

Summary
We demonstrates that DNA methylation alterations at multiple genes, including those in the TH1-TH2 pathways and some novel genes, are associated with cow's milk allergy (CMA), which shed new light on the epigenetic underpinnings of CMA.
Keywords: DNA methylation, cow's milk allergy, epigenome-wide association
To the editor
IgE-mediated cow's milk allergy (CMA), which affects 2-3% of young children, is among the most common types of food allergy (FA). The molecular mechanisms underlying the development, persistence, and resolution of CMA remain largely unknown. Several recent studies have suggested that epigenetic alterations are involved in allergy development.1, 2 DNA methylation (DNAm), a type of epigenetic mechanism, regulates gene expression. Alterations in DNAm play a key role in T-cell differentiation and in maintaining TH1/TH2 balance,1, 2 and thus may offer a potential mechanistic explanation for the natural history of CMA. However, to date, research on the epigenetics of FA,3 or CMA in particular,4 has been very limited. Most of the available studies are based on small sample sizes (<60 children), and were not replicated in an independent sample.
We performed the first epigenome-wide association study (EWAS) of CMA in U.S. children using a two-stage approach. In the discovery stage, we measured DNAm at 485,512 genomic loci in whole blood samples from 106 Caucasian children with CMA (cases) and 76 non-allergic and non-atopic Caucasians (controls) using the Illumina HumanMethylation450 arrays. All of the cases and controls were enrolled by the Chicago Food Allergy Study using identical protocols. We then sought to validate the top significant DNAm loci in two independent replication samples including: 1) childhood whole blood samples of 25 Caucasian children (5 CMA cases and 20 controls) from the same Chicago Food Allergy Study (Chicago replication sample), and 2) cord blood samples from 140 African-American children (8 cases and 132 controls) from the Boston Birth Cohort (Boston replication sample) (see Methods in the online Repository). Population characteristics of the discovery and replication samples are presented in Tables E1 and E2, respectively.
CMA cases were defined if the children met the following criteria: 1) a convincing history of symptoms indicative of an allergic reaction within 2 hours of ingestion of cow's milk; and 2) clear evidence of sensitization defined as having a specific IgE (sIgE) ≥ 0.35 kU/L to cow's milk and/or a positive skin prick test to cow's milk with mean wheal diameter ≥ 3 mm greater than the saline control. After quality control steps (see Methods in the online Repository), 435,642 CpG sites were available for subsequent data analyses. We fit a linear regression model for ComBat-transformed M value at each CpG site as a function of CMA status, adjusting for potential confounders (see Methods in the online Repository). Bonferroni correction was applied to account for multiple testing (p<1.15E-07).
In the discovery stage, we identified 575 significant autosomal differentially methylated positions (DMPs) (469 located within 385 genes and 106 intergenic): 568 hypomethylated and 7 hypermethylated in CMA cases compared to controls (p<1.15E-07, Fig 1). The majority of these 575 DMPs showed a small DNAm change between cases and controls, and only five DMPs had an absolute mean DNAm difference ≥ 5% (Fig 1 & Table 1). On the X-chromosome, DMP cg16737869 met our p-value significance threshold, but with a modest DNAm change between cases and controls (2%).
Fig. 1. Volcano plot showing the significance and magnitude of autosomal DNA methylation changes associated with cow's milk allergy (CMA).
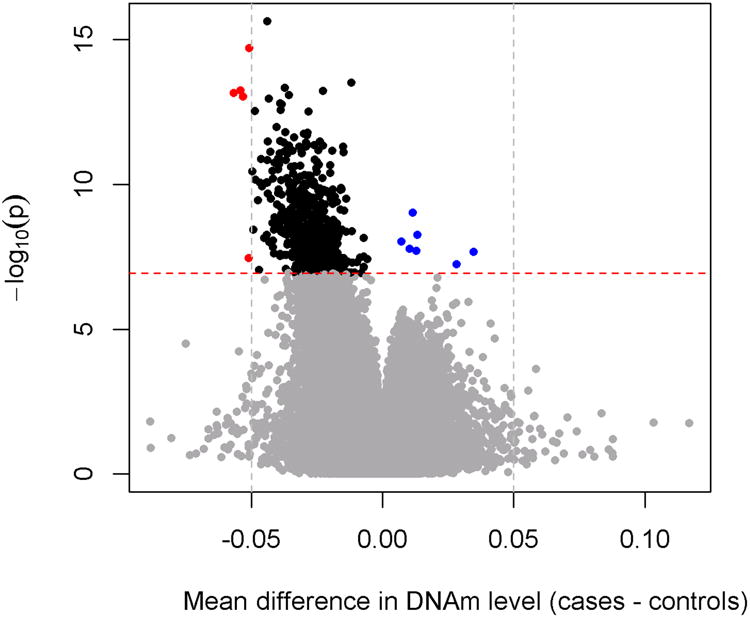
Mean differences in DNA methylation (cases-controls) are plotted on the x-axis and corresponding -log10(p-value)s are plotted on the y-axis. Each point represents a measured CpG site. Black and blue points represent CpG sites significantly hypomethylated and hypermethyated in CMA cases, respectively. Red points represent those significant CpG sites with DNA methylation difference >5% between cases and controls. The red dashed horizontal line represents the genome-wide significance threshold (p=1.15E-07).
Table 1. The top significant differentially methylated positions and their associations with cow's milk allergy in the discovery and replication samples.
| DMPs | CHR | Gene | Location | Chicago Discovery | Chicago Replication | Boston Replication | |||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||
| βdiffa | p-valueb | βdiffa | p-valuec | βdiffa | p-valued | ||||
| DMPs with absolute DNAm difference ≥ 5% in cases vs controls | |||||||||
| cg11770323 | 13 | NDFIP2 | Intron | -5.7 | 2.0E-15 | -4.0 | 0.005* | -0.9 | 0.811 |
| cg16409452 | 14 | EVL | 3′UTR | -5.4 | 5.6E-14 | -7.3 | 0.042 | -1.4 | 0.322 |
| cg18550847 | 14 | EVL | 3′UTR | -5.3 | 9.6E-14 | -7.4 | 0.034* | -1.9 | 0.187 |
| cg09377531 | 8 | TRAPPC 9 | Intron | -5.1 | 7.0E-14 | -7.8 | 0.004* | -1.5 | 0.260 |
| cg15090899 | 6 | RPS6KA2 | Exon | -5.1 | 3.6E-08 | -7.1 | 0.384 | -2.1 | 0.811 |
| DMPs with annotated genes relevant to TH1-TH2 pathway | |||||||||
| cg16386158 | 2 | IL1RL1 | TSS1500 | -3.8 | 1.7E-13 | -8.8 | 0.007* | -2.4 | 0.041 |
| cg08404225 | 3 | IL5RA | 5′UTR | -3.7 | 9.3E-10 | -5.5 | 0.062 | -2.2 | 0.001* |
| cg13316148 | 2 | STAT4 | Intron | -3.2 | 6.3E-09 | -5.2 | 0.013* | -0.4 | 0.301 |
| cg26787239 | 5 | IL4 | TSS1500 | -2.6 | 6.7E-09 | -4.8 | 0.006* | -1.5 | 0.042 |
| cg06040872 | 17 | CCL18 | Intron | -3.2 | 4.0E-09 | -5.2 | 0.018* | -0.6 | 0.256 |
CHR: chromosome; DMP: differentially methylated position; UTR: untranslated region. TSS: translation starting site.
Mean methylation difference between cases and controls.
p-value was calculated based on linear regression models with ComBat-transformed M value at each DMP as the outcome, adjusted for age, gender, breastfeeding, parental history of food allergy, estimated cell composition, and genetic ancestry.
p-value was calculated based on the linear regression model with ComBat-transformed M value at each DMP as the outcome, adjusted for cell composition and age. Further adjustment for gender and parental history of allergy did not significantly change the associations.
p-value was calculated based on the linear regression model with ComBat-transformed M-value at each DMP as the outcome, adjusted for cell composition, gender, parental history of food allergy, and gestational age group.
FDR (false discovery rate) < 5%
In parallel with individual DMP analyses, we performed a genome-wide screen to identify differentially methylated regions (DMRs) using a “bump hunting” approach. A marginally significant DMR in the 3′ untranslated region of the Enah/Vasp-like (EVL) gene was identified, which was hypomethylated in CMA cases compared to controls (family-wise error rate=0.085, Fig E1a). This DMR included five highly correlated DMPs (r > 0.8, Fig E1b) that were all significantly associated with CMA at p <1.15E-07, two of which having a DNAm difference >5% between cases and controls.
We then performed KEGG pathway enrichment analysis using WebGestalt on the 386 annotated genes that contained at least one significant DMP associated with CMA. Three enriched pathways were identified, including “Butirosin and neomycin biosynthesis”, “Starch & sucrose metabolism” and “Fructose and mannose metabolism”, all of which shared three genes (hexokinase 1 -3) that are critical to glucose metabolism (Table E3). This result supports our previous findings on the significant associations between gestational diabetes and increased risk of food sensitization.5
Replications
Among the 576 DMPs associated with CMA, 10 loci meeting the following criteria were selected for replication: 1) significant DMPs with an absolute DNAm difference >5% (n=5); and/or 2) significant DMPs that annotated to genes relevant to the TH1-TH2 pathway (n=5) (Table 1). In the Chicago replication sample, 7 DMPs were validated for their associations with CMA at a false discovery rate (FDR) <0.05 (Table 1). In the Boston replication sample, only DMP cg08404225 in the IL5RA gene was significantly hypomethylated in CMA cases relative to the normal controls after FDR correction (FDR=0.01). The associations between the 8 validated DMPs and CMA in the replication samples were in the same directions as were identified in the discovery sample (Table 1, Fig E2).
The biological significance of the 8 validated DMPs was explored with a web tool, EpiExplorer. As shown in Table E4, DMP cg11770323 overlapped with the DNaseI hypersensitive site; DMP cg18550847 was within a CpG island; and DMPs cg13316148, cg08404225 and cg26787239 overlapped with the enhancers and with lamina-associated domains (LAD). We also found that DMPs cg16386158, cg13316148, cg26787239 and cg11770323 co-localized with the CTCF-binding sites (Table E4). Overall, these data indicate that DNAm alteration of these loci may be biologically relevant, but further validation is needed.
This study not only confirmed that DNAm alterations in TH1-TH2 balance (i.e., those annotated to IL1RL1, IL5RA, IL4, CCL18 and STAT4 genes) are associated with CMA, it also revealed some novel candidates for CMA. Among the 8 validated DMPs, three DMPs, all with absolute mean DNAm >5% between cases and controls, were annotated to NDFIP2 (cg11770323), EVL (cg18550847) and TRAPPC9 (cg09377531), respectively. These three genes have not previously been implicated in allergy, but their role may be biologically plausible. NDFIP2, one of the IL-4 regulated genes, can promote interferon gamma production via polarized human TH1 lymphocytes.6 NDFIP2 can also activate E3 ubiquitin ligases, which may play an important role in preventing allergic diseases. EVL is one of the IL-13-regulated genes.7 The analyses from GIANT8 showed that the EVL and IL4 genes are highly co-expressed in basophils. TRAPPC9 encodes a protein that likely plays a role in NF-kappa-B signaling, a pathway that is relevant to the immune system. Further studies are needed to explore the role of these annotated genes in CMA.
The two replication analyses in this study, although with limited sample sizes, have provided additional information. Replication in the Chicago sample, where the cases were all allergic to both peanut and cow's milk and the non-CMA controls were all allergic to peanut only, suggested that the replicated DNAm associations may be specific to CMA; while replication in the Boston sample, where DNAm was measured in cord blood from African-American samples, indicate that the replicated DNAm alterations between cases and controls may occur in-utero and may be common across race groups.
Several limitations are noted. First, we used whole blood DNA to obtain DNAm profiling. Second, questions remain regarding how to interpret small DNAm differences between CMA and controls, mostly <5%. One possibility is that such small changes in whole blood may be reflective of larger changes in a specific cell type, as demonstrated in a previous study9. Third, DNAm levels of the identified DMPs were not validated using other techniques such as pyrosequencing, and no functional assays were conducted in this study. However, previous studies have demonstrated that the 450K array correlates well with direct pyrosequencing.10
In conclusion, this was the first EWAS of CMA in U.S. children and it clearly demonstrated that methylation alterations in specific gene loci are associated with CMA. Findings from this study warrant additional validation and functional studies, and, if confirmed, may help to improve our understanding of the epigenetic mechanisms underlying the development and resolution of CMA and offer novel targets for prediction, prevention and treatment of CMA.
Supplementary Material
Fig E1. A genomic cluster in the EVL gene that hypomethylated in CMA children compared to normal controls. 1a). The upper panel shows DNA methylation levels on the y-axis and genomic location on the x-axis. Each point represents the methylation level at a specific CpG site for each CMA case (in blue) and control (in black). The lower panel displays the methylation difference between cases and controls for the six involved CpG sites (cg26415437, cg16409452, cg14084609, cg06756385, cg18550847 and cg01000631);* p < 1.15E-07 for the methylation difference between cases and controls. 1b). DNA methylation correlation for the genomic cluster shown in fig E1a. The measure of correlation coefficient (r) among each pair of CpG is shown graphically, with blue representing high positive correlation.
Fig E2. Scatter plots of DNA methylation levels at 8 CMA-associated DMPs that were validated in at least one replication sample. (A) Plot for each DMP in the discovery sample (n=182); (B) Plot for each DMP in the Chicago replication sample; (C) Plot for each DMP in the Boston replication sample. The red segment denotes the median DNA methylation levels within each subset of subjects.
Table E1. Population characteristics of the 182 Caucasian children included in the discovery stage
Table E2. Population characteristics of the Caucasian replication sample from the Chicago Food Allergy Study and of the African-American replication sample from the prospective BBC birth cohort
Table E3. KEGG pathways with enrichment of genes that exhibit differential DNA methylation in children with CMA
Table E4. Biological relevancea of the eight CMA-associated DMPs that were validated in at least one replication sample.
Acknowledgments
We gratefully acknowledge the individuals and families who participated in the Chicago Food Allergy Study and in the Boston Birth Cohort. This research was supported in part by grants from the Bunning Family and their family foundations, Sacks Family Foundation Fund, Food Allergy Research and Education (FARE), the National Institute of Allergy and Infectious Diseases (NIAID, PI: X.Wang, U01AI090727 from the Consortium of Food Allergy Research, R56AI080627 and R21AI088609) and Department of Defense (PI: X.Wang, W81XWH-10-1-0123). Dr Keet is partly supported by NIAID (K23AI103187). Dr Hui-Ju Tsai is partly supported by grants from National Science Council (101-2314-B-400-009-MY2) and Ministry of Science and Technology (103-2314-B-400-004-MY3).
Footnotes
Conflict of interest: The authors have declared that no conflicting interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hong X, Wang X. Early life precursors, epigenetics, and the development of food allergy. Semin Immunopathol. 2012;34:655–69. doi: 10.1007/s00281-012-0323-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Su RC, Becker AB, Kozyrskyj AL, Hayglass KT. Epigenetic regulation of established human type 1 versus type 2 cytokine responses. J Allergy Clin Immunol. 2008;121:57–63. doi: 10.1016/j.jaci.2007.09.004. e3. [DOI] [PubMed] [Google Scholar]
- 3.Martino D, Dang T, Sexton-Oates A, Prescott S, Tang ML, Dharmage S, et al. Blood DNA methylation biomarkers predict clinical reactivity in food-sensitized infants. J Allergy Clin Immunol. 2015;135:1319–28 e12. doi: 10.1016/j.jaci.2014.12.1933. [DOI] [PubMed] [Google Scholar]
- 4.Berni Canani R, Paparo L, Nocerino R, Cosenza L, Pezzella V, Di Costanzo M, et al. Differences in DNA methylation profile of Th1 and Th2 cytokine genes are associated with tolerance acquisition in children with IgE-mediated cow's milk allergy. Clin Epigenetics. 2015;7:38. doi: 10.1186/s13148-015-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar R, Ouyang F, Story RE, Pongracic JA, Hong X, Wang G, et al. Gestational diabetes, atopic dermatitis, and allergen sensitization in early childhood. J Allergy Clin Immunol. 2009;124:1031–8 e1. doi: 10.1016/j.jaci.2009.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lund RJ, Loytomaki M, Naumanen T, Dixon C, Chen Z, Ahlfors H, et al. Genome-wide identification of novel genes involved in early Th1 and Th2 cell differentiation. J Immunol. 2007;178:3648–60. doi: 10.4049/jimmunol.178.6.3648. [DOI] [PubMed] [Google Scholar]
- 7.Scotton CJ, Martinez FO, Smelt MJ, Sironi M, Locati M, Mantovani A, et al. Transcriptional profiling reveals complex regulation of the monocyte IL-1 beta system by IL-13. J Immunol. 2005;174:834–45. doi: 10.4049/jimmunol.174.2.834. [DOI] [PubMed] [Google Scholar]
- 8.Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, et al. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet. 2015;47:569–76. doi: 10.1038/ng.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang L, Willis-Owen SA, Laprise C, Wong KC, Davies GA, Hudson TJ, et al. An epigenome-wide association study of total serum immunoglobulin E concentration. Nature. 2015 doi: 10.1038/nature14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roessler J, Ammerpohl O, Gutwein J, Hasemeier B, Anwar SL, Kreipe H, et al. Quantitative cross-validation and content analysis of the 450k DNA methylation array from Illumina, Inc. BMC Res Notes. 2012;5:210. doi: 10.1186/1756-0500-5-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig E1. A genomic cluster in the EVL gene that hypomethylated in CMA children compared to normal controls. 1a). The upper panel shows DNA methylation levels on the y-axis and genomic location on the x-axis. Each point represents the methylation level at a specific CpG site for each CMA case (in blue) and control (in black). The lower panel displays the methylation difference between cases and controls for the six involved CpG sites (cg26415437, cg16409452, cg14084609, cg06756385, cg18550847 and cg01000631);* p < 1.15E-07 for the methylation difference between cases and controls. 1b). DNA methylation correlation for the genomic cluster shown in fig E1a. The measure of correlation coefficient (r) among each pair of CpG is shown graphically, with blue representing high positive correlation.
Fig E2. Scatter plots of DNA methylation levels at 8 CMA-associated DMPs that were validated in at least one replication sample. (A) Plot for each DMP in the discovery sample (n=182); (B) Plot for each DMP in the Chicago replication sample; (C) Plot for each DMP in the Boston replication sample. The red segment denotes the median DNA methylation levels within each subset of subjects.
Table E1. Population characteristics of the 182 Caucasian children included in the discovery stage
Table E2. Population characteristics of the Caucasian replication sample from the Chicago Food Allergy Study and of the African-American replication sample from the prospective BBC birth cohort
Table E3. KEGG pathways with enrichment of genes that exhibit differential DNA methylation in children with CMA
Table E4. Biological relevancea of the eight CMA-associated DMPs that were validated in at least one replication sample.