

Cryo-EM structures illustrate a novel mechanism for Vps4-mediated disassembly of ESCRT-III filaments.
Keywords: vps4 structure, cryoEM, ESCRT III, ESCRT pathway, AAA+ ATPases
Abstract
Vps4 is a member of AAA+ ATPase (adenosine triphosphatase associated with diverse cellular activities) that operates as an oligomer to disassemble ESCRT-III (endosomal sorting complex required for transport III) filaments, thereby catalyzing the final step in multiple ESCRT-dependent membrane remodeling events. We used electron cryo-microscopy to visualize oligomers of a hydrolysis-deficient Vps4 (vacuolar protein sorting-associated protein 4) mutant in the presence of adenosine 5′-triphosphate (ATP). We show that Vps4 subunits assemble into an asymmetric hexameric ring following an approximate helical path that sequentially stacks substrate-binding loops along the central pore. The hexamer is observed to adopt an open or closed ring configuration facilitated by major conformational changes in a single subunit. The structural transition of the mobile Vps4 subunit results in the repositioning of its substrate-binding loop from the top to the bottom of the central pore, with an associated translation of 33 Å. These structures, along with mutant-doping experiments and functional assays, provide evidence for a sequential and processive ATP hydrolysis mechanism by which Vps4 hexamers disassemble ESCRT-III filaments.
INTRODUCTION
The endosomal sorting complexes required for transport (ESCRTs) are a collection of evolutionarily conserved proteins that “catalyze” a neck-severing reaction leading to membrane budding away from the cytoplasm (1). ESCRTs function in a wide range of crucial biological processes, including multivesicular body biogenesis (2), budding of HIV (3, 4), and cytokinesis (5). Central to the function of ESCRT are the ESCRT-III complex and the oligomeric AAA+ ATPase (adenosine triphosphatase) Vps4 (vacuolar protein sorting-associated protein 4) (6). Site-specific adaptor proteins recruit subunits of the ESCRT-III complex to different membrane structures, where they assemble into helical filaments that can produce negative curvature and induce membrane scission (7). Vps4 catalyzes the disassembly of membrane-bound ESCRT-III filaments (8), and its function is essential for all ESCRT-dependent biological processes (9–11). To understand the mechanism of Vps4 function, we sought to determine electron cryo-microscopy (cryo-EM) structures of reconstituted Vps4 oligomers.
RESULTS
Although oligomerization is a prerequisite for Vps4 activity, a stable oligomer that is suitable for structural studies has proven to be elusive using wild-type protein. This is because adenosine 5′-triphosphate (ATP) hydrolysis is rapid and results in weak subunit association and oligomer disassembly. To overcome this problem, we explored the oligomerization properties of an ATP hydrolysis–deficient mutant, Vps4E233Q (12). Using size exclusion chromatography and multiangle light scattering (MALS) analysis, we found that Vps4E233Q is a monomer that oligomerizes into a hexamer in the presence of ATP (fig. S1), similar to what has been proposed for the wild-type protein (13, 14). It has been shown that wild-type Vps4 can form a fully active hetero-oligomer with Vps4E233Q (15), and thus, that this mutant is a suitable alternative for cryo-EM studies of the Vps4 oligomer.
Vps4 oligomers suspended in vitreous ice for cryo-EM assume a highly preferred orientation that could not be alleviated with varying buffer conditions. To address this major limitation, we applied a novel strategy that involved merging cryo-EM projections recorded at tilts of 0° and 45° of the specimen stage, thereby sampling all missing views and eliminating three-dimensional (3D) reconstruction artifacts (figs. S2 and S3). This approach was critical to identifying and separately refining 3D reconstructions of two distinct conformers of Vps4 oligomers. Both conformers are asymmetric hexamers, representing 54 and 25% of the particles, with resolutions of 6.1 and 6.7 Å, respectively (Fig. 1, A and B, and fig. S4). We further used molecular dynamics flexible fitting (MDFF) (16) to dock the subunit crystal structure of Vps4 (17) into the cryo-EM density, thereby obtaining pseudoatomic resolution models for each oligomer configuration (Fig. 1C, figs. S5 to S7, movie S1). In one conformer (Fig. 1A), six Vps4 subunits are organized on an approximate helical path into an open ring. In the other conformer (Fig. 1B), subunit A is found rotated ~37° by pivoting on the β domain, resulting in a 33 Å downward translation of its large ATPase subdomain and formation of a closed ring (Fig. 1D). Comparison between the two structures shows that the remaining five subunits (B to F) in Vps4 occupy nearly identical positions.
Fig. 1. Vps4 hexamers in open and closed conformations.
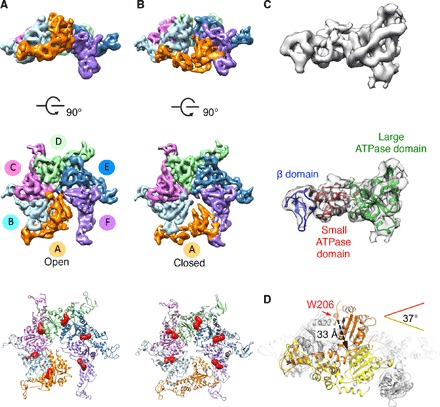
(A and B) Vps4 hexamer in the open (A) and closed (B) conformations. Top and middle: Side and top views, respectively, of cryo-EM density maps. Bottom: Top view of structural models. Difference densities (red) corresponding to ATP are shown at the same threshold cutoff. (C) Flexibly fit crystal structure of Vps4 subunit into the corresponding EM density region (subunit D, closed conformation). (D) Structural transitions between the open and closed Vps4 conformers. The conformers are aligned on the basis of subunits B to F (gray) and shown from the side of subunit A in the open (orange) and closed (yellow) conformations.
The Vps4 hexamer is maintained by three regions of intersubunit interactions (Fig. 2A). The first interface lies between helix α1 in the large ATPase subdomain and a surface patch formed by the small ATPase subdomain and the β domain of a neighboring subunit. The second interface houses the canonical nucleotide-binding site, where a neighboring subunit provides the arginine finger residues (288RR289) that are critical for ATP hydrolysis (6) (fig. S8). These two subunit interfaces have been predicted on the basis of their homology to other AAA+ ATPases and have been experimentally confirmed (18). However, in addition, we observe a novel third interface involving a C-terminal helix α11 of one subunit that interacts with an interhelical loop (between α10 and α11) of a neighboring subunit. Mutation of two highly conserved residues, Arg414 and Phe432, led to a significant decrease in protein oligomerization and ATPase activity (fig. S9), supporting the functional relevance of this third interface. In the open hexamer structure, all three interfaces exist between all subunit pairs except F/A. In the closed structure, the three interfaces exist in four subunit pairs (B/C, C/D, D/E, and E/F), whereas only the first interface is maintained in subunit pairs A/B and F/A (Fig. 2B). Thus, the structural transition of subunit A from the open to closed conformation involves breaking the interactions at interfaces II and III in A/B, followed by establishing interface I in F/A.
Fig. 2. Subunit interfaces within the Vps4 hexamer.
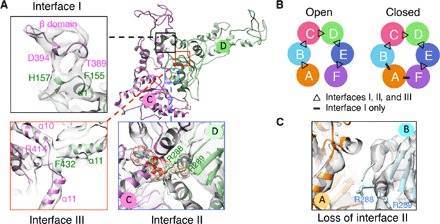
(A) Ribbon representation of two neighboring Vps4 subunits with inserts showing details of the three interfaces (I to III). (B) Schematic diagram showing the presence of the three interfaces between different subunit pairs in the two Vps4 hexamer conformers. (C) Only partial ATP-binding sites are formed at the A/B subunit interface of the closed conformer.
Difference mapping between each hexamer structure model and the corresponding cryo-EM 3D map reveals clear density attributed to ATP in the nucleotide-binding pockets of 5 of the 6 Vps4 subunits (Fig. 1, A and B). The only exception is subunit A in the closed conformer, where no significant density was observed, providing the first strong indication that subunit A reflects different nucleotide-binding states in the two structures.
Furthermore, subunit A in the closed conformer adopts a much more compact configuration between its large and small ATPase subdomains compared to all other subunits in both conformers (fig. S10). For subunits A to E in the open conformer and subunits B to E in the closed conformer, the arginine finger residues from the neighboring subunit are in close contact with the nucleotide density, suggesting that these subunits are in the ATP-bound prehydrolysis state (Fig. 2A). For subunit A in the closed conformer, the neighboring arginine finger residues are far away from the nucleotide-binding pocket, suggesting that the mobile member of the Vps4 hexamer is in a posthydrolysis state (Fig. 2C). This interpretation agrees with the lack of nucleotide density in the binding pocket.
Given the unique and distinct conformational states displayed only by subunit A in the two conformers, we hypothesized that active hydrolysis in the hexamer occurs only at the A/B interface. To confirm this, we conducted mutant complementation experiments. As shown previously (15), addition of Vps4E233Q to wild-type Vps4 does not inhibit, but instead stimulates, ATP hydrolysis (Fig. 3A), suggesting that the mutant forms heterohexamers with wild-type Vps4. On the other hand, the arginine finger mutant of Vps4 (Vps4R288A/R289A) cannot oligomerize by itself and therefore has no ATPase activity (19). However, on the basis of our structures, Vps4R288A/R289A can form an active heterohexamer with Vps4E233Q if and only if the arginine finger mutant occupies the position of subunit A in the open conformer (Fig. 3B). As the arginine finger mutant was titrated with Vps4E233Q, robust ATPase activity was observed, although neither mutant alone is active (Fig. 3A) (12).
Fig. 3. Number of subunits required for ATP hydrolysis and ESCRT-III disassembly.
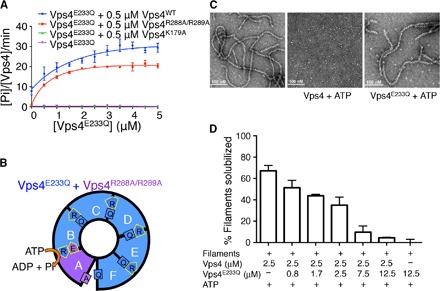
(A) Titration of Vps4E233Q into one of the Vps4 variants (Vps4WT, Vps4R288A/R289A, Vps4K179A, or Vps4E233Q, all fixed at 0.5 μM). ATPase activity was measured as micromolar inorganic phosphate released per micromolar Vps4 variant per minute. (B) Cartoon illustrating how mixing of Vps4E233Q and Vps4R288A/R289A can produce an active heterohexamer. (C) Electron micrograph of negative-stained filaments of Vps24-Vps2 chimera, before (left) and after incubation with wild-type Vps4 (middle) or Vps4E233Q (right) in the presence of ATP. (D) Disassembly of Vps24-Vps2 filaments by 2.5 μM Vps4WT as Vps4E233Q is titrated into the reaction mixture. Error bars are SD of results from three independent repeats.
Consistent with our prediction that only one arginine finger mutant can be incorporated into the heterohexamer, the ATPase activity increases and plateaus as the molar ratio between Vps4R288A/R289A and Vps4E233Q approaches 1:5 (Fig. 3A). A similar trend was also observed for wild-type Vps4, suggesting that only one of the six subunits in the Vps4 hexamer may be active at any time. To gain a more quantitative assessment, we mixed different ratios of wild-type Vps4 and Vps4E233Q together, while maintaining the total concentration of Vps4 constant. Assuming that wild-type and mutant subunits incorporate randomly into a hexameric ring, the concentration of the heterohexamer ensemble composed of different numbers of wild-type and mutant subunits will be determined according to a binomial distribution that varies as a function of the molar ratio between the two subunits. Our results show that the experimental activity curve is in good agreement with the theoretical activity curve predicted for one wild-type subunit per hexamer being sufficient for full Vps4 hexamer ATPase activity (fig. S11), reinforcing the finding that only one subunit is active at any given time.
Although ATP hydrolysis per se only requires one active subunit, disassembly of ESCRT-III filaments may require more than one active subunit in the hexamer. To examine this possibility, we used an established ESCRT-III disassembly assay for filaments formed by a Vps24-Vps2 chimera (Fig. 3C and fig. S12, A and B) (20, 21). In the presence of ATP, a large fraction of Vps24-Vps2 proteins are in the soluble fraction when incubated with wild-type Vps4 (Fig. 3D and fig. S12C). Adding increasing amounts of Vps4E233Q mutant to a fixed amount of wild-type Vps4 gradually decreased the ability of Vps4 to disassemble Vps24-Vps2 filaments (Fig. 3D and fig. S12D). The results are in stark contrast to the stimulatory effect of Vps4E233Q on ATP hydrolysis, suggesting that processive engagement of multiple active subunits in a Vps4 hexamer is required for effective disassembly of ESCRT-III filaments.
DISCUSSION
Although we cannot completely rule out a probabilistic model, our analysis is in line with a sequential model of ATP hydrolysis by Vps4. The two hexamer structures suggest that interactions at the A/B interface weaken upon ATP hydrolysis and that subunit A establishes a new interaction with subunit F before dissociating from B (Fig. 1D). In the ensuing new open conformer, subunit B assumes the role of the active subunit to undergo hydrolysis. With the active subunit alternating in a cyclic order, every subunit needs to wait for another five ATP hydrolysis events before it assumes the right position for catalysis.
Earlier studies have shown that active disassembly of ESCRT-III filaments requires the presence of loops lining the central pore of the Vps4 hexamer. Only one ESCRT-III subunit can bind to one Vps4 hexamer via the pore loops at any time (20–22), and the cryo-EM structures provide an explanation for this behavior. The pore loops stack sequentially to form a steep helical ladder (Fig. 4A), with pore loop 1 of subunit A being the only one exposed at the top of the ladder in the open conformation. In the closed conformation, pore loop 1 of subunit A translates by about 33 Å and relocates to the bottom, leaving pore loop 1 of subunit B exposed at the top of the ladder (Fig. 4B). Thus, only one pore loop 1 is exposed at a time and is in position to engage ESCRT-III. We postulate that the interaction of ESCRT-III with pore loops of subunit A induces a conformational change that accelerates ATP hydrolysis, which, in turn, results in the observed conformational change from open to closed, providing the necessary mechanical force for ESCRT-III disassembly (Fig. 4C). As has been suggested, the interaction between ESCRT-III and pore loops promotes unfolding of ESCRT-III and its dissociation from the filament (20). The cryo-EM structures suggest that the translocation of pore loops coupled with sequential ATP hydrolysis within the Vps4 hexamer provides the structural basis for ESCRT-III disassembly.
Fig. 4. Model of Vps4-mediated ESCRT-III disassembly.
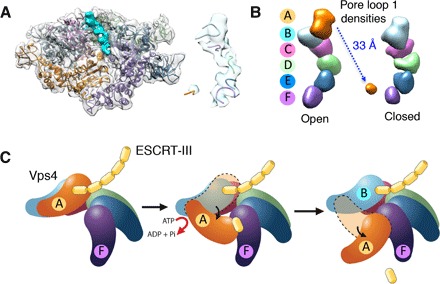
(A) Cryo-EM 3D map and fitted structure model of Vps4 hexamer in the closed conformation, with density corresponding to the stacked pore loop 1 (cyan). The zoom-in view (right) shows the map and model for pore loop 1 densities from the six Vps4 subunits. (B) Comparison of pore loop 1 density segmentation between the open and closed conformations. (C) Model of Vps4-mediated ESCRT-III disassembly. Vps4 forms an open hexamer in the presence of ATP. Binding of one ESCRT-III subunit to the pore loops of subunit A stimulates its ATPase activity. Upon hydrolysis, subunit A dissociates from subunit B and establishes a new contact with subunit F through a closed ring hexamer intermediate. Eventually, a new open Vps4 hexamer is formed, where A repositions by ~33 Å and becomes the new F whereas B becomes the new A. The ESCRT-III subunit bound to A will experience the mechanical force accompanying the conformational change of the subunit in the hexamer.
In conclusion, the structures described here provide crucial insights into the oligomerization of Vps4 into functional hexamers. The visualization of distinct conformers, supported by biochemical and functional analysis of mutants, reveals an elegant sequential ATP hydrolysis mechanism in the context of a stable asymmetric hexamer. The observed conformational changes appear compatible with the disassembly of ESCRT-III filaments in a processive and coordinated fashion (Fig. 4C and movie S2). These findings set the stage for further structural studies of Vps4 hexamers in association with substrate and regulatory proteins.
MATERIALS AND METHODS
Plasmid construction, protein expression, and purification
Saccharomyces cerevisiae Vps4 was expressed in Escherichia coli Rosetta (DE3) cells using a modified pET-21a vector with a His8 tag inserted before the protein-coding region. Site-directed mutagenesis was performed using a standard polymerase chain reaction (PCR) mutagenesis protocol (Stratagene). The codons used to introduce the point mutations for Vps4 were K179A (AAA→GCG), E233Q (GAA→CAG), R288A (AGA→GCG), R289A (AGA→GCG), C317S (TGC→AGC), C376S (TGC→AGC), R414A (CTG→GCG), and F432A (TTT→GCG). The S. cerevisiae Vps241–179–Vps2181–232 (Vps24-Vps2) construct was generated by overlapping PCR. The chimeric Vps24-Vps2 protein was expressed in E. coli Rosetta (DE3) cells using a modified pET-28b vector, with a small ubiquitin-like modifier (SUMO) protein tag inserted between a His6 tag and the protein-coding region.
Vps4, Vps4E233Q, Vps4K179A, Vps4R288A/R289A, Vps4R414A, Vps4F432A, Vps4R414A/F432A, Vps4E233Q/R414A, Vps4E233Q/F432A, and Vps4E233Q/R414A/F432A were purified using a protocol, as previously described (23). Briefly, bacterial cells were grown to midlog phase in LB medium at 37°C and induced with 0.2 mM isopropyl β-d-1-thiogalactopyranoside for an additional 16 to 20 hours at 16°C. Harvested cells were lysed with sonication in buffer A {25 mM tris-HCl (pH 8.0), 300 mM NaCl, 5 mM 2-mercaptoethanol, and phenylmethylsulfonyl fluoride [10 μg/ml (w/v)]}. Cell lysate was cleared by centrifugation, and supernatant was loaded onto a Ni2+–nitrilotriacetic acid affinity column. Bound protein was washed with buffer A and eluted with buffer A supplemented with 250 mM imidazole. Fractions were pooled, and tobacco etch virus or Ubl-specific protease 1 was added to cleave off the His8 tag or His6-SUMO tag. After dialysis against a buffer containing 50 mM tris-HCl (pH 8.0) and 25 mM NaCl, the digested protein mixture was passed through a second Ni2+–nitrilotriacetic acid column to remove undigested protein and the tag. Proteins were further purified by ion exchange chromatography on a Source Q column (GE Healthcare). Vps4 protein used for cryo-EM analysis was further purified by gel filtration on a HiLoad Superdex 200 (GE Healthcare) column equilibrated with buffer containing 50 mM tris (pH 7.5), 100 mM KCl, and 5 mM MgCl2. Vps24-Vps2 was purified in the same way as with Vps4. Following a Source Q column affinity step, peak fractions containing untagged Vps24-Vps2 were pooled and further purified in buffer containing 20 mM tris (pH 8.0) and 100 mM NaCl on a HiLoad Superdex 75 (GE Healthcare) column.
Size exclusion chromatography and MALS analysis
Size exclusion chromatography was performed with a Superdex 200 10/300 GL (GE Healthcare) column to compare the elution profiles of wild-type and mutant Vps4 in the presence or absence of ATP. The column was equilibrated in 50 mM tris-HCl (pH 7.5), 100 mM KCl, and 5 mM MgCl2, with or without 0.1 mM ATP. Vps4E233Q, Vps4E233Q/R414A, Vps4E233Q/F432A, and Vps4E233Q/R414A/F432A were analyzed at a concentration of 30 to 50 μM (fig. S9).
The average molecular weight of Vps4E233Q was determined by separation using a WTC-050S5 column (Wyatt Technology Corporation) with an ÄKTAmicro column (GE Healthcare) and by analysis using a DAWN HELEOS II MALS detector and Optilab rEX differential refractive index detector using ASTRA VI software (Wyatt Technology Corporation). The molecular weight was calculated from the Raleigh ratio based on the static light scattering and the corresponding protein concentration of a selected peak (fig. S1).
Cryo-EM specimen preparation and data acquisition
Protein samples were concentrated to 16.5 mg/ml and incubated for 10 min with 2 mM ATP. Sample (3 μl) at a concentration of 1 mg/ml was loaded onto glow-discharged QUANTIFOIL R2/2 200 mesh grids and flash-frozen using Vitrobot (FEI Mark IV). The samples were visualized at liquid nitrogen temperature on a Titan Krios electron microscope (FEI) operating at 300 kV. Cryo-EM images were recorded at a nominal magnification of ×29,000 using a K2 Summit direct electron detector (Gatan Inc.) in counted mode, corresponding to a pixel size of 1 Å per pixel with a dose rate of ~6.0 electrons Å−2 s−1. Vps4 oligomers were mostly observed in relatively thick ice, with apparent preferred top-down orientations. To compensate for the preferred particle orientation, images were taken at both tilt angles of 0° and 45° using defocus values ranging from −3 to −5 μm. The total exposure time was 8.0 s, and intermediate frames were recorded in 0.2-s intervals, resulting in an accumulated dose of ~48 electrons/Å2 and a total of 40 frames per micrograph.
Cryo-EM image processing and 3D reconstructions
A total of 976 and 936 cryo-EM images tilted at 0°and 45°, respectively, were recorded during multiple microscope sessions. Dose-fractionated image stacks were binned 2 × 2 and were subjected to whole-frame motion correction using MotionCor2 (http://biorxiv.org/content/early/2016/07/04/061960). The power spectra of motion-corrected images were evaluated to remove micrographs showing resolution lower than ~8 Å, resulting in a working data set of 834 untilted and 856 tilted micrographs. A sum of the total 8-s frames in each image stack was integrated with dose filter applied using MotionCor2, and the integrated images were used for single-particle extraction and analysis. Contrast transfer function (CTF) parameters were determined by CTFFIND3 and CTFTILT (24) for untilted and tilted micrographs, respectively. The defocus value for each individual tilted particle was assigned according to its location in the corresponding micrograph, calculated by CTFTILT on the basis of micrograph rotating axis and angle.
A total of 183,658 particles were interactively selected using e2boxer.py (25). Two-dimensional and 3D classifications were performed on a binned data set with a pixel size of 2 Å and a box size of 128 × 128 pixels using RELION 1.3 (26). In the first step, a rough 3D map was generated ab initio by e2initial.py (25) using the 2D class averages as input and without enforcing any symmetry (step 1, fig. S3). Particle projections were subjected to 3D classification using the 60 Å filtered initial model as reference to sort out homogeneous subsets or different particle conformers. The classification was executed in eight classes, starting with an angular sampling of 7.5°, followed sequentially by angular samplings of 1.8° and 0.9° that were combined with local angular searches, yielding one well-defined class resembling the closed conformation (yellow map in step 2, fig. S3). To improve particle partitioning, the closed conformation map was low-pass–filtered to a resolution of 40 Å and was used as an initial model to repeat the 3D classification into eight classes (step 3, fig. S3). This process resulted in reconstructions representing closed and open hexamer conformations as well as apparent pentamers. After removing particles belonging to poorly defined classes (shaded maps in fig. S3), we performed an additional cycle of 3D classification into six classes using a 40 Å low-pass–filtered map of the closed conformation as reference (step 4, fig. S3). This process resulted in better-defined closed and open hexamers, as well as pentamer maps. Particle projections belonging to closed hexamer classes (orange maps in step 4, fig. S3) were pooled together and subjected to autorefinement in RELION 1.3 (26). The open conformation hexamers and pentameric particles were grouped together and subjected to one more cycle of 3D classification into two classes using the open hexamer conformation map as the initial model. This step resulted in well-defined open hexamer and pentamer classes (step 5, fig. S3) that were separately subjected to autorefinement and reconstruction. A soft mask covering the entire reconstruction volume was only applied at the last refinement cycle, producing final maps with global resolutions of 6.1 and 6.7 Å for closed (99,175 particles; 54%) and open (45,915 particles; 25%) hexamer conformations, respectively. About 20% (38,569 particles) of Vps4 oligomers were classified as apparent pentamers with a five-subunit configuration that is nearly identical to its counterpart in the hexamer structures.
Reported resolutions are based on the gold standard Fourier shell correlation (FSC) using the criterion of 0.143 (fig. S4) (27). High-resolution noise substitution was used to correct for the effects of soft masking on the FSC curves. All density maps were corrected for the modulation transfer function of the K2 Summit direct electron detector and then sharpened by applying a negative B-factor that was estimated using postprocessing in RELION 1.3 (26). Local resolution was determined using ResMap (28), with half-reconstructions as input maps.
Molecular dynamics flexible fitting
The program Situs (29) was used for rigid body fitting of six independent monomer structures [Protein Data Bank (PDB) ID: 2QPA] (17) into the cryo-EM density maps. Rigid body fitting was done successively for each of the six monomers comprising the molecular complex. After each monomer was fitted into the map, the simulated map of the newly added monomer was subtracted from the original map, and the next monomer was fit into the residual map. This process was repeated until all six monomers were accommodated into the density map. The six monomers were then simultaneously refined against the original map using the multifragment refinement program collage from the Situs program package (29) to ensure that steric clashes between the monomers were eliminated. The resulting model provided the starting structure in the subsequent flexible fitting. The same rigid body fitting procedure was used for both closed and open conformations.
The starting monomer structure used in the rigid body fitting procedure was derived from chain A of the crystal structure of Vps4 with bound ATP (PDB ID: 2QPA) (17), which has the same sequence as the proteins used in the present study. A phosphate group was added to transform adenosine 5′-diphosphate (ADP) into ATP, and Mg2+ was added to each monomer structure. The positions for both Mg2+ and the phosphate group were determined from the crystal structure of S. cerevisiae Vps4 in the presence of adenosine 5′-O-(3-thiotriphosphate (PDB ID: 3EIH) (18) after aligning it with 2QPA on the basis of the composite protein structures.
MDFF version 0.2 (16) was used to further refine our starting hexamer models via targeted flexible fitting into the cryo-EM density. These calculations were set up using VMD version 1.9.2 (30), and molecular dynamics was run using NAMD version 2.9 (31) with the CHARMM27 (32, 33) force field. For this process, the pore loop 1 region (residues 204 to 210 from all six subunits) of the hexamer structure that resulted from rigid body fitting was mutated into alanines, because the original side chains in this region were trapped in a local energy minimum. The mutated hexamer structure was solvated in a TIP3P (34) water box for each starting complex (closed or open conformation). The water boxes had dimensions of 180.1 × 179.6 × 178.4 Å for the closed conformation and 179.2 × 184.7 × 176.4 Å for the open conformation. The size of the water box was obtained by adding 25 Å to the size of the hexamer along each dimension. K+ and Cl− ions were added to the system to maintain charge neutrality, as well as an excess ion concentration of 0.10 M KCl: 384 K+ and 330 Cl− ions were added for the closed conformation, and 388 K+ and 334 Cl− ions were added for the open conformation. To diminish any effect of overfitting, restraints were added to maintain the protein secondary structure, cis peptide bonds, and chirality (35). For the protein secondary structure, harmonic restraint forces were applied on dihedral angles, bond angles, and hydrogen bonds between amide nitrogens and carbonyl oxygens, with force constants of 200 kcal mol−1 rad−2, 20 kcal mol−1 rad−2, and 20 kcal mol−1 Å−2, respectively. The harmonic restraint force constants used for cis peptide bond angles and chiral sites are 200 and 50 kcal mol−1 rad−2, respectively. The MDFF protocol used here was run in four phases. In the first phase, the system was minimized and equilibrated in a constant volume and temperature ensemble for 10 ps at a temperature of 300 K using Langevin dynamics with a damping coefficient of 5 ps−1. The time step used was 1 fs. The cutoff distance for nonbonded interactions was 10 Å, with a switching distance at 9 Å. The particle mesh Ewald (36) method was used to calculate long-range electrostatic interactions. Steering forces from the cryo-EM density map were not used in the first phase. Starting from the second phase, a steering force from the map was added. The scaling factors were 0.3 kcal mol−1 in the second phase and 1 kcal mol−1 in the third phase [a value around 0.3 kcal mol−1 corresponds to a 10- to 15-pN force per carbon atom (16)]. The other molecular dynamics settings in both the second and the third phases were the same as those used in the equilibrium part of the first phase. These two phases were carried out for 1 ns, wherein the structure had converged as measured both by root mean square deviation (RMSD) with respect to initial structure and by the cross-correlation coefficient between the structure and the map (fig. S6). In the fourth phase, the steering force scaling factor was increased to 3 kcal mol−1, and the system was minimized for 2000 steps. The same MDFF procedure was repeated three times with different starting velocities for both closed and open conformations. The three resulting structures from the three replicate runs had similar cross-correlation coefficients: 0.845, 0.845, and 0.846 for the closed conformation and 0.886, 0.886, and 0.886 for the open conformation. The backbone RMSDs between the resulting structures were within 1.5 Å for both closed and open conformations. One structure, which had the most of its backbones inside the Coulomb potential map, was selected out of the three resulting structures for each conformation. The selected structures were further inspected visually for their fit into the map, and small regions of the structures comprising fragments of the N-loop and core loop were manually refined into density using the interactive molecular dynamics feature in MDFF.
ATPase assay
A modified malachite green assay (23) was used to measure the ATPase activity of various Vps4 proteins. Briefly, proteins used in the assay were dialyzed against the ATPase buffer [50 mM tris-HCl (pH 7.5), 100 mM KCl, and 5 mM MgCl2]. Vps4 (25 μl) or the mixture of Vps4 and different mutants was incubated in buffer with 2 mM ATP (final concentration) for 10 min at 37°C. Malachite green reagent (80 μl) was then added, and the reaction was quenched with the addition of 10 μl of 32% (w/v) sodium citrate. Absorbance at 620 nm (A620) was measured on a SpectraMax M5 microplate reader (Molecular Devices). To account for background ATP hydrolysis, signal from an identically treated sample lacking protein was subtracted. Each experiment was repeated three times.
Sedimentation analysis of filament disassembly
Vps24-Vps2 chimeric filaments were assembled by concentrating the protein to at least 350 μM and by incubating at 4°C overnight. Filament formation was confirmed by negative-stain EM imaging (37) and ultracentrifugation of protein samples in a TLA120.1 rotor at 60,000 rpm for 40 min at 10°C. After ultracentrifugation, the pellet and supernatant were diluted with equal volumes of SDS–polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, and equal volumes of supernatant and pellet fractions were analyzed on a 15% SDS-PAGE and visualized by Coomassie staining. The intensity of bands was quantified with ImageJ.
For the filament disassembly assay, overnight polymerized filaments were diluted to 3.5 μM and mixed with 2.5 μM Vps4 or the mixture of Vps4 and Vps4E233Q at different ratios, in the presence of 2 mM ATP or ADP in a buffer with 50 mM tris-HCl (pH 7.5), 100 mM KCl, and 5 mM MgCl2. Reaction mixtures (300 μl) were incubated at 37°C for 1 min and then quenched at 0°C by adding EDTA to a final concentration of 50 mM. Samples were then subjected to ultracentrifugation and analyzed as described above. Each experiment was repeated three times.
Supplementary Material
Funding
This work was supported by NIH R01 GM095769 (to Z.X.; “Structural basis of membrane scission: Regulation of the Vps4 ATPase complex,” from 1 January 2011 to 31 March 2015) and R01 DK090165 (to G.S.; “Architectural basis of leptin transmembrane signaling,” from 01 April 2011 to 31 July 2017). C.B. was funded by the NIH grant GM037554 and the NSF grant CHEM1506273. Author contributions: E.Z.G. prepared the Vps4 samples and performed biochemical characterization. M.S. collected the cryo-EM data and obtained 3D reconstructions. X.D. refined the structural models based on cryo-EM maps. J.T.T. helped perform filament disassembly using negative stain. Y.L. performed MALS analysis and contributed in the early stage of the project. Z.X. directly supervised all the biochemistry experiments. G.S. directly supervised the cryo-EM analysis. C.L.B. supervised MDFF. Z.X. and G.S. supervised the overall project design and execution. M.S., E.Z.G., X.D., C.L.B., Z.X., and G.S. wrote the manuscript, and all authors contributed to its final version. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The Vps4 hexamer cryo-EM maps in the closed and open conformations are deposited in the Electron Microscopy Data Bank under accession codes EMD-8587 and EMD-8588, respectively.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1700325/DC1
fig. S1. Vps4 oligomerizes into a hexamer in the presence of ATP.
fig. S2. Cryo-EM images of Vps4 oligomer.
fig. S3. Flow chart of particle classification and 3D map reconstruction.
fig. S4. Map resolution estimation and projection angle distribution.
fig. S5. Fitting of the Vps4 hexamer structure into the cryo-EM map.
fig. S6. Molecular dynamics flexible fitting.
fig. S7. Comparison of the crystal structure and Vps4 hexamer subunit structures.
fig. S8. Sequence alignments of Vps4 proteins from S. cerevisiae, Schizosaccharomyces pombe, Caenorhabditis elegans, Drosophila melanogaster, and Homo sapiens.
fig. S9. Residues at subunit interface III are important for Vps4 oligomerization and ATPase activity.
fig. S10. Structural comparison of Vps4 subunits in the open and closed conformations.
fig. S11. One wild-type subunit per hexamer is sufficient to maintain full Vps4 hexamer ATPase activity.
fig. S12. Filament disassembly activity of Vps4.
movie S1. Molecular dynamic flexible fitting into Vps4 open and closed cryo-EM maps.
movie S2. Morphing motion between Vps4 open and closed models.
REFERENCES AND NOTES
- 1.Hanson P. I., Cashikar A., Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 28, 337–362 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Katzmann D. J., Babst M., Emr S. D., Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 106, 145–155 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Garrus J. E., von Schwedler U. K., Pornillos O. W., Morham S. G., Zavitz K. H., Wang H. E., Wettstein D. A., Stray K. M., Côté M., Rich R. L., Myszka D. G., Sundquist W. I., Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107, 55–65 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Martin-Serrano J., Zang T., Bieniasz P. D., HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7, 1313–1319 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Carlton J. G., Martin-Serrano J., Parallels between cytokinesis and retroviral budding: A role for the ESCRT machinery. Science 316, 1908–1912 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Hanson P. I., Whiteheart S. W., AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 6, 519–529 (2005). [DOI] [PubMed] [Google Scholar]
- 7.McCullough J., Colf L. A., Sundquist W. I., Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 82, 663–692 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hill C. P., Babst M., Structure and function of the membrane deformation AAA ATPase Vps4. Biochim. Biophys. Acta 1823, 172–181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stuchell-Brereton M. D., Skalicky J. J., Kieffer C., Anne Karren M., Ghaffarian S., Sundquist W. I., ESCRT-III recognition by VPS4 ATPases. Nature 449, 740–744 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Kieffer C., Skalicky J. J., Morita E., De Domenico I., Ward D. M., Kaplan J., Sundquist W. I., Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev. Cell 15, 62–73 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 11.Obita T., Saksena S., Ghazi-Tabatabai S., Gill D. J., Perisic O., Emr S. D., Williams R. L., Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449, 735–739 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Babst M., Wendland B., Estepa E. J., Emr S. D., The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17, 2982–2993 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schöneberg J., Lee I.-H., Iwasa J. H., Hurley J. H., Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol. 18, 5–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monroe N., Han H., Gonciarz M. D., Eckert D. M., Karren M. A., Whitby F. G., Sundquist W. I., Hill C. P., The oligomeric state of the active Vps4 AAA ATPase. J. Mol. Biol. 426, 510–525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies B. A., Azmi I. F., Payne J., Shestakova A., Horazdovsky B. F., Babst M., Katzmann D. J., Coordination of substrate binding and ATP hydrolysis in Vps4-mediated ESCRT-III disassembly. Mol. Biol. Cell 21, 3396–3408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trabuco L. G., Villa E., Mitra K., Frank J., Schulten K., Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–683 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao J., Xia H., Yoshino-Koh K., Zhou J., Xu Z., Structural characterization of the ATPase reaction cycle of endosomal AAA protein Vps4. J. Mol. Biol. 374, 655–670 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonciarz M. D., Whitby F. G., Eckert D. M., Kieffer C., Heroux A., Sundquist W. I., Hill C. P., Biochemical and structural studies of yeast Vps4 oligomerization. J. Mol. Biol. 384, 878–895 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue M., Kamikubo H., Kataoka M., Kato R., Yoshimori T., Wakatsuki S., Kawasaki M., Nucleotide-dependent conformational changes and assembly of the AAA ATPase SKD1/VPS4B. Traffic 9, 2180–2189 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Yang B., Stjepanovic G., Shen Q., Martin A., Hurley J. H., Vps4 disassembles an ESCRT-III filament by global unfolding and processive translocation. Nat. Struct. Mol. Biol. 22, 492–498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han H., Monroe N., Votteler J., Shakya B., Sundquist W. I., Hill C. P., Binding of substrates to the central pore of the Vps4 ATPase is autoinhibited by the microtubule interacting and trafficking (MIT) domain and activated by MIT interacting motifs (MIMs). J. Biol. Chem. 290, 13490–13499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott A., Chung H. Y., Gonciarz-Swiatek M., Hill G. C., Whitby F. G., Gaspar J., Holton J. M., Viswanathan R., Ghaffarian S., Hill C. P., Sundquist W. I., Structural and mechanistic studies of VPS4 proteins. EMBO J. 24, 3658–3669 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vild C. J., Xu Z., Vfa1 binds to the N-terminal microtubule interacting and trafficking (MIT) domain of Vps4 and stimulates its ATPase activity. J. Biol. Chem. 289, 10378–10386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mindell J. A., Grigorieff N., Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Scheres S. H. W., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenthal P. B., Henderson R., Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chacón P., Wriggers W., Multi-resolution contour-based fitting of macromolecular structures. J. Mol. Biol. 317, 375–384 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K., Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M., All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Mackerell A. D. Jr, Feig M., Brooks C. L. III, Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 35.Schreiner E., Trabuco L. G., Freddolino P. L., Schulten K., Stereochemical errors and their implications for molecular dynamics simulations. BMC Bioinformatics 12, 190 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. H., A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995). [Google Scholar]
- 37.Peisley A., Skiniotis G., 2D projection analysis of GPCR complexes by negative stain electron microscopy. Methods Mol. Biol. 1335, 29–38 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1700325/DC1
fig. S1. Vps4 oligomerizes into a hexamer in the presence of ATP.
fig. S2. Cryo-EM images of Vps4 oligomer.
fig. S3. Flow chart of particle classification and 3D map reconstruction.
fig. S4. Map resolution estimation and projection angle distribution.
fig. S5. Fitting of the Vps4 hexamer structure into the cryo-EM map.
fig. S6. Molecular dynamics flexible fitting.
fig. S7. Comparison of the crystal structure and Vps4 hexamer subunit structures.
fig. S8. Sequence alignments of Vps4 proteins from S. cerevisiae, Schizosaccharomyces pombe, Caenorhabditis elegans, Drosophila melanogaster, and Homo sapiens.
fig. S9. Residues at subunit interface III are important for Vps4 oligomerization and ATPase activity.
fig. S10. Structural comparison of Vps4 subunits in the open and closed conformations.
fig. S11. One wild-type subunit per hexamer is sufficient to maintain full Vps4 hexamer ATPase activity.
fig. S12. Filament disassembly activity of Vps4.
movie S1. Molecular dynamic flexible fitting into Vps4 open and closed cryo-EM maps.
movie S2. Morphing motion between Vps4 open and closed models.