

Abstract
Huntington disease (HD) is a late onset ultimately fatal neurodegenerative disorder caused by a CAG triplet repeat expansion in the Huntingtin gene which was discovered in 1993. The PHAROS study is a unique observational study of 1001 individuals at risk for HD who had not been previously tested for HD and who had no plans to do so. In this cohort, 104 (10%) individuals changed their minds and chose to be tested during the course of the study but outside of the study protocol.
Baseline behavioral scores, especially apathy, were more strongly associated with later genetic testing than motor and chorea scores, particularly among subjects with expanded CAG repeat length. In the CAG expanded group, those choosing to be tested were older and had more chorea and higher scores on the behavioral section of the UHDRS at baseline than those not choosing to be tested. Following genetic testing, 56% of subjects with CAG < 37 had less depression when compared to prior to testing, but depression generally stayed the same or increased for 64% of subjects in the expanded group. This finding suggests that approaches to testing must continue to be cautious, with appropriate medical, psychological and social support.
Keywords: Genetic testing, Huntington disease, observational trial
Graphical abstract
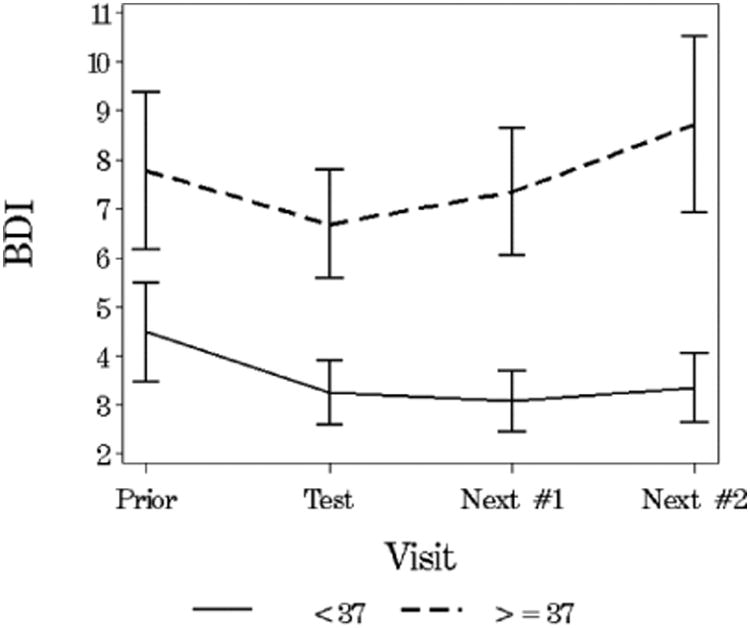
Mean (SE) Beck Depression Score Before and After the Visit at Which Testing Was Disclosed (N = 61)
Introduction
Huntington disease (HD) is a relatively rare, highly penetrant, inherited neurodegenerative disorder with a prevalence of 4.1 – 7.5 cases per 100,000 in the Caucasian population (1). The characteristic signs of HD include abnormal movements, cognitive decline often leading to dementia, and a variety of psychiatric disorders, most notably depression. These clinical features gradually emerge around the age of 40, although the onset of the disease has been seen in children as young as 2 and adults as old as 70 (2). Death normally occurs between ten and seventeen years after onset (3). It is estimated that some 30,000 individuals in the United States and Canada have clinically manifest HD, and an additional 150,000 are at 50:50 risk of having inherited a mutated HTT gene (4, 5).
HD is inherited in an autosomal dominant pattern such that each child of an affected parent has a 50:50 chance of having inherited the HD gene; men and women are equally likely to be affected. In 1983, genetic markers linked to the HD gene were first identified, allowing some individuals at risk, if their family structure permitted, to establish whether or not they would develop HD using a process known as linkage analysis (6). In 1986, centers at Massachusetts General Hospital in Boston and The Johns Hopkins Hospital in Baltimore began offering pre-symptomatic testing for HD on an experimental basis. Testing protocols included psychiatric screening, a neurological examination, mandatory counseling, informed consent, results given in person and long term follow-up and excluded testing individuals under the age of 18 years. Preliminary findings began to appear in the literature suggesting that testing for HD could be done safely when administered in the context of these structured research protocols (7, 8, 9).
In 1993, the underlying genetic cause of HD was discovered to be a cytosine-adenine-guanine (CAG) trinucleotide repeat expansion in the huntingtin (HTT) gene on chromosome 4 (Huntington Disease Collaborative Research Group, 1993). This discovery meant that the presence or absence of the HD gene mutation could be determined in any at-risk individual by direct gene testing for the expanded CAG repeat, without regard to the participation of other family members. Studies done prior to the finding of the HD mutation suggested that there would be high rates of utilization for predictive testing in individuals at risk for HD (10, 11, 12, 13, 14). However, current estimates indicate that only 12-15% of adults at risk have elected to pursue so-called pre-symptomatic testing (15, 16), although a recent report suggests that uptake may be increasing in some countries (17).
Most of the information we have about individuals who have undergone pre-symptomatic testing for the mutated HTT gene comes from the test centers themselves which, by necessity, can only collect data from individuals who present for testing (7, 15, 17,18, 19, 20, 21, 22, 23). Previous studies indicate that the majority of those choosing to be tested are women, with percentages ranging from 57.6% to 66.7% (24, 4, 25, 26, 22, 17, 23). The mean age of adults requesting testing may be trending lower with time, from a high of 40.5 (26) to 36.4 (22) to 30 (17). Few differences in psychological outcomes between those who received a positive result (expanded CAG repeat) compared to a negative result (non-expanded CAG repeat) have been reported (27) and most appear to be related more to psychological adjustment prior to testing than the test result itself. Although the duration of follow up differs, ranging from 3 to 10 years, no direct catastrophic outcomes have been reported as a consequence of pre-symptomatic testing for the HD gene.
While generally concurring with the long term effects of testing, Hayden and Bombard (28) examined the psychosocial effects of testing and focused primarily on the 5-10% of people who enter predictive testing programs already manifesting signs and symptoms of this disease. They report that their clinical experience suggests that, at least in the short term, these candidates who receive results indicating an increased risk undergo a difficult period of adjustment with some experiencing a prolonged period of considerable emotional distress (29). Factors associated with a difficult adjustment include a past history of psychiatric disorder and adverse responses to other difficult life events such as the loss of a spouse or change in employment status.
In preparation for future clinical trials, the Huntington Study Group, a consortium of HD researchers, conducted a study of individuals at risk for HD. This study, the Prospective Huntington At-Risk Observational Study (PHAROS), was designed to establish whether experienced clinicians can reliably identify emerging clinical signs and symptoms that indicate the earliest onset of HD in clinically unaffected individuals at risk for carrying the HD gene who have chosen not to undergo pre-symptomatic genetic testing. The PHAROS study, therefore, presents a unique research setting to examine factors related to the decisions by at-risk research participants who had at enrollment chosen not to pursue genetic testing and had indicated no plans to do so in the future but who subsequently changed their mind about testing (30).
The purpose of this report is to examine the factors associated with the decision to be tested by PHAROS research participants who changed their minds and opted to undergo pre-symptomatic HD genetic testing, compared with those who still chose not to be tested.
Materials and Methods
Between July 1999 and January 2004, 1001 clinically unaffected adults at risk for HD who had chosen not to undergo predictive genetic testing for the presence or absence of the mutated HTTgene were enrolled in this non-interventional, observational, longitudinal study. Study visits included the Unified Huntington's Disease Rating Scale (UHDRS) (31); the Beck Depression Inventory (32); medical history; surveys of attitudes, perceptions, and beliefs regarding HD; neuropsychological testing; and a one-time research analysis at enrollment for the HD CAG expansion. A central basis of consent was that individual and potentially identifiable results of CAG analysis would never be disclosed to anyone, neither investigators, clinicians, research participants nor their families. Participants returned for research visits about every nine months and committed to at least three years of observation, a period that was subsequently extended for an additional 7 years through consent amendments. All sites received institutional review board approval of the protocols and consent procedures.
Genotype Assessment
Coded venous blood samples from research participants were sent to the DNA laboratory of the Molecular Neurogenetics Unit at Massachusetts General Hospital where CAG repeat analysis was performed under the direction of Marcy MacDonald, PhD, using previously described techniques (33). A bar code system de-identified the samples, thereby preventing anyone from gaining access to or linking individually identifiable genetic and clinical data. All participants and site personnel were blinded to the results.
Clinical Assessment
At the initial evaluation, a site coordinator and site investigator (SI) obtained a comprehensive medical history and physical examination, including the UHDRS. An independent rater (IR) at each site served strictly as a motor examiner, interacting with research participants only to assess the motor component of the UHDRS. A key item on the motor examination requires the rater to assign a level of ‘diagnostic confidence of HD’. A rating of 4, motor abnormalities that are unequivocal signs of HD with ≥ 99% confidence (phenoconversion), has been found to have good reliability among independent raters (34). The UHDRS behavioral assessment consists of eleven items (depressed mood, low self-esteem/guilt, anxiety, suicidal thoughts, disruptive or aggressive behavior, irritable behavior, perseverative/obsessional thinking, compulsive behavior, delusions, hallucinations, apathy), each of which is evaluated for frequency and severity on scales of 0 (never or almost never/none) to 4 (very frequently/severe); a total behavioral score was calculated as frequency times severity, summed over the eleven items, and individual questions were coded as impaired if either frequency or severity was ≥ 2. The cognitive portion of the UHDRS includes a test of category fluency (number of words beginning with a specified letter), the symbol digit modalities test (pairing of symbols and numbers), and the Stroop interference test (identification of words and colors), with lower scores indicating worse impairment. The functional portion includes total functional capacity (TFC), which measures the subject's overall functioning in the areas of employment, finances, domestic chores, activities of daily living, and need for care. The UHDRS includes a question asking if the participant since the last visit had obtained HD gene testing outside of the research protocol.
The Life Experiences Survey is a 57-item, self-report measure that allows respondents to indicate life events that they have experienced in the last year. Participants rate the impact of each event, and the numbers of negative and positive events can be calculated (35).
Statistical Methods
Subject characteristics were compared by genetic testing status using Kruskal-Wallis tests or chi-square tests, as appropriate. Pre-specified potential predictors of time to genetic testing were evaluated using separate Cox proportional hazards models, adjusted for age and gender, and then creating a combined model that included only those measures that were significant in individual models. Similar Cox proportional hazards models were also used to evaluate the UHDRS functional and cognitive scores and the individual items on the UHDRS behavioral score. Among subjects who had changed their minds and decided to be tested, scores were compared by CAG status at the visit immediately prior to and at the one where the testing was reported using Kruskal-Wallis tests, and changes between these two visits were assessed using analysis of covariance on the ranks, with scores from the prior visit included in the models. The same method was also used to evaluate depression at the two visits following the one where testing was reported.
A case-control subset was used to compare participants who opted to undergo genetic testing with those who did not, at the visit prior to the one where the testing was reported. Participants who had obtained genetic testing outside of the study were matched with participants who were never tested based on four factors: visit number when genetic testing was first reported, prior visit number, gender, and CAG repeat length (<37 vs ≥37), three controls to one case. Conditional logistic regression was used to assess the effects of pre-specified predictors on the decision to obtain testing before the next visit.
Using the case-control cohort and Kruskal-Wallis tests, numbers of negative and of positive life events experienced in the year that subjects obtained genetic testing were compared with those of matched subjects at the same visit who were not tested, separately by CAG status.
Results
Out of the original study cohort of 1001 unaffected adults at risk for inheriting the HD gene, 104 (10%) opted to obtain genetic testing outside of the PHAROS research study. While the overall cohort consisted of approximately 1/3 CAG expanded and 2/3 CAG non-expanded individuals, close to what would be predicted (28), half of those who opted for testing had an expanded CAG repeat length (≥ 37) and half did not. The mean time to testing from study baseline was 4.3 (2.4) years, with a range of 1 to 10 years.
For these analyses, four participants lacking CAG data and 14 who had manifested signs of HD at study baseline were excluded (28), creating an analysis cohort of N=983 (Figure 1). Subject baseline characteristics for this cohort are shown in Table 1. The median age was 42.1 years; 69% were women and 35% had an expanded CAG repeat. During the study, 101 (10%) participants in this cohort chose to undergo HD gene testing outside of the study protocol. Fifty-one (8%) participants with non-expanded CAG repeat length were tested, compared with 50 (14%) participants in the expanded group (p=0.0014, chi-square test). Among the 101 participants tested, 73 (72%) were women, 65 (70%) were married, and the mean (SD) age at testing was 47.3 (7.5) years.
Figure 1.

Flow diagram of ascertainment of study cohort.
Table 1. Baseline Characteristics by Genetic Testing Status (N = 983).
| Never Tested (N = 882) | Tested Later (N = 101) | All (N = 983) | P-Value | |
|---|---|---|---|---|
| Age at Baseline | 41.7 (35.4, 47.3) | 42.9 (38.3, 48.6) | 42.1 (35.7, 47.4) | 0.07 |
| Education | 15 (13, 16) | 16 (13, 17) | 15 (13, 16) | 0.25 |
| UHDRS Motor Score | 1 (0, 3) | 1 (0, 4) | 1 (0, 3) | 0.79 |
| UHDRS Chorea Score | 0 (0,0) | 0 (0,0) | 0 (0,0) | 0.0527 |
| Beck Depression Score | 3 (1,6) | 3 (1,6) | 3 (1,6) | 0.32 |
| UHDRS Behavioral Score (Frequency × Severity) | 4 (0, 10) | 6 (2, 13) | 4 (0, 10) | 0.0127 |
| Female Gender | 608 (69%) | 73 (72%) | 681 (69%) | 0.49 |
| CAG ≥ 37 | 295 (33%) | 50 (50%) | 345 (35%) | 0.0014 |
| Married | 573 (70%) | 65 (70%) | 638 (70%) | 0.98 |
Values shown are medians (25%, 75% percentiles) or N (%), as appropriate.P-values indicate the significance levels for comparisons by genetic testing status using Kruskal-Wallis tests or chi-square tests, as appropriate.
Compared with the 882 participants who were not tested, those tested did not differ significantly by age at the study baseline, gender, or marital status. Participants were not significantly different by testing status on the UHDRS motor or Beck depression scores at baseline, but those who were later tested scored significantly higher (more impaired) on the baseline UHDRS behavioral score (p=0.013, Kruskal-Wallis test) than those who were never tested. Among participants with CAG < 37, there were no significant differences at baseline between those who were later tested and those who were not tested. However, among participants with CAG ≥ 37, those later tested were significantly older (p=0.0048) and had significantly more severe chorea scores (p=0.0318) and behavioral scores (p=0.0307) than those who were not tested (Table S1).
Individual Cox proportional hazards models revealed that CAG status and the behavioral score were significant predictors of time to genetic testing, and both retained significance in a combined model (Table 2). When these models were run separately by CAG status, the behavioral score was a significant predictor only in the CAG expanded group (p=0.0222) (Table S2).
Table 2. Baseline Characteristics to Predict Time to Genetic Testing (N = 983).
| Hazard Ratios, Individual Models | Hazard Ratios, Combined Model | |||
|---|---|---|---|---|
| HR (95% CI) | P-Value | HR (95% CI) | P-Value | |
| CAG ≥ 37 | 2.04 (1.38, 3.02) | 0.0004 | 1.91 (1.28, 2.85) | 0.0015 |
| UHDRS Motor Score | 1.03 (0.98, 1.08) | 0.31 | --- | --- |
| UHDRS Chorea Score | 1.12 (0.98, 1.28) | 0.10 | --- | --- |
| Beck Depression Score | 1.03 (0.99, 1.06) | 0.16 | --- | --- |
| UHDRS Behavioral Score (Frequency × Severity) | 1.03 (1.01, 1.04) | 0.0046 | 1.02 (1.00, 1.04) | 0.0250 |
Values shown are hazard ratios (95% confidence intervals) from Cox proportional hazards models. The hazard ratios are per point of the scale; for example, a motor score hazard ratio of 1.03 means that, for each one point increase in the motor score, the chance of getting tested increased by 3%. All models were adjusted for age and gender. The combined model includes only those measures that were significant in the individual models.
Responses to individual questions on the UHDRS behavioral score showed that participants subsequently tested were significantly more likely than those not tested to have low esteem, increased irritability, and more apathy at baseline. In an adjusted analysis, apathy was a particularly strong predictor in the expanded group (p=0.0004) with 30% of subjects with baseline apathy obtaining testing, compared to 11% of subjects who were not apathetic at baseline (Table S3). In a Cox model to predict time to testing, adjusted for age, gender and CAG status, apathy remained a significant predictor of time to testing (p=0.0258) (data not shown).
Among the UHDRS functional and cognitive scores, total functional capacity and the symbol digit modalities test were significant in separate models; the symbol digit modalities score remained significant (p=0.0098) in a combined model that included age, gender, CAG status and the UHDRS behavioral score (data not shown).
Eighty-seven of the 101 tested subjects had scores from the visit immediately prior to the visit at which the testing status was made known to the investigator. Scores for these subjects at the prior visit were compared by CAG status with those at the testing disclosure visit (Table 3). At both the visit prior to testing and the testing disclosure visit, the Beck depression scores were not significantly different between the two CAG status groups. However, after testing, among those in the CAG non-expanded group, more than half (56%) had less depression than before testing while levels of depression stayed the same or increased in 44% of subjects. In the CAG expanded group, 36% had less depression, and 64% had the same or increased levels of depression following testing (p=0.07). Among tested subjects in the CAG expanded group, depression scores appeared to increase at subsequent visits following testing, while scores remained lowered in the CAG non-expanded group (Figure 2). Compared with the CAG non-expanded group, behavior scores were higher in the CAG expanded group both before (p=0.07) and after (p=0.0379) testing. However, the changes were similar in the two groups (p=0.31).
Table 3. Characteristics of Tested Subjects by CAG Status (N = 87).
| CAG <37 (N = 47) | CAG ≥37 (N = 40) | P-Value | |
|---|---|---|---|
| SCORES AT PRIOR VISIT | |||
| UHDRS Motor Score | 0 (0, 3) | 3 (0, 12) | 0.0009 |
| UHDRS Chorea Score | 0 (0, 0) | 1 (0, 4) | 0.0005 |
| Beck Depression Score | 3 (1, 8) | 5 (1, 13) | 0.71 |
| UHDRS Behavioral Score (Frequency × Severity) | 3 (0, 10) | 5.5 (2, 19.5) | 0.07 |
| SCORES AT VISIT WHEN TESTING WAS DISCLOSED | |||
| UHDRS Motor Score | 1 (0, 2) | 5 (2, 14.5) | <0.0001 |
| UHDRS Chorea Score | 0 (0, 0) | 0 (0, 3.5) | 0.0016 |
| Beck Depression Score | 3 (0, 5) | 3 (1, 10.5) | 0.12 |
| UHDRS Behavioral Score (Frequency × Severity) | 2 (0, 6) | 7 (0, 13) | 0.0379 |
| CHANGES FROM PRIOR VISIT | |||
| Change in UHDRS Motor Score | 0 (0, 0) | 0 (-1, 4) | 0.0016 |
| Change in UHDRS Chorea Score | 0 (0, 0) | 0 (0, 1) | 0.17 |
| Change in Beck Depression Score | -1 (-3, 0) | 0 (-2, 3) | 0.0286 |
| Change in UHDRS Behavioral Score (Frequency × Severity) | 0 (-4, 2) | -0.5 (-6, 2.5) | 0.31 |
Values shown are medians (25%, 75% percentiles). P-values indicate the significance levels for comparisons by CAG status using Kruskal-Wallis tests for the scores at each visit, and analysis of covariance on the ranks, including the prior visit score, was used for the change scores.
Figure 2. Mean (SE) Beck Depression Score Before and After the Visit at Which Testing Was Disclosed (N = 61).
Analyses to predict genetic testing at the next visit utilized the case-control cohort, and the results from separate conditional logistic regression models are shown in Table 4. In a combined model, both higher motor scores and higher Beck depression scores were marginally significant predictors of opting for testing before the next visit, compared with never-tested participants (p=0.07 and p=0.06, respectively).
Table 4. Predict Genetic Testing before Next Visit, Case-Control Analysis (N = 86 cases and 258 controls): Scores at Prior Visit.
| Median (25th, 75th Percentiles) | Hazard Ratios | |||||
|---|---|---|---|---|---|---|
| Not Tested | Tested | HR (95% CI) | P-Value | |||
| CAG <37 N = 141 | CAG ≥37 N = 117 | CAG <37 N = 47 | CAG ≥37 N = 39 | |||
| UHDRS Motor Score | 1 (0, 3) | 2 (0, 5) | 0 (0, 3) | 3 (0, 12) | 1.06 (1.01, 1.10) | 0.0077 |
| UHDRS Chorea Score | 0 (0, 0) | 0 (0, 2) | 0 (0, 0) | 1 (0, 4) | 1.13 (1.01, 1.26) | 0.0338 |
| Beck Depression Score | 1.5 (0, 5) | 2.5 (1, 6) | 3 (1, 8) | 5 (1, 13) | 1.05 (1.02, 1.09) | 0.0044 |
| UHDRS Behavioral Score (Frequency × Severity) | 1 (0, 5) | 3 (0, 12) | 3 (0, 10) | 6 (2, 21) | 1.03 (1.01, 1.05) | 0.0185 |
Cases (tested) and controls (not tested) were matched on testing visit, prior visit, gender, and CAG status(< 37 or >= 37). Values shown are medians (25%, 75% percentiles) and hazard ratios (95% confidence intervals) from separate conditional logistic regression models. The hazard ratios are per point of the scale; for example, a motor score hazard ratio of 1.06 means that, for each one point increase in the motor score, the chance of getting tested increased by 6%.
In similar analyses by CAG status, higher Beck depression scores were the only significant predictor of testing before the next visit for subjects with CAG less than 37, whereas higher depression, motor and chorea scores were all significant predictors for CAG ≥ 37 subjects (Table S4).
Among subjects in the case-control cohort, numbers of negative and numbers of positive life events reported at the visit at which the testing was disclosed were not significantly different when compared with matched subjects at the same visit who had not been tested. We also looked at life events among tested subjects at the visit at which testing was disclosed and the first and second follow-up visits and there were no significant differences in positive or negative life events for those with CAG expanded test results and those without.
Discussion
Most information about individuals who obtain genetic testing comes from testing centers, which obtain data only from those who present for testing. The PHAROS study is unique in having the ability to compare participants who obtain genetic testing with those who do not in a cohort that was followed longitudinally over several years and whose CAG status was concealed from participants and investigators.
As in most studies of at-risk individuals undergoing testing, the majority of those choosing to be tested were women (72%). Although there has been speculation about why women choose to be tested at a greater rate compared with men, few studies have examined this in detail and we know little about why this is so.
Although at enrollment participants in the PHAROS study did not intend to undergo genetic testing for the HD gene, 10% subsequently changed their minds and obtained testing independent of the study, in keeping with the relatively low uptake of testing reported in clinical practice and in other studies. This suggests that decision-making about genetic testing is fluid, can change over time, and is likely to be different for different people. There also may be subtle changes in their cognition or mood that are causing such individuals to re-consider testing.
In this cohort of individuals who expressly did not desire genetic testing at the time of their entry into the study, 101 individuals decided subsequently to be tested outside of the study protocol. Their changed decision to be tested may be based on a growing awareness of emerging symptoms that may be related to disease onset as evidenced by the fact that those in the CAG expanded group had both higher chorea scores and higher behavioral scores, specifically higher scores on low self-esteem, increased irritability and apathy. Alternatively, for the CAG non-expanded group, the decision to be tested might be based on a growing awareness that symptoms are not developing and the wish to eliminate uncertainty.
Although the actual number of individuals choosing to be tested is almost identical, it is important to note that a greater percentage of CAG-expanded individuals chose to be tested 50 (14%) vs in the non-expanded group 51 (8%). In addition, among CAG expanded individuals who were tested, 19 (38%) developed manifest signs of HD during the study, compared with 42 (14%) of CAG expanded individuals who were not tested. However, our analyses have demonstrated that worsening motor and chorea scores did not appear to be the sole motivation for testing. Instead, depression, cognitive impairment, and behavioral scores (particularly apathy) seemed to be motivating factors. As apathy is considered one of the symptoms of HD that is most distressing to caregivers, it is interesting to speculate on what role pressure from family members to be tested may have played (36) This finding also suggests that for those with an expanded CAG, their decision to be tested may be based on a gradual realization of emerging symptoms, a realization that may have been brought to their attention by the fact that they were having a neurological examination approximately every 9 months as a condition of study participation. While this is not a situation at all similar to that of most people coming for testing, one may surmise that a certain percentage of the population at risk will chose to be tested because they feel that they are becoming symptomatic. If so, a neurological examination prior to testing remains an important part of the testing process. In addition, HD researchers need to be aware of this possibility as they consider entering gene positive individuals into clinical trials in which regular neurological examinations are required as a part of the study protocol.
Post-testing measures showed that while depression was not increased for the CAG expanded group immediately after testing, over time, depression increased in those who learned that they had an expanded CAG repeat, whereas subjects in the non-expanded group became less depressed. This finding is in contrast with most published studies showing an increase in depression immediately after testing for those with an expanded CAG repeat with a return to baseline levels within 1-6 months (36) but is consistent with outcomes found in other studies focusing on individuals who come for testing and are exhibiting symptoms suggesting that this group may be especially vulnerable should their test indicate an expanded CAG repeat. Although the data are limited, it appeared that genetic testing did not affect subsequent life experiences in the short term.
This research cohort may differ in some ways from individuals who are not research participants and choose gene testing at clinical sites. They are well educated with most having at least some college. They are clearly engaged in HD research, agreeing to return for study visits every nine months for several years. And, as a neurological examination was an integral part of the study visit, they were forced to be more mindful of emerging symptoms.
In summary, in PHAROS, for those who subsequently opted for testing, this decision appears to be motivated by changes in mood, most notably depression, subtle changes in cognitive function, and apathy. The fact that depression increased post testing in those with expanded CAG repeats, supports a continuing cautious approach to testing, requiring appropriate medical, psychological and social support. In addition, the fact that those who had an expanded CAG repeat had higher chorea scores also suggests that a neurological examination remains an important part of the pre-symptomatic HD testing protocol.
Acknowledgments
Support: NIH (# 2 RO1 HG002449-06), National Human Genome Research Institute, National Institute of Neurological Disorders and Stroke
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
References
- 1.Folstein SE. Huntington's Disease a Disorder of Families. Johns Hopkins University Press; 1989. [Google Scholar]
- 2.Shoulson I, Young AB. Milestones in Huntington disease. Mov Disord. 2011;26(6):1127–1133. doi: 10.1002/mds.23685. [DOI] [PubMed] [Google Scholar]
- 3.Harper PS, editor. Huntington Disease Major Problems in Neurology. Vol. 22. W.B. Saunders Company Ltd; London: 1991. [Google Scholar]
- 4.Harper PS. The epidemiology of Huntington disease. Hum Genet. 1992;89(4):365–376. doi: 10.1007/BF00194305. [DOI] [PubMed] [Google Scholar]
- 5.Biglan KM, Shoulson I. Huntington's disease. In: Jankovic J, Tolosa E, editors. Parkinson's Disease and Movement Disorders. 4th. Lippincot Williams and Wilkins; 2002. pp. 212–227. [Google Scholar]
- 6.Gusella JF, Wexler NS, Conneally PM, et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature. 1983;306(5940):234–238. doi: 10.1038/306234a0. [DOI] [PubMed] [Google Scholar]
- 7.Brandt J, Quaid KA, Folstein SE, et al. Pre-symptomatic diagnosis of delayed-onset disease with linked DNA markers. The experience in Huntington's disease. JAMA. 1989;261(21):3108–3114. [PubMed] [Google Scholar]
- 8.Meissen GJ, Myers RH, Mastromauro CA, et al. Predictive testing for Huntington's disease with use of a linked DNA marker. N Engl J Med. 1988;318(9):535–542. doi: 10.1056/NEJM198803033180903. [DOI] [PubMed] [Google Scholar]
- 9.Quaid KA, Brandt J, Folstein SE. The decision to be tested for Huntington's disease. JAMA. 1987;257(24):3362. [PubMed] [Google Scholar]
- 10.Kessler S, Field T, Worth L, et al. Attitudes of persons at risk for Huntington disease toward predictive testing. Am J Med Genet. 1987;26(2):259–270. doi: 10.1002/ajmg.1320260204. [DOI] [PubMed] [Google Scholar]
- 11.Markel DS, Young AB, Penney JB. At-risk persons' attitudes toward pre-symptomatic and prenatal testing of Huntington disease in Michigan. Am J Med Genet. 1987;26(2):295–305. doi: 10.1002/ajmg.1320260207. [DOI] [PubMed] [Google Scholar]
- 12.Mastromauro C, Myers RH, Berkman B. Attitudes toward pre-symptomatic testing in Huntington disease. Am J Med Genet. 1987;26(2):271–282. doi: 10.1002/ajmg.1320260205. [DOI] [PubMed] [Google Scholar]
- 13.Meissen GJ, Berchek RL. Intended use of predictive testing by those at risk for Huntington disease. Am J Med Genet. 1987;26(2):283–293. doi: 10.1002/ajmg.1320260206. [DOI] [PubMed] [Google Scholar]
- 14.Tyler A, Morris M, Lazarou L, et al. Pre-symptomatic testing for Huntington's disease in Wales 1987-90. Br J Psychiatry. 1992;161:481–488. doi: 10.1192/bjp.161.4.481. [DOI] [PubMed] [Google Scholar]
- 15.Tassiker RJ, Marchall PK, Liebeck TA, et al. Predictive and pre-natal testing for Huntington disease in Australia: results and challenges encountered during a 10-year period. Clin Genet. 2006;70(6):480–489. doi: 10.1111/j.1399-0004.2006.00701.x. [DOI] [PubMed] [Google Scholar]
- 16.Morrison PJ, Harding-Lester S, Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin Genet. 2011;80(3):281–286. doi: 10.1111/j.1399-0004.2010.01538.x. [DOI] [PubMed] [Google Scholar]
- 17.Sizer E, Haw T, Wessels TM, et al. The utilization and outcome of diagnostic, predictive, and prenatal testing for Huntington disease in Johannesburg, South Africa. Genet Test Mol Biomarkers. 2012;16(1):58–62. doi: 10.1089/gtmb.2011.0007. [DOI] [PubMed] [Google Scholar]
- 18.Almqvist EW, Bloch M, Brinkman R, et al. A worldwide assessment of the frequency of suicide, suicide attempts, or psychiatric hospitalization after predictive testing for Huntington disease. Am J Hum Genet. 1999;64(5):1293–1304. doi: 10.1086/302374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meiser B, Dunn S. Psychological impact of genetic testing for Huntington's disease: an update of the literature. J Neurol Neurosurg Psychiatry. 2000;69(5):574–578. doi: 10.1136/jnnp.69.5.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robins Wahlin TB. To know or not to know: a review of behavior and suicidal ideation in preclinical Huntington disease. Patient Educ Couns. 2007;65(3):279–287. doi: 10.1016/j.pec.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Tibben A. Predictive testing for Huntington disease. Brain Res Bull. 2007;72(2-3):165–171. doi: 10.1016/j.brainresbull.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 22.Dufrasne S, Roy M, Galvez M, et al. Experience over fifteen years with a protocol for predictive testing for Huntington disease. Mol Genet Metab. 2011;102(4):494–504. doi: 10.1016/j.ymgme.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Scuffham TM, MacMillan JC. Huntington disease: Who seeks pre-symptomatic genetic testing, why and what are the outcomes. J Genet Couns. 2014;23:754–761. doi: 10.1007/s10897-013-9678-z. [DOI] [PubMed] [Google Scholar]
- 24.Hayden MR. Huntington's chorea. Springer-Verlag; 1981. [Google Scholar]
- 25.Creighton S, Almqvist EW, MacGregor D, et al. Predictive, prenatal and diagnostic genetic testing for Huntington disease: the experience in Canada from 1987 to 2000. Clin Genet. 2003;63:462–475. doi: 10.1034/j.1399-0004.2003.00093.x. [DOI] [PubMed] [Google Scholar]
- 26.Trembeth M, Tassiker R, Collins V, et al. Fifteen years of experience in predictive testing for Huntington disease in a single testing center in Victoria, Australia. Genet Med. 2006;8(11):673–680. doi: 10.1097/01.gim.0000245633.97952.f1. [DOI] [PubMed] [Google Scholar]
- 27.Crozier S, Robertson N, Dale M. The psychological impact of predictive genetic testing for Huntington disease: A systematic review of the literature. J Genet Couns. 2015;24(1):29–29. doi: 10.1007/s10897-014-9755-y. [DOI] [PubMed] [Google Scholar]
- 28.Hayden MR, Bombard Y. Psychosocial effects of predictive testing for Huntington's disease. Adv Neur. 2005;96:226–239. [PubMed] [Google Scholar]
- 29.Bloch M, Adam S, Wiggins S, et al. Predictive testing for Huntington disease in Canada: the experience of those receiving an increased risk. Am J Med Genet. 1992;42:499–507. doi: 10.1002/ajmg.1320420416. [DOI] [PubMed] [Google Scholar]
- 30.Huntington Study Group PHAROS Investigators. At risk for Huntington disease: The PHAROS (Prospective Huntington At-Risk Observational Study) Cohort Enrolled. Arch Neurol. 2006;63(7):991–996. doi: 10.1001/archneur.63.7.991. [DOI] [PubMed] [Google Scholar]
- 31.Kieburtz K, et al. Unified Huntington's Disease Rating Scale: Reliability and Consistency. Mov Dis. 1996;11(2):136–142. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- 32.Beck AT, Ward CH, Mendelson M, et al. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- 33.MacDonald ME, Ambrose CM, Duyao MP, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72(6):971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 34.Paulson J. Early detection of Huntington disease. Future Neurology. 2010;5:85–104. doi: 10.2217/fnl.09.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarason IG, Johnson JH, Siegel JM. Assessing the impact of life changes: development of the life experiences survey. J Consult Clinic Psychol. 1978;46(5):932–946. doi: 10.1037//0022-006x.46.5.932. [DOI] [PubMed] [Google Scholar]
- 36.Boyle PA, Malloy PF, Salloway S, et al. Executive Dysfunction and apathy predict functional impairment in Alzheimer disease. Am J Geriatr Psychiatry. 2003;11:214–221. [PubMed] [Google Scholar]