

Abstract
Diabetic retinopathy is the leading cause of blindness in working-age adults. It is caused by oxygen starvation in the retina inducing aberrant formation of blood vessels that destroy retinal architecture. In humans, vitreal stromal cell–derived factor–1 (SDF-1) concentration increases as proliferative diabetic retinopathy progresses. Treatment of patients with triamcinolone decreases SDF-1 levels in the vitreous, with marked disease improvement. SDF-1 induces human retinal endothelial cells to increase expression of VCAM-1, a receptor for very late antigen–4 found on many hematopoietic progenitors, and reduce tight cellular junctions by reducing occludin expression. Both changes would serve to recruit hematopoietic and endothelial progenitor cells along an SDF-1 gradient. We have shown, using a murine model of proliferative adult retinopathy, that the majority of new vessels formed in response to oxygen starvation originate from hematopoietic stem cell–derived endothelial progenitor cells. We now show that the levels of SDF-1 found in patients with proliferative retinopathy induce retinopathy in our murine model. Intravitreal injection of blocking antibodies to SDF-1 prevented retinal neovascularization in our murine model, even in the presence of exogenous VEGF. Together, these data demonstrate that SDF-1 plays a major role in proliferative retinopathy and may be an ideal target for the prevention of proliferative retinopathy.
Introduction
Diabetic retinopathy is a major cause of blindness among Americans under the age of 65. There are approximately 16 million diabetics in the United States, with nearly 8 million having some form of diabetic retinopathy. Diabetes is caused when the body can no longer produce enough insulin or is not able to utilize the insulin produced. Without insulin, blood sugar levels cannot be regulated, and an increase of blood glucose levels occurs. Prolonged high levels of blood glucose in diabetic patients destroy the small blood vessels in the eye. As the vessels are damaged, vascular permeability increases, resulting in fluid leakage into the surrounding tissue, often resulting in a swelling. When swelling occurs in the macula of the eye (the area of the retina responsible for sharp central vision), vision can often become distorted. This condition is called macular edema. Further vessel deterioration results in poor blood flow and the onset of ischemia, or oxygen starvation. Ischemia promotes new blood vessel proliferation in an attempt to restore blood flow. Vision loss during this proliferative stage of diabetic retinopathy is caused by aberrant neovascularization resulting in newly formed blood vessels intruding into the vitreous of the eye (referred to as preretinal neovascularization). These new vessels destroy the normal retinal architecture and may hemorrhage, easily causing bleeding into the eye, which ultimately impairs vision (1).
The mechanisms governing this aberrant neovascularization during diabetic retinopathy are still being elucidated. We have recently demonstrated in 2 murine models of ocular neovascularization that adult HSCs function as hemangioblasts, producing both blood cells and the circulating endothelial progenitor cells (EPCs) that give rise to new blood vessels in the eye (2, 3). CD34+ cells, which are highly enriched for human HSCs, from umbilical cord blood also produce new blood vessels in a murine xenograft adaptation of our model (4). In this study, we used a unique murine model that induces adult-onset retinal neovascularization that closely mimics the pathology of neovascularization observed in diabetic humans. Retinal neovascularization in the adult mouse requires the administration of exogenous VEGF in addition to ischemic injury to promote new vessel formation. We have also shown that chronic vascular injury alone can be sufficient to induce EPC production by adult HSCs (5). The cytokine VEGF is a major inducer of angiogenesis and the resultant migration of EPCs (6). Within the retina, VEGF expression is increased in response to ischemia to promote vascular repair. VEGF induces vascular permeability and protease production and promotes endothelial cell migration and proliferation — key steps in angiogenesis. VEGF is widely recognized as a potential therapeutic target for regulating angiogenesis (7, 8). We were interested in investigating other cytokines/chemokines that may work in conjunction with VEGF to promote the recruitment of endothelial progenitors from remote locations such as the bone marrow into the ischemic retina. We examined the role of stromal cell–derived factor–1 (SDF-1) in the process of retinal neovascularization. SDF-1 is the predominant chemokine that mobilizes HSCs and EPCs (9–11). SDF-1 has been shown to be upregulated in many damaged tissues as part of the injury response and is thought to call stem/progenitor cells to promote repair (12). We have shown that SDF-1 levels increase in diabetics with proliferative diabetic retinopathy (PDR) and that SDF-1 may play an important role in the migration of HSC-derived EPCs to the site of vascular injury by regulating molecules important in the injury/repair response. Exogenous SDF-1 can also substitute for exogenous VEGF to drive retinal neovascularization in our murine model. Furthermore, blocking SDF-1 function can prevent neovascularization and may serve as an important advancement in the treatment of ocular disease such as diabetic retinopathy.
Results
Measurement of SDF-1 in patients with varying severity of diabetic retinopathy.
Previously we demonstrated that HSCs can be a major source of EPCs (2). In the present study, we postulated that SDF-1 plays a key role in the recruitment of these progenitors to sites of vascular injury to produce new blood vessels. We further hypothesized that retinal ischemia results in increased SDF-1 expression. Our data suggest that vascular permeability may be increased by angiogenic factors, such as SDF-1 and VEGF produced in response to ischemia. The increased permeability would allow for a portion of the SDF-1 produced by the damaged retina to leak into the vitreous of the eye. SDF-1 leaking into the vitreous may create an artificially high SDF-1 concentration gradient due to the relative lack of proteases within the vitreous (13). New vessel growth would be directed into the vitreous by the SDF-1 gradient. We postulated that if our hypothesis is correct, the addition of SDF-1 protein in the eye should augment preretinal neovascularization within the vitreous. Conversely, blocking SDF-1 activity in the eye should abrogate preretinal neovascularization within the vitreous.
To test our first hypothesis, we obtained vitreous samples from 46 patients undergoing treatment for diabetic macular edema (DME) with and without PDR. Forty-four of the 46 patients were type 2 diabetics. Vitreous samples from nondiabetic patients having vitrectomy for non-PDR–related conditions were used as controls. ELISA were performed in a masked fashion to measure SDF-1 levels in the vitreous samples. Once the ELISA data were compiled, the samples were matched with patients. Samples were divided into 4 categories based on the severity of disease: control samples (n = 8 eyes) and those from patients with DME but no current PDR (n = 30 eyes), DME with PDR (n = 20 eyes), and neovascularization of the iris (NVI), representing the most fulminate version of the disease (n = 4 eyes). As predicted by our hypothesis, SDF-1 increased with the severity of the patients’ diabetic retinopathy (Figure 1). SDF-1 was undetectable by ELISA (sensitivity 18 pg/ml) in vitreous samples from control patients. Patients with fulminate NVI averaged more than 1,000 pg/ml of SDF-1 in their vitreous, or at least 50-fold the level found in normal eyes. Patients with DME and proliferating diabetic retinopathy had an average of more than 200 pg/ml SDF-1, while those with only DME had an average of 75 pg/ml SDF-1 in their vitreous. These results demonstrate that SDF-1 concentrations increase in the vitreous of patients with macular edema and diabetic retinopathy and that SDF-1 concentration correlates with disease severity (P < 0.005).
Figure 1.

SDF-1 expression increases with severity of diabetic retinopathy. SDF-1 concentration in human patients with increasing severity of proliferative diabetic retinopathy. Human SDF-1–specific ELISAs were performed in triplicate on vitreous samples from patients with various stages of DME without (n = 30) or with (n = 20) PDR. The most fulminate stage of the disease is represented by patients with NVI (n = 4). Control vitreous samples (n = 8) were obtained from nondiabetic patients being treated for other ailments. All control samples were below the level of detection by ELISA (18 pg/ml SDF-1).
Corticosteroid treatment reduces SDF-1 levels.
Corticosteroids have been used for decades to suppress intraocular inflammation and to reduce blood vessel leakage (14, 15). Triamcinolone or Kenalog (the commercial name for triamcinolone acetonide; Bristol-Meyers Squibb) has been used intravitreally in 2 recent studies on DME and has been shown to decrease breakdown of the blood-retina barrier with a significant improvement in visual acuity (16, 17). The mechanism by which triamcinolone achieves a therapeutic benefit remains unknown. We hypothesized that triamcinolone may reduce the expression of SDF-1 by damaged tissue. To test this hypothesis, we assayed vitreous samples from our 46 patients after they received treatment for DME by intravitreal administration of 4 mg of triamcinolone in 0.2 ml balanced salt solution. In select patients with mild disease, repeat intravitreal taps were performed 1 month after treatment. Patients with more severe disease such as NVI received multiple triamcinolone treatments, with intravitreal samples obtained at each treatment. The vitreous samples were withdrawn, according to the standard of therapy, prior to every triamcinolone injection to ensure maintenance of normal ocular pressure. These vitreous samples were from the same patients we previously assayed prior to treatment (Figure 1). After triamcinolone treatment the patients showed a uniform drop in SDF-1 levels in the vitreous to near the limits of detection (Figure 2). This suggests that reducing SDF-1 levels and the subsequent recruitment of circulating EPCs may be one of the mechanisms of action for triamcinolone. Unfortunately, triamcinolone has serious side effects. Nearly one-third of triamcinolone-treated patients develop glaucoma that requires treatment to prevent additional vision loss (18). Therefore, more targeted therapies, such as directly blocking SDF-1 activity, may provide optimized patient care.
Figure 2.

SDF-1 concentrations in the vitreous of human patients after treatment with triamcinolone. All patients were treated with at least 1 round of triamcinolone (4 mg) injections intravitreally. Vitreous samples were obtained 1 month after treatment. Human SDF-1–specific ELISAs were performed in triplicate on the vitreous samples. The results are presented according to the severity of the patients’ original disease. DME only, n = 30; DME + PDR, n = 20. The most fulminate stage of the disease is represented by patients with NVI (n = 4). Control vitreous samples (n = 8) were obtained from nondiabetic patients being treated for other ailments. All control samples were below the level of detection by ELISA (18 pg/ml SDF-1).
The role of SDF-1 in neovascularization.
SDF-1 is one of the primary chemokines responsible for the homing of HSCs to the bone marrow (19). SDF-1 expression is induced by a wide variety of cell types in response to stimuli such as stress and injury (12, 20). SDF-1 signals through its only known receptor, CXCR-4, a transmembrane G protein–coupled receptor. VEGF induces increased CXCR-4 (21) expression from endothelial cells, while SDF-1 induces VEGF expression in cells that are both hematopoietic and endothelial in origin (22, 23). Chemotaxis assays have shown that purified endothelial progenitor cells migrate along an SDF-1 concentration gradient in vitro (24–26). Retinal endothelial cells were a more relevant cell type when testing whether SDF-1 has any effect in our unique murine model of ischemic retinopathy. We have shown by ELISA that an increase in SDF-1 expression results in a significant increase (P < 0.007) of VCAM-1 on retinal endothelial cells (Figure 3). An increase in VCAM-1 plays an important role in HSC homing to and mobilization from the bone marrow by allowing for firm adhesion to the activated bone marrow endothelium (27). We also studied the effects SDF-1 had on retinal endothelial cells and on gap junction proteins. We have shown by Western blot analysis that as SDF-1 levels are increased, the expression of occludin by retinal endothelial cells is decreased. Occludin is a gap junction protein responsible for tight junctions between endothelial cells, which prevents leakage of vessel contents into the surrounding tissue (Figure 4). These data suggest that SDF-1 acts at several key steps in the process of ischemic repair, such as recruitment of EPCs from the marrow, increasing of VCAM-1 expression to promote EPC adhesion, and decreasing of tight junctions to allow EPCs to extravagate to the site of ischemia.
Figure 3.

SDF-1 increases VCAM-1 expression on ECs. HRECs upregulate VCAM-1 in response to SDF-1. HRECs were isolated from 2 separate donors: a 43-year-old donor and a 53-year-old donor. The HRECs were cultured in endothelial growth medium (EGM) containing 10% FCS for 3 weeks in order to establish superconfluent cultures. Control superconfluent HREC cultures were treated with reduced serum medium or continued in EGM. Test superconfluent HREC cultures were treated with increasing concentrations of SDF-1 protein in reduced serum medium. All treatments were for 48 hours. Cells were harvested in extraction buffer, and equal quantities of total protein were used in ELISAs to check for the expression of VCAM-1. No changes in VCAM-1 expression were seen in either control group. Therefore, results were normalized to the combined average of both control groups and are expressed as percent of control. Increasing levels of SDF-1 upregulates the expression of VCAM-1 on HRECs.
Figure 4.

SDF-1 reduces occludin expression on ECs. HRECs were isolated from 2 separate donors: (A) a 43-year-old donor and (B) a 53-year-old donor. The HRECs were cultured in EGM containing 10% FCS for 3 weeks in order to establish tight cellular junctions. The test cultures were treated for 2 days with either EGM with reduced serum (RS) of 1%, or with EGM and 1% FCS plus either 0.1 nM SDF-1 or 100 nM SDF. Cells were harvested in extraction buffer and equal quantities of total protein separated by SDS-polyacrylamide gels followed by transfer to nitrocellulose and immunoblotted for occludin and β-actin (loading control) levels.
SDF-1 enhances neovascularization in ischemic retinopathy.
In order to support our hypothesis that SDF-1 is significant in the progression of proliferative retinopathy, we tested whether the administration of exogenous recombinant SDF-1 (rSDF-1) protein (R&D Systems) could promote neovascularization. To test this hypothesis, we used our murine model, which mimics the pathology seen in PDR in an adult mouse. The model requires the administration of growth factor and injury. The model allows us to tag new vessel formation with GFP+ cells and serves as an important tool in investigating the underlying mechanisms of proliferative retinopathy. There is no evidence that cell fusion plays a role in the development of functional blood vessels in our system, but this important point is still being investigated. The basic model has been previously described (2). The model was modified by replacing the administration of recombinant adeno-associated virus–VEGF (rAAV-VEGF) with the administration of rSDF-1 protein at a concentration of 75 pg/μl within the vitreous. The 75 pg/μl dose was chosen to match the lowest concentration of SDF-1 found the vitreous of patients with proliferative diabetic retinopathy (Figure 1). Weekly injections were performed up to 4 weeks after laser injury in order to sustain the concentration of SDF-1 in the vitreous. Exogenous SDF-1 was able to enhance GFP+ HSC-derived EPC migration and incorporation into the sites of ischemic injury (Figure 5). We also observed the recruitment of a large population of GFP+ cells that were incorporated outside of the retinal vasculature. They could be inflammatory cells, such as neutrophils, that have an increase in their migratory response toward SDF-1 (28) due to the administration of exogenous rSDF-1 protein, but the time of analysis makes this unlikely. Exogenous rSDF-1 protein may have also recruited a surplus of retinal astrocytes, which provide a template for the promotion of retinal angiogenesis (29, 30).
Figure 5.
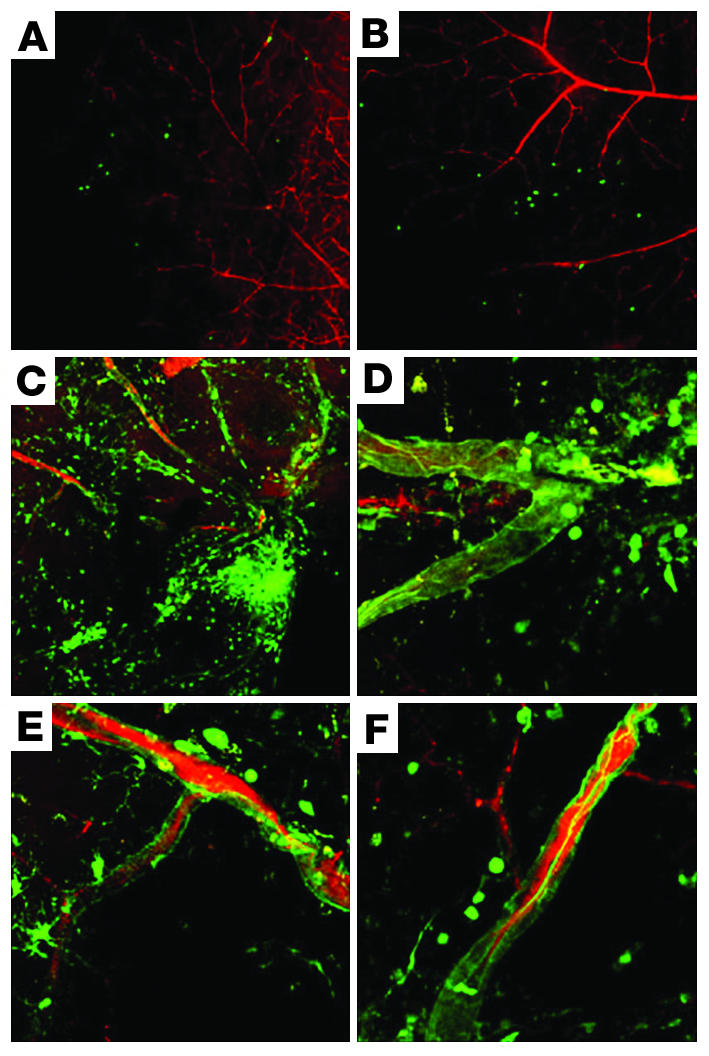
Recombinant SDF-1 protein enhances HSC-derived EPC migration and incorporation in sites of ischemia. Animals were perfused with a red fluorescent dye (RITC-dextran; Sigma-Aldrich) to delineate the vasculature. New blood vessels incorporated GFP+ HSC progeny, thereby forming areas of green/yellow fluorescence. GFP+ progeny suggestive of astrocytes or glia are also seen incorporated outside of the vasculature. (A and B) Left, or untreated, eyes of 2 C57BL/6.gfp mice that were treated in their right eyes to induce retinal ischemia with administration of exogenous rSDF-1 protein (75 pg/μl) and without exogenous AAV-VEGF. Note the lack of recruitment and incorporation of transplanted gfp+ HSC progeny in the control untreated left eyes. (C–F) Right, or treated, eyes of 4 representative C57BL/6.gfp mice (including the right, treated eyes of the animals in A and B), in which retinal ischemia was induced and which were injected intravitreally with rSDF-1 protein as a replacement for the rAAV-VEGF used in our standard model. Compare Figure 8 – where rAAV-VEGF was used – in which model control eyes show similar recruitment and incorporation of transplanted GFP+ HSCs. Magnification, ×10 (A and B), ×20 (C), ×63 (D–F).
Prevention of neovascularization in ischemic retinopathy.
We next challenged the postulate that blocking SDF-1 should reduce retinal neovascularization from HSC-derived EPCs by blocking their recruitment to the site of injury. To test this hypothesis, we once again used our unique murine model, as described above. To abrogate SDF-1 activity, we injected a cohort of 10 long-term engrafted animals with an SDF-1–specific blocking antibody in PBS (R&D Systems) into the vitreous at the time of laser injury. The injections were designed to yield a final antibody concentration of 1 μg/μl in the vitreous. Weekly booster injections of SDF-1–blocking antibody were given intravitreally during the ischemic repair phase. Two control cohorts of 10 animals, all with equivalent hematopoietic engraftment, received either no intravitreal injections (model control) or weekly intravitreal mock antibody injections with a PBS + IgG isotype control antibody. The 2 control cohorts yielded similar levels of HSC-derived contributions to retinal neovascularization. This indicated that the isotype control antibody had no effect on HSC-derived neovascularization (Figure 6). Strikingly, the cohort treated with SDF-1–blocking antibody produced almost no HSC-derived blood vessels in response to VEGF bolus and ischemia injury. Confocal microscopy images from 4 independent test retinas are shown for each of the cohorts (Figure 6). Green or yellow fluorescence of vessels indicate the presence of HSC-derived endothelium (2). Purely red fluorescence of vessels, like those seen in the cohort that received antibody and the negative control eyes, indicates no HSC-derived contributions. The negative control eyes in all experiments are untreated left eyes of the animals used in the model. Therefore, they represent the background level of HSC contribution to undamaged vessels after a bone marrow transplant. None of the 10 animals that received injections of SDF-1–blocking antibody had significant GFP+ HSC-derived contributions to the retinal vasculature described above that was seen in the control eyes.
Figure 6.

Anti–SDF-1 antibody prevents retinal neovascularization by HSC-derived circulating endothelial progenitors. All micrographs are merged confocal images of retinal flat mounts. Animals were perfused with a red fluorescent dye (RITC-dextran; Sigma-Aldrich) to delineate the vasculature. New blood vessels incorporate GFP+ HSC progeny, thereby forming areas of green/yellow fluorescence. Model control: Right, or treated, eyes from 4 representative C57BL/6.gfp animals in which retinal ischemia was induced according to our standard model. GFP+ progeny suggestive of astrocytes or glia are also seen incorporated outside of the vasculature. Negative control: Left, or untreated, eyes of the same 4 C57BL/6.gfp mice in which retinal ischemia was induced according to our standard model in their right eyes. Note the lack of recruitment and incorporation of transplanted gfp+ HSC progeny. Mock injections: Right, or treated, eyes from 4 representative C57BL/6.gfp mice in which retinal ischemia was induced according to our standard model with the added step of intravitreal injection with PBS containing an isotype control antibody to a final concentration of 1 μg/μl. Note the similar recruitment and incorporation of transplanted GFP+ HSC, as in model control. Anti–SDF-1, 1×, 4 injections: Right, or treated, eyes from 4 representative C57BL/6.gfp mice in which retinal ischemia was induced according to our standard model with the added step of intravitreal injection with PBS containing an anti–SDF-1 antibody to a final concentration of 1 μg/μl. Note the absence of newly formed GFP+ HSC in the vascular tufts.
The lack of GFP+ HSC-derived contribution to the injured eyes that received anti–SDF-1 treatment could result from the fact that HSC/EPC-derived contribution to neovascularization was blocked, while new vessels were formed from local endothelial cell proliferation. Alternatively, all new vessel formation could be stopped by the treatment. The confocal imaging analysis suggested the latter result when the individual images from differing focal planes along the z axis used to form the merged images were viewed separately. The remaining red vessels observed in the anti–SDF-1–treated eyes appeared to be the preexisting vessels of the retina. No new preretinal vessels were observed (in the model, it is these newly formed preretinal vessels that are GFP+). To confirm, we performed cross-sectional histological analysis of treated versus nontreated control eyes to better assess total neovascularization (Figure 7). Results are shown for 3 animals from each cohort of 10. Cross sections of untreated left eyes show normal histology (Figure 7, normal retina). All of the eyes that underwent treatment according to the standard model (Figure 7, model control) exhibited severe preretinal neovascularization, as shown by the gross disruption of the retinal architecture, in response to VEGF administration and retinal ischemia. We have previously shown that these are the GFP+ vessels in our model (2). None of the anti–SDF-1–treated eyes exhibited retinal neovascularization, and all retained a retinal architecture (Figure 7, anti–SDF-1) similar to that of a normal retina. The anti–SDF-1–treated retinas showed disruption to the normal architecture of retina, reflective of damage caused by poorly repaired ischemic injury. These results clearly demonstrate that treating the eye with anti–SDF-1–blocking antibody prevents retinal neovascularization in spite of the viral overexpression of VEGF-189. This suggests that SDF-1 is the more critical proangiogenic factor in our model.
Figure 7.

Cross-sectional analysis of retinal architecture. Normal retina: H&E staining of cross sections from 3 untreated C57BL/6.gfp eyes showing normal retina morphology. Model control: H&E staining of cross sections of 3 C57BL/6.gfp eyes that underwent the neovascularization model showing clearly disrupted retinal architecture and new vessel formation. Anti–SDF-1, 1×, 4 injections: H&E staining of cross sections of 3 C57BL/6.gfp eyes that underwent the neovascularization model and were treated with 1 μg/μl anti-SDF-1 antibody. Note the similar morphology of the control C57BL/6 and anti–SDF-1 antibody–treated cross sections. Magnification, ×20.
Titration of SDF-1 antibody.
Our initial treatment regime used multiple rounds of antibody injection at what was estimated to be a saturating concentration based on the manufacturer’s use suggestions. To test the overall effectiveness of the antibody treatment, we treated 2 additional cohorts (n = 10). The first treated cohort received 1 log less antibody per injection (0.1 μg/μl) with 4 weekly injections beginning the day after laser-induced ischemia, as described above. The second treated cohort received only a single injection, 1 day after laser coagulation, at the original antibody concentration (1 μg/μl). Both test cohorts, along with a normal model control cohort, were then allowed to recover for 1 month prior to analysis. Both of the new treatments proved as effective at blocking retinal neovascularization as our original regime (Figure 8). The control cohort exhibited a large degree of GFP+ HSC-derived neovascularization in their injured eyes, with no GFP+ contributions in their uninjured eyes (Figure 8, left 2 columns). Both treated cohorts showed greatly decreased GFP+ HSC-derived contribution to the injured, anti–SDF-1–injected eyes. Almost no GFP+ contributions were seen in the vasculature (Figure 8, right 2 columns). This suggests that easily achievable SDF-1 antibody concentrations may provide effective preventative treatment for diseases such as proliferative retinopathy.
Figure 8.

Anti–SDF-1 antibody titration. Model control: Fluorescence confocal micrograph of 3 C57BL/6.gfp retinas that underwent retinal ischemia. Negative control: Left, untreated, eyes of 3 C57BL/6.gfp mice that underwent retinal ischemia. Anti–SDF-1, 0.1×, 4 injections: Three C57BL/6.gfp mice that underwent retinal ischemia and were injected with 0.1 μg/μl final concentration of anti–SDF-1 antibody, a 10-fold decrease from the original concentration, intravitreally once a week for 4 weeks. Anti–SDF-1, 1×, one injection: Three C57BL/6.gfp mice that underwent retinal ischemia and were injected with 1 μg/μl final concentration of anti–SDF-1 antibody intravitreally the day after injury.
Discussion
When PDR is left untreated, about 60% of patients become blind in one or both eyes within 5 years. For 3 decades, laser photocoagulation has been the mainstay in the management of diabetic retinopathy (31). Laser treatment for PDR breaks down the blood-retinal barrier and can cause or worsen DME (31, 32). Surgical treatment of PDR and DME results in reduction of visual acuity and is not always effective. Corticosteroid treatment can lessen the impact of macular edema and PDR but also has serious side effects, such as glaucoma, that require additional treatment (33). A highly selective therapy that could prevent new vessel formation within the vitreous without serious side effects would represent a significant improvement over the current standard of care for proliferative retinopathy.
There are currently 2 large-scale phase III clinical trials underway for the treatment of AMD and DME. Both involve blocking the activity of VEGF by binding to it and inhibiting its signaling with its receptor (34, 35). The preliminary studies have been very promising, with an increase of visual acuity in approximately 26% of the patients who have been treated. Though anti-VEGF treatments may be a great advancement in alleviating the effects of ocular diseases, there may be other cytokines/chemokines that, when blocked, may improve visual acuity by augmenting anti-VEGF treatments. Our clinical data provide what we believe to be the first evidence that SDF-1 may play a major role in the pathology of both macular edema and proliferative retinopathy. Our murine data have also shown that SDF-1 is both necessary and sufficient to promote the incorporation of bone marrow–derived endothelial cells within an ischemic retina. Blocking SDF-1 activity in our murine model completely abrogated recruitment of HSC-derived endothelial precursors and local endothelial cell–driven ischemic repair, thus effectively preventing preretinal neovascularization. In our murine model system, an acute injury promotes a proliferative retinopathy that has a pathology of preretinal neovascularization similar to that seen in the proliferative stages of diabetic retinopathy in humans. The assumption and caveat is that similar pathologies result from similar mechanisms. This assumption may not hold absolutely true. Given the lack of a true animal model for diabetic retinopathy, we cannot fully validate our assumption short of performing clinical trials. However, our patient data strongly correlates with the murine model and further suggests that targeting SDF-1 may serve as a safe, alternative approach in treating proliferative retinopathies.
Blocking SDF-1 activity within the vitreous via immunoglobulin injections or other means could potentially provide such an improved treatment for PDR and DME. Single antibody injections have already been shown to be effective for up to 1 month in our murine model. We are currently testing how long a single antibody injection can provide effective preventive therapy in this model. Antibodies are stable proteins that should be able to persist for extended times in the relatively protease-free environment of the vitreous (13). Since the eye is self-contained, high antibody concentrations are easy to achieve and maintain. We are also hopeful that we can titer the amount of SDF-1–blocking antibody to such a point where destructive preretinal neovascularization is prevented, while allowing ischemic repairs within the retina. Our murine model of adult preretinal neovascularization is currently being used to define other means for blocking SDF-1 activity and to better define the kinetics of HSC/EPC contributions to angiogenesis.
In summary, out data suggest that SDF-1 may be a key player in angiogenesis and in the progression of proliferative retinopathy. SDF-1 clearly has the potential to give EPC the directional cues necessary to reach sites of ischemia. SDF-1 can increase the expression of VCAM on endothelial cells, which suggests that SDF-1 may promote firm adhesion of HSC-derived endothelial cells to the vasculature endothelium and may also facilitate the migration and homing of the EPC. SDF-1 appears to have an impact on the ability of gap junction proteins to form tight junctions, making it possible for EPCs to enter sites of ischemia. Our clinical data show that the corticoid steroid triamcinolone decreases the severity of diabetic retinopathy. Triamcinolone may be working in part by reducing the levels of SDF-1, as shown by ELISA. Unfortunately, triamcinolone treatment comes with serious side effects, such as glaucoma. Our murine data suggest that as few as 1 intravitreal injection of a blocking antibody to SDF-1 can work to block neovascularization in our acute injury model for up to 1 month. These data suggest that using antibodies to block SDF-1 activity may provide a safe and effective alternative treatment for ischemic diseases, such as PDR and DME.
Methods
Cell isolation.
Enriched GFP+ HSCs were isolated from ubiquitous GFP-expressing donors (Jax Mice; The Jackson Laboratory) by cell sorting, as previously described (2). Human retinal endothelial cells (HRECs) were cultured in DMEM as previously described (36). CD34+, Flk-1+ circulating human EPCs were isolated from PBMCs by flow cytometry using a BD FACSVantage SE System.
Triamcinolone treatment.
Vitreous samples were obtained at the time of vitreous aspiration for treatment with triamcinolone in 46 patients with DME. Vitreous samples from nondiabetic patients who had undergone vitrectomy surgery for macular pucker and epiretinal membrane were used as controls. All patients received the standard treatment for DME with removal of 0.2 cc of liquid vitreous and injection of 4 mg (0.1–0.2 cc) of triamcinolone. Triamcinolone was injected through the pars plana, with the remaining volume replaced with a balanced salt solution. Vitreous aspirates that were collected were frozen at –20°C until analysis.
Measurement of intravitreal SDF-1 levels in patients.
We obtained vitreous samples at the time of vitreous aspiration prior to and during treatment for DME with triamcinolone in diabetic patients. Patients were classified with respect to the status of their diabetic retinopathy, gender, and duration of diabetes. All patients protocols and consents were fully reviewed and approved by the University of Florida Institutional Review Board.
Levels of SDF-1 were measured using a commercially available ELISA (R&D Systems). Each sample (0.05 cc) was run in triplicate and compared with a standard curve. All samples were assigned a random number and run without knowledge of the donor’s disease or treatment status. Once the data were compiled, the sample classifications were revealed. The mean concentration was determined per sample and per group classification. Data were analyzed by both a χ2 and rank statistical analysis to determine significance.
Isolation of protein from SDF treated cells.
The HREC cultures were washed twice with ice-cold PBS (BioWhittaker Inc.) and scraped in lysis buffer (20 mM Tris-HCl [Bio-Rad Laboratories Inc.], 1 mM EDTA [Sigma-Aldrich], 255 mM sucrose [Fisher Scientific International], 1% Igepal CA-630 [Sigma-Aldrich], 1% protease inhibitor cocktail [Sigma-Aldrich]). The lysed cells were sonicated (Model 100 Sonic Dismembrator; Fisher Scientific International) for 2 seconds and centrifuged (5415D Eppendorf; Fisher Scientific International) at 16,300 g for 5 minutes at 4°C. The pellet was discarded and the amount of protein was determined using a bicinchoninic acid protein assay kit (Pierce Biotechnology Inc.).
ELISA for VCAM-1.
HRECs were cultured beyond confluence for 3 weeks to establish tight cellular junctions. Triplicate HREC cultures were then treated for 48 hours with varying concentrations of SDF-1 and total protein extracts were prepared as indicated above. Each triplicate assay was repeated 3 times. Equal amounts of protein were used for VCAM-1 ELISAs according to the manufacturer’s instructions (R&D Systems).
Western blot analysis of occludin levels in SDF-treated cells.
A total of 50 μg of total protein was blotted to a nitrocellulose membrane (Millipore Corp.) and a Western blot analysis performed according to the manufacturer’s instructions. For occludin detection, the membrane was incubated with a 1:125 dilution of a rabbit polyclonal anti-occludin antibody (Zymed Laboratories Inc.). Following occludin detection, the membrane was also used to detect β-actin protein levels using a 1:5,000 dilution of mouse monoclonal anti–β-actin antibody (Sigma-Aldrich) and an HRP-conjugated anti–mouse IgG secondary antibody (Sigma-Aldrich). The protein bands were visualized with an ECL Western Blot Detection Kit (Amersham Biosciences Ltd.). Standard molecular weight markers (Bio-Rad Laboratories Inc.) served to verify the molecular size of occludin at 65 kDa and of β-actin at 42 kDa. Analysis of occludin and β-actin protein levels was performed using Image analysis software (Scion Corp.).
Generation of adult retinal neovascularization mouse model.
All animal procedures were approved by of the University of Florida Animal Care and Use Committee. The basic model has been previously described (2). In brief, C57BL/6.gfp radiation chimeric mice (n = 70) were generated by irradiating C57BL/6 mice with 9.50 Gy followed by retinal orbital sinus injection of highly enriched HSCs (Sca-1+c-kit+Lin–) from GFP+ donor mice. Durable hematopoietic reconstitution was confirmed by flow cytometry 3–4 months after transplant. Once reconstitution was confirmed, chimeric mice were matched for engraftment level (greater than 80% donor-derived PBMCs), then injected intravitreally with adeno-associated virus–VEGF (AAV-VEGF) into the right eye. One month was allowed for peak VEGF expression, at which time the mice underwent retinal vessel photocoagulation of half the optic disc of the right, or test, eye with an argon laser. Venous occlusions were accomplished using laser parameters of 1-second duration, 50-mm spot size, and 100- to 150-mW intensity.
Administration of blocking antibodies.
Immediately following laser treatment, cohorts (n = 10 per cohort) of chimeric mice underwent intravitreal injections into the right, or test, eye. Mice were anesthetized, and an anti–SDF-1 antibody (MAB 310; R&D Systems) or PBS plus isotype control was injected intravitreally (1 μl total volume) to achieve a final effective concentration of 1 μg/μl or 0.1 μg/μl of antibody in the vitreous. Some cohorts received additional booster injections weekly for 4 weeks.
Murine data collection and analysis.
Four weeks after induction of retinal ischemia, mice were euthanized and their eyes enucleated. For confocal visualization, mice (n = 60) were perfused with rhodamine isothiocyanate-conjugated dextran in phosphate-buffered formaldehyde (pH 7.4). Immediately afterward, eyes were removed and retinas dissected and mounted flat using Vectashield mounting media (Vector Laboratories) for confocal imagining. Confocal microscopy was performed with a Leica TCS SP2 system with spectral analysis software for data collection and analysis (Leica Microsystems). For retina sectioning, mice (n = 20) were perfused and eyes enucleated and fixed in 4% paraformaldehyde, and frozen sections were prepared. The sections were stained with H&E.
Acknowledgments
We thank Tim Vaught and Doug Smith for confocal imaging and the Scott and Grant laboratories for additional technical support. This work was supported by NIH grants HL70813 and HL70738 (to E.W. Scott), EY12601 (to M.B. Grant), and a grant from the Juvenile Diabetes Foundation. S.M. Guthrie was supported by NIH Training Grant T32AR07603. E.W. Scott is a Leukemia and Lymphoma Society Scholar.
Footnotes
Nonstandard abbreviations used: AAV, adeno-associated virus; DME, diabetic macular edema; EPC, endothelial progenitor cell; HREC, human retinal endothelial cell; NVI, neovascularization of the iris; PDR, proliferative diabetic retinopathy; rAAV, recombinant adeno-associated virus; rSDF-1, recombinant SDF-1; SDF-1, stromal cell–derived factor–1.
Conflict of interest: E.W. Scott is a cofounder of RegenMed Inc., currently a virtual start-up company that plans to market stem cell–based therapies.
References
- 1.Kaufmann, P., and Alm, A. 2003. Adler’s physiology of the eye. 10th edition. Mosby. St. Louis, Missouri, USA. 876 pp.
- 2.Grant MB, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat. Med. 2002;8:607–612. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- 3.Sengupta N, et al. The role of adult bone marrow-derived stem cells in choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2003;44:4908–4913. doi: 10.1167/iovs.03-0342. [DOI] [PubMed] [Google Scholar]
- 4.Cogle CR, et al. Adult human hematopoietic cells provide functional hemangioblast activity. Blood. 2004;103:133–135. doi: 10.1182/blood-2003-06-2101. [DOI] [PubMed] [Google Scholar]
- 5.Guthrie, S.M., Curtis, L.M., Mames, R.N., Simon, G.G., and Scott, E.W. 2004. The nitric oxide pathway modulates hemangioblast activity of adult hematopoietic stem cells. Blood. doi:10.1182/blood-2004-09-3415. [DOI] [PubMed]
- 6.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 7.Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO. Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retin. Eye Res. 2003;22:1–29. doi: 10.1016/s1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 8.Aiello LP, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc. Natl. Acad. Sci. U. S. A. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ceradini DJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 10.Hattori K, Heissig B, Rafii S. The regulation of hematopoietic stem cell and progenitor mobilization by chemokine SDF-1. Leuk. Lymphoma. 2003;44:575–582. doi: 10.1080/1042819021000037985. [DOI] [PubMed] [Google Scholar]
- 11.Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J. Exp. Med. 2002;195:1145–1154. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatch H, Zhang D, Jorgensen MS, Petersen BE. SDF-1/CXCR-4: A possible mechanism for bone marrow derived hepatic oval cell recruitment to the rat liver. Cloning Stem Cells. 2002;4:339–352. doi: 10.1089/153623002321025014. [DOI] [PubMed] [Google Scholar]
- 13.Sjostrand J, Karlsson JO, Andersson AK. Changes in the soluble protein of the human vitreous in vitreoretinal disease. Acta Ophthalmol. 1992;70:814–819. doi: 10.1111/j.1755-3768.1992.tb04893.x. [DOI] [PubMed] [Google Scholar]
- 14.Tano Y, Chandler D, Machemer R. Treatment of intraocular proliferation with intravitreal injection of triamcinolone acetonide. Am. J. Ophthalmol. 1980;90:810–816. doi: 10.1016/s0002-9394(14)75196-7. [DOI] [PubMed] [Google Scholar]
- 15.McCuen BW, 2nd, Bessler M, Tano Y, Chandler D, Machemer R. The lack of toxicity of intravitreally administered triamcinolone acetonide. Am. J. Ophthalmol. 1981;91:785–788. doi: 10.1016/0002-9394(81)90013-1. [DOI] [PubMed] [Google Scholar]
- 16.Jonas JB, Kreissig I, Sofker A, Degenring RF. Intravitreal injection of triamcinolone for diffuse diabetic macular edema. Arch. Ophthalmol. 2003;121:57–61. [PubMed] [Google Scholar]
- 17.Martidis A, et al. Intravitreal triamcinolone for refractory diabetic macular edema. Ophthalmology. 2002;109:920–927. doi: 10.1016/s0161-6420(02)00975-2. [DOI] [PubMed] [Google Scholar]
- 18.Jonas JB, Kreissig I, Degenring R. Intraocular pressure after intravitreal injection of triamcinolone acetonide. Br. J. Ophthalmol. 2003;87:24–27. doi: 10.1136/bjo.87.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J. Exp. Med. 2002;195:1145–1154. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazarini F, Tham TN, Casanova P, Arenzana-Seisdedos F, Dubois-Dalcq M. Role of the alpha-chemokine stromal cell-derived factor (SDF-1) in the developing and mature central nervous system. Glia. 2003;42:139–148. doi: 10.1002/glia.10139. [DOI] [PubMed] [Google Scholar]
- 21.Salcedo R, et al. Vascular endothelial growth factor and basic fibroblast growth factor induce expression of CXCR4 on human endothelial cells: In vivo neovascularization induced by stromal-derived factor-1alpha. Am. J. Pathol. 1999;154:1125–1135. doi: 10.1016/s0002-9440(10)65365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kijowski J, et al. The SDF-1-CXCR4 axis stimulates VEGF secretion and activates integrins but does not affect proliferation and survival in lymphohematopoietic cells. Stem Cells. 2001;19:453–466. doi: 10.1634/stemcells.19-5-453. [DOI] [PubMed] [Google Scholar]
- 23.Neuhaus T, et al. Stromal cell-derived factor 1alpha (SDF-1alpha) induces gene-expression of early growth response-1 (Egr-1) and VEGF in human arterial endothelial cells and enhances VEGF induced cell proliferation. Cell Prolif. 2003;36:75–86. doi: 10.1046/j.1365-2184.2003.00262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mirshahi F, et al. SDF-1 activity on microvascular endothelial cells: consequences on angiogenesis in in vitro and in vivo models. Thromb. Res. 2000;99:587–594. doi: 10.1016/s0049-3848(00)00292-9. [DOI] [PubMed] [Google Scholar]
- 25.Salvucci O, et al. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood. 2002;99:2703–2711. doi: 10.1182/blood.v99.8.2703. [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi J, et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation. 2003;107:1322–1328. doi: 10.1161/01.cir.0000055313.77510.22. [DOI] [PubMed] [Google Scholar]
- 27.Rafii S, Heissig B, Hattori K. Efficient mobilization and recruitment of marrow-derived endothelial and hematopoietic stem cells by adenoviral vectors expressing angiogenic factors. Gene Ther. 2002;9:631–641. doi: 10.1038/sj.gt.3301723. [DOI] [PubMed] [Google Scholar]
- 28.Suratt BT, et al. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood. 2004;104:565–571. doi: 10.1182/blood-2003-10-3638. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Stone J. Role of astrocytes in the control of developing retinal vessels. Invest. Ophthalmol. Vis. Sci. 1997;38:1653–1666. [PubMed] [Google Scholar]
- 30.Otani A, et al. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nat. Med. 2002;8:1004–1010. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- 31.The Diabetic Retinopathy Study Research Group. Preliminary report on effects of photocoagulation therapy. The Diabetic Retinopathy Study Research Group. Am. J. Ophthalmol. 1976;81:383–396. doi: 10.1016/0002-9394(76)90292-0. [DOI] [PubMed] [Google Scholar]
- 32.Wilson CA, et al. Treatment with intravitreal steroid reduces blood-retinal barrier breakdown due to retinal photocoagulation. Arch. Ophthalmol. 1992;110:1155–1159. doi: 10.1001/archopht.1992.01080200135041. [DOI] [PubMed] [Google Scholar]
- 33.Jaffe EA, Hoyer LW, Nachman RL. Synthesis of von Willebrand factor by cultured human endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 1974;71:1906–1909. doi: 10.1073/pnas.71.5.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, et al. Selection and analysis of an optimized anti-VEGF antibody: crystal structure of an affinity-matured Fab in complex with antigen. J. Mol. Biol. 1999;293:865–881. doi: 10.1006/jmbi.1999.3192. [DOI] [PubMed] [Google Scholar]
- 35.Ruckman J, et al. 2’-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998;273:20556–20567. doi: 10.1074/jbc.273.32.20556. [DOI] [PubMed] [Google Scholar]
- 36.Grant MG, Guay C. Plasminogen activator production by human retinal endothelial cells of nondiabetic and diabetic origin. Invest. Ophthalmol. Vis. Sci. 1991;32:53–64. [PubMed] [Google Scholar]