

Abstract
Intermolecular functionalizations of aliphatic C–H bonds offer unique strategies for the synthesis and late-stage derivatization of complex molecules, but the chemical space accessible remains limited. Herein, we report a transformation significantly expanding the chemotypes accessible via C–H functionalization. The C–H xanthylation proceeds in useful chemical yields with the substrate as limiting reagent using blue LEDs and an easily prepared N-xanthylamide. The late-stage functionalizations of complex molecules occur with high levels of site selectivity, and a variety of common functionality is tolerated in the reaction. This approach capitalizes on the versatility of the xanthate functional group via both polar and radical manifolds to unlock a wide array of C–H transformations previously inaccessible in synthesis.
Graphical Abstract
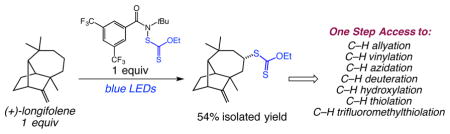
Unactivated carbon-hydrogen (C–H) bond functionalization is a powerful tool for chemical synthesis, enabling an array of new approaches to the construction and late-stage derivatization of complex molecules.1 In drug discovery and development, C–H functionalization offers tools to facilitate the preparation of structural analogs of targets with enhanced structure-activity relationships (SAR) or other desired physicochemical properties without de novo approaches.2 Owing to the plethora of different C–H bonds present in most organic molecules, great effort has been devoted to the site-selective and predictable functionalization of C–H bonds in complex targets. Intramolecular, or substrate-directed C–H functionalizations, are widely used in chemical synthesis, as these reactions benefit from exquisite site selectivity and improved kinetics provided by the functionality present in the substrate. However, this approach is inherently limited by the requisite functionality and lacks generality in applications to diverse structures. The development of site-selective, intermolecular C–H functionalizations of isolated aliphatic C–H bonds is a significantly more daunting challenge, as these processes lack the advantages of intramolecular reactions.
Important recent studies have sought to address this challenge and have provided a number of practical methods for intermolecular C–H functionalizations using substrate as limiting reagent with good efficiency. For example, transformations achieving the oxidation3, azidation4, and halogenation5 of isolated aliphatic C–H bonds have appeared with good site selectivities, functional group compatibilities, and notable application to the derivatization of complex molecules and natural products. However, the limited set of molecular functionality accessible via these strategies restricts the chemical space and subsequent utility of C–H functionalizations. Examples of desirable transformations with no general, practical approach include the vinylation, allylation, thiolation, hydroxylation, trifluoromethylthiolation, and deuteration of isolated aliphatic C–H bonds. The development of strategies capable of achieving these goals would greatly enhance the power of intermolecular C–H functionalization in complex synthesis and late-stage functionalization. We reasoned that the sheer number and diversity of these functional groups would render a single synthetic approach to achieve these transformations impractical. Instead, we envisioned an alternative strategy involving the development of a new intermolecular aliphatic C–H functionalization that could enable facile access to a wide array of functional groups via the established chemistry of a single intermediate.
Previous efforts from our group culminated in the development of site-selective C–H halogenations of unactivated aliphatic C–H bonds using N-haloamides.6 These reactions use a bulky, electron-poor amidyl radical for site-selective C–H halogenations with substrate as limiting reagent, enhancing their utility in complex synthesis. We hypothesized that the combination of the efficiency and site selectivity of our approach with the transfer of a single, highly versatile functional group would generate a new, diversity-oriented approach to C–H functionalization (Figure 1).
Figure 1.

Approaches to site-selective, intermolecular C–H functionalization.
Upon surveying functional groups for transfer to maximize molecular diversity, the xanthate (dithiocarbonate) group appeared ideally suited for this purpose. Zard and others have demonstrated the versatility of alkyl xanthates in radical-mediated synthesis to access an impressive array of valuable functionality.7 The diverse array of aliphatic C–H functionalizations potentially accessible via alkyl xanthates includes many unknown transformations (e.g., C–H to C–S).8 Therefore, we hypothesized that a site-selective aliphatic C–H xanthylation would significantly increase the power of intermolecular C–H functionalization in synthesis and facilitate new late-stage modifications of complex molecules.9
Since the xanthate functionality participates in radical-mediated group transfer processes, we targeted the preparation of N-xanthylamide 1 as an initial goal. We anticipated that a successful aliphatic C–H xanthylation using 1 would display the notable site selectivity and chemoselectivity of our prior C–H halogenation, as both processes would involve similar amidyl radical intermediates. The synthesis of 1 via the direct N-xanthylation of amides with strong base was unsuccessful,10 but we were able to develop a new approach that avoids the use of strongly basic conditions and is also amenable to large-scale preparation: the reaction of the parent N-chloroamide of 1 with inexpensive potassium ethyl xanthate provided facile access to shelf-stable N-xanthylamide 1 on decagram scale.11
With key reagent 1 in hand, we commenced our studies of the aliphatic C–H xanthylation with a number of simple hydrocarbons using the substrate as limiting reagent to ensure reaction practicality (Figure 2).12 Reactions of 1 with cycloalkanes in PhCF3 using blue LED irradiation provided alkyl xanthates 2–5 in good yields (59–85%). A competition experiment between cyclohexane and d12-cyclohexane indicated a kinetic isotope effect of 6.3, comparable to our C–H halogenations and consistent with irreversible C–H abstraction by an amidyl radical. The C–H xanthylation of n-hexane favored the methylene sites with ksecondary/kprimary (ks/kp) ~11 after correcting for the number of hydrogen atoms, with a preference for the 2-position. The xanthylation of norbornane provided 7 as the single exo diastereomer in 49% yield. The sterically dictated site selectivity of 1 is clear from the reactions of adamantane and trans-decalin–the functionalization of adamantane greatly favored the less hindered 3° C–H sites, providing xanthate 8 as a single product in 70% yield. The reaction of trans-decalin proceeded with a high methylene/methine site selectivity (ks/kt = >99).
Figure 2.

Products of C–H xanthylation. Yields refer to NMR yield with hexamethyldisiloxane (HMDS) as an internal standard or GC yield with dodecane as an internal standard. *Isolated yield.
We next surveyed a range of heterocycles and functionalized acyclic substrates to study the site selectivity and functional group compatibility of the C–H xanthylation (Figure 2). Xanthylation adjacent to heteroatoms was efficient across a number of substrates, with hyperconjugation likely assisting in the site selectivity. For example, the xanthylations of tetrahydrofuran and 1,4-dioxane delivered 10 and 11, respectively, in useful yields. Although reagent 1 possesses similar C–H bonds adjacent to oxygen, no products resulting from such abstraction were observed. The reactions of important nitrogen heterocycles were successful, as demonstrated by the functionalizations of 2-methoxypyridines to afford 12 and 13 in addition to N-methyl pyrrole giving 14. These substrates demonstrate that the system tolerates nitrogen functionality that can pose a challenge to metal-oxo catalysts. The electron withdrawing N-phthalimide functionality of linear substrate 15 dictates selective functionalization of the distal methylene sites, with a δ selectivity of 64% (68% combined yield). In addition, the xanthylation of 15-crown-5 afforded 16 in good yield (70%), providing a new approach to the derivatization of crown ethers. The electronic site selectivity of the C–H xanthylation across a number of acyclic compounds (17–20) indicates a preference for reaction at the most electron rich methylene sites, consistent with our previous studies involving amidyl radicals. These C–H xanthylations proceed in good overall yields, with site selectivities comparable (or superior) to those of most metal-catalyzed processes.
We were keenly interested in ascertaining the potential for site-selective C–H xanthylations of complex molecules owing to the range of structures accessible via the xanthate functional group. A number of diverse complex natural products and drug precursors were studied in this context (Figure 3). The C–H xanthylations of the terpenoids (+)–sclareolide and (−)–ambroxide delivered xanthates 21 and 22 in 55% and 80% yield, respectively.13 The xanthylation reaction is amenable to scale-up, delivering xanthate 21 in 54% yield on gram-scale. In both cases we observed functionalization at a single site, with (+)–sclareolide reacting at the sterically most accessible, electron-rich methylene site and (−)–ambroxide reacting at the position activated by hyperconjugation. Functionalization of a precursor to the topical retinoid differin occurred at the most accessible tertiary C–H site, giving xanthate 23 in 51% yield. Notably, this xanthylation succeeds in the presence of an electron rich aromatic ring, which would likely pose a major challenge for alternative strategies involving oxidizing intermediates. The reaction of 5α-cholestane occurred on the steroidal A-ring, with a 3:1 site selectivity favoring the C3 position to give product 24; this selectivity is remarkable since the substrate contains seven tertiary C–H bonds and 13 methylene sites for functionalization with no inherent substrate electronic bias. Steroid trans-androsterone acetate underwent xanthylation to afford C6 and C2 functionalization products 25 in a 1:1 ratio as single diastereomers (56% combined yield). We next questioned whether electronic deactivation of the steroidal A-ring would enable site-selective functionalization of the B-ring. Steroid 5α-androstanedione–with ketone deactivation of the A-ring–exhibited perfect diastereoselectivity and site selectivity on the B-ring, delivering xanthate 26 as a single product in 44% yield. As a comparison, the C–H oxidation of this substrate with a number of Fe-catalysts proceeded with poor site selectivity involving several methylene and methine positions,3e highlighting the unique capabilities of our system in the steric and electronic differentiation of the C–H sites.14 As a final challenge, we examined the C–H xanthylation of the terpenoid (+)–longifolene, a classic target for studies in organic synthesis. This substrate poses a significant chemoselectivity challenge for alternative C–H functionalizations involving oxidizing intermediates, as the alkene is known to undergo facile oxidation and skeletal rearrangement.15 The xanthylation of (+)–longifolene with reagent 1 under solvent-free conditions provides xanthate 27 as a single diastereomer with excellent site selectivity. The regioselectivity of the reaction can be rationalized by selective C–H abstraction of the less hindered ring system away from the quaternary centers in the substrate, and the mild conditions of the C–H xanthylation minimize any undesired alkene functionalization.
Figure 3.

Aliphatic C–H xanthylations of complex molecules. Yields refer to isolated yields. *NMR yield.
We view the aliphatic C–H xanthylation herein as a platform technology serving to unlock an array of important C–H transformations, including many with no synthetic precedent. Our approach exploits the impressive versatility of alkyl xanthates in a broad range of radical-mediated as well as polar bond-forming reactions. An initial demonstration of this approach is outlined in Figure 4. For example, the product of C–H vinylation, 28, is readily accessed from differin precursor xanthate 23 via radical coupling with ethyl styryl sulfone.16 Lewis acid-mediated addition of bistrimethylsilyl thymine to (−)–ambroxide xanthate 22 delivers N-alkyl thymine derivative 29 in high yield (87%), highlighting the utility of the xanthate group in polar reactions.17
Figure 4.

Aliphatic C–H transformations of complex substrates. Yields refer to isolated yields.
The selective deuteration of aliphatic C–H bonds represents another attractive yet undeveloped transformation. Such a process would expedite the preparation of isotopically labeled compounds for mechanistic and metabolic studies and offer an attractive route to deuterated drugs with enhanced pharmacokinetic properties.18 The reaction of amino acid derivative xanthate 20 with CD3OD using Et3B/O2 initiation provides deuterated product 30 in good yield (71%, 85% deuterium incorporation).19 Thus, the xanthylation-deuteration sequence constitutes a formal, site-selective monodeuteration of aliphatic C–H bonds, which, to our knowledge, does not exist.
Oxidations of unactivated aliphatic C–H bonds are likely the most studied intermolecular C–H transformation, but over-oxidation of alcohols to ketones is a major concern.20 A functional group interconversion from an alkyl xanthate could enable control of the product oxidation state, and the mild conditions of our approach would allow C–H oxidation of substrates containing oxidation-sensitive functionality. We have developed a xanthate to hydroxyl group interconversion, constituting a formal C–H hydroxylation using xanthylamides. The reaction of C6-functionalized steroidal xanthate 25 with persistent radical TEMPO and tris(trimethylsilyl)silane followed by mild reduction of the intermediate alkoxyamine delivered alcohol 31 in good yield (56%).21 Alternatively, oxidation of the alkoxyamine using mCPBA selectively yields a ketone product (59% yield).22 This approach allows complete reagent control of the product oxidation state, offering an attractive solution to major challenges in aliphatic C–H oxidation.
The trifluoromethylthiol group has become invaluable in the preparation of bioactive compounds owing to its high electronegativity and ability to modulate the lipophilicity of drug molecules.23 Recent reports of aliphatic C–H trifluoromethylthiolation using AgSCF3 and persulfate oxidants have emerged, but are generally selective for tertiary C–H bonds and often require excess substrate to obtain synthetically useful yields.24 We have developed simple conditions to introduce the trifluoromethylthiol group from alkyl xanthates using an easily accessible reagent developed by Shen, as shown in the preparation of 32.25 This provides a general approach to formal aliphatic C–H trifluoromethylthiolation using substrate as limiting reagent, appropriate for late-stage functionalizations of complex substrates.
The thiolene addition is useful for bioconjugation, offering a bioorthogonal alternative to azide-alkyne cycloadditions, with additional applications in polymer synthesis and materials science.26 In this light, a site-selective, aliphatic C–H thiolation would enable a formal thiolene from alkane starting materials. The C–H xanthylation is ideal for such a transformation: simple work-up with an amine base provides thiols in excellent yield. As a demonstration of this approach, we have studied the thiolene coupling of (+)–longifolene xanthate 27 with a glucose-derived allyl glycoside. Following quantitative conversion of 27 to its thiol derivative, photochemical thiolene delivered glycoconjugated (+)–longifolene adduct 33 in 62% yield. Considering the mild conditions and functional group compatibility of the C–H xanthylation and the broad utility of the thiolene process, we expect this tactic will offer major opportunities in the conjugation of complex molecules. We would also note that while Figure 4 offers a number of attractive avenues for molecular derivatization via C–H xanthylation, this is certainly not comprehensive. Alkylation, alkynylation16, and acylation reactions27, among others, are also possible using alkyl xanthates.
Applying different transformations to a single xanthate substrate provides facile access to a wide range of derivatives via a C–H diversification approach. As an example, (+)–sclareolide xanthate 21 is converted to seven different derivatives in a single step following the initial C–H xanthylation (Figure 4). This highlights an additional unique feature of our approach–not only does the xanthylation unlock new aliphatic C–H transformations, it also facilitates the synthesis of diverse analogs via a simple switch of reagent in the product elaboration rather than through the application of a new C–H functionalization.
The versatility of the current aliphatic C–H xanthylation–in addition to our previous efforts in C–H halogenation–clearly highlights the unique capabilities of functionalized amides in enabling a broad range of practical aliphatic C–H transformations. These studies also demonstrate the potential for enhanced site selectivity and functional group compatibility with respect to other aliphatic C–H functionalizations. We anticipate that these characteristics will lead to powerful applications in molecular derivatization for the synthesis and study of functionalized molecules in a number of contexts.
Supplementary Material
Acknowledgments
Financial support was provided by the ACS PRF (55108-ND1), Award R01 GM 120163 from the National Institute of General Medical Sciences, an Eliel Award (W.L.C.), and a Royster Fellowship (C.G.N.).
Footnotes
The authors declare no competing financial interest.
Supporting Information. Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; (b) Newhouse T, Baran PS. Angew Chem Int Ed. 2011;50:3362. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) White MC. Science. 2012;335:807. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 2.Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW. Chem Soc Rev. 2016;45:546. doi: 10.1039/c5cs00628g. [DOI] [PubMed] [Google Scholar]
- 3.(a) Brodsky BH, Du Bois J. J Am Chem Soc. 2005;127:15391. doi: 10.1021/ja055549i. [DOI] [PubMed] [Google Scholar]; (b) Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (c) Chen MS, White MC. Science. 2010;327:566. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]; (d) Gormisky PE, White MC. J Am Chem Soc. 2013;135:14052. doi: 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]; (e) Canta M, Font D, Gómez L, Ribas X, Costas M. Adv Synth Catal. 2014;356:818. [Google Scholar]; (f) Font D, Canta M, Milan M, Cussó O, Ribas X, Klein Gebbink RJM, Costas M. Angew Chem Int Ed. 2016;55:5776. doi: 10.1002/anie.201600785. [DOI] [PubMed] [Google Scholar]
- 4.(a) Huang X, Bergsten TM, Groves JT. J Am Chem Soc. 2015;137:5300. doi: 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]; (b) Sharma A, Hartwig JF. Nature. 2015;517:600. doi: 10.1038/nature14127. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang X, Groves JT. ACS Catal. 2016;6:751. [Google Scholar]
- 5.(a) Liu W, Groves JT. J Am Chem Soc. 2010;132:12847. doi: 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]; (b) Liu W, Huang X, Cheng M-J, Nielsen RJ, Goddard WA, III, Groves JT. Science. 2012;337:1322. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]; (c) Bloom S, Pitts CR, Miller DC, Haselton N, Holl MG, Urheim E, Lectka T. Angew Chem Int Ed. 2012;51:10580. doi: 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]; (d) Halperin SD, Fan H, Chang S, Martin RE, Britton R. Angew Chem Int Ed. 2014;53:4690. doi: 10.1002/anie.201400420. [DOI] [PubMed] [Google Scholar]
- 6.(a) Schmidt VA, Quinn RK, Brusoe AT, Alexanian EJ. J Am Chem Soc. 2014;136:14389. doi: 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]; (b) Quinn RK, Könst ZA, Michalak SE, Schmidt Y, Szklarski AR, Flores AR, Nam S, Horne DA, Vanderwal CD, Alexanian EJ. J Am Chem Soc. 2016;138:696. doi: 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Quiclet-Sire B, Zard SZ. Chem Eur J. 2006;12:6002. doi: 10.1002/chem.200600510. [DOI] [PubMed] [Google Scholar]; (b) Quiclet-Sire B, Zard SZ. Pure Appl Chem. 2011;83:519. [Google Scholar]; (c) Quiclet-Sire B, Zard SZ. Beilstein J Org Chem. 2013;9:557. doi: 10.3762/bjoc.9.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.There are a limited number of reports of C–H thiolations and sulfoxidations of simple hydrocarbons, generally with substrate in large excess: Du B, Jin B, Sun P. Org Lett. 2014;16:3032. doi: 10.1021/ol5011449.Zhao J, Fang H, Han J, Pan Y, Li G. Adv Synth Catal. 2014;356:2719. doi: 10.1002/adsc.201400032.Ferguson RR, Crabtree RH. J Org Chem. 1991;56:5503.Ishii Y, Matsunaka K, Sakaguchi S. J Am Chem Soc. 2000;122:7390.
- 9.Although there has been a sole report of an aliphatic C–H xanthylation, reactions were limited to the functionalization of ethereal and hydrocarbon solvents with substrate in large excess: Sato A, Yorimitsu H, Oshima K. Chem Asian J. 2007;2:1568. doi: 10.1002/asia.200700251.
- 10.Gagosz F, Moutrille C, Zard SZ. Org Lett. 2002;4:2707. doi: 10.1021/ol026221m. [DOI] [PubMed] [Google Scholar]
- 11.See the Supporting Information for reaction details.
- 12.See Table S1 in the Supporting Information for optimization.
- 13.For products 13, 16, and 22, isolated yields are lower than 1H NMR yields owing to challenges in product isolation.
- 14.Oxidation at the C6 site of cholestanol acetate has been reported in very low yield (1.6%): Barton DHR, Göktürk AK, Morzycki JW, Motherwell WB. J Chem Soc Perkin Trans I. 1985:583.
- 15.Dev S. Acc Chem Res. 1981;14:82. [Google Scholar]
- 16.Bertrand F, Quiclet-Sire B, Zard SZ. Angew Chem Int Ed. 1999;38:1943. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1943::AID-ANIE1943>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 17.Jean-Baptiste L, Yemets S, Legay R, Lequeux T. J Org Chem. 2006;71:2352. doi: 10.1021/jo052528y. [DOI] [PubMed] [Google Scholar]
- 18.Harbeson SL, Tung RD. Ann Rep Med Chem. 2011;46:403. [Google Scholar]
- 19.Allais F, Boivin J, Nguyen VT. Beilstein J Org Chem. 2007;3:46. doi: 10.1186/1860-5397-3-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For a selective C–H trifluoroacetoxylation that overcomes this challenge, see: Asensio G, Mello R, González-Núnez ME, Castellano G, Corral J. Angew Chem Int Ed. 1996;35:217.
- 21.For a related reaction using a dithiocarbamate, see: Grainger RS, Welsh EJ. Angew Chem Int Ed. 2007;46:5377. doi: 10.1002/anie.200701055.
- 22.See the Supporting Information for reaction details.
- 23.Landelle G, Panossian A, Leroux FR. Curr Top Med Chem. 2014;14:941. doi: 10.2174/1568026614666140202210016. [DOI] [PubMed] [Google Scholar]
- 24.(a) Guo S, Zhang X, Tang P. Angew Chem Int Ed. 2015;54:4065. doi: 10.1002/anie.201411807. [DOI] [PubMed] [Google Scholar]; (b) Wu H, Xiao Z, Wu J, Guo Y, Xiao JC, Liu C, Chen QY. Angew Chem Int Ed. 2015;54:4070. doi: 10.1002/anie.201411953. [DOI] [PubMed] [Google Scholar]
- 25.Shao X, Xu C, Lu L, Shen Q. Acc Chem Res. 2015;48:1227. doi: 10.1021/acs.accounts.5b00047. [DOI] [PubMed] [Google Scholar]
- 26.(a) Azagarsamy MA, Anseth KS. ACS Macro Lett. 2013;2:5. doi: 10.1021/mz300585q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoyle CE, Bowman CN. Angew Chem Int Ed. 2010;49:1540. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 27.Kim S, Song HJ, Choi TL, Yoon JY. Angew Chem Int Ed. 2001;40:2524. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.