

Abstract
The synthesis of the first terminal group 9 hydrazido(2−) complex, Cp*IrN(TMP) (6) (TMP = 2,2,6,6-tetramethylpiperidine) is reported. Electronic structure and X-ray diffraction analysis indicate that this complex contains an Ir-N triple bond, similar to Bergman’s seminal Cp*Ir(NtBu) imido complex. However in sharp contrast to Bergman’s imido, 6 displays remarkable redox noninnocent reactivity owing to the presence of the Nβ lone pair. Treatment of 6 with MeI results in electron transfer from Nβ to Ir prior to oxidative addition of MeI to the iridium center. This behavior opens the possibility of carrying out facile oxidative reactions at a formally IrIII metal center via a hydrazido(2−)/isodiazene valence tautomerization.
Keywords: Redox Noninnocence, Iridium Complexes, Hydrazide Ligand
Graphical abstract

Late transition metal-ligand multiple bonding plays a significant role in many organic transformations, such as C-H bond functionalization[1–3] and group transfer reactions.[4] Consequently, the field has experienced substantial growth in the past 25 years: several imido and oxo complexes of the late transition metals (groups 8-10) have been synthesized,[5–12] but examples of heteroatom functionalized multiply-bound ligands, such as terminal hydrazido(2−) and alkoxyimidos, are extremely limited.[13] Such terminal moieties are attractive synthetic targets because they are important intermediates along N2 reduction cycles, and in other catalytic reactions where they can serve as “built-in” oxidants via cleavage of the N-heteroatom bond.[14–16] Furthermore, terminal hydrazidos have the potential to access “redox noninnocent”[17] manifolds through contribution from the β-N lone pair (hydrazido(2−)/1,1-isodiazene resonance),[18] which may allow for redox reactions that would otherwise inaccessible without such metal-ligand cooperativity.
While several early and mid transition metal hydrazido(2−) complexes[19–25] have been synthesized via salt metathesis, protonolysis, or activation of N2.[13,26] However, there are no isolated examples of terminal late transition metal (groups 9 and 10) hydrazido(2−) complexes, and only a single example has been proposed as an intermediate in catalysis. In this reported example, Lu implicates a Co hydrazido(2−) as an intermediate in N2 functionalization.[27] The closest isolated analogue to a group 9 hydrazido(2−) was reported by Hartwig, who isolated the isomeric 1,1-isodiazene complex (PCP)IrNNC5H10.[28] In this case, the neutral 1,1-isodiazene electronic structure must dominate because the hydrazido(2−) form would violate the “oxo wall” of the square planar ligand field. This is borne out in the structural parameters and reactivity of the complex, which has a short N-N bond (1.244(16) Å), long Ir-N bond (2.008(11) Å), and reactivity that mirrors that of free aminonitrenes. As a result of the dearth of group 9 examples, we sought to synthesize iridium hydrazido(2−) complexes and investigate their electronic structure and reactivity. Herein, we report the first example of a terminal hydrazido(2−) complex of a late transition metal and report preliminary reactivity that demonstrates the unique redox noninnocent behavior of the terminal moiety, as well as the synthesis and differential reactivity of related hydrazido dimers.
The synthesis for Cp*-based iridium hydrazidos is reported in Scheme 1. Following Glueck and Bergman’s synthesis of Cp*IrNtBu,[29] initial attempts to synthesize a monomeric hydrazido using lithium 1,1-diphenylhydrazide or lithium 1,1-dimethylhydrazide resulted in dimeric products [Cp*Ir(μ2-N2Ph2)]2 (1) and [Cp*Ir(μ2-N2Me2)]2 (2). Similarly, treatment of [Cp*IrCl2]2 with N-aminopiperidine followed by double dehydrohalogenation of the resulting Ir hydrazine adduct 3 with two equivalents of KOtBu affords a third dimeric product [Cp*Ir (μ2-NNC5H10)]2 (4). Dimerization of analogous imido complexes with small N-substituents is well established.[30,31] In order to increase the steric profile of the β-N substituent, reactions were attempted with 1-amino-2,2,6,6-tetramethylpiperidine (NH2TMP). Treatment of [Cp*IrCl2]2 with two equivalents of LiNHTMP yielded a deep red solution of Cp*IrCl(NHTMP) (5). Interestingly, unlike the other hydrazides, 5 was unable to be dehydrohalogenated by further equivalents of LiNHTMP under standard reaction conditions. However, addition of KOtBu resulted in the formation of the monomeric terminal hydrazido(2−) Cp*IrNTMP (6) as a gold-colored solid in high yield.
Scheme 1.

Synthesis of compounds 1-5.
The crystal structures of compounds 1, 2, 4, and 6 are shown in Figure 1. The structure of 6 contains two independent molecules within the asymmetric unit. The bonding metrics confirm 6 as a terminally-bound hydrazido(2−) that contains an Ir-N triple bond. The Ir–N bond distances (1.714(3) Å and 1.721(3) Å) are similar to the Ir–N distance observed in Bergman’s Ir-N triple bonded Cp*IrNtBu complex (1.712(7) Å). The Nα–Nβ distances (1.340(5) Å and 1.342(4) Å) are significantly shorter than in the dimeric structures 1,2 and 4 (1.384 Å, 1.43 Å and 1.413 Å, respectively). This is consistent with strong π-donation to Ir from Nα in 6, which reduces Nα–Nβ lone pair repulsion. A significant difference in the Ir–Nα–Nβ bond angles exists between the two independent molecules of 6 (178.3(3)° and 166.8(3)°); however, Parkin and Bercaw have argued that steric/crystal packing factors contribute more to the crystalographic bond angles in metal-N multiple bonds than the hybridization at N.[32] In contrast, the bonding metrics of 6 are significantly different from Hartwig’s 1,1-isodiazene complex (PCP)IrNNC5H10, which contains a long Ir–N distance, 2.008(11) Å, a short Nα–Nβ distance, 1.244(16) Å, and an Ir–Nα–Nβ bond angle of 135.1(12)°.[28]
Figure 1.

Thermal ellipsoid (50% probablilty) drawings of a) [Cp*Ir(N2Ph2)]2 (1)) b) [Cp*Ir(N2Me2)]2·LiCl (2) c) [Cp*Ir(NPip)]2 (4) and d) Cp*IrNTMP (6) Hydrogen atoms, solvent molecules, half a tetrameric unit of 2, and a crystallographically independent molecule of 6 have been omitted for clarity.
A qualitative MO description of the relevant π orbitals of 6 is shown in Figure 2. In contrast to normal metal imidos, the Nβ lone pair mixes with one of the Ir-N π bonds, which results in a bonding and antibonding combination.[18] This removes the degeneracy of the Ir-N π-bonds and yields a HOMO comprised of Ir-Nα π-bonding and Nα-Nβ π-antibonding interactions. This antibonding contribution raises the energy of the HOMO relative to the LUMO, which is the Ir-Nα π-antibonding orbital that is out of plane with the β-nitrogen lone pair. DFT calculations (M06-L/MWB60 on Ir; 6-31G(d) on C and H; 6-311+G(2df,p) on N) provide a HOMO-LUMO energy gap of 409 nm, which is in good agreement with the experimental UV-vis of 6, which shows an absorbance shoulder at 400 nm. The resonance Raman spectrum (514.5 nm excitation) shown in Figure 3 also agrees with the DFT calculations, enhancing N–N, Ir–N, and C–C stretching frequencies predicted for the HOMO, and the N–N and Ir–N frequencies are in excellent agreement with related third row metal hydrazidos.[18]
Figure 2.

Qualitative MO diagram of compound 6 showing relevant π-bonding and antibonding combinations of Ir d-orbitals and diazene π-orbitals, as well as the DFT calculated HOMO and LUMO.
Figure 3.

Resonance Raman spectrum of Cp*IrNTMP (6), showing features attributed to N–N (811, 1315 cm−1), Ir–N (1157 cm−1), and C–C (1365 cm−1) bond stretching frequencies.
With terminal and bridged Ir hydrazides in hand, we were interested in exploring the differential reactivity of the series in comparison to Bergman’s terminal Ir imido. A summary of [Cp*IrNR]x reactivity with CO is shown in Scheme 2. Exposure of a C6D6 solution of 6 to an atmosphere to CO yields a deep red solution, which over time results in the formation of Cp*Ir(CO)2 and several organic byproducts, which were identified to be decomposition products of free N-(2,2,6,6-tetramethylpiperidyl)nitrene.[33] In this case, we propose that coordination of CO leads to charge transfer from the hydrazido Nβ lone pair to the Ir, formally reducing it from IrIII to IrI. Then, the resulting 1,1-isodiazene can be displaced by CO, forming Cp*Ir(CO)2 and the free nitrene, which decomposes in solution. In comparison, Cp*IrNtBu and other related imido complexes react with CO to form η2-bound organoisocyanates,[11] Thus, unlike normal terminal imidos, the nitrogen ligand of 6 undergoes CO-induced tautomerization rather than reacting directly with CO. Reactions of 6 with other L donors such as PMe3 or outer-sphere oxidants such as [Fc]+[PF6]-also produce product mixtures from diazene decomposition, and traces of the analogous reduced product Cp*Ir(PMe3)2 were detectable by NMR. In contrast, the Ir-N bond in the dimeric hydrazidos behaves similarly to bridging imidos of group 9.[30] For example, 1 reacts with an atmosphere of CO to give the eventual generation of CO-inserted product 7, which contains two inequivalent Ir centers. The crystal structure of 7 is shown in Figure 4.
Scheme 2.

Reactions of CO with Cp*IrNTMP (6) (top), Bergman’s Cp*IrNtBu (middle) and [Cp*Ir(N2Ph2)]2 (1) (bottom).
Figure 4.
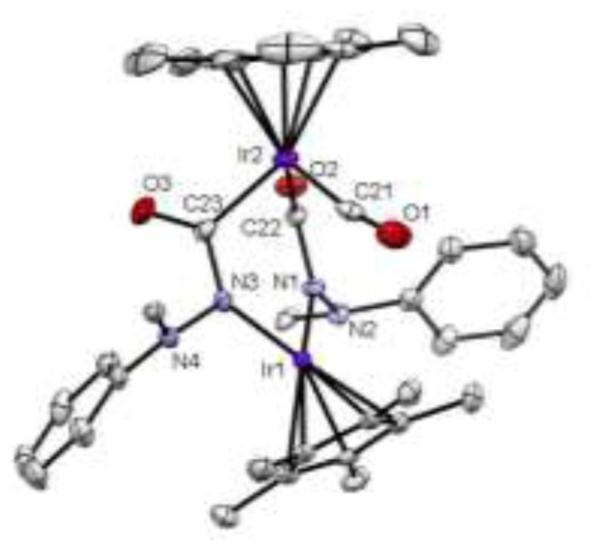
Thermal ellipsoid (50% probability) drawing of compound 7. Hydrogen atoms have been omitted and two phenyl rings have been reduced to ipso carbon for clarity. Select bond lengths (Å) and angles (°): Ir1–N1, 2.020(3); Ir1–N3, 2.022(3); N1–N2, 1.429(4); N3–N4, 1.418(4); Ir2–C21, 1.842(4); Ir2–C22, 2.044(4); Ir2–C23, 2.046(4); C21–O1, 1.143(5); C22–O2, 1.222(4); C23–O3, 1.225(5); Ir2–C21–O1, 170.3(4).
Reaction of terminal hydrazidos with electrophiles also yields differential and remarkable reactivity. In the case of 6, one could envision an electrophile such as MeI reacting in one of three ways: (1) direct reaction with the HOMO (predominantly Nβ lone pair), (2) attack of the electrophile by the Ir metal center in an SN2-like fashion, or (3) reaction across the Ir-N bond in analogy to Cp*IrNtBu, which ultimately gives [tBuNMe3]+I− and [Cp*IrI2]2. Remarkably, the reaction of 6 with stoichiometric MeI produces complex 8, the apparent product of nucleophilic attack by an IrIII center on MeI (Scheme 3). 8 is isolated as a mixture of disastereomers, both of which can be crystallized. Their solid-state structures are reported in Figure 5.
Scheme 3.

Reaction of Cp*IrNTMP (6) with methyl iodide to produce compound 8 with the loss of methane. The proposed mechanism for the formation of 8 is also shown, showing the concerted nucleophilic attack and hydrazido(2−) oxidation to form intermediate A. When using ethyl iodide, competitive β-H elimination allows for the formation of ethylene as well as compound 8.
Figure 5.

Thermal ellipsoid (50% probability) drawings of both diasteromers of compound 8. Hydrogen atoms and one crystallographically independent molecule in 8b have been omitted for clarity. Select bond lengths (Å) and angles (°) for 8a: Ir1–N1, 1.936(6); N1–N2, 1.249(8); Ir1–C11, 2.107(7); Ir1–I1, 2.6881(8); Ir1–N1–N2, 121.5(5); C12–N2–C16, 122.7(4).
The structure of 8 shows an elongated Ir–N bond distance of 1.939(6) Å as well as a shortened Nα–Nβ distance of 1.249(8) Å, indicating double bond character between the nitrogens. Thus, the structural parameters of 8 demonstrate a formal oxidation of the hydrazido(2−) moiety to the neutral isodiazene ligand, similar to Hartwig’s isolated isodiazene complex. This is the first instance of reagent-induced redox isomerism in a hydrazido(2−) moiety, where the Nβ lone pair ultimately provides an electron reservoir for reactivity at Ir.
The proposed mechanism for the formation of 8 is shown in Scheme 3. MeI undergoes nucleophilic attack by Ir with concurrent electron transfer from the Nβ to Ir. This “redox noninnocent” electron transfer from hydrazide(2−) to isodiazene helps to avoid the intermediacy of the IrV oxidation state. The resulting IrIII methyl isodiazene structure A can then undergo σ-bond metathesis with a proximal piperidine methyl group to form the cyclometallated product 8, with concomitant loss of methane. The reaction of 6 with CD3I in C6D6 carried out in a sealed J-Young tube shows a peak for CD3H, confirming the production of methane. At this stage, the exact nature of the tautomerization mechanism and its degree of concertedness remain ambiguous: the arrow-pushing designation in Scheme 3 is used simply to indicate that tautomerization occurs while the complex is undergoing reactivity.
Complex 8 can also be formed from the reaction of 6 with ethyl iodide. The kinetics of the reaction are slower, as would be expected for an SN2-type nucleophilic attack by Ir.[34] In addition to the expected ethane byproduct, ethylene is also observed by 1H NMR of the reaction mixture. The generation of ethylene presumably occurs through a competitive β-H elimination from the ethyl intermediate A followed by subsequent intramolecular σ-bond metathesis of the resulting iridium hydride with the piperidine methyl group.
In summary, monomeric iridium terminal hydrazido(2−) complexes can be synthesized using bulky, alkyl 1,1-organohydrazines. Cp*IrNTMP displays a short, linear Ir–N linkage indicative of triple bonding. The Ir–Nα–Nβ moiety displays remarkable redox non-innocent behavior in all characterized cases; reactions with neutral CO results in the formally reduced Cp*Ir(CO)2, while reactions with electrophiles results in a net oxidation of the ligand rather than the metal center. We are currently exploring redox catalytic applications of this new moiety on Ir and related late transition metal centres.
Supplementary Material
Table 1.
Select bond distances (Å) and angles (°) for compounds 1,2,4 and 6.
| Bond | 1[a] | 2[a] | 4[a] | 6[b] |
|---|---|---|---|---|
| Ir-Nα | 1.982 | 1.99 | 1.983 | 1.718 |
| Nα–Nβ | 1.384 | 1.43 | 1.413 | 1.341 |
| Ir-Cpcentroid | 1.836 | 1.837 | 1.846 | 1.842 |
| Ir-Nα–Nβ | 129.8 | 126.1 | 127.1 | 172.3 |
Metrics listed are average values for both Ir centers.
Metrics listed for compound 6 are average values for two crystallographically independent monomers.
Acknowledgements
Financial support was provided by the University of Minnesota (start up funds) and the ACS Petroleum Research Fund (ACS-PRF 54225-DNI3). The Bruker-AXS D8 Venture diffractometer was purchased through a grant from NSF/MRI (1224900) and the University of Minnesota. Equipment purchases for the NMR facility were supported through a grant from the NIH (S10OD011952) with matching funds from the University of Minnesota. The Minnesota Supercomputing Institute (MSI) at the University of Minnesota provided resources that contributed to the research results reported within this paper. We would like to thank Kari Kusler for assistance with DFT calculations, as well as Dr. Victor Young for help with crystal structure solutions.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Kleespies ST, Oloo WN, Mukherjee A, Que L. Inorg. Chem. 2015;54:5053–64. doi: 10.1021/ic502786y. [DOI] [PubMed] [Google Scholar]
- [2].Lyaskovskyy V, Suarez AIO, Lu H, Jiang H, Zhang XP, de Bruin B. J. Am. Chem. Soc. 2011;133:12264–73. doi: 10.1021/ja204800a. [DOI] [PubMed] [Google Scholar]
- [3].Perry RH, Cahill TJ, Roizen JL, Du Bois J, Zare RN. Proc. Natl. Acad. Sci. 2012;109:18295–18299. doi: 10.1073/pnas.1207600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gao G-Y, Jones JE, Vyas R, Harden JD, Zhang XP. J. Org. Chem. 2006;71:6655–6658. doi: 10.1021/jo0609226. [DOI] [PubMed] [Google Scholar]
- [5].Dai X, Kapoor P, Warren TH. J. Am. Chem. Soc. 2004;126:4798–9. doi: 10.1021/ja036308i. [DOI] [PubMed] [Google Scholar]
- [6].Jenkins DM, Betley TA, Peters JC. J. Am. Chem. Soc. 2002;51:11238–11239. doi: 10.1021/ja026852b. [DOI] [PubMed] [Google Scholar]
- [7].Cowley RE, Bontchev RP, Sorrell J, Sarracino O, Feng Y, Wang H, Smith JM. J. Am. Chem. Soc. 2007;129:2424–2425. doi: 10.1021/ja066899n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang L, Liu Y, Deng L. J. Am. Chem. Soc. 2014;136:15525–15528. doi: 10.1021/ja509731z. [DOI] [PubMed] [Google Scholar]
- [9].Du J, Wang L, Xie M, Deng L. Angew. Chemie Int. Ed. 2015;54:12640–12644. doi: 10.1002/anie.201505937. [DOI] [PubMed] [Google Scholar]
- [10].Mindiola DJ, Hillhouse GL. J. Am. Chem. Soc. 2001;123:4623–4624. doi: 10.1021/ja010358a. [DOI] [PubMed] [Google Scholar]
- [11].Geer AM, Tejel C, López JA, Ciriano MA. Angew. Chemie Int. Ed. 2014;53:5614–5618. doi: 10.1002/anie.201400023. [DOI] [PubMed] [Google Scholar]
- [12].Cowley RE, Holland PL. Inorg. Chem. 2012;51:8352–61. doi: 10.1021/ic300870y. [DOI] [PubMed] [Google Scholar]
- [13].Anderson JS, Cutsail GE, Rittle J, Connor BA, Gunderson WA, Zhang L, Hoffman BM, Peters JC. J. Am. Chem. Soc. 2015;137:7803–7809. doi: 10.1021/jacs.5b03432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schofield AD, Nova A, Selby JD, Manley CD, Schwarz AD, Clot E, Mountford P. J. Am. Chem. Soc. 2010;132:10484–97. doi: 10.1021/ja1036513. [DOI] [PubMed] [Google Scholar]
- [15].Gehrmann T, Lloret Fillol J, Scholl SA, Wadepohl H, Gade LH. Angew. Chemie Int. Ed. 2011;50:5757–5761. doi: 10.1002/anie.201101070. [DOI] [PubMed] [Google Scholar]
- [16].Herrmann H, Lloret Fillol J, Wadepohl H, Gade LH. Angew. Chemie Int. Ed. 2007;46:8426–8430. doi: 10.1002/anie.200703938. [DOI] [PubMed] [Google Scholar]
- [17].Lyaskovskyy V, de Bruin B. ACS Catal. 2012;2:270–279. [Google Scholar]
- [18].Tonks IA, Durrell AC, Gray HB, Bercaw JE. J. Am. Chem. Soc. 2012;134:7301–4. doi: 10.1021/ja302275j. [DOI] [PubMed] [Google Scholar]
- [19].Clulow AJ, Selby JD, Cushion MG, Schwarz AD, Mountford P. Inorg. Chem. 2008;47:12049–62. doi: 10.1021/ic801735c. [DOI] [PubMed] [Google Scholar]
- [20].Tonks IA, Bercaw JE. Inorg. Chem. 2010;49:4648–56. doi: 10.1021/ic1004193. [DOI] [PubMed] [Google Scholar]
- [21].Banerjee S, Odom AL. Dalt. Trans. 2008:2005–2008. doi: 10.1039/b718968k. [DOI] [PubMed] [Google Scholar]
- [22].Patel S, Li Y, Odom AL. Inorg. Chem. 2007;46:6373–81. doi: 10.1021/ic700426s. [DOI] [PubMed] [Google Scholar]
- [23].Li Y, Shi Y, Odom AL. J. Am. Chem. Soc. 2004;126:1794–803. doi: 10.1021/ja038320g. [DOI] [PubMed] [Google Scholar]
- [24].Danopoulos AA, Wilkinson G, Williams DJ. J. Chem. Soc. Dalt. Trans. 1994:907. [Google Scholar]
- [25].Bustos C, Manzur C, Carrillo D, Robert F, Gouzerh P. Inorg. Chem. 1994;33:1427–1433. [Google Scholar]
- [26].Yandulov DV, Schrock RR. J. Am. Chem. Soc. 2002;124:6252–6253. doi: 10.1021/ja020186x. [DOI] [PubMed] [Google Scholar]
- [27].Siedschlag RB, Bernales V, Vogiatzis KD, Planas N, Clouston LJ, Bill E, Gagliardi L, Lu CC. J. Am. Chem. Soc. 2015;137:4638–41. doi: 10.1021/jacs.5b01445. [DOI] [PubMed] [Google Scholar]
- [28].Huang Z, Zhou JS, Hartwig JF. J. Am. Chem. Soc. 2010;132:11458–60. doi: 10.1021/ja1053835. [DOI] [PubMed] [Google Scholar]
- [29].Glueck DS, Wu J, Hollander FJ, Bergman RG. J. Am. Chem. Soc. 1991;113:2041–2054. [Google Scholar]
- [30].Tejel C, Ciriano MA, Jiménez S, Passarelli V, López JA. Inorg. Chem. 2008;47:10220–10222. doi: 10.1021/ic801703d. [DOI] [PubMed] [Google Scholar]
- [31].Dobbs D, Bergman RG. Organometallics. 1994;13:4594–4605. [Google Scholar]
- [32].Parkin G, Van Asselt A, Leahy DJ, Whinnery L, Hua NG, Quan RW, Henling LM, Schaefer WP, Santarsiero BD, Bercaw JE. Inorg. Chem. 1992;31:82–85. [Google Scholar]
- [33].Hinsberg WD, Schultz PG, Dervan PB. J. Am. Chem. Soc. 1982;104:766–773. [Google Scholar]
- [34].Labinger JA. Organometallics. 2015;34:4784–4795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.