

Abstract
Phosphoinositides are rapidly turning-over phospholipids that play key roles in intracellular signaling and modulation of membrane effectors. Through technical refinements we have improved sensitivity in the analysis of the phosphoinositide PI, PIP, and PIP2 pools from living cells using mass spectrometry. This has permitted further resolution in phosphoinositide lipidomics from cell cultures and small samples of tissue. The technique includes butanol extraction, derivatization of the lipids, post-column infusion of sodium to stabilize formation of sodiated adducts, and electrospray ionization mass spectrometry in multiple reaction monitoring mode, achieving a detection limit of 20 pg. We describe the spectrum of fatty-acyl chains in the cellular phosphoinositides. Consistent with previous work in other mammalian primary cells, the 38:4 fatty-acyl chains dominate in the phosphoinositides of the pineal gland and of superior cervical ganglia, and many additional fatty acid combinations are found at low abundance. However, Chinese hamster ovary cells and human embryonic kidney cells (tsA201) in culture have different fatty-acyl chain profiles that change with growth state. Their 38:4 lipids lose their dominance as cultures approach confluence. The method has good time resolution and follows well the depletion in < 20 s of both PIP2 and PIP that results from strong activation of Gq-coupled receptors. The receptor-activated phospholipase C exhibits no substrate selectivity among the various fatty-acyl chain combinations.
1. Introduction
This paper analyzes three major classes of phosphoinositides and compares the profiles of their fatty-acyl chains in several cell types. Phosphoinositide phospholipids are low-abundance membrane lipids [1] used for signaling and as identity labels for different cellular membranes. They participate in membrane trafficking, cell polarity, cytoskeletal regulation, and ion channel function [2–4]. Thus far, almost all biological studies have focused on signaling by the phosphorylated inositol ring that forms the polar head group of the lipid. The unphosphorylated parent compound phosphatidylinositol (PI) gives rise to seven phosphorylated forms when lipid kinases phosphorylate the inositol combinatorially on the 3-, 4-, and 5-positions. Each form is a signal for proteins that recognize this specific headgroup. For example, phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), accumulates uniquely in and identifies the plasma membrane (PM) where it localizes or activates numerous membrane proteins. Some PM ion channels require PI(4,5)P2 for full function [4]. We know little about the biological importance of the fatty acids attached to these lipids.
Quantitative analysis of phosphoinositide species across multiple cell types and conditions can help reveal biological and pathophysiological mechanisms. The most powerful method today uses liquid chromatography followed by mass spectrometry (MS). As instruments improved, good results came from a combination of high performance liquid chromatography (HPLC) with electrospray ionization (ESI) and tandem mass spectrometry with multiple reaction monitoring (HPLC-ESI-MS/MS), achieving initial lower detection limits for PIP and PIP2 ranging from 50 to 250 pmoles [5, 6]. However, for phosphoinositides, the many negative charges along with their relatively low abundance relative to other lipids presented challenges for MS measurement. Derivatization, neutralizing the negative charges by methylation, greatly improved the detection to 1 pmole [7]. Here we continue with methodological refinements including adding sodium after the HPLC column to achieve detection of 20 fmoles (~20 pg) of PIP and PIP2 applied to the column. This facilitated quantitative monitoring of rare fatty-acyl species and use of small samples.
Past work revealed that different major membrane phospholipid classes contain different combinations of fatty-acyl chains [8–16]. The distribution of acyl chains can differ between tissues [17], it can change with dietary fatty acids [18], and it changes with disease mutations or knockout of lipid metabolizing enzymes [5, 15]. The significance of having different acyl species has yet to be elucidated except that arachidonyl lipids are essential as precursors for the many signaling pathways based on arachidonic acid, and in a broad sense, the chain length and unsaturation affect fluidity and melting temperature of the lipid bilayer. The combination of fatty-acyl chains in phosphoinositides is unique. In primary mammalian cells, 50–80% of the phosphoinositide molecules seem to be stearoyl/arachidonyl (in the sn-1 and sn-2 positions, respectively) designated C18:0/C20:4 (the number of carbons:number of double bonds) or 38:4 for the whole molecule [5, 6, 17, 19]. Flies, yeast, and Dictyostelium each use different dominant acyl species in their phosphoinositides; they are not dominantly 38:4 [5, 14, 20, 21]. Our high detection sensitivity broadens the spectrum of rarer fatty-acyl chains detectable in phosphoinositides of primary mammalian cells and cell lines and leads to new insights.
2. Materials and methods
2.1. Reagents and Lipid Standards
Modified Ringers Solution without HEPES buffer or glucose (160 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2) was brought to pH 7.4 with NaOH immediately prior to the extraction of lipid. Butanol, methanol, chloroform, dichloromethane, and acetonitrile (Fisher) were all of mass spectrometry grade. Sodium formate and HCl were from Sigma, and trimethylsilyldiazomethane (TMS-DM, 2.0 M in diethyl ether or hexanes) from Sigma-Aldrich and Acros. The synthetic lipids were ammonium salts of 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-4′,5′-bisphosphate), (36:2 PI(4,5)P2); 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-4′-phosphate), (36:2 PI(4)P); and 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol), (36:2 PI) from Avanti Polar Lipids. The three lipid analytical internal standards were ammonium salts of 1-heptadecanoyl-2-(5Z,8Z,11Z,14Z-eicosatetraenoyl)-sn-glycero-3-phospho-(1′-myo-inositol-4′,5′-bisphosphate) [17:0, 20:4 PI(4,5)P2]; 1-heptadecanoyl-2-(5Z,8Z,11Z,14Z-eicosatetraenoyl)-sn-glycero-3-phospho-(1′-myo-inositol-4′-phosphate) [17:0, 20:4 PI(4)P]; and 1-heptadecanoyl-2-(5Z,8Z,11Z,14Z-eicosatetraenoyl)-sn-glycero-3-phospho-(1′-myo-inositol) [17:0, 20:4 PI] from Avanti Polar Lipids (LIPID MAPS MS Standards).
2.2. Cells, animals, and sample preparation
We analyzed lipids in cell lines and primary cells. Experiments with cell lines used Chinese-hamster-ovary (CHO)-M1 cells (85–95% confluent), a line of CHO cells stably expressing M1 muscarinic receptors [22], and tsA201 cells (transformed human embryonic kidney cells), 40% or 85–95% confluent as indicated [3]. For each sample, cultured cells on one 35 mm tissue culture dish were gently washed twice with modified Ringers solution (2 × 1 ml). In most cases, 1 ml of ice-cold modified Ringers solution was added to remove cells from the dish, and cells were spun down (0.3–3 million) at 12,000 G for 3 min at 4°C and resuspended in 40 μl ice-cold molecular-grade water. In other cases, 1 ml of ice-cold methanol/1 M HCl (1:1 v/v) was added to the dishes instead of 1 ml modified Ringers solution to terminate cellular reactions, and then cells were scraped, removed to a centrifuge tube, and taken to dryness under N2 prior to resuspension in 40 μl ice-cold water. For primary tissues, the pineal glands and superior cervical ganglia (SCG) were obtained from Male Sprague–Dawley rats (7–12 weeks old) euthanized by CO2 during the morning of their circadian light cycle according to guidelines approved by the University of Washington Institutional Animal Care and Use Committee. For each sample, five glands or two ganglia were cut into small pieces before being suspended in 40 μl ice-cold water. For both cell lines and primary tissues, the internal analytical standards (17:0–20:4 PI(4)P and PI(4,5)P2, 10 ng) were added to each suspension in water. To normalize for cell numbers, 10% of each sample was removed before lipid extraction, and genomic DNA was extracted with GeneJet Genomic DNA Purification Kit (Thermo Scientific(TM), USA) according to the manufacturer’s instructions. Total genomic DNA amounts were quantified by spectrophotometry and used for normalization of lipid signal intensities.
2.3. Modified lipid extraction, derivatization, and analysis by liquid chromatography and mass spectrometry
The results of this paper required refinements of lipid extraction, liquid chromatography, and mass spectroscopic analysis that permitted a higher resolution analysis of endogenous phosphoinositides. We provide a detailed description of the development and validation of our new methods in the Supplemental Methods and Data section. We simply list the resulting protocol steps in brief here. Phosphatidylinositol lipid analytical internal standards were added to the water-suspended biological samples. Endogenous lipids with the internal standards were then isolated from each sample by extraction with acidic n-butanol and chloroform [23–27]. Phosphate negative charges were neutralized by methylation with trimethylsilyldiazomethane (TMS-DM) [7]. We introduce the symbol “D” for “derivatized” in the following notation for the fully methylated phosphoinositides: D-PI for PI-CH3; D-PIP for PIP-(CH3)3; and D-PIP2 for PIP2-(CH3)5. An aliquot of the derivatized sample was separated on a C4 column in an acetonitrile formic acid gradient and, for the figures in the main text, monitored by a Waters XEVO TQ-S MS/MS in multiple reaction monitoring mode (MRM) using electrospray and positive ion mode. An important step was to infuse Na formate into the post-column eluate before entry into the mass spectrometer. This promoted formation of positively charged sodiated adducts in the MS machine. The molecular fragments chosen for quantitative analysis are described and validated in the Supplemental Methods and Data. This method showed high sensitivity and many well-resolved peaks for lipid species in the PI, PIP, and PIP2 classes, as well as peaks for phosphatidylserine, phosphatidylcholine and phosphatidic acid. However since we included internal standards only for PI, PIP, and PIP2, we do not quantify the others further here.
2.4. Data Analysis
Peak areas for lipid species and standards were quantitated by integrating curves like those in Fig. S1 using Waters Quanlynx software. For absolute calibrations and comparisons of different samples, peak areas were normalized to the synthetic 17:0, 20:4 PIP2, PIP, and PI internal standards for calibration and lipid-collection efficiency and further corrected for cell number using total genomic DNA. For rat tissues we could use the standard 2N genomic size, but for human tsA201 cells, which are hypotriploid, we assumed a 2.5N genome size, subject to some error. Statistics are given as mean ± SEM. Student’s t test was used to test for statistical significance. p values <0.05 were considered significant.
3. Results
3.1. Calibration shows improved limits of detection
To determine absolute lipid amounts and sensitivity we calibrated signal intensities using the synthetic analytical 37:4 lipid standards. These 37:4 lipids are not naturally occurring. The PI, PIP, and PIP2 standards were spiked into samples containing in addition approximately 1 million tsA201 cells, then extracted and derivatized. Varying amounts were injected on the UPLC column and peak areas were measured (Fig. 1). The lines plot the peak area of 37:4 PI, PIP, and PIP2 standards measured versus the predicted amount of standard in the injected sample. The lowest points plotted correspond to injection of 0.02 ng of 37:4 standards onto the column. This provided signals that were easily resolved, so our detection limit is below 20 pg lipid species per injection (~20 fmole), a significant improvement over previous reports.
Fig. 1.
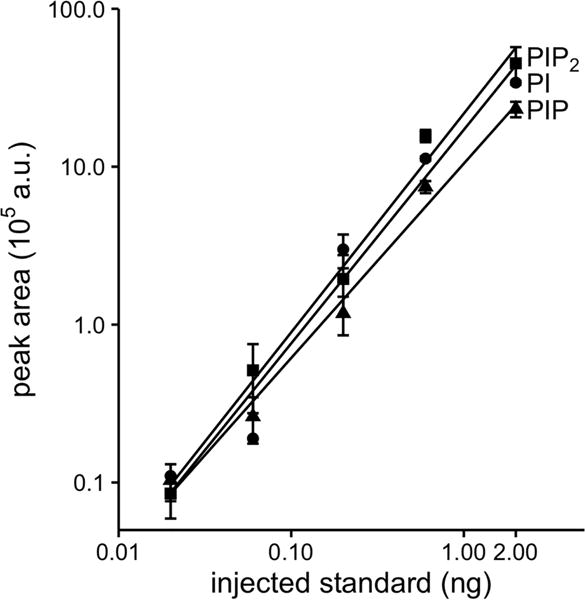
Calibration curves shown as responses to analytical internal standards for 37:4 PI, PIP, and PIP2 spiked into 106 tsA201 cells. Cells combined with internal standards were extracted and derivatized. Different quantities of extract were injected, specified as nanograms of the standard actually injected onto the C4 UPLC column. We used post-column sodium infusion (50 μM at 5 μl/min) and monitored the effluent with the Waters Xevo TQ MS/MS run in MRM mode using the fragmentation transitions identified in Fig. S3. (n = 3)
3.2. Fatty-acid composition of phosphoinositides in resting pineal gland
Our method provided sufficient added resolution to detect many low-abundance phosphoinositide species in cell extracts. We illustrate with extracts from rat pineal glands. Lipids of glands with spiked-in analytical standards were extracted, derivatized, and analyzed in MRM mode. Results were normalized to standards and then within each group, to the extracted DNA–equivalent to the number of cells. Like other cell types that have been studied, the pineal gland has much more of PI than of the downstream phosphorylated products PIP and PIP2 (Table 1). Recall here that mass spectrometry normally does not distinguish regioisomers, so for example under PIP are included PI(3)P, PI(4)P, and PI(5)P, of which PI(4)P is likely to be most abundant. Similarly, mass spectrometry typically reports only the sums of the carbon atoms and of the double bonds in the lipid side chains rather than differentiating the exact formulas of the individual chains. As is conventional in this field, we refer to such results as “fatty-acid composition,” and we tacitly assume that the composition 38:4 primarily reflects stearoyl/archidonyl side chains. Fig. 2 plots the relative abundance of fatty-acyl side chains (total carbons:double bonds) within the PI, PIP, and PIP2 pools. Note that the vertical scale here is in logarithmic units to emphasize the wide dynamic range of detection, 4-orders of magnitude for abundant PI. For each phosphoinositide class, we chose to optimize detection for 13 or 14 species plus the standard. The species chosen, not entirely the same for each phosphoinositide, were all determined during the same 17-min run. As reported for several other native tissues, by far the most abundant fatty acid combination in native pineal glands was 38:4 in each phosphoinositide class. The relative abundance of 38:4 was 86 ± 1%, 75 ± 4%, and 66 ± 8% for PI, PIP, and PIP2 (n = 3). However, several other unsaturated 38:x compositions were present (38:1, 38:2: 38:3–second most abundant, 38:4–most abundant, and 38:5), and among the rarer species were 36:1, 36:2, 36:3, and 36:4 as well as 40:4 and 40:5. Overall, lipids with multiple double bonds were favored. The rough similarity of fatty-acid compositions seen for the three phosphoinositide groups would be expected if the bulk of these lipid pools remains in a dynamic and rapid precursor-product relationship through continual actions of lipid kinases and lipid phosphatases.
Table 1.
Percentage of PI, PIP, and PIP2 relative to total phosphoinositides in different cell types
| Cell or tissue | PI | PIP | PIP2 | Number of experiments: |
|---|---|---|---|---|
| Pineal gland | 96.9 ± 1.6% | 0.7 ± 0.3% | 2.4 ± 1.3% | 3 |
| SCG | 96.2 ± 1.2% | 1.8 ± 0.6% | 2.1 ± 0.6% | 4 |
| tsA201 | 96.9 ± 0.9% | 1.5 ± 0.3% | 1.7 ± 0.7% | 7 |
| CHO | 90.5 ± 0.9% | 2.0 ± 0.1% | 7.5 ± 0.9% | 3 |
Measured with post-column infusion of 50 μM sodium formate. Each column includes the sum of all measured species of that lipid. tsA201 and CHO cells were harvested at 85–95% confluency.
Fig.2.
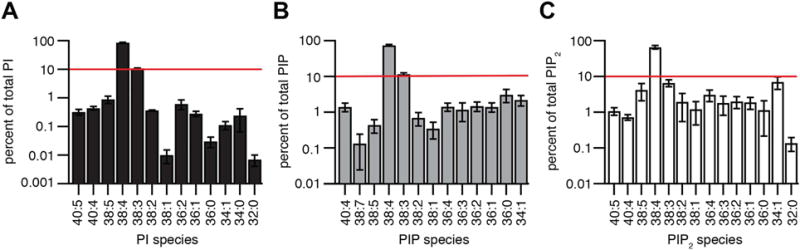
Fatty-acyl chains in phosphoinositide lipids of rat pineal glands. Mass spectrometry analysis of the fatty-acid composition of D-PI, D-PIP, and D-PIP2 in derivatized extracts from untreated pineal glands expressed as percent of the total pool of each major lipid type. Horizontal red lines are at 10% abundance. Note that bars are drawn here on a logarithmic scale to emphasize the broad dynamic range of detection of minor species and the dominance of 38:4 fatty acids in all three phosphoinositide classes. Each result is pooled from 5 pineal glands with three repeats (n = 3). Values are displayed as mean ± SEM.
3.3. Comparison of different cells and an effect of growth conditions
A similar analysis was repeated on three other cell types: native rat superior cervical ganglia (SCG), cultured tsA201 cells, and cultured CHO cells. Like the pineal gland, each shows a strong predominance of PI over PIP and PIP2 (Table 1). The fatty-acid chain distributions are plotted in Fig. 3, this time on more familiar linear coordinates. The SCG phosphoinositides resemble those of the pineal gland with 38:4 fatty acids dominating (29–54%), 38:3 near the 10 % level, and the additional presence of several 36:x species, especially in PIP. In contrast, for CHO and tsA201 cells the pattern is quite different. The total of 34:x species exceeds that of 36:x species, 38:4 is not dominant, and there was less polyunsaturation. These cultured cells were harvested at 85–95% confluency as we have ordinarily prepared them for electrophysiology [3], and the result was robust over several repeats. We then compared an earlier stage of tsA201-cell cultures that would be in a logarithmic growth phase with the late-stage confluent cultures closer to contact inhibition. The early-stage cultures were much more like primary cells with 38:4 fatty-acids dominant and higher degrees of unsaturation. There was a progression towards more saturation of phosphoinositide fatty acids in late stages. If the cells were harvested at low confluency (40–70%), the ratio of 38:4 to 36:1 fatty acid chains was 3.8 ± 0.5 for D-PI, 2.2 ± 0.2 for D-PIP, and 1.7 ± 0.1 for D-PIP2. In contrast, if the cells were ≥ 90% confluent, these ratios fell to 0.4 ± 0.1, 0.3 ± 0.1, and 0.4 ± 0.1, respectively (Fig. 4). The ratios of 36:2 to 36:1 and of 38:4 to 38.1, 38.2 and 38.3 (Fig. 4) fell as well. Thus, the predominance of arachidonyl and polyunsaturated side chains in phosphoinositides was lost as cells reached confluency. These changes were especially profound for PI, the parent phosphoinositide that is about 50 times more abundant than PIP and PIP2 (Table 1).
Fig. 3.
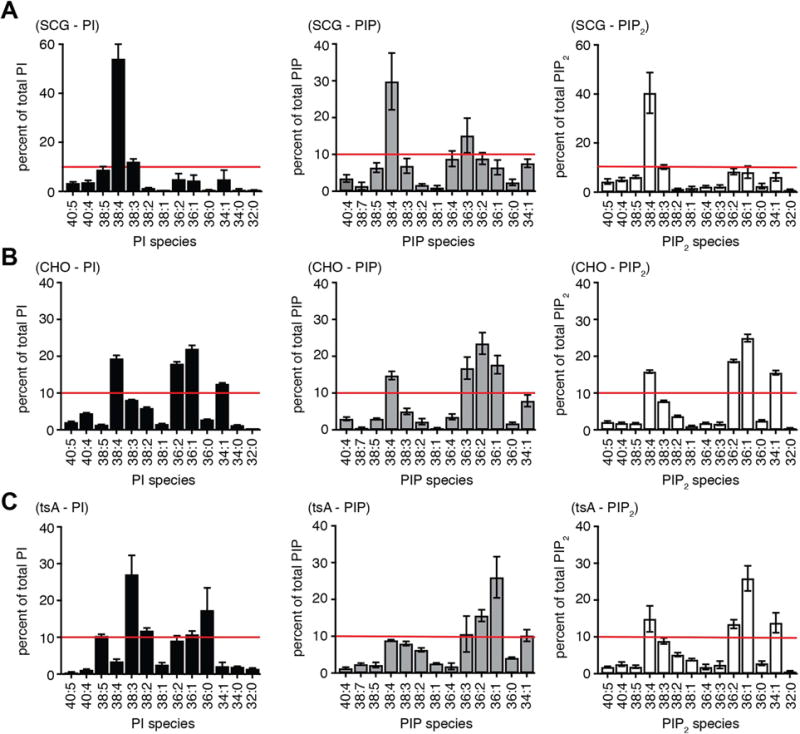
Fatty acid compositions of phosphoinositides in SCG neurons compared with CHO and tsA201 cell lines. Experimental protocol and display are as for Fig. 2, but the results are shown on linear axes here. Red lines are at 10% abundance. (A) Superior cervical ganglia: data from four independent extractions, two ganglia per extraction. (B) CHO-M1 cells: ~106 cells harvested at 85–95% confluency (n = 3). (C) tsA-201 cells: ~106 cells harvested at 85–95% confluency (n = 7)
Fig. 4.
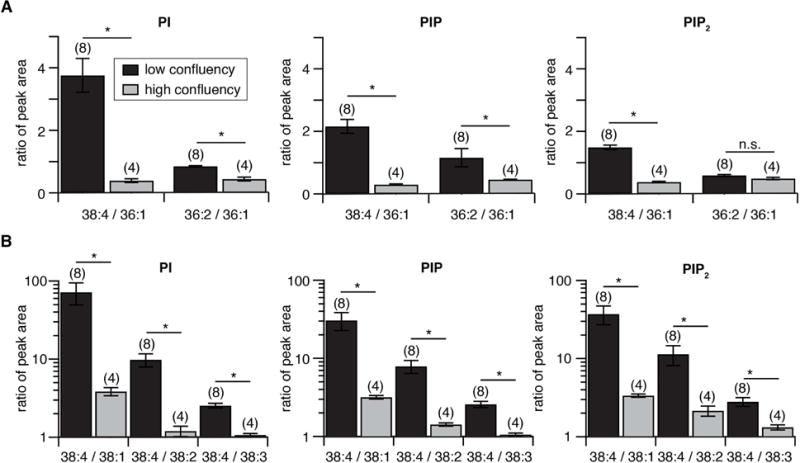
Fatty-acid unsaturation of phosphoinositides decreases as cultured cells become confluent. Phosphatidylinositides were extracted from tsA201-cells harvested at 40–70% confluency (“low confluency,” black) or >90% confluency (“high confluency,” gray) and analyzed in MRM mode for different fatty-acid compositions. (A) Bar graphs show ratio of peak areas (more-saturated:less-saturated). for 38:4 versus 36:1 and 36:2 versus 36:1 in PI, PIP, and PIP2. (B) Similar comparisons of the ratios of 38:4 versus 38:1, 38:2, and 38:3. Numbers in brackets indicate number of experiments. *: p <0.05 (t-test).
3.4. Quantitation of phosphoinositides
We now illustrate lipid mass calculations. Samples to be analyzed were routinely spiked with 10 ng of two analytical internal standards (PIP 37:4; PIP2 37:4) and sometimes also PI 37:4, prior to extraction and derivatization. Then, 2% of the final sample was typically injected over the LC/MS equating to 200 pg or approximately 200 fmoles of internal standard per run. For each extraction, we calculated the quantity of 38:4 PIP2 in a million cells as follows: 10 ng [the amount of lipid standard] × (area of peak for 38:4 D-PIP2)/{(# of millions of cells) × (area of peak for internal 37:4 D-PIP2 standard)}. For the pineal, SCG, and tsA201 cells, the results were 78 ± 40, 49 ± 14, and 10 ± 3 ng/106 cells, respectively. Here we chose the one lipid species (38:4) that most closely resembles the internal standard. If we made the assumption that PIP2 species with chain lengths different from 38:4 are recorded with a similar efficiency (see e.g., [5, 28], we could estimate the aggregate pool size for all PIP2 species using their amplitudes relative to 38:4 in graphs like Fig. 2 and 3. For pineal, SCG, and tsA201 cells as before, the results were 119, 93, and 64 ng total PIP2/106 cells. Note that 38:4 is not the dominant fatty acid combination for tsA201 cells, so the conversion from 38:4 to total for this cell type involved the largest correction. Repeating the same kind of calculations but for total PIP using the 37:4 PIP internal standard and the same assumptions gave roughly 1.2- to 3-fold higher numbers: 260, 304, and 76 ng total PIP/106 cells. Such calculations have to be regarded as approximate considering that there are numerous assumptions and noting that for the two primary tissues they average over several cell types and sizes. Now we can estimate the PIP2 density in pinealocytes, which are the fairly uniform dominant cell type of the pineal gland and whose average plasma membrane surface area (2,000 μm2) we know from their 20 pF membrane capacitance [29]. There is approximately 114 fmole of PIP2 per cell, which is 69 × 106 molecules per cell making an estimated density of 34,000 PIP2/μm2 of plasma membrane in pinealocytes.
3.5. Receptor-induced changes in phosphoinositides
Stimulation of Gq-coupled G protein coupled receptors (GqPCRs), initiates phosphoinositide breakdown by phospholipase Cβ (PLCβ). To quantitate the resulting phosphoinositide depletion by mass spectrometry, we activated stably expressed muscarinic type-1 receptors (M1R) in CHO-M1 cells with a saturating concentration of the agonist oxotremorine (Oxo-M, 10 μM). Stimulating this GqPCR initiates cleavage of plasma membrane PI(4,5)P2 into the canonical second messengers inositol (1,4,5)-trisphosphate (IP3) and diacylglycerol (DAG) accompanied by background metabolic restoration of PI(4,5)P2 from the PI(4)P and PI pools (Fig. 5A). Responses from different dishes of cells were normalized by the spiked-in analytical standards and by the measured genomic DNA. We wanted to determine the extent of depletion of PIP and PIP2 and the profile of lipid species during agonist applications for times up to 60 s. The relative abundance of the phosphoinositide types (D-PI, D-PIP, and D-PIP2) and their fatty acid compositions in resting, 85–95% confluent CHO cells, are shown in (Table 1 and Fig. 3B). Activation of the muscarinic receptor had little effect on the size of the total cellular PI pool, whereas the total PIP and PIP2 pools declined markedly in parallel for each time point (Fig. 5B). Overall, the PIP and PIP2 pools were reduced by ~80% following 60 s of Oxo-M, reflecting net receptor-induced depletion of PI(4,5)P2 by PLCβ. The parallel decline of PIP most likely reflects ongoing re-equilibration with the declining plasma membrane PIP2 pool.
Fig. 5.
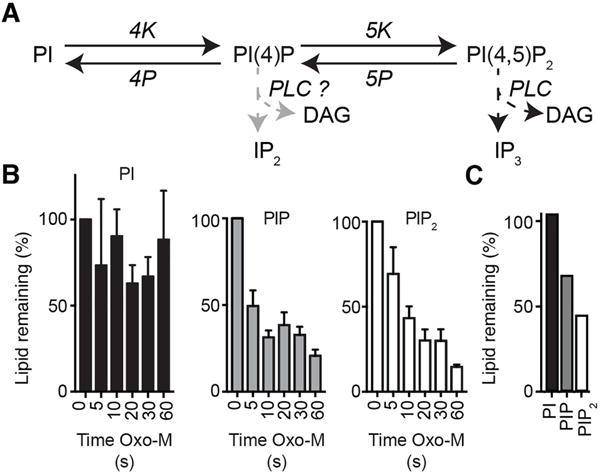
UPLC mass spectrometry detects cellular depletion in total PIP and PIP2 pools following activation of phospholipase C (PLC) in CHO-M1 cells. (A) Schematic of cellular phosphoinositide metabolism and the actions of PLC. (B) Summary histograms of the total PI, PIP, and PIP2 signals in control (0 s) and following 5 – 60 s oxotremorine-M (Oxo-M, 10 μM) to activate a stably expressed M1R receptor in the CHO cells (n = 5). Bars are sums of all fatty-acid species in their sodiated and protonated forms. (C) Changes in phosphoinositide lipids of rat pineal glands after stimulation by norepinephrine showing percent remaining in PI, PIP, and PIP2 pools after a 60 min incubation with 1 μM norepinephrine at 37°C compared to untreated control.
Pinealocytes, the dominant cell type of the pineal gland, normally secrete the night hormone melatonin continuously in response to continuous norepinephrine signaling in parallel through α1 and β1 adrenergic receptors throughout the night [30]. The α1 receptors are GqPCRs that cleave PI(4,5)P2 by activating PLCβ. To see whether a net phosphoinositide depletion occurs in long-term stimulation of whole pineal glands, we measured the lipid content of pineal glands and the changes with adrenergic stimulation. We have already seen the ratio of total phosphoinositides (D-PI:D-PIP:D-PIP2) and the fatty-acyl chain composition of the three lipid classes for unstimulated pineal glands in Table 1 and Fig. 3. Similar to what we saw with muscarinic action on the CHO cell line, when pineal glands were incubated in 1 μM norepinephrine, this time for a whole hour, their PIP2 and PIP pools were partially depleted (not as extensively), and PI was unchanged (Fig. 5C). Thus noradrenergic stimuli deplete membrane PIP2 significantly in the (whole) pineal gland.
These experiments also suggest a test for a possible preference of PLCβ for lipid substrates with specific fatty-acyl chains. One could imagine that helping to keep 38:4 phosphoinositides most abundant in primary cells, PLCβ might have a preference to deplete non-38:4 lipids. However, we did not find much evidence for such selectivity. As Fig. 2C showed, the 38:4 species is present in 66 ± 8% (n = 3) of the total pineal gland PIP2. One hour of NE reduces the mass of PIP2 to 0.4 relative to control (Fig. 5C); nevertheless we found that the percentage of 38:4 lipid is still 63% of total PIP2. This test can be criticized as allowing too much time (1 h) for compensatory re-equilibration with the larger PI pool, so we looked also at the 60-s kinetic experiments with CHO cells harvested at 85–95% confluency. Their resting fatty-acyl profiles were shown in Fig. 3B. We asked whether during Oxo-M treatment 38:4 PIP2 declined more slowly than 34:1, 36:0, 36:1, 36:2, 38:1, or 38:3 (Fig. 6). However, the ratio of 38:4 to these other PIP2 species is not markedly changed during the kinetic experiment with the exception that for two of these comparisons, there might have been a transient drop for the first 5-s point only, and for most, there might have been a very slow upward drift. Overall, these experiments provide little evidence that 38:4 PIP2 is spared or depleted significantly by PLCβ over the other PIP2 species. Any effect is small.
Fig. 6.
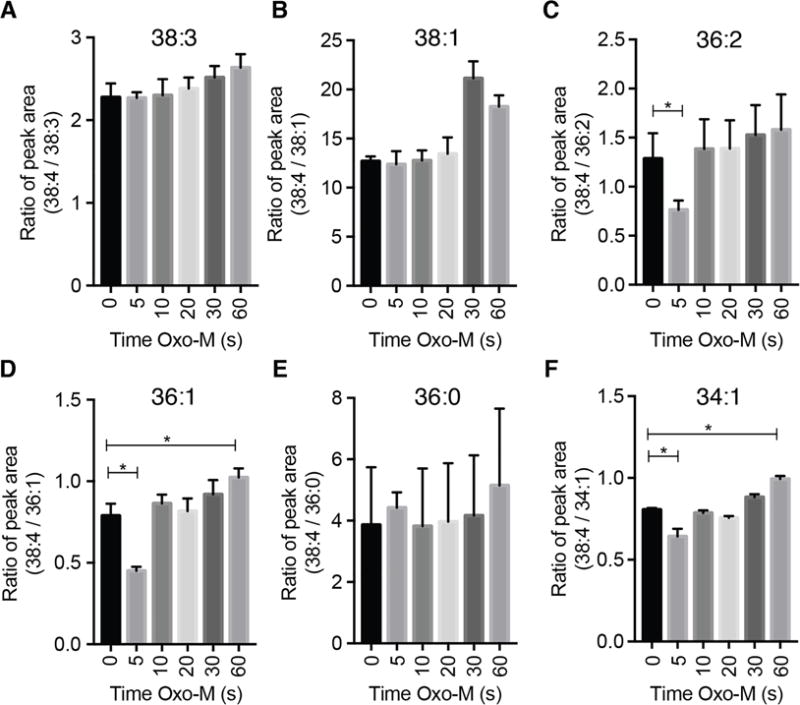
Relative constancy of fatty-acyl chain distribution in PIP2 as PIP2 is being depleted by Oxo-M-activated PLC. Lipids are analyzed in CHO-M1 cells at time points during activation of M1 muscarinic receptors by 10 μM Oxo-M (n = 5). Plotted are the ratios of 38:4 lipids to the indicated species. Same experiment as in Fig. 5B.
To study dynamics of cellular free phosphoinositides, cell biologists often monitor the localization of overexpressed fluorescent biosensors that bind to specific phospholipids. Analogously, electrophysiologists can monitor the ion current amplitude in PI(4,5)P2-sensitive ion channels such as KCNQ. These indirect proxies for free phosphoinositide are excellent for kinetics at the single-cell level although they provide neither absolute nor relative measures of different lipids in the cell. We wanted to compare the responses of the mass spectrometry method to both the optical and the electrical real-time methods. We start with the commonly used PI(4,5)P2 biosensor, the fluorescently labeled pleckstrin homology domain of PLCδ1 (PHPLCδ1) [31, 32]. In resting cells, this biosensor accumulates preferentially at the PM where PI(4,5)P2 is localized (Fig. 7A, left image). Here, activation of M1R with Oxo-M increased the amount of cytoplasmic biosensor (Fig. 7A, middle image) reflecting the hydrolysis and net depletion of plasma membrane PI(4,5)P2, which releases bound fluorescent PHPLCδ1 to the cytoplasm. After removal of agonist, the probe slowly returns to the membrane, reflecting net resynthesis of PI(4,5)P2 from PI(4)P and PI. Changes in fluorescence intensity in a cytoplasmic region of interest are often followed to monitor changes of PI(4,5)P2. The time course of PHPLCδ1 translocation is shown in Fig. 7B during a 60 s exposure to agonist. By 20 s, PI(4,5)P2 depletion has reached a steady state, and over a much longer time it recovers (Fig. 7A, right image). Rather than use this confocal translocation method, our lab often uses photometry to measure loss of Förster resonance energy transfer (FRET) between pairs of probes [33] or patch clamp to measure the decline of ion current in PI(4,5)P2-dependent KCNQ ion channels (e.g. [34]. One potential advantage is that both of these signals come directly from the PM. Fig. 7C compares the time courses reported by these PM-directed methods, PH-probe FRET in CHO-M1 cells (this paper) and KCNQ channels expressed in tsA201 cells [34] with the cytoplasmic fluorescence increase by the confocal translocation method (CHO-M1 cells) from Fig. 7B and the total D-PIP2 decline (CHO-M1 cells) measured by our MS method. The mass spectrometry data are in close agreement with the optical and electrophysiological methods. The published electrophysiology [34] showed that transfection with PHPLCδ1 slightly slows the rate of decline of KCNQ currents during Oxo-M treatment (Fig. 7C), and our new mass spectrometry reveals that transfection with the PH probe increases the total masses of PIP2 and PIP in the cell by ~30% (Fig. 7D; see Discussion). This demonstrated concordance of the total PIP2 mass with the specific PM PIP(4,5)P2 signals agrees with the conventional notion that most of the PIP2 in the cell is plasma membrane PI(4,5)P2.
Fig. 7.
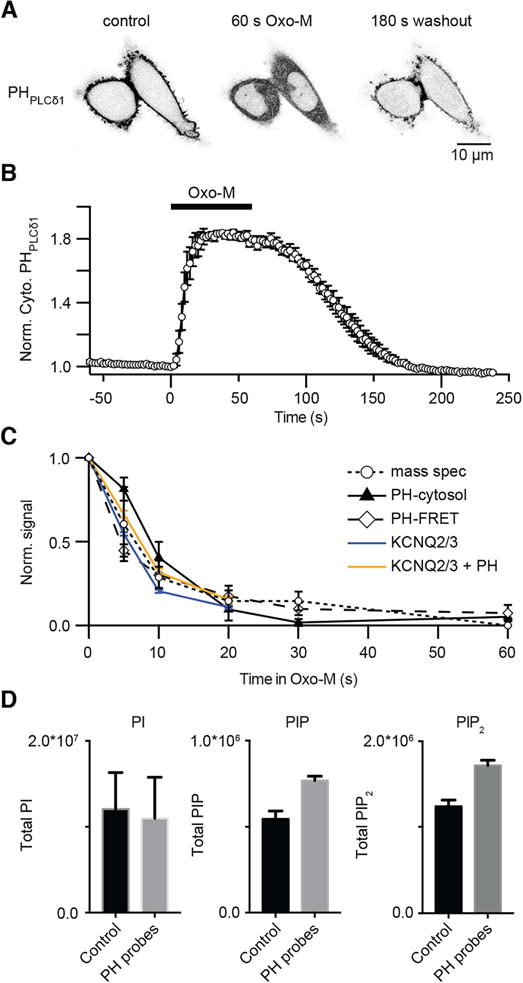
Comparison of four methods for monitoring kinetics of PIP2 depletion in cultured cells. (A) Inverted confocal micrographs from two CHO-M1 cells expressing the fluorescent YFP-PH PLCδ1 biosensor (dark in this negative image) before (Control), after 60-s in Oxo-M (10 μM), and after 180 s of washout. (B) Normalized time courses of YFP-PH PLCδ1 fluorescence from a region of interest in the cytoplasm, with application of Oxo-M (10 μM), a confocal experiment as in (A) (mean ± SEM, n = 7). Up in the graph indicates PIP2 depletion at the PM. (C) Normalized time courses of PIP2 depletion following 60 s application of Oxo-M (10 μM) as measured by three methods in CHO-M1 cells: mass spectrometry (open circles, n = 5), cytosolic translocation of YFP-PH PLCδ1 (filled triangles from (B)), and FRET measurements of RFP-PH PLCδ1 probes (open diamonds, n = 5). These data are compared with the time course of KCNQ current amplitude in receptor-transfected tsA201 cells (orange and blue lines) (published KCNQ data from Jensen et al, 2009). For KCNQ currents, one group of cells was cotransfected with RFP-PHPLCδ1 (orange line) and another was not (blue line). (D) Summary comparison of total resting PI, PIP, and PIP2 measured by mass spectrometry in CHO-M1 cells untransfected (black) or transfected with RFP-PH PLCδ1 probes (gray) (n = 3).
4. Discussion
Through technical refinements we have achieved greater sensitivity in the analysis of phosphoinositide lipids from living cells. We described the distributions of summed fatty-acyl chains in the phosphoinositides of several cell types, finding that they can be different for different cells and for different growth states. We calculated the absolute number of phosphoinositide molecules per cell. We compared the rapid real-time kinetics of receptor-induced depletion of phosphoinositides as reported by mass spectrometry with those reported by PH-domain optical indicators and by a PI(4,5)P2-dependent ion channel. We found no systematic preference of the PLCβ enzyme for substrates with different fatty-acyl chains. Each of these points is now discussed.
4.1. Measurement technique
The lipid analysis requires preparation of the lipid sample, chromatography, and mass spectrometry. For lipid preparation, we reverted to butanol extraction of an acidified sample [23–27]. Then we adapted the protocol of Clark and coworkers [7] who used TMS-DM to methylate phosphoinositide phosphate groups and improve volatility. We took the lipid extract to dryness prior to addition of 20 μl of TMS-DM and used 60 min incubation, in contrast to the Clark procedure where 50 μl TMS-DM is added to the final organic extract solution and incubated for 10 min prior to quenching. Subsequently, and key to our method was to promote the formation of sodiated adducts. Hsu and coworkers had added LiOH to the mixture for infusion into the ESI source of a Finnigan TSQ-7000 triple stage quadrupole spectrometer to generate lithiated adducts of phosphatidylcholine and later extended this technique to include phosphatidylethanolamines [35, 36] and by others to polyphosphoinositides [37]. The post-column infusion option of the Waters Xevo TQ bypassed the need for adding sodium to the mobile phase. This avoids effects on the chromatography and minimizes the sodium included in chromatographic runs so as not to coat the source. Therefore we infused Na formate post column continuously, immediately before the sample entered the MS machine. The sodium adducts of the phosphoinositides varied inversely with the number of phosphates on the inositol ring and the sodiated forms fragmented at collision energies suitable for MRM quantitative analysis. The parent sodiated adducts fragmented with a different fragmentation pattern. The dominant fragments from sodiated derivatized phosphoinositides were the sodiated headgroups rather than the diacylglycerol fragments seen with protonated adducts. Altogether, with these methods, we achieved a detection limit below 20 pg (~20 fmole) injected into the MS machine. We have used these methods in several recent papers [38–40].
In this paper we compared relatively abundant PI with less abundant PIP and PIP2 (Fig. 2), but did not discuss still rarer PIP3. At rest, when no lipid 3-kinase stimulus is applied, the PIP3 abundance would be expected to be only a few percent that of PIP2, near the detection floor of our usual experiments that analyze 2% of the derivatized extract from 106 cells. Included in all our experiments was analysis for 38:4, 36:1, and 36:2 PIP3, but without any PIP3 analytical standard. All those species were in the noise level except for protonated (M+H+) 38:4, for which the putative signals were at the 0.6 to 2% level compared with 38:4 PIP2. We would have to stimulate lipid 3-kinases, increase the number of cells extracted, or increase the injected sample fraction to raise the putative PIP3 signal in resting cells into a reliable range.
While our paper was under review, a new mass spectrometry method for polyphosphoinositides was published [37]. The technique was shotgun lipidomics that did not use liquid chromatography but rather multiple sequential extractions, methyl derivatization, and 5-min continuous infusion into a nanospray triple-quadrupole mass spectrometer running both in positive ion mode and in negative ion mode. The extraction, derivatization, and mass spectrometry were different from ours. They found that lithiated adducts could be used, giving results like ours with sodiated adducts. This nice work offered solutions to two important problems: (i) ability to distinguish PI(3)P from PI(4) and PI(5)P (together) and PIP(3,5)P2, PIP(3,4)P2, and PIP(4,5)P2 from each other (from their different patterns of methylation and hypermethylation), and (ii) ability to identify the individual fatty acid chains (in negative ion mode) rather than the sum of the two fatty acids. A histogram they provide suggests that there is much more PI(4,5)P2 than PI(3,5)P2 or PI(3,4)P2 in mouse brain cortex, and that the 38:4 lipids are most abundant and made up from 18:0 and 20:4 fatty acids both as anticipated. Unlike our results in pineal and SCG, their histogram suggests that 36:1 is the second most abundant lipid combination in polyphosphoinositides of brain cortex. Their extraction technique did not permit comparison with PI species or neutral lipids in the same samples.
4.2. Fatty-acyl chain composition
Consistent with previous work on other mammalian primary cells, we found that 38:4 fatty acids dominated in the phosphoinositides of the rat pineal gland and of rat superior cervical ganglia (SCG). Many other fatty-acyl combinations were also present but at lower abundance (38:4 >> 38:1, 38:2, 38:3 38:5 > 36:1, 36:2, 36:3, 36:4, 40:4, and 40:5). A dominance of 38:4 (>60%) in phosphoinositides is reported for rodent or human brain, heart, intestine, stomach, kidney, liver, pancreas, spleen, lung, skeletal muscle, primary neutrophils, and fibroblasts [5, 7, 17, 19]. This would make phosphoinositides good precursors for arachidonic acid signaling including by endocannabinoids such as 2-arachidonyl glycerol. The dominance of this unique fatty-acyl combination in phosphoinositides contrasts sharply with the situation in pools of phosphatidylserine and phosphatidylcholine (and in some cases, phosphatidylethanolamine) in the same mammalian tissues [17, 19]. In these other classes of phospholipids, no one fatty-acyl combination stands out prominently, and 38:4 is always less abundant than several others. The approximate fatty-acid uniformity of phosphoinositides suggests that much of cellular PI, PIP, PIP2, and PIP3 acts as a single rapidly interconverting metabolic pool. Three interesting hypotheses, each with partial support, have been discussed for the “purity” of the fatty acid chains unique to phosphoinositides [19]: (i) There is strong selection favoring 38:4 diacylglycerol when PI is being made at the level of diacylglycerol kinase, cytidine diphosphate-diacylglycerol synthase, or PI synthase. (ii) There is ongoing “correction” or remodeling of existing fatty acid content of phosphoinositides by phospholipases followed by acyl-transfer reactions such as lysophosphatidylinositol-acyltransferase. (iii) There are small substrate preferences at the level of one or more lipid kinases and lipid phosphatases that after hundreds of cycles of inositol phosphorylation and dephosphorylation cumulatively result in a selective increase of abundance of 38:4. In this paper we looked for evidence that PLCβ has a substrate preference for or against 38:4 lipids. Had there been a bias, the bars (ratios) of Fig. 6 should have shown a progressive increase or decrease with time. If there is any trend it might be towards a slow increase of the residual 38:4, but any net effect is too subtle to be statistically reliable or perhaps biologically meaningful.
There are several circumstances where 38:4 lipids are not the majority species in phosphoinositides. One clear variation is across taxonomy. Fly-head phosphoinositides are dominated by 36:2 and 36:3 [14]; yeast has 32:1 > 36:1 [5], and Dictyostelium has ether-linked 34:1 lipids (plasmanyl inositides, [21]. In 6 tissues of a marine fish, the PI contains 40 ± 5% of 38:4 lipids, and in chicken eggs, 49% [41, 42], so possibly all vertebrates use 38:4, but the taxonomic range for 38:4 dominance in phosphoinositides seems not yet well delimited. Surprisingly, transformed cell lines in culture, even though they are mammalian, have diminished 38:4 in their phosphoinositides. We showed this here for nearly confluent CHO and tsA201 cells (both having 36:1 and 36:2) [38], and a similar “arachidonic acid deficiency” has been reported before for RAW264.7 cells, MCF10a breast cancer cells, and HeLa cells [7, 16, 18]. Then we found in rapidly growing, 40% confluent tsA201 cells, that 38:4 is in fact dominant in the phosphoinositides, as it is in mammalian primary cells, and that the shift to shorter chain and less unsaturated fatty acids develops subsequently. Irrespective of the underlying mechanisms, this points to a need to control for cell density when measuring phosphoinositides in lipid extracts from cells grown in tissue culture or when studying phospholipid-dependent physiological functions. Since cell density impacts a number of cell functions, a possible connection to membrane lipids deserves further study. In our culture conditions, we are talking about a time scale of only a few days so the fatty acid composition can change within 24 hours. Cells that are nearly confluent (low 38:4) are replated at ~10% density. They grow to 40% confluence (high 38:4) in 2 days and to near confluence (low 38:4) in one more day. Since fresh medium is supplied only at the time of replating, the cultures might run low on some metabolites or lipid precursors that favor long fatty acid chains and unsaturation. Alternatively there may be signals related to contact inhibition, growth, or “aging” that change lipid metabolism. Such losses of the more highly unsaturated fatty-acyl chains could result in more rigid membranes in confluent cells and possibly could be linked to the reduction in growth rate. The dynamic nature of the lipid pools is evidenced in other kinds of experiments. We have shown that the total mass of PIP and PIP2 can more than double in tsA201 cells exposed overnight to a culture medium containing elevated levels of myo-inositol and transfected with the Na+-coupled myo-inositol transporter SMIT1 [40]. Nasuhoglu et al. [43] have reported that PIP and PIP2 can increase in 3 min upon exposure of cells to hypertonicity. Both in RAW264.7 cells and in flies, adding polyunsaturated fatty acids to the diet will increase the prevalence of unsaturated acyl chains in glycerophospholipids [14, 18]. The relative amounts of different phosphoinositide classes are changed in cells expressing mutant or transfected lipid phosphatases [5, 38]. Finally rapid changes of phosphoinositide signaling, inositol phosphate abundance, and membrane viscosity occur during the cell cycle perhaps through an increase of cells in G0/G1, and conversely, the cell cycle may be altered by phosphoinositides [44–49]. All these results emphasize that not only can the phosphorylation state of the inositol headgroup change in seconds during activity, but also the fatty-acid side chains can change at least within hours.
4.3. Quantitation
Through use of internal standards and normalization by DNA content, it was possible to make our phosphoinositide measurements quantitative. In one already published study with this method [40], the ability to normalize to DNA gave us confidence to state that treatment with myo-inositol plus transfection with SMIT1 more than doubled the mass of PIP and PIP2 per cell. In the present paper, we have calculated for pineal, SCG, and tsA201 cells, 119, 93, and 64 ng total PIP2/106 cells and 260, 304, and 76 ng total PIP/106 cells. These numbers compare well with estimates of 130 ng of PIP and 110 ng of PIP2 per 106 cells in Balb/c/3T3 cells by monoclonal antibody assay of lipid extracts [50]. Averaging, they correspond to 110 and 55 × 106 molecules of PIP and PIP2 per cell, respectively. For pinealocytes we were able to translate this into a surface density of 34,000 PIP2/μm2 of plasma membrane. This compares reasonably with PIP2 densities discussed in the literature [1]: Hilgemann [51] estimates >20,000/μm2, we have assumed 15,000/μm2 in previous modeling [52], and Xu et al. [53] assumed 4,000/μm2. For determining the mole fraction of PIP2 molecules, we can estimate the total number of phospholipids that would fit on 1 μm2 of one leaflet of a lipid bilayer. Assuming a standard area per phospholipid head group of 0.68 nm2 (68 Å2), one leaflet of a bilayer can accommodate 1.47 × 106 phospholipids/μm2 (The area is for dioleyoylphosphatidylcholine bilayers [54].). If only 60% of the inner leaflet area were available for lipids because of the presence of membrane proteins, for a density of 34,000 PIP2/μm2, the PIP2 mole fraction would be very approximately 0.04 in the cytoplasmic leaflet of the plasma membrane. About 33% of them would be free and in continuous rapid equilibrium with a larger pool that is bound to various PM molecules [52, 55].
Now we briefly consider quantitative aspects of expressing of PHPLCδ1 probes. In earlier quantitative work with tsA201 cells, we estimated that in a typical transfection, cells would express the equivalent of 6,400 PH-probe molecules/μm2 of plasma membrane and that about 50% of them would be bound to the PM in complex with PI(4,5)P2. Thus a fraction of the cellular PI(4,5)P2 would become buffered by PH domains. Since the rates of enzymes of phosphoinositide metabolism are governed by the free phosphoinositide levels, the expectation would be that they will restore the normal setpoint of free lipid even when an additional portion of the PI(4,5)P2 is bound to PH probes. Thus methods like mass spectrometry measuring total PIP2 would report elevated total amounts as in Fig 7D, and methods measuring the rate of depletion by PLC like the KCNQ experiments of Fig. 7C would show kinetic slowing of depletion. In qualitative agreement, both methods suggest a 30% augmentation of total PIP2, and they are consistent with the expectation that a modest portion of the available PI(4,5)P2 becomes bound to PHPLCδ1. The mass spectrometry experiments show unexpectedly that the total PIP also rises with PH-probe expression. That would suggest that there has been some compensatory increase of PI-4-kinase or slowing of PI(4)P-4-phosphatase rates.
4.4. Depletion by receptor activation
In a previous HPLC study we found that 60 s of Oxo-M strongly depleted PIP2 and PIP levels in CHO-M1 cells [22]. As others observed using radioactive labeling in SH-SY5Y cells and HEK cells, strong activation of PLCβ allows net hydrolysis of PI(4,5)P2 as well as loss of PI(4)P faster than they can be resynthesized [56, 57]. The same idea for PI(4,5)P2 at the PM has been documented many times using the fluorescent PHPLCδ1 translocation or FRET reporters [31, 34, 53, 58]. Operationally, using mass spectrometry, we can now assert that 75–80% of all forms of cellular PIP2 and PIP can be hydrolyzed in <20 s when PLCβ is activated at the PM. The deep depletion of PIP2 implies that most PIP2 in the cell is the PI(4,5)P2 regioisomer rather than PI(3,4)P2 or PI(3,5)P2. The depth and speed of the decline of the monophosphate PIP imply that nearly all cellular pools of PIP communicate with the plasma membrane PI(4,5)P2 in <20 s and they are likely comprised mainly of the PI(4)P regioisomer. Thus, even though a significant fraction of cellular PI(4)P is associated with the Golgi, that pool is in remarkably rapid metabolic exchange with PM pools of PI(4,5)P2 (c.f. [59]). We found that the kinetics of PIP2 depletion reported by three methods, mass spectroscopy, PHPLCδ1 translocation or FRET at the PM, and KCNQ ion channels were virtually the same. Some authors suggest that the PHPLCδ1 probe might give artifacts because it binds to IP3 in addition to PI(4,5)P2 ([60]. The Tubby probe has been called more specific and preferable [58, 61]. In our experience with several cell types, these two probes give the same time courses in response to muscarinic receptor stimulation [39]. The agreement of mass spectrometry with the other methods reinforces the ability of mass spectrometry faithfully to follow phosphoinositide changes over time periods as short as 5–10 s. With the small cells we use, the time course of PH-probe translocation is apparently not greatly delayed by a need to diffuse a few micrometers into the cytoplasm, a problem that does become significant in larger cells such as oocytes (c.f. [62]) Similarly, with pineal glands we observed that norepinephrine, the major sympathetic night signal for melatonin production, induces ~50% depletion of PIP2 and PIP. The experiment shows in this tissue both that the endogenous PLCβ can be activated enough to induce significant net PI(4,5)P2 depletion and that the endogenous α1 adrenergic receptors are not fully desensitized by exposure to high norepinephrine concentration for as long as an hour.
4.5. Pros and cons of methods
We end by comparing the advantages of the mass spectrometry method with the advantages of using pleckstrin homology domain indicators or ion channels to measure PI(4,5)P2. Only the MS method can give total absolute lipid pool sizes (mass), and only the optical and electrical signals can relate to subcellular localization. The MS method is best with tens of thousands of cells but can be referred to absolute standards and has the clear advantage of a broader dynamic range and linearity starting from a defined zero. The optical signal, by contrast, starts from a significant non-zero level and varies with the concentration and saturation of the probe, but it is attractive because it offers a continuous real-time record with even single cells, often can distinguish individual regioisomers readily, and it uses much simpler apparatus at much less expense. Optical methods readily distinguish local lipid depletion in part of a single cell and not in others [63], and they can track asynchronous oscillations in individual cells that would be obscured by averaging responses from thousands of cells. They can monitor onset and recovery nondestructively from a stimulus in the same specimen. The electrical methods are similar to the optical method. The mass spectrometric analysis cannot be beat for more absolute quantitation and is the only method that reports fatty-acyl chain composition.
Supplementary Material
Highlights.
Refined quantitative mass spectrometry of cell phosphoinositides
Resolution of many fatty-acyl chain combinations in phosphoinositides
Dominance of 38:4 lipids in mammalian primary cells but not in cultured cells
Change of fatty-acyl profile with culture conditions
Fast kinetics (<10 s) of phosphoinositide depletion after receptor activation
Acknowledgments
We would like to thank Marco Hennrich for helpful comments, the School of Pharmacy Mass Spectrometry Center for assistance, and Lea M. Miller for technical help. This work was supported by National Institutes of Health grants R37NS008174 and R01GM-83913 (to BH), and the Wayne E. Crill Endowed Professorship.
Abbreviations
- DAG
diacylglycerol
- D-PI, D-PIP, D-PIP2
derivatized PI, PIP, and PIP2
- IP3
inositoltrisphosphate
- MRM
multiple reaction monitoring
- PI
phosphatidylinositol
- PIP
phosphatidylinositol phosphate
- PIP2
phosphatidylinositol bisphosphate
- PLC
phospholipase C
- SCG
superior cervical ganglion
- TQ
triple quadrupole
Appendix A. Supplementary data
Supplementary figures and tables to this article can be found online at http://dx.doi.org/…………….
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. 2013;93:1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shewan A, Eastburn DJ, Mostov K. Phosphoinositides in cell architecture. Cold Spring Harb Perspect Biol. 2011;3:a004796. doi: 10.1101/cshperspect.a004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falkenburger BH, Jensen JB, Dickson EJ, Suh BC, Hille B. Phosphoinositides: lipid regulators of membrane proteins. The Journal of physiology. 2010;588:3179–3185. doi: 10.1113/jphysiol.2010.192153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hille B, Dickson EJ, Kruse M, Vivas O, Suh BC. Phosphoinositides regulate ion channels. Biochim Biophys Acta. 2015;1851:844–856. doi: 10.1016/j.bbalip.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wenk MR, Lucast L, Di Paolo G, Romanelli AJ, Suchy SF, Nussbaum RL, Cline GW, Shulman GI, McMurray W, De Camilli P. Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat Biotechnol. 2003;21:813–817. doi: 10.1038/nbt837. [DOI] [PubMed] [Google Scholar]
- 6.Haag M, Schmidt A, Sachsenheimer T, Brugger B. Quantification of Signaling Lipids by Nano-Electrospray Ionization Tandem Mass Spectrometry (Nano-ESI MS/MS) Metabolites. 2012;2:57–76. doi: 10.3390/metabo2010057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark J, Anderson KE, Juvin V, Smith TS, Karpe F, Wakelam MJ, Stephens LR, Hawkins PT. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat Methods. 2011;8:267–272. doi: 10.1038/nmeth.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Traynor-Kaplan AE, Thompson BL, Harris AL, Taylor P, Omann GM, Sklar LA. Transient increase in phosphatidylinositol 3,4-bisphosphate and phosphatidylinositol trisphosphate during activation of human neutrophils. J Biol Chem. 1989;264:15668–15673. [PubMed] [Google Scholar]
- 9.Stephens LR, Hughes KT, Irvine RF. Pathway of phosphatidylinositol(3,4,5)-trisphosphate synthesis in activated neutrophils. Nature. 1991;351:33–39. doi: 10.1038/351033a0. [DOI] [PubMed] [Google Scholar]
- 10.Rusten TE, Stenmark H. Analyzing phosphoinositides and their interacting proteins. Nat Methods. 2006;3:251–258. doi: 10.1038/nmeth867. [DOI] [PubMed] [Google Scholar]
- 11.Guillou H, Stephens LR, Hawkins PT. Quantitative measurement of phosphatidylinositol 3,4,5-trisphosphate. Methods Enzymol. 2007;434:117–130. doi: 10.1016/S0076-6879(07)34007-X. [DOI] [PubMed] [Google Scholar]
- 12.Sauer K, Huang YH, Lin H, Sandberg M, Mayr GW. Phosphoinositide and inositol phosphate analysis in lymphocyte activation. Curr Protoc Immunol. 2009 doi: 10.1002/0471142735.im1101s87. Chapter 11. Unit11 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones DR, Ramirez IB, Lowe M, Divecha N. Measurement of phosphoinositides in the zebrafish Danio rerio. Nat Protoc. 2013;8:1058–1072. doi: 10.1038/nprot.2013.040. [DOI] [PubMed] [Google Scholar]
- 14.Randall AS, Liu CH, Chu B, Zhang Q, Dongre SA, Juusola M, Franze K, Wakelam MJ, Hardie RC. Speed and sensitivity of phototransduction in Drosophila depend on degree of saturation of membrane phospholipids. J Neurosci. 2015;35:2731–2746. doi: 10.1523/JNEUROSCI.1150-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson KE, Kielkowska A, Durrant TN, Juvin V, Clark J, Stephens LR, Hawkins PT. Lysophosphatidylinositol-acyltransferase-1 (LPIAT1) is required to maintain physiological levels of PtdIns and PtdInsP2 in the mouse. PLoS One. 2013;8:e58425. doi: 10.1371/journal.pone.0058425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung J, Nakatsu F, Baskin JM, De Camilli P. Plasticity of PI4KIIIα interactions at the plasma membrane. EMBO Rep. 2015;16:312–320. doi: 10.15252/embr.201439151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hicks AM, DeLong CJ, Thomas MJ, Samuel M, Cui Z. Unique molecular signatures of glycerophospholipid species in different rat tissues analyzed by tandem mass spectrometry. Biochim Biophys Acta. 2006;1761:1022–1029. doi: 10.1016/j.bbalip.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Rouzer CA, Ivanova PT, Byrne MO, Milne SB, Marnett LJ, Brown HA. Lipid profiling reveals arachidonate deficiency in RAW264.7 cells: Structural and functional implications. Biochemistry. 2006;45:14795–14808. doi: 10.1021/bi061723j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Souza K, Epand RM. Enrichment of phosphatidylinositols with specific acyl chains. Biochim Biophys Acta. 2014;1838:1501–1508. doi: 10.1016/j.bbamem.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Doignon F, Laquel P, Testet E, Tuphile K, Fouillen L, Bessoule JJ. Requirement of Phosphoinositides Containing Stearic Acid To Control Cell Polarity. Mol Cell Biol. 2016;36:765–780. doi: 10.1128/MCB.00843-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark J, Kay RR, Kielkowska A, Niewczas I, Fets L, Oxley D, Stephens LR, Hawkins PT. Dictyostelium uses ether-linked inositol phospholipids for intracellular signalling. EMBO J. 2014;33:2188–2200. doi: 10.15252/embj.201488677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horowitz LF, Hirdes W, Suh BC, Hilgemann DW, Mackie K, Hille B. Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol. 2005;126:243–262. doi: 10.1085/jgp.200509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bjerve KS, Daae LN, Bremer J. The selective loss of lysophospholipids in some commonly used lipid-extraction procedures. Anal Biochem. 1974;58:238–245. doi: 10.1016/0003-2697(74)90463-1. [DOI] [PubMed] [Google Scholar]
- 24.Bremer J. Carnitine in Intermediary Metabolism. The Biosynthesis of Palmitylcarnitine by Cell Subfractions. J Biol Chem. 1963;238:2774–2779. [PubMed] [Google Scholar]
- 25.Shaikh NA. Assessment of various techniques for the quantitative extraction of lysophospholipids from myocardial tissues. Anal Biochem. 1994;216:313–321. doi: 10.1006/abio.1994.1047. [DOI] [PubMed] [Google Scholar]
- 26.Ivanova PT, Cerda BA, Horn DM, Cohen JS, McLafferty FW, Brown HA. Electrospray ionization mass spectrometry analysis of changes in phospholipids in RBL-2H3 mastocytoma cells during degranulation. Proc Natl Acad Sci U S A. 2001;98:7152–7157. doi: 10.1073/pnas.131195098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meade CJ, Turner GA, Bateman PE. The role of polyphosphoinositides and their breakdown products in A23187-induced release of arachidonic acid from rabbit polymorphonuclear leucocytes. The Biochemical journal. 1986;238:425–436. doi: 10.1042/bj2380425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang M, Wang C, Han X. Selection of internal standards for accurate quantification of complex lipid species in biological extracts by electrospray ionization mass spectrometry-What, how and why? Mass Spectrom Rev. 2016;9999:1–22. doi: 10.1002/mas.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu H, Dickson EJ, Jung SR, Koh DS, Hille B. High membrane permeability for melatonin. J Gen Physiol. 2016;147:63–76. doi: 10.1085/jgp.201511526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simonneaux V, Ribelayga C. Generation of the melatonin endocrine message in mammals: a review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol Rev. 2003;55:325–395. doi: 10.1124/pr.55.2.2. [DOI] [PubMed] [Google Scholar]
- 31.Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- 33.van der Wal J, Habets R, Varnai P, Balla T, Jalink K. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J Biol Chem. 2001;276:15337–15344. doi: 10.1074/jbc.M007194200. [DOI] [PubMed] [Google Scholar]
- 34.Jensen JB, Lyssand JS, Hague C, Hille B. Fluorescence changes reveal kinetic steps of muscarinic receptor-mediated modulation of phosphoinositides and Kv7.2/7.3 K+ channels. J Gen Physiol. 2009;133:347–359. doi: 10.1085/jgp.200810075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu FF, Bohrer A, Turk J. Formation of lithiated adducts of glycerophosphocholine lipids facilitates their identification by electrospray ionization tandem mass spectrometry. J Am Soc Mass Spectrom. 1998;9:516–526. doi: 10.1016/S1044-0305(98)00012-9. [DOI] [PubMed] [Google Scholar]
- 36.Hsu FF, Turk J. Characterization of phosphatidylethanolamine as a lithiated adduct by triple quadrupole tandem mass spectrometry with electrospray ionization. Journal of mass spectrometry: JMS. 2000;35:595–606. doi: 10.1002/(SICI)1096-9888(200005)35:5<595::AID-JMS965>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 37.Wang C, Palavicini J, Wang M, Chen L, Yang K, Crawford P, Han X. Comprehensive and Quantitative Analysis of Polyphosphoinositide Species by Shotgun Lipidomics Revealed Their Alterations in db/db Mouse Brain. Anal Chem. 2016;88:12317–12144. doi: 10.1021/acs.analchem.6b02947. [DOI] [PubMed] [Google Scholar]
- 38.Dickson EJ, Jensen JB, Vivas O, Kruse M, Traynor-Kaplan AE, Hille B. Dynamic formation of ER-PM junctions presents a lipid phosphatase to regulate phosphoinositides. J Cell Biol. 2016;213:33–48. doi: 10.1083/jcb.201508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kruse M, Vivas O, Traynor-Kaplan A, Hille B. Dynamics of Phosphoinositide-Dependent Signaling in Sympathetic Neurons. J Neurosci. 2016;36:1386–1400. doi: 10.1523/JNEUROSCI.3535-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai G, Yu H, Kruse M, Traynor-Kaplan A, Hille B. Osmoregulatory inositol transporter SMIT1 modulates electrical activity by adjusting PI(4,5)P2 levels. Proc Natl Acad Sci U S A, ( 2016 doi: 10.1073/pnas.1606348113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka T, Iwawaki D, Sakamoto M, Takai Y, Morishige J, Murakami K, Satouchi K. Mechanisms of accumulation of arachidonate in phosphatidylinositol in yellowtail. A comparative study of acylation systems of phospholipids in rat and the fish species Seriola quinqueradiata. Eur J Biochem. 2003;270:1466–1473. doi: 10.1046/j.1432-1033.2003.03512.x. [DOI] [PubMed] [Google Scholar]
- 42.Pacetti D, Boselli E, Hulan HW, Frega NG. High performance liquid chromatography-tandem mass spectrometry of phospholipid molecular species in eggs from hens fed diets enriched in seal blubber oil. J Chromatogr A. 2005;1097:66–73. doi: 10.1016/j.chroma.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 43.Nasuhoglu C, Feng S, Mao J, Yamamoto M, Yin HL, Earnest S, Barylko B, Albanesi JP, Hilgemann DW. Nonradioactive analysis of phosphatidylinositides and other anionic phospholipids by anion-exchange high-performance liquid chromatography with suppressed conductivity detection. Anal Biochem. 2002;301:243–254. doi: 10.1006/abio.2001.5489. [DOI] [PubMed] [Google Scholar]
- 44.de Laat SW, van der Saag PT, Shinitzky M. Microviscosity modulation during the cell cycle of neuroblastoma cells. Proc Natl Acad Sci U S A. 1977;74:4458–4461. doi: 10.1073/pnas.74.10.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han JK, Fukami K, Nuccitelli R. Reducing inositol lipid hydrolysis, Ins(1,4,5)P3 receptor availability, or Ca2+ gradients lengthens the duration of the cell cycle in Xenopus laevis blastomeres. J Cell Biol. 1992;116:147–156. doi: 10.1083/jcb.116.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dobos GJ, Wu XR, Traynor-Kaplan A. A product of phosphatidylinositol-3 kinase is elevated in dividing HT29 colonic epithelial cells. FEBS Lett. 1993;324:143–146. doi: 10.1016/0014-5793(93)81381-9. [DOI] [PubMed] [Google Scholar]
- 47.Balla T, Sim SS, Baukal AJ, Rhee SG, Catt KJ. Inositol polyphosphates are not increased by overexpression of Ins(1,4,5)P3 3-kinase but show cell-cycle dependent changes in growth factor-stimulated fibroblasts. Mol Biol Cell. 1994;5:17–27. doi: 10.1091/mbc.5.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barker CJ, Wright J, Hughes PJ, Kirk CJ, Michell RH. Complex changes in cellular inositol phosphate complement accompany transit through the cell cycle. The Biochemical journal. 2004;380:465–473. doi: 10.1042/BJ20031872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poli A, Billi AM, Mongiorgi S, Ratti S, McCubrey JA, Suh PG, Cocco L, Ramazzotti G. Nuclear Phosphatidylinositol Signaling: Focus on Phosphatidylinositol Phosphate Kinases and Phospholipases C. J Cell Physiol. 2016;231:1645–1655. doi: 10.1002/jcp.25273. [DOI] [PubMed] [Google Scholar]
- 50.Fukami K, Takenawa T. Quantitative changes in polyphosphoinositides 1,2-diacylglycerol and inositol 1,4,5-trisphosphate by platelet-derived growth factor and prostaglandin F2α. J Biol Chem. 1989;264:14985–14989. [PubMed] [Google Scholar]
- 51.Hilgemann DW. Local PIP2 signals: when, where, and how? Eur J Physiol. 2007;455:55–67. doi: 10.1007/s00424-007-0280-9. [DOI] [PubMed] [Google Scholar]
- 52.Falkenburger BH, Dickson EJ, Hille B. Quantitative properties and receptor reserve of the DAG and PKC branch of Gq-coupled receptor signaling. J Gen Physiol. 2013;141:537–555. doi: 10.1085/jgp.201210887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu C, Watras J, Loew LM. Kinetic analysis of receptor-activated phosphoinositide turnover. J Cell Biol. 2003;161:779–791. doi: 10.1083/jcb.200301070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White SH, King GI. Molecular packing and area compressibility of lipid bilayers. Proc Natl Acad Sci U S A. 1985;82:6532–6536. doi: 10.1073/pnas.82.19.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Golebiewska U, Nyako M, Woturski W, Zaitseva I, McLaughlin S. Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Molecular biology of the cell. 2008;19:1663–1669. doi: 10.1091/mbc.E07-12-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willars GB, Nahorski SR, Challiss RA. Differential regulation of muscarinic acetylcholine receptor-sensitive polyphosphoinositide pools and consequences for signaling in human neuroblastoma cells. J Biol Chem. 1998;273:5037–5046. doi: 10.1074/jbc.273.9.5037. [DOI] [PubMed] [Google Scholar]
- 57.Balla A, Kim YJ, Varnai P, Szentpetery Z, Knight Z, Shokat KM, Balla T. Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIα. Molecular biology of the cell. 2008;19:711–721. doi: 10.1091/mbc.E07-07-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nelson CP, Nahorski SR, Challiss RA. Temporal profiling of changes in phosphatidylinositol 4,5-bisphosphate, inositol 1,4,5-trisphosphate and diacylglycerol allows comprehensive analysis of phospholipase C-initiated signalling in single neurons. J Neurochem. 2008;107:602–615. doi: 10.1111/j.1471-4159.2008.05587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dickson EJ, Jensen JB, Hille B. Golgi and plasma membrane pools of PI(4)P contribute to plasma membrane PI(4,5)P2 and maintenance of KCNQ2/3 ion channel current. Proc Natl Acad Sci U S A. 2014;111:E2281–2290. doi: 10.1073/pnas.1407133111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hirose K, Kadowaki S, Tanabe M, Takeshima H, Iino M. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science. 1999;284:1527–1530. doi: 10.1126/science.284.5419.1527. [DOI] [PubMed] [Google Scholar]
- 61.Hughes S, Marsh SJ, Tinker A, Brown DA. PIP2-dependent inhibition of M-type (KV7.2/7.3) potassium channels: direct on-line assessment of PIP2 depletion by Gq-coupled receptors in single living neurons. Eur J Physiol. 2007;455:115–124. doi: 10.1007/s00424-007-0259-6. [DOI] [PubMed] [Google Scholar]
- 62.Grimm SS, Isacoff EY. Allosteric substrate switching in a voltage-sensing lipid phosphatase. Nat Chem Biol. 2016;12:261–267. doi: 10.1038/nchembio.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Idevall-Hagren O, Dickson EJ, Hille B, Toomre DK, De Camilli P. Optogenetic control of phosphoinositide metabolism. Proc Natl Acad Sci USA. 2012;109:E2316–E2323. doi: 10.1073/pnas.1211305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.