale of local concentration changes and the change of SR Ca content within tens of seconds.
This review will focus on the development of structural mathematical models of cardiac Ca dynamics. While of importance for select cardiac pathologies, in general the role of mutation and protein dysfunction or dysregulation will not be treated in detail here, nor will the study of targets or putative drug therapies for Ca handling dysfunction take a major role. Discussion of mitochondrial electrodynamics and signaling, though offering critical insight, will not be treated here and have been reviewed elsewhere,11 nor will the explicit roles of oxidative stress or CaMKII signaling.12,13 Others have also offered excellent reviews of modeling of myocardial Ca at different spatial scales from the perspective of parameter sensitivity analysis,14 which will not be the primary focus at present. Instead, the present review will orient the reader broadly towards the development of models of subcellular Ca handling in cardiomyocytes. We will place specific focus on progress in recent years in terms of multi-scale modeling employing resolved spatial models of subcellular calcium machinery. A review of the state-of-the-art will be followed by a review of emergent insights into calcium-dependent etiologies in heart disease and, finally, will offer a perspective on future directions for related computational modeling and simulation efforts.
State-of-the-art: established and emerging methods
Mathematical modeling of the physiology of both atrial and ventricular cardiomyocytes is a broad field including several mathematical formalisms. For the convenience of the less-familiar reader, we here offer a brief review of a few common computational modeling terms used in the present review. Something which is discretely valued is constant over a spatial or temporal interval, while one that is continuous would have smoothly varying values in time and space; binary in this context refers to an all-or-nothing phenomenon. A compartment refers to a restricted virtual space in the mathematical model which represents a volume considered to be separate from a common or bulk space in the interior of the cell. A deterministic model refers to a system wherein no randomness is involved in calculation of its future states. A deterministic model will thus always produce the same output from a given input, while stochasticity refers to event or system that includes uncertainty in outputs because of random variation in one or more inputs over time. Markov processes are stochastic processes that satisty the property of “memorylessness;” the concept is that one can make predictions for the future based only on the present state, with the future and past as independent. Monte Carlo simulations are used to model the probability of different outcomes in processes that cannot easily be predicted because of stochastic inputs. In a single trial, a range of values (probability distribution) is substituted for a number for any factor that has inherent uncertainty; with many, many trials, each using a different set of random values, a complete picture of probable outcomes is obtained. The probability density function (arising from statistics) is a function used to specify the probability of a random variable falling within a particular range of values, as opposed to taking on any one value.
Models of subcellular calcium handling: an overview
While models of ECC in cardiomyocytes developed over the past 30 years have been reviewed completely elsewhere,15,16 a brief orientation follows. Movement and dynamics of Ca ions in the dyad have often been described by assigning continuously valued Ca concentrations to one or more dyadic compartments; several models have been based upon deterministic representations (without stochasticity) wherein CRUs were lumped into a single “common pool.”17–19 As the RyRs in this common pool are activated via influx of Ca through LCCs, the strong positive feedback of CICR ensures that the pooled release units activate completely, resulting in coordinated, binary calcium release. Ventricular cardiomyocytes, however, display a graded response to trigger Ca in vivo: as the amount of trigger Ca from LCCs goes up, so does release via the SR through the RyRs. While some of the deterministic computational models do simulate graded release, these require an artificial mechanism to do so.8 Stern20 demonstrated that high-gain graded release could not be simulated via common-pool means, but that modeling CRU including local CICR triggers and recruitment of a neighborhood cluster of RyRs permitted graded response; this was also later demonstrated in a more physiological model.21 Thus, a sufficient increase in dyadic subspace Ca following local LCC opening causes apposed RyRs to open; specifically, then, stochastic recruitment of neighboring CRU results in graded release: the so-called “local control”-type model. These stochastic properties of the system can be reduced to a representative Markov model based on principles of timescale decomposition. The RyRs and LCCs are often modeled via Markov state models, as reviewed elsewhere16 (see Figure 2 for examples of four- and seven-state models, respectively).
Figure 2.
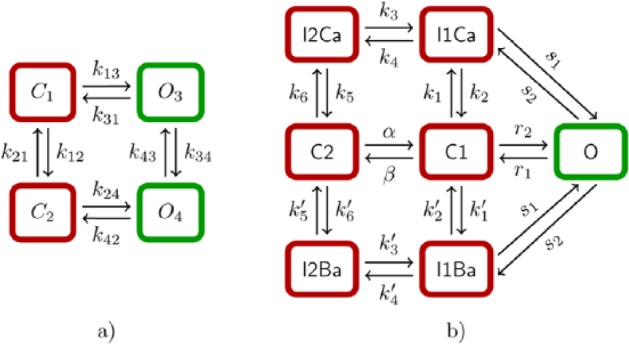
(a) A four-state Markov model for ryanodine receptors (RyRs). If the horizontal and vertical transitions are independent of each other (k13 = k24, k31 = k42 and k12 = k34; k21 = k43), then the model can be expressed with the Hodgkin–Huxley formalism. (b) A seven-state Markov model for L-type Ca channels (LCCs), from Mahajan et al.92
Local control models of Ca release wherein interactions between LCCs and RyRs are simulated stochastically are able to reconstruct the key physiological phenomena of both graded Ca release and high CICR gain,22–25 but at a high computational cost relative to common-pool models. Approaches have sought to reduce the cost levied by stochastic simulation; a breakthrough model presented a general analytical approach for deriving simplified models of local control of CICR.26 The resulting coupled LCC–RyR gating model successfully reproduced the LCC response to voltage-clamp stimuli, and the inactivation of LCCs with and without Ca release from the SR found in experiments, at reduced computational cost. In another non-spatially resolved model, a probability density function approach was used to replace the costly Monte Carlo simulations necessary to model local control via stochastic Markov processes. The method derived coupled advection–reaction equations relating the time-dependent probability density of subsarcolemmal subspace and junctional SR Ca concentration ([Ca]) conditioned on the CRU state. Modeling CRU activity using this probability density approach avoids resolution of precise spatial aspects of global Ca signaling, but represents heterogeneous local Ca signals in a population of dyadic subspaces and junctional SR depletion domains with reasonable accuracy.27 Both approaches to modeling local control of ECC produce high-gain Ca release that is graded with changes in membrane potential, phenomena not exhibited by common pool models. Other non-spatially resolved models of local control have linked the processes of subcellular Ca machinery to whole cell behavior.28,29
Pseudo-spatial and spatially-resolved models of subcellular calcium processes
The local nature of the ECC control mechanism was not clear until the discovery of Ca sparks by Cheng et al.30 In recent decades, there has been much additional evidence that the control of CICR is contingent upon local Ca in the immediate vicinity of the CRU, rather than on the whole-cell Ca.31,32 This tight, local regulation of CICR is made possible by the clustering of LCCs and RyRs into discrete couplons, rendering them sensitive to local rather than global Ca. While Ca sparks represent the “unit” of Ca release from large arrays of RyRs in the CRU, Ca “blinks” represent the Ca depletion signal produced in the terminal cisternae of the jSR. Furthermore, so-called quarky Ca release observes release events smaller than sparks at a level substructural to the CRU.33 In other words, macroscopic Ca release events are intrinsically controlled by the type and number of individual LCCs and RyRs in the CRU, the relative spatial localization of the two channel types and geometry of this functionally significant nanodomain, as well as the organization of the CRUs at the cell level.
An early model of Sobie et al.23 consisted of a single LCC closely apposed to a cluster of RyRs in a region of jSR. While simple, the stochastic current descriptions for RyRs and LCCs sought to lend insight to the question of Ca spark termination via the novel “sticky cluster” model, meaning a model that incorporated the “sticky” cooperativity between adjacent RyRs. Next efforts employed explicitly spatially-resolved single CRUs; one study to investigate the effects of action potential prolongation in a model of murine HF used a rectangular space with dimensions 100 nm × 100 nm and a dyadic cleft space of 12 nm between the t-tubular and SR membranes, analysis revealed that Ca spark amplitude and rise time were highly dependent on the number of activated channels (LCCs and RyRs) and their packing within the CRU, though not very sensitive to other cleft dimensions.34 More detailed was a study by our groups which developed a three-dimensional computational model of a single dyad, modeled as a cylindrical disk,35,36 with CRUs of two sizes: 25 or 100 RyRs per CRU, with RyRs positioned in a highly regular two-dimensional lattice grid.6 In a follow-up study on whether RyR function promoted slowing of Ca release in murine HF, the sticky cluster model was re-parameterized37 and, rather than incorporate a highly regular RyR geometry in the CRU, was extended to include subclusters in a CRU in addition to one “mother cluster.” All subclusters were connected with the mother cluster through diffusion but not with each other, and Ca sparks were initiated by opening one RyR in the mother cluster.38
Computational models have additionally moved towards the level of the whole myocyte, generally stepping up dimension and complexity in geometry. Tao et al.39 used a schematic model of a cluster of coupled RyRs in a cardiac ventricular cell of length of 150 µm, with 2 µm spatial resolution40 to study intracellular Ca alternans (see Section 3.3) by coupling model elements via Ca diffusion between neighboring cytoplasmic and network SR spaces. The more complex model of Rovetti et al.41 developed a quasi-two-dimensional spatially-distributed Ca cycling model via network of 100 by 100 CRU which included a network SR (nSR) domain and a myoplasmic domain coupled via SR Ca release and uptake. The model comprises a CRU network coupled via Ca diffusion in each domain. Each CRU contained a jSR diffusively connected to the nSR, and a dyadic space diffusively connected to the myoplasm, as well as stochastic LCCs (5 channels per CRU) and RyRs (100 channels per CRU). Gaur et al.42 developed an initial multi-scale model of a spatially distributed mammalian ventricular myocyte consisting of 10,000 diffusively coupled CRU, with the number of stochastic LCCs and RyRs in a dyad as 15 and 100, respectively, to investigate how microscopic changes in dyadic properties including detubulation in HF, can affect whole-cell behavior. A recent model of ECC in the mouse cardiac ventricular myocyte, developed to further elucidate the physiologic consequences of leaky RyRs, included a three-dimensional spatial implementation of the same group’s local control model, representing a single sarcomere centered on a z-line and containing equally distributed CRU (inter-CRU distance is 600 nm).43 Cannell and Laver44,45 have recently employed with others a cylindrical model of the dyad with a t-tubule at the center to investigate control and termination of CICR via the SR; importantly, this model permits calcium gradients within the CRU.
Others have used similar approaches to examine local Ca in the atrial myocyte. The observation of variable t-tubule density in atrial myocytes46 has not yet been taken into account in several models; instead, models have assumed wave-like CICR propagation of Ca into the cell interior. Given cell-type potential differences in ultrastructure, the “z-plane” distribution of RyRs was modeled radially, rather as a regular grid in one study, to monitor the three-dimensional diffusion of Ca along a portion of the cell.47 Another model of the human atrial cardiomyocyte48 included a spatial representation of Ca-handling based on longitudinal division into ~2µm wide segments, and in the transverse division into ~1µm long domains, as based on the model of Grandi et al.49
Physiologically detailed models of subcellular Ca cycling including a three-dimensional network of ~20,000 CRU at the level of the whole ventricular cell have been developed by Restrepo and colleagues.50–54 The model and its iterations include a three-dimensional network of 19,305 (65 × 27 × 11) CRUs with CRU spacing at 1.84 µm (longitudinal) and 0.9 µm (transverse direction), corresponding to a ventricular cell of dimension ~ 120 × 25 × 10 μm3. CRUs are coupled via Ca diffusion in the cytosolic space and SR. Each CRU contains five subvolumes: the nSR, jSR, dyadic space or proximal space, a submembrane space, and a cytosolic space. While spatially resolved, each CRU incorporates a cluster of 100 RyR channels and 10 LCCs, both simulated using random Markov transitions: cluster size is fixed, other aspects of CRU geometry not taken into account, and the CRU is considered a common-pool with respect to [Ca] gradients. Others have used a similar approach, but simplified the three-dimensional problem to a two-dimensional model of a cardiac myocyte,55 similar to the work by Izu et al.56 Recently, Sato et al.57 expanded upon the work of Restrepo et al. with RyR clusters of a few to several hundred RyRs, varying the number of functional RyRs in a single cluster, diffusion within the SR network, diffusion between network and junctional SR, cytosolic Ca diffusion, SERCA uptake activity, and RyR open probability. To address the need for explicit representation of CRU geometry, Hake et al.58 developed novel tools to generate computational geometry from electron tomographic images and created a detailed computational model of a single CRU. Ca diffusion was modeled within the SR and the cytosol to examine the effects of localization and density of the NCX, SERCA, and CSQN. Others have developed high-resolution imaging and analysis approaches to measure the three-dimensional distribution of immunolabeled proteins with confocal microscopy in cardiomyocytes as both RyRs and t-tubular system distributions show large variation from the simple grid geometries assumed in previous work; this three-dimensional RyR cluster distribution has been used to construct a model of stochastic Ca dynamics in a myocyte.59 Figure 3 offers an overview of the development of models of pseudo-spatial and spatially-resolved models of subcellular calcium release.
Figure 3.
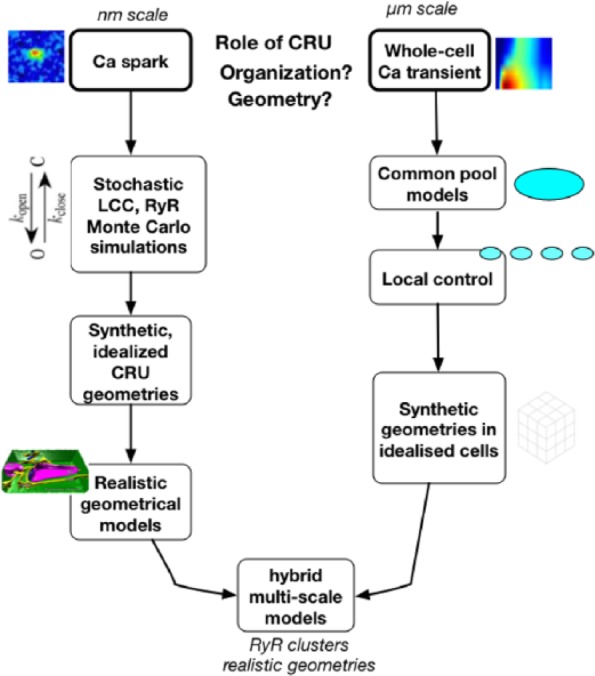
An overview of the development of spatially resolved models of the cardiac myocyte. Modeling of subcellular processes emerged from the need to understand the link between the Ca spark, calcium release unit (CRU) organization and geometry, and whole-cell emergent behavior. Modeling of Ca sparks at the level of the CRU have employed stochastic ryanodine receptors (RyRs) and L-type Ca channels (LCCs) and Monte Carlo simulation in first synthetic and idealized, and, more recently, real geometries, to offer insight into spark dynamics, including spark termination. Concomitantly, whole-cell calcium dynamics modeling began with common-pool representations, replaced by local-control models and sticky-cluster modeling including the effects of RyR cooperativity on Ca release. These models have been stacked into repeating units to approximate whole-cell function, first with generic geometries in idealized cells, to reconcile observed phenomena such as graded release with high gain and cellular alternans. Incompletely resolved questions include how much LCC current is actually required for RyR activation, the phenomena of long-lasting “embers,” and how CRU structure collectively impacts waves, subtle aspects of CRU recruitment and modifies the stability of the dynamical range in which the CRU/couplon ensemble is operating. (Image reproduced from Hake et al.58 with permission.)
Multi-scale modeling of calcium-driven processes in cardiac electrophysiology
Computational models at present seek to combine the data emergent from newer imaging technologies to gain multi-scale insight into calcium-driven processes. Spatial point process statistics techniques were recently developed to simulate the spatial distribution of RyR clusters, combining confocal-scale (~200 nm) data of RyR clusters with three-dimensional electron microscopy data (~30 nm) of myofibrils and mitochondria, both collected from adult rat left ventricular myocytes. This hybrid-scale spatial model was employed in reaction–diffusion Ca simulations during the rising phase of the CaT.60 Another recent study introduced a new concept for a multi-scale mathematical model of CICR and whole cardiomyocyte electrophysiology. This incorporated stochastic simulation of individual LCCs and RyRs, spatially detailed concentration dynamics in dyadic clefts, rabbit membrane potential dynamics, and a system of partial differential equations (PDEs) for intracellular and SR free Ca as well as Ca-buffering, and resolved concentration gradients from the level of the dyad to the whole-cell level by using a quasi-static approximation in CRU.61
Emergent results: insights into Ca-handling mechanisms
Mechanistic insight into normal function
Multi-scale computational models of subcellular Ca handling have offered a wealth of mechanistic insight into the cardiomyocytes’ function in the normal heart. To illustrate, models of stochastic molecular signaling between LCCs and RyRs describing known features of dyad geometry, including key dyadic proteins and electrodiffusive movement of individual Ca ions enabled investigation of how local Ca signaling is influenced by the dyad structure. The geometry of the individual RyR may indeed function to restrict the diffusion of and to “funnel” Ca ions to activation-binding sites on the RyRs, increasing RyR open probability and ECC gain.24,62 Furthermore, models incorporating realistic CRU distributions permitted Ca waves that spread axially along the cell at observed velocities, demonstrating that spatial features of the CRU distribution on multiple length scales critically affects intracellular Ca dynamics.59 A recent study showed that RyRs positioned axially helped propagate Ca waves in the axial direction.63 Greenstein et al.29 generalized an earlier analytical approach for deriving simplified mechanistic models of CICR to formulate an integrative model of the canine cardiac myocyte, which was used to study the role of local redundancy in LCC channel gating and the role of dyad configuration on ECC. Simulations suggested that the characteristic steep rise in ECC gain observed at hyperpolarized potentials is a result of increased functional coupling between LCCs and RyRs.
Calcium sparks and calcium waves
Modeling and simulation has permitted detailed insight into Ca spark generation in normal function. The Williams et al. model for calcium dynamics in the rat ventricular myocyte includes 20,000 CRU consisting of clusters of stochastically gated RyRs.64 The model resolves the multiple single opening events that do not result in a Ca spark (~3,000 per cell per second), and also shows that a single RyR release event can trigger others via CICR and that once ~12% of RyRs are open, a spark usually follows. The termination of a calcium spark has been an active research topic for many years with several hypotheses proposed; the reader is referred to an excellent recent review.65 The spatial arrangement of RyRs within clusters has a major influence on the frequency of Ca sparks. The probability of a Ca spark occurring when a single RyR in the cluster opens spontaneously can be predicted from the precise spatial arrangement of the RyRs; function follows directly from structure, in this case. A computational model of the dyadic cleft which specified the spatial localization of LCCs and RyRs revealed that reaction specificity and efficiency are regulated by microdomain geometry as well as the physical separation of signaling molecules into functional complexes. Both Ca spark amplitude and rise time were found to be highly dependent on the concentration of activated channels in the CRU microdomain and on the intermembrane separation channel packing (Koh, 2006).34 Previous model studies also have predicted that the duration of the spark is determined by the local CRU geometry, as well as the localization and density of Ca-handling proteins.58 It has been newly demonstrated that long-lasting Ca sparks emerge as a collective dynamical behavior of a network of diffusively coupled CRU; there exists an optimal range of RyR open probability favoring long-lasting sparks.54 Another recent study employed a spatially resolved mathematical model of subcellular Ca cycling to examine how Ca spark duration is influenced by the number of functional RyRs in a junctional cluster and other SR Ca-handling properties; if the number of RyRs is under a certain threshold, it is difficult to maintain consecutive openings and stochastic attrition terminates Ca release while, if the number of RyRs in a cluster is too large, the depletion of Ca from the jSR terminates release. It was found that protracted Ca release events require small RyR clusters and sufficiently rapid intra-SR Ca diffusion.57
Intracellular Ca waves are a form of Ca signaling executed in many cell types and can occur in cardiac myocytes during Ca overload.30 Spontaneous increase in [Ca] can occur at a single or multiple CRU within a cell and can lead to propagation throughout the myoplasm in a wave-like pattern. Cheng et al.30,66 speculated that Ca waves arose from the collective firing of Ca sparks; that Ca released in a dyad can diffuse and trigger Ca release in adjacent dyads, forming traveling waves. The nature of this propagating mechanism means that a wave travels at a finite velocity. This is therefore dissimilar to the CaT evoked by an action potential, which is a whole-cell release event, coordinated by depolarization-activated Ca entry through LCCs.67 Ca waves are a natural consequence of CICR; the evolution of Ca wave models reflects the growth of knowledge of ECC in muscle. The appearance of Ca waves may lead to whole-cell depolarization and triggering of an action potential, as these may activate inward currents such as that carried by the NCX.68 Thus, there exists a putative link between Ca waves and triggered activity leading to initiation of dangerous arrhythmias; experimental studies have demonstrated that abnormal Ca cycling is a critical factor in the development of focal excitations.69 Spatially-resolved cell models have shown that the time needed to form cluster of sufficient size to elicit a Ca wave, as well as the critical cluster size, becomes smaller as SR Ca load and diastolic myoplasmic Ca increases.52 A separate study in a similar model effected sensitivity analyses to study physiological parameter effects on global Ca waves: computed results were in agreement with confocal microscopy imaging, and found that the current flow amplitude through the CRU affected dynamic properties of Ca waves more significantly than the duration of this current, and that longitudinal and transverse separation of CRU affected the longitudinal velocity and amplitude of Ca waves significantly.55
Calcium alternans
Cellular CaT alternans are beat-to-beat alternations in the peak cytosolic calcium concentration exhibited by cardiac cells during rapid electrical stimulation or under pathological conditions. CaT alternans promote action potential duration alternans, which have been linked to the onset of life-threatening ventricular arrhythmias. Ca alternans in cardiac myocytes have been shown in many experimental studies, and the mechanisms remain incompletely understood. The ability to link microscopic properties of CRU to whole cell behavior is thus a powerful tool to investigate the arrhythmogenic role of abnormal Ca dynamics in cardiac disease. A study employing the multi-scale model of Restrepo et al.50 showed that luminal (SR-side) gating of the RyRs mediated by CSQN can cause calcium transient alternans regardless of the steepness of the release–load relationship: alternans were caused by a beat-to-beat alternation in the number of refractory RyR channels and could occur with or without diastolic SR calcium content alternans. The same group showed that ion channel stochasticity at the level of a single CRU can influence the whole-cell alternans. Depending on the sign and magnitude of Ca–voltage coupling, Ca alternans can be spatially synchronized or desynchronized, and in or out of phase with action potential duration alternans. Calcium alternans can, for instance, be spatially synchronized but out of phase with action potential duration alternans.51 The concurrent model of Tao et al.39 found that alternans of systolic Ca were generated by propagating Ca waves sustained through alternation of SR Ca content, implicating additional mechanisms for intracellular Ca alternans in addition to refractoriness of LCCs or RyRs under rapid pacing. Rovetti et al.41 showed that Ca alternans emerge as a collective behavior of Ca sparks, determined by the CRU network, via three Rs: “randomness” (of Ca spark activation), “refractoriness” (of a CRU after a Ca spark), and “recruitment” (Ca sparks inducing Ca sparks in adjacent CRUs). Nivala et al.53 later employed the ventricular myocyte couplon network model to study how SR Ca load and other physiological parameters, such as RyR sensitivity, SR uptake rate, NCX current, and Ca buffering affect Ca alternans in the context of the 3R theory, and found that alternans only occurs for an intermediate range of the SR Ca load, and the underlying mechanism can be explained via its effects on the three Rs (randomness, recruitment, and refractoriness).
Understanding disease: insights into mechanisms
Atrial fibrillation and atrial arrhythmia
Electrical, structural, and Ca handling remodeling contribute to the perpetuation and progression of AF. Evidence has suggested a role for Ca leak and spontaneous SR Ca release events at several stages of disease progression,70–73 and arrhythmogenic Ca waves resulting from heterogeneities in subcellular Ca alternans have been implicated as a mechanism in atrial dysrhythmia.74 Studying structure–function relationships and aberrant calcium handling in AF is complicated, however, by the fact that there are significant species-based differences in ultrastructure; whereas many large mammals, including humans, evince t-tubules in atrial cells,75,76 smaller mammals may not, and regional differences introduce further heterogeneity. The potential import of the t-tubular network in large mammal atrial EC coupling, despite the likely profound importance in subcellular Ca dynamics in these cells, has largely been overlooked thus far, and is an active subject in emerging modeling approaches at date of publication. Some human atrial myocytes do lack t-tubules (and thus compose an easily discretized structure as a starting point) and contain SR of both the junctional and non-junctional types, both of which have RyRs. An innovative mathematical modeling approach allowing detailed characterization of Ca movement within the three-dimensional volume of an atrial myocyte, displaying the centripetal Ca waves that occur within atrial myocytes during EC coupling and demonstrated that altering the strength of Ca release, RyR refractoriness, the magnitude of initiating stimulus, or the introduction of stochastic Ca channel activity could cause the nucleation of proarrhythmic traveling Ca waves.47 An influential combined experimental-computational study in human atrial cardiomyocytes from patients in sinus rhythm or with paroxysmal AF (pAF) showed increases in SR Ca leak and incidence of delayed after-depolarizations in pAF, underpinned by increased inactivation (phosphorylation) of the SERCA inhibitor protein phospholamban in pAF (thus, increased SERCA function), and increased RyR expression and single-channel open probability. Computational modeling indicated that both RyR dysregulation and enhanced SERCA promoted SR Ca leak and spontaneous SR Ca-release events, causing delayed afterdepolarization and potential triggered activity in pAF.48 In addition, while Torres et al.77 developed a model including the spatial arrangement of the sarcolemma including t-tubular system in ventricular myocytes, their findings were explained by a modified local control model, which constrained the region of regenerative activation of non-junctional RyR clusters, which may prove useful for describing ECC in AF cardiac myocytes, with a sparse t-system More recent work into RyR cluster fragmentation and redistribution in persistent AF78 motivate the further use of computational models to elucidate the role of structural disturbance in atrial dysrhythmia.
Heart failure
The abnormalities in Ca handling occur at nearly every point of Ca cycling in the failing heart cell, including activation and termination of SR Ca release, diastolic SR Ca leak, and SR Ca uptake.79 Cardiomyocytes from failing hearts exhibit a characteristic slowing of the rising phase of the CaT and additionally exhibit spatially nonuniform or dyssynchronous SR Ca release. A combined experimental/computational study from our groups used a computational model of the dyad to investigate the contribution of AP prolongation in a murine model of HF; ultimately, the study found that dyssynchronous Ca release in HF mouse myocytes does not result from electrical remodeling, but rather other factors, such as t-tubule reorganization (Figure 1B, right panel),6 as the longer murine action potential in HF resulted in increase SR Ca content, offsetting the desynchronizing effect of the extended action potential in this species. Furthermore, related work established that synchrony of cardiomyocyte Ca release is not only determined by t-tubule organization but also by the interplay between RyR sensitivity and SR Ca content. Cardiomyocytes from failing hearts also exhibit slow, dyssynchronous CaTs resulting from a subset of Ca sparks with slow kinetics.38 Slow sparks may occur at intact dyads: slow sparks are predicted to result from reorganization of CRUs in HF (Figure 1A, right panel). In addition to impaired contraction, this aberrant intracellular Ca cycling in HF has been implicated in both triggered and reentrant arrhythmias.80 The model of Gaur and Rudy42 was used to investigate how changes in microscopic dyadic properties, including detubulation in HF, affect whole-cell behavior. They found that increased dyadic volume and reduced LCCs/RyRs decrease ECC gain and can cause asynchrony of SR Ca release; when CSQN function is decreased, interdyad coupling increases diastolic Ca release activity to form Ca waves and long-lasting Ca release events. A recent study using a spatially-resolved ventricular myocyte model (see section 2) investigated effects of t-tubule disruption and other HF remodeling factors (CRU refractoriness, CRU coupling, and RyR leakiness) on Ca alternans. While others have seen that detubulation reduces the likelihood of sparks,6 in this model, disruption removed LCCs from the associated CRUs, and resulted in orphaned RyR clusters, providing increased opportunity for spark-induced Ca sparks to occur.2 The authors found that this t-tubular disruption promoted Ca alternans by two distinct mechanisms (1) (with normal SERCA function) by both CRU refractoriness and inter-CRU coupling, and (2) in the context of down-regulated SERCA, alternans was caused by an SR Ca load-dependent mechanism, independent of CRU refractoriness. The authors concluded that the mechanisms of Ca alternans for normal and down-regulated SERCA are different, and that t-tubular disruption promotes Ca alternans by both mechanisms, which may contribute to alternans at different stages of HF.81
Future directions: continued advances in imaging
Multi-scale meshes are key for computational studies of structure–function relationships in ECC. An early study aimed at developing an approach for spatial reconstruction of structures involved in calcium handling reconstructed clusters of RyRs together with the sarcolemma as based on dual labeling and three-dimensional confocal imaging of myocytes, leading to three-dimensional stacks of cross-sections; digital image processing was applied to deconvolve, filter, and segment image stacks.82 Clearly, advancing computational models via spatially resolved approaches now relies in part upon advances in experimental and imaging technologies which permit leaps in computational models.83
Advances in microscopic imaging technologies such as serial block face scanning electron microscopy (SBF-SEM) now allow description of new micro-domain in cardiomyocytes.84,85 Three-dimensional electron microscopy technologies such as electron tomography have been able to determine realistic nanogeometries of membrane junctions (dyads and peripheral junctions) and associated t-tubules. Labeling with antibodies has allowed examination of the three-dimensional distribution of RyRs with confocal microscopy,86 revealing couplon geometries later used in detailed computational models.44,45 At the same time, super-resolution light microscopy has gone beyond the diffraction limit to determine the distribution of smaller dyadic molecules, such as LCCs, at unprecedented resolutions, offering insight into the central machinery controlling cardiac ECC via calcium signaling.87 Computational models built upon such technologies are able to furnish unprecedented insight.58
Correlated light and electron microscopic (CLEM) imaging is a powerful method wherein each imaging mode provides unique information for dissecting cell and tissue function at high resolution.88 Other recent approaches have combined fluorescence resonance energy transfer (FRET), simulated-annealing (a form of combinatorial optimization), cryo-electron microscopy, and crystallographic data to locate a biosensor peptide bound to RyR Ca channels89 and have targeted a new sensitive Ca biosensor to the junctional space, where it co-localized with t-tubules and RyRs, allowing selective visualization and measurement of nanodomain Ca dynamics in intact cells.90 These multi-scale experimental and imaging approaches will offer mechanistic insights into CRU RyR operations in health and in disease states, and additionally offer potential for future inclusion in mechanistic computational modeling. Furthermore, emerging super-resolution single-molecule localization microscopy (SMLM) techniques offer an order of magnitude improvement over resolution of conventional fluorescence light microscopy; nanometer-scale distributions of multiple molecular targets can be resolved. In conjunction with the next generation of electron microscopy, SMLM has allowed the visualization and quantification of intricate t-tubule morphologies within large areas of muscle cells at unprecedented levels of detail, as recently reviewed.91 Novel and emerging imaging methods will enable the incorporation of detailed subcellular structural and functional information into the next generation of computational models (Figure 4), providing entirely new insights into the ion dynamics underpinning excitation and contraction in the heart, as well as the ways in which the system can fail in cardiac disease.
Figure 4.

Representation of the next generation of subcellular computational models, from left: super-resolution light microscopy permits resolution of the morphology of ryanodine receptor (RyR) clusters, which can be incorporated into synthetic geometries of the calcium release unit (CRU). Using these synthetic geometries, one can easily and systematically alter the distribution of RyRs, the shape and volume of the junctional SR (jSR) and network SR (nSR), and the cleft volume, and begin to analyze the different contributions quantitatively, permitting query into how spark fidelity is affected by RyR density, by cluster breakup, by cleft height, or by small and narrow jSR (local depletion of Ca). On the other hand (from right), these synthetic geometries neglect the potentially important role played by detailed and realistic CRU structures, which can now be obtained from electron tomography. (Images reproduced from Hake et al.58 with permission.)
Summary and conclusions
In the present review, we have focused on the development of structural models of cardiac Ca dynamics, introducing the reader to the development of models of subcellular Ca handling in cardiomyocytes, with specific focus on progress in recent years. Computational modeling and simulation have helped to uncover the extent to which macroscopic Ca release events are intrinsically controlled by the type and number of individual LCCs and RyRs in the CRU, the relative spatial localization of the two channel types and the geometry of this functionally significant nanodomain, as well as the role of CRU organization at the cell level.
Computational models have additionally moved towards the level of the whole myocyte, generally stepping up dimension and complexity in geometry. Both subcellular models investigating spark dynamics at the level of the single CRU and physiologically detailed models of whole-cell subcellular Ca cycling including networks thousands of CRU have been developed, at present seeking to combine the data emergent from newer imaging technologies to gain multi-scale insight into calcium-driven processes. This research has offered new knowledge into Ca spark termination, and, most recently, long-lasting Ca sparks. Structural distribution of CRU have additionally been shown to affect dynamics of Ca waves. Biophysical models have furthermore been employed to show that Ca alternans emerges as a collective behavior of Ca sparks, determined by the CRU network, via randomness of Ca spark activation, refractoriness of a CRU after a Ca spark, and recruitment, inducing Ca sparks in adjacent CRUs. Other computational strategies have offered new insight in the context of AF, wherein research revealed the role of RyR cluster fragmentation and redistribution on Ca remodeling in persistent AF, as well as HF, where the role of t-tubular disruption on electrical abnormalities (alternans) has been studied.
Finally, novel labeling and imaging techniques now permit selective visualization at the nanodomain in intact cardiac myocytes, offering needed intelligence into Ca operations in health and disease, as well as a platform for the next generation of mechanistic computational models. These emerging multi-modality experimental methods will enable incorporation of detailed subcellular structural as well as functional information, providing powerful new computational tools for insight into the dynamics underpinning excitation and contraction in the heart.
Footnotes
Peer Review:4 peer reviewers contributed to the peer review report. Reviewers reports totaled 1623 words, excluding any confidential comments to the academic editor.
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project has received funding from the European Union’s Horizon 2020 research and innovation programme (Consolidator grant, WEL) under grant agreement No 647714. The authors also graciously acknowledge funding support from the Research Council of Norway via the Center for Cardiological Innovation Center for Research-Based Innovation SFI 2011-2019.
References
- 1. Franzini-Armstrong C, Protasi F, Tijskens P. The assembly of calcium release units in cardiac muscle. Ann NY Acad Sci 2005; 1047: 76–85. [DOI] [PubMed] [Google Scholar]
- 2. Song LS, Sobie EA, McCulle S, et al. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA 2006; 103(11): 4305–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pinali C, Bennett H, Davenport JB, et al. Three-dimensional reconstruction of cardiac sarcoplasmic reticulum reveals a continuous network linking transverse-tubules: this organization is perturbed in heart failure. Circulat Res 2013; 113(11): 1219–1230. [DOI] [PubMed] [Google Scholar]
- 4. Louch WE, Bito V, Heinzel FR, et al. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res 2004; 62(1): 63–73. [DOI] [PubMed] [Google Scholar]
- 5. Louch WE, Mørk HK, Sexton J, et al. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol 2006; 574(2): 519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Louch WE, Hake J, Jølle GF, et al. Control of Ca2+ release by action potential configuration in normal and failing murine cardiomyocytes. Biophys J 2010; 99(5): 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Øyehaug L, Loose KØ, Jølle GF, et al. Synchrony of cardiomyocyte Ca(2+) release is controlled by T-tubule organization, SR Ca(2+) content, and ryanodine receptor Ca(2+) sensitivity. Biophys J 2013; 104(8): 1685–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. II. Afterdepolarizations, triggered activity, and potentiation. Circulat Res 1994; 74(6): 1097–1113. [DOI] [PubMed] [Google Scholar]
- 9. Wang SQ, Song LS, Lakatta EG, et al. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature 2001; 410(6828): 592–596. [DOI] [PubMed] [Google Scholar]
- 10. Zima AV, Picht E, Bers DM, et al. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circulat Res 2008; 103(8): e105–e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maack C, O’Rourke B. Excitation–contraction coupling and mitochondrial energetics. Basic Res Cardiol 2007; 102(5): 369–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Limbu S, Hoang-Trong T, Prosser B, et al. Modeling local X-ROS and calcium signaling in the heart. Biophys J 2015; 109(10): 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foteinou PT, Greenstein JL, Winslow RL. Mechanistic investigation of the arrhythmogenic role of oxidized CaMKII in the heart. Biophys J 2015; 109(4): 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee YS, Liu OZ, Sobie EA. Decoding myocardial Ca2+ signals across multiple spatial scales: a role for sensitivity analysis. J Mol Cell Cardiol 2013; 58: 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Williams GSB, Smith GD, Sobie EA, et al. Models of cardiac excitation-contraction coupling in ventricular myocytes. Math Biosci 2010; 226(1): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jafri MS. Models of excitation–contraction coupling in cardiac ventricular myocytes. Meth Mol Biol 2012; 910: 309–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jafri MS, Rice JJ, Winslow RL. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys J 1998; 74(3): 1149–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shannon TR, Wang F, Puglisi J, et al. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J 2004; 87(5): 3351–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Faber GM, Silva J, Livshitz L, et al. Kinetic properties of the cardiac L-type Ca2+ channel and its role in myocyte electrophysiology: a theoretical investigation. Biophys J 2007; 92(5): 1522–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J 1992; 63(2): 497–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rice JJ, Jafri MS, Winslow RL. Modeling gain and gradedness of Ca2+ release in the functional unit of the cardiac diadic space. Biophys J 1999; 77(4): 1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cannell MB, Soeller C. Numerical analysis of ryanodine receptor activation by L-type channel activity in the cardiac muscle diad. Biophys J 1997; 73(1): 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sobie EA, Dilly KW, dos Santos Cruz J, et al. Termination of cardiac Ca(2+) sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J 2002; 83(1): 59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanskanen AJ, Greenstein JL, Chen A, et al. Protein geometry and placement in the cardiac dyad influence macroscopic properties of calcium-induced calcium release. Biophys J 2007; 92(10): 3379–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Louch WE, Land S, Niederer SA. Strange bedfellows: biologists and mathematical modelers tie the knot on cardiomyocyte calcium homeostasis. Drug Discov Today Disease Models 2014; 14: 11–16. [Google Scholar]
- 26. Hinch R, Greenstein JL, Tanskanen AJ, et al. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys J 2004; 87(6): 3723–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Williams GSB, Huertas MA, Sobie EA, et al. A probability density approach to modeling local control of calcium-induced calcium release in cardiac myocytes. Biophys J 2007; 92(7): 2311–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Greenstein JL, Winslow RL. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys J 2002; 83(6): 2918–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Greenstein JL, Hinch R, Winslow RL. Mechanisms of excitation–contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophys J 2006; 90(1): 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science (New York, NY) 1993; 262(5134): 740–744. [DOI] [PubMed] [Google Scholar]
- 31. López-López JR, Shacklock PS, Balke CW, et al. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science (New York, NY) 1995; 268(5213): 1042–1045. [DOI] [PubMed] [Google Scholar]
- 32. Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science (New York, NY) 1995; 268(5213): 1045–1049. [DOI] [PubMed] [Google Scholar]
- 33. Brochet DXP, Xie W, Yang D, et al. Quarky calcium release in the heart. Circulat Res 2011; 108(2): 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koh X, Srinivasan B, Ching HS, et al. A 3D Monte Carlo analysis of the role of dyadic space geometry in spark generation. Biophys J 2006; 90(6): 1999–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soeller C, Cannell MB. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys J 1997; 73(1): 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hake J, Lines GT. Stochastic binding of Ca2+ ions in the dyadic cleft; continuous versus random walk description of diffusion. Biophys J 2008; 94(11): 4184–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramay HR, Liu OZ, Sobie EA. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc Res 2011; 91(4): 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Louch WE, Hake J, Mørk HK, et al. Slow Ca2+ sparks de-synchronize Ca2+ release in failing cardiomyocytes: evidence for altered configuration of Ca2+ release units? J Mol Cell Cardiol 2013; 58: 41–52. [DOI] [PubMed] [Google Scholar]
- 39. Tao T, O’Neill SC, Diaz ME, et al. Alternans of cardiac calcium cycling in a cluster of ryanodine receptors: a simulation study. Am J Physiol Heart Circulat Physiol 2008; 295(2): H598–H609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen-Izu Y, McCulle SL, Ward CW, et al. Three-dimensional distribution of ryanodine receptor clusters in cardiac myocytes. Biophys J 2006; 91(1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rovetti R, Cui X, Garfinkel A, et al. Spark-induced sparks as a mechanism of intracellular calcium alternans in cardiac myocytes. Circulat Res 2010; 106(10): 1582–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gaur N, Rudy Y. Multiscale modeling of calcium cycling in cardiac ventricular myocyte: macroscopic consequences of microscopic dyadic function. Biophys J 2011; 100(12): 2904–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wescott AP, Jafri MS, Lederer W, et al. Ryanodine receptor sensitivity governs the stability and synchrony of local calcium release during cardiac excitation-contraction coupling. J Mol Cell Cardiol 2016; 92: 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laver DR, Kong CHT, Imtiaz MS, et al. Termination of calcium-induced calcium release by induction decay: An emergent property of stochastic channel gating and molecular scale architecture. J Mol Cell Cardiol 2013; 54(1): 98–100. [DOI] [PubMed] [Google Scholar]
- 45. Cannell M. Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys J 2013; 104(10): 2149–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frisk M, Koivumäki JT, Norseng PA, et al. Variable t-tubule organization and Ca2+ homeostasis across the atria. Am J Physiol Heart Circulat Physiol 2014; 307(0407): H609–H620. [DOI] [PubMed] [Google Scholar]
- 47. Thul R, Coombes S, Roderick HL, et al. Subcellular calcium dynamics in a whole-cell model of an atrial myocyte. Proc Natl Acad Sci USA 2012; 109(6): 2150–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Voigt N, Heijman J, Wang Q, et al. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 2014; 129(2): 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grandi E, Pandit SV, Voigt N, et al. Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circulat Res 2011; 109(9): 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Restrepo JG, Weiss JN, Karma A. Calsequestrin-mediated mechanism for cellular calcium transient alternans. Biophys J 2008; 95(8): 3767–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Restrepo JG, Karma A. Spatiotemporal intracellular calcium dynamics during cardiac alternans. Chaos (Woodbury, NY) 2009; 19(3): 037115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nivala M, Ko CY, Nivala M, et al. Criticality in intracellular calcium signaling in cardiac myocytes. Biophys J 2012; 102(11): 2433–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nivala M, Qu Z. Calcium alternans in a couplon network model of ventricular myocytes: role of sarcoplasmic reticulum load. Am J Physiol Heart Circulat Physiol 2012; 303(3): H341–H352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Song Z, Karma A, Weiss JN, et al. Long-lasting sparks: multi-metastability and release competition in the calcium release unit network. PLoS Computat Biol 2016; 12(1): e1004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen X, Guo L, Kang J, et al. Calcium waves initiating from the anomalous subdiffusive calcium sparks. J R Soc Interface 2014; 11(91): 20130934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Izu LT, Means SA, Shadid JN, et al. Interplay of ryanodine receptor distribution and calcium dynamics. Biophys J 2006; 91(1): 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sato D, Shannon TR, Bers DM. Sarcoplasmic reticulum structure and functional properties that promote long-lasting calcium sparks. Biophys J 2016; 110(2): 382–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hake J, Edwards AG, Yu Z, et al. Modelling cardiac calcium sparks in a three-dimensional reconstruction of a calcium release unit. J Physiol 2012; 590(18): 4403–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Soeller C, Jayasinghe ID, Li P, et al. Three-dimensional high-resolution imaging of cardiac proteins to construct models of intracellular Ca2+ signalling in rat ventricular myocytes. Experimental Physiol 2009; 94(5): 496–508. [DOI] [PubMed] [Google Scholar]
- 60. Rajagopal V, Bass G, Walker CG, et al. Examination of the effects of heterogeneous organization of RyR clusters, myofibrils and mitochondria on Ca2+ release patterns in cardiomyocytes. PLoS Computat Biol 2015; 11(9): e1004417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vierheller J, Neubert W, Falcke M, et al. A multiscale computational model of spatially resolved calcium cycling in cardiac myocytes: from detailed cleft dynamics to the whole cell concentration profiles. Front Physiol 2015; 6: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Winslow RL, Tanskanen A, Chen M, et al. Multiscale modeling of calcium signaling in the cardiac dyad. Ann NY Acad Sci 2006; 1080: 362–375. [DOI] [PubMed] [Google Scholar]
- 63. Macquaide N, Tuan HTM, Hotta JI, et al. Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc Res 2015; 108(3): 387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Williams GSB, Chikando AC, Tuan HTM, et al. Dynamics of calcium sparks and calcium leak in the heart. Biophys J 2011; 101(6): 1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hoang-Trong TM, Ullah A, Jafri MS. Calcium sparks in the heart: dynamics and regulation. Res Rep Biol 2015; 6: 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cheng H, Lederer MR, Xiao RP, et al. Excitation=-contraction coupling in heart: new insights from Ca2+ sparks. Cell Calcium 1996; 20(2): 129–140. [DOI] [PubMed] [Google Scholar]
- 67. Izu LT, Wier WG, Balke CW. Evolution of cardiac calcium waves from stochastic calcium sparks. Biophys J 2001; 80(1): 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bers DM. Cardiac excitation–contraction coupling. Nature 2002; 415(6868): 198–205. [DOI] [PubMed] [Google Scholar]
- 69. Wehrens XHT, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol 2005; 67: 69–98. [DOI] [PubMed] [Google Scholar]
- 70. Hove-Madsen L, Llach A, Bayes-Genís A, et al. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 2004; 110(11): 1358–1363. [DOI] [PubMed] [Google Scholar]
- 71. Neef S, Dybkova N, Sossalla S, et al. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circulat Res 2010; 106(6): 1134–1144. [DOI] [PubMed] [Google Scholar]
- 72. Voigt N, Li N, Wang Q, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012; 125(17): 2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Greiser M, Schotten U. Dynamic remodeling of intracellular Ca2+ signaling during atrial fibrillation. J Mol Cell Cardiol 2013; 58(1): 134–142. [DOI] [PubMed] [Google Scholar]
- 74. Blatter LA, Kockskämper J, Sheehan KA, et al. Local calcium gradients during excitation–contraction coupling and alternans in atrial myocytes. J Physiol 2003; 546(1): 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Richards MA, Clarke JD, Saravanan P, et al. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am J Physiol Heart Circulat Physiol 2011; 301(5): H1996–H2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dibb KM, Clarke JD, Eisner DA, et al. A functional role for transverse (t-) tubules in the atria. J Mol Cell Cardiol 2013; 58: 84–91. [DOI] [PubMed] [Google Scholar]
- 77. Torres NS, Sachse FB, Izu LT, et al. A modified local control model for Ca2+ transients in cardiomyocytes: junctional flux is accompanied by release from adjacent non-junctional RyRs. J Mol Cell Cardiol 2014; 68: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Macquaide N, Bito V, Sipido KR. Measuring sarcoplasmic reticulum Ca2+ content, fractional release, and Ca2+ buffering in cardiac myocytes. Cold Spring Harbor Protocols 2015; 4: 403–407. [DOI] [PubMed] [Google Scholar]
- 79. Zima AV, Bovo E, Mazurek SR, et al. Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch 2014; 466(6): 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Aistrup G, Balke C, Wasserstrom J. Arrhythmia triggers in heart failure: The smoking gun of I dysregulation. Heart Rhythm 2011; DOI: 10.1016/j.hrthm.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 81. Nivala M, Song Z, Weiss JN, et al. T-tubule disruption promotes calcium alternans in failing ventricular myocytes: mechanistic insights from computational modeling. J Mol Cell Cardiol 2015; 79: 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sachse FB, Savio-Galimberti E, Goldhaber JI, et al. Towards computational modeling of excitation-contraction coupling in cardiac myocytes: reconstruction of structures and proteins from confocal imaging. In Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing, 2009; pp. 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hake J, Kekenes-Huskey PM, McCulloch AD. Computational modeling of subcellular transport and signaling. Curr Opin Struct Biol 2014; 25: 92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hayashi T, Martone ME, Yu Z, et al. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci 2009; 122(7): 1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pinali C, Kitmitto A. Serial block face scanning electron microscopy for the study of cardiac muscle ultrastructure at nanoscale resolutions. J Mol Cell Cardiol 2014; 76: 1–11. [DOI] [PubMed] [Google Scholar]
- 86. Soeller C, Crossman D, Gilbert R, et al. Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc Natl Acad Sci USA 2007; 104(38): 14958–14963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Das T, Hoshijima M. Adding a new dimension to cardiac nano-architecture using electron microscopy: coupling membrane excitation to calcium signaling. J Mol Cell Cardiol 2013; 58: 5–12. [DOI] [PubMed] [Google Scholar]
- 88. Ellisman MH, Deerinck TJ, Shu X, et al. Picking faces out of a crowd: genetic labels for identification of proteins in correlated light and electron microscopy imaging. Meth Cell Biol 2012; 111: 139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Svensson B, Oda T, Nitu FR, et al. FRET-based trilateration of probes bound within functional ryanodine receptors. Biophys J 2014; 107(9): 2037–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shang W, Lu F, Sun T, et al. Imaging Ca2+ nanosparks in heart with a new targeted biosensor. Circulat Res 2014; 114(3): 412–420. [DOI] [PubMed] [Google Scholar]
- 91. Jayasinghe ID, Clowsley AH, Munro M, et al. Revealing t-tubules in striated muscle with new optical super-resolution microscopy techniquess. Eur J Translat Myol 2015; 25(1): 4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mahajan A, Sato D, Shiferaw Y, et al. Modifying L-type calcium current kinetics: consequences for cardiac excitation and arrhythmia dynamics. Biophys J 2008; 94(2): 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]