

Summary
Tauopathies are neurodegenerative disorders that affect distinct brain regions, progress at different rates, and exhibit specific patterns of tau accumulation. The source of this diversity is unknown. We previously characterized two tau strains that stably maintain unique conformations in vitro and in vivo, but did not determine the relationship of each strain to parameters that discriminate between tauopathies such as regional vulnerability or rate of spread. We have now isolated and characterized 18 tau strains in cells based on detailed biochemical and biological criteria. Inoculation of transgenic tau P301S (PS19) mice with these strains causes strain-specific intracellular pathology in distinct cell types and brain regions, and induces different rates of network propagation. In this system, strains alone are sufficient to account for diverse neuropathological presentations, similar to those that define human tauopathies. Further study of these strains can thus establish a structural logic that governs these biological effects.
Introduction
Tauopathies are a diverse group of neurodegenerative diseases characterized by clinical heterogeneity, progressive deposition of tau protein aggregates in characteristic brain regions, and distinct cellular pathologies (Lee et al., 2001). The etiology of this clinical and pathological diversity is unknown, but may hold the key to accurate diagnosis, prognosis, and therapy. Tauopathies include Alzheimer's disease (AD), frontotemporal dementias (FTDs), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and others (Lee et al., 2001). Most tauopathies are sporadic, but dominantly inherited mutations in the MAPT gene, which encodes the tau protein, lead to specific FTD syndromes (Hutton et al., 1998). Disease-associated mutations enhance the ability of tau to form amyloids, which are ordered protein assemblies rich in cross beta sheet structure (Knowles et al., 2014), and support a causal role for tau aggregation in pathogenesis (Barghorn et al., 2000).
In AD, the most common tauopathy (Lee et al., 2001), tau amyloid deposition occurs in an orderly fashion, beginning in the transentorhinal cortex, spreading to synaptically connected regions such as the hippocampus, and eventually moving to more distant regions of the neocortex (Braak and Braak, 1991). Multiple studies have now documented tau aggregate uptake, “seeding” (i.e. aggregate serving as a template for the conversion of monomer to a fibrillar form), and transfer of aggregates among cultured cells (Frost et al., 2009a; Guo and Lee, 2011; Holmes et al., 2013; Nonaka et al., 2010). Experimental evidence suggests that “propagation,” or the movement of tau aggregates between connected neurons with seeding of tau monomer in recipient cells, mediates this progression in vivo (Sanders et al., 2016; Walker and Jucker, 2015). Importantly, injection of tau aggregates into mice that express human tau protein induces tau pathology that spreads outwards along known brain networks (Clavaguera et al., 2009; Iba et al., 2013). Transgenic mice that limit the expression of tau to the entorhinal cortex also show spread of tau pathology to distant, connected brain regions (de Calignon et al., 2012; Liu et al., 2012). Together, these studies suggest that propagation of an aggregated state underlies the progression of tau pathology. These observations match the established mechanisms of propagation of pathological prion protein (PrP) (Prusiner, 1998).
The pathology of tauopathies occurs in distinct brain regions (Arnold et al., 2013), involves disparate brain networks (Raj et al., 2012; Zhou et al., 2012), and features unique tau inclusions in various cell types (Kovacs, 2015). Individuals may develop rapid or slow neurodegeneration even within the same syndrome (Armstrong et al., 2014; Thalhauser and Komarova, 2011). The basis of these diverse disease patterns is unknown.
We initially observed that tau adopts multiple, stably propagating conformers in vitro, and speculated that structural variation in amyloids could underlie different tauopathies (Frost et al., 2009b). We subsequently determined that tau forms discrete prion “strains” that propagate with remarkable fidelity through living systems (Sanders et al., 2014). The concept of prion strains derived from a realization that PrP prions can induce distinct transmissible spongiform encephalopathies with reproducible incubation times and patterns of neuropathology (Collinge and Clarke, 2007). It is now clear that PrP prion strains derive from different PrP amyloid conformations, and produce predictable incubation times and neurodegenerative phenotypes upon serial passage in vivo. Moreover, distinct PrP strains probably account for the myriad features of individual PrP prion diseases (Collinge et al., 1996). The concept of a strain as a stably propagating structure that induces a specific and reproducible phenotype is critical, as it anticipates and enables mechanistic, diagnostic, and therapeutic insights based on knowledge of a defined molecular assembly (Sanders et al., 2016).
In addition to the distinct morphologies of tau fibrils and isoform composition of amyloid deposits in tauopathies (Lee et al., 2001), prior studies have suggested that unique tau amyloid structures might account for some aspects of clinical variation. Injection of homogenate from different tauopathy brains into a mouse model that expresses full-length human tau induced pathology that closely resembled that of the human source cases (Clavaguera et al., 2013). In a similar study, tau aggregates purified either from AD or CBD brains induced distinct patterns of tau pathology that affected different cell populations in transgenic mice that express 1N4R tau with a P301S mutation (PS19) (Boluda et al., 2015). However, both works relied upon a limited number of patient samples that likely contain a heterogeneous mixture of tau aggregate conformations (Sanders et al., 2014). Thus, the structure and biochemical properties of injected tau aggregates could not be well defined, making it impossible to directly link an amyloid structure to pathology.
Like PrP, tau forms bona fide prion strains that propagate in cells and animals (Sanders et al., 2014). We have now isolated 18 putative tau prion strains derived from recombinant, mouse, or human sources. We have studied them extensively in vivo, and find that they can account for diverse and predictable patterns of neuropathology. This work thus develops a framework to understand the relationship of tau prion structure to distinct tauopathy syndromes.
Results
Generation of a library of tau strains
We previously created a monoclonal HEK293 cell line (Clone 1/DS1) that stably expresses the repeat domain (amino acids 244-372) of 2N4R tau with two disease-associated mutations (P301L and V337M), which allows us to indefinitely propagate tau prion strains derived from a variety of sources (Sanders et al., 2014). We treated DS1 cells with tau aggregates from diverse recombinant, mouse, and human brain samples (Figure 1A). We isolated and amplified 90 monoclonal cell lines that stably propagated tau aggregates and froze them for later study. Following preliminary analyses by several assays (inclusion morphology, limited proteolysis, and seeding by tau split-luciferase complementation, as described previously (Sanders et al., 2014)), we isolated 18 putatively distinct strains (DS2-DS19; see Figure S1A for origin of each strain). These strains featured several striking differences in their subcellular distribution of aggregated tau (Figure 1B): a single, large juxtanuclear inclusion (Ordered: DS3, 10, 14, 19), prominent nuclear inclusions (Speckles: DS4, 5, 7, 8, 9, 12, 16, 17, 18), aggregated tau that failed to organize into ordered inclusions (Disordered: DS11, 13), fibril-like ribbons of aggregated tau throughout the cytoplasm (Threads: DS6, 15), and one strain that sectored with time, reverting from the aggregated state to the soluble diffuse state (Mosaic: DS2). Importantly, with the exception of the mosaic strain DS2, every daughter cell featured the same inclusion morphology even after months of passage, suggesting that each monoclonal cell line stably propagated a single strain.
Figure 1. Generation and characterization of a tau prion strain library.
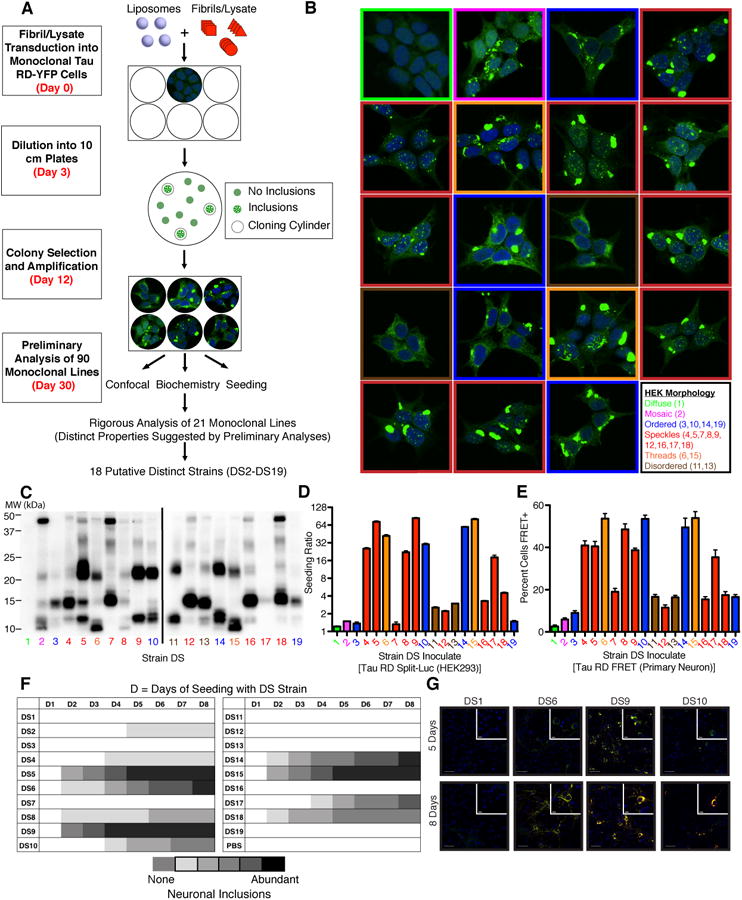
(A) A monoclonal HEK293 line stably expressing tau RD(P301L/V337M) (“LM”)-YFP (DS1) was treated with diverse sources of fibrillar tau seeds. 90 monoclonal lines that stably propagated tau inclusions were derived and characterized by the indicated metrics. 18 strains are differentiated based on their unique properties in the indicated assays. See Figure S1A for origin of inoculates used to derive each strain.
(B) Several tau inclusion phenotypes were identified in the monoclonal strains: mosaic (magenta), ordered (blue), speckles (red), threads (orange), and disordered (brown). With the exception of the mosaic phenotype, these inclusion phenotypes stably propagate to daughter cells over months of division. A negative control cell line (DS1) features diffuse tau (green). See Figure S2F-H for data regarding stability of specific strains upon passage into DS1.
(C) Limited proteolysis using pronase differentiates the protected fibrillar cores in individual tau strains. Unique “fingerprints” along with other metrics indicated structurally distinct tau prion strains. See Figure S1B for pronase digestion of strains diluted with HEK lysate.
(D) Seeding activity of strains in a split-luciferase assay. A tau RD(P301S) split-luciferase assay based on enzymatic complementation following aggregation demonstrates differences in strain seeding activities following introduction into the cytoplasm using lipofectamine. Seeding ratio indicates luminescence relative to sham treatment. Biological quadruplicates with saturating quantities of lysate were averaged. Error bars represent S.E.M. for biological quadruplicates.
(E) Strain seeding activities replicate in primary neurons expressing tau RD. Primary hippocampal neurons expressing tau RD(P301S)-CFP and tau RD(P301S)-YFP were treated with lysates derived from various strains. After 96 hours, neurons were fixed and the percentage of cells featuring seeded aggregates was determined by FRET flow cytometry. Error bars represent S.E.M. for biological quintuplicates.
(F) Strain seeding activities replicate in primary neurons expressing full-length tau. Primary hippocampal neurons expressing 1N4R tau(P301S)-YFP were exposed to lysates from each strain and the extent of seeding was semi-quantitatively determined at various time points (D =number of days) based on the extent of visible YFP puncta (0-5: 0 = none; 5 = abundant inclusions). Strains show variable lag times and extent of seeding, which correlates with the split-luciferase complementation assay.
(G) Strains differentially induce the formation of insoluble tau aggregates in primary neurons. Triton X-100 was used to remove soluble tau and primary neurons were stained for conformationally altered tau (MC1) five or eight days following seeding. Strains show significant differences in seeding of aggregation in neurons. This parallels differences in the split-luciferase complementation assay. Scale bars represent 50 μm for the wide view and 10 μm for the inset images. See Figure S1C for representative images for all strains.
To examine whether the tau inclusions were composed of structurally distinct tau amyloids, we used limited proteolysis, an assay previously shown to differentiate prion strains derived from PrP (Bessen and Marsh, 1994) and tau (Sanders et al., 2014). We thus determined a “fingerprint” for each putative strain based on the regions of tau protected from digestion by pronase (Figure 1C). These digestion patterns were stable even upon dilution with HEK lysate (Figure S1B), confirming their independence from the amount of aggregated tau present in each sample. This assay suggested that while cell lines with different inclusion morphologies always propagated different conformations, inclusion morphology alone could not discriminate all strains.
Tau strains show unique seeding profiles in dividing cells and neurons
Next, we examined the ability of each putative strain to seed tau monomer using a cell line that expresses tau RD(P301S) fused to the N or C terminus of click beetle green luciferase enzyme (Mirbaha et al., 2015; Sanders et al., 2014). When saturating concentrations of lysate were transduced directly into the cytoplasm of these cells, induction of luminescence ranged from 0.3 to 80-fold increase in seeding at saturation vs. background, termed the “seeding ratio” (Figure 1D). We observed no association between inclusion morphology and seeding. For example the four ordered strains showed different seeding activity (DS3, 10, 14, 19). The relative seeding abilities of individual tau strains were largely recapitulated when lysates were applied to primary hippocampal neurons expressing tau RD(P301S) FRET biosensor proteins (Holmes et al., 2014) (Figure 1E). We also observed these relative differences upon exposure of strains to primary neurons expressing full-length 1N4R P301S-YFP, suggesting common effects on either truncated or full-length tau. Strains also showed different lag phases to induce inclusions in neurons that express full-length tau (Figure 1F-G, S1C).
Seeding activity correlates with toxicity in dividing cells
To investigate the relationship between seeding activity and toxicity, we first performed a detailed titration of cell lysates (30 pg to 10 μg) from the 18 putatively distinct strains. We determined the EC50 and peak seeding ratio for each strain using the tau split-luciferase complementation assay. Strains differentially seeded tau monomer in cell culture as reflected by their peak seeding ratios (Figure 2A,B). Different strains displayed >10× range for EC50 (DS9: 287 ng; DS3, 4908 ng) (Figure 2B). Peak seeding and EC50 strongly anti-correlated (Figure 1C), suggesting that peak seeding accurately reflects a strain's potency in triggering tau aggregation.
Figure 2. Seeding activity, but not insoluble tau, correlates with strain toxicity in vitro.

(A) Strains have large differences in seeding of monomeric tau as determined by a tau split-luciferase complementation assay. Strain lysates were transduced into tau RD(P301S) split-luciferase cells, seeding ratios relative to sham treatment were determined, and titration curves were plotted using non-linear regression with a one-phase decay fit. Curves are plotted on two separate graphs for clarity. Error bars represent S.E.M for biological quadruplicates.
(B) Based on titration curves in the tau split-luciferase complementation assay, the EC50, inflection point, and peak seeding ratio were determined for each strain. The inflection point represents the amount of lysate required to achieve a 50% increase in luminescence relative to sham treatment.
(C) Peak seeding significantly correlates with EC50s for the strain library in the tau split-luciferase complementation assay.
(D) Strains display significant differences in toxicity. Strains were transduced in biological triplicates into cells overexpressing both tau RD(LM)-CFP and tau RD(LM)-YFP. After 72 hours, equivalent numbers of aggregate-containing (FRET+) cells were sorted for each condition by FRET flow cytometry. For the negative control (DS1), aggregate-negative (FRET-) cells were sorted. Sorted cells were allowed to proliferate in technical sextuplicates for 1 week. Aggregate-positive (FRET+) and aggregate-negative (FRET-) cells were then quantified by flow cytometry. The presence of FRET- cells in certain conditions reflects the fact that some strains lose the aggregated state with cell division. Technical sextuplicates were averaged for each biological replicate. Error bars represent S.E.M. of biological triplicates.
(E) Aggregate-positive (FRET+) cells were quantified and plotted after one week of growth. This highlights the variable growth defects in aggregate-containing cells. Error bars represent S.E.M. of biological triplicates.
(F) Toxicity correlates with seeding activity. The number of aggregate-positive (FRET+) cells for a strain was plotted against its peak seeding ratio in the tau split-luciferase complementation assay. Strains that seed more efficiently are associated with reduced growth of aggregate-positive cells. See Figure S2A and B for data indicating the correlation between a strain's toxicity, EC50, and inflection point in the seeding assay.
(G) Sedimentation analysis of strains. Lysates were ultracentrifuged and tau as well as a loading control protein (cofilin) were probed in the total, supernatant, and pellet fractions (Tot = total, Sup = supernatant, Pel = pellet). Blots are representative of biological quadruplicates.
(H) Strains feature the majority of tau in the insoluble fraction. Densitometric analysis of tau in the total, supernatant, and pellet fractions was used to calculate supernatant to total ratios (a higher ratio indicates a smaller proportion of tau in the insoluble pellet). Error bars represent S.E.M. of biological quadruplicates.
(I) Densitometric analyses highlight variation in insoluble tau in the various strains. Error bars represent S.E.M. of biological quadruplicates. See Figure S2C for quantification of tau in the total fractions.
(J) Lack of correlation between insoluble tau and seeding activity as measured by peak seeding ratio. See Figure S2D for data indicating lack of correlation between total tau and seeding.
(K) Lack of correlation between total tau and seeding activity. See Figure S2E for data indicating lack of correlation between total tau and toxicity.
We then compared the toxicity of each putative strain to their seeding potential. We generated a cell line (LM10) that expresses a mutant tau RD FRET pair (CFP/YFP) at high levels, and assessed the growth potential of cells propagating various strains after first isolating aggregate-positive cells by FRET FACS (Holmes et al., 2014). Several ordered and mosaic strains (DS2, 3, 10, 19) lost the aggregated state with repeated cell division (“sectored”) (Figure 2D). All others stably propagated the aggregated state, but exhibited growth defects relative to LM10 cells that lacked tau aggregates (Figure 2E). Strains that sectored, a possible correlate of low seeding, were the least toxic. All three seeding metrics (peak seeding, EC50, and inflection point) correlated with inhibition of growth (Figure 2F, Figure S2A-B). In other words, strains that seeded more efficiently were significantly more toxic to cells that express high levels of monomeric tau.
We next performed sedimentation analyses to determine the level of soluble, insoluble, and total tau for all 18 putative strains (Figure 2G). While each strain (DS2-19) contained the majority of tau in the insoluble fraction (Figure 2G-H), different strains featured variable levels of insoluble (Figure 2I) and total (Figure S2C) tau. Neither total nor insoluble tau levels correlated with seeding (Figure 2J, S2D) or toxicity (Figure 2K, S2E). Thus, structural differences among strains, rather than soluble/insoluble tau levels per se, account for seeding activity and toxicity in dividing cells.
Diversity of pathology induced by tau strains
We hypothesized that to account for variation in tauopathies, individual tau strains should produce a wide array of pathological phenotypes in vivo. To test whether these putative strains can produce such diversity, we inoculated cell lysate from each line (DS1-DS19) into the PS19 mouse model that expresses 1N4R tau with the FTDP-17-associated P301S mutation from the prion promoter (Yoshiyama et al., 2007) (Figure 3A) and examined tau pathology induced eight weeks after inoculation (Figure 3B-J).
Figure 3. Tau prion strains induce diverse patterns of hippocampal tau pathology.
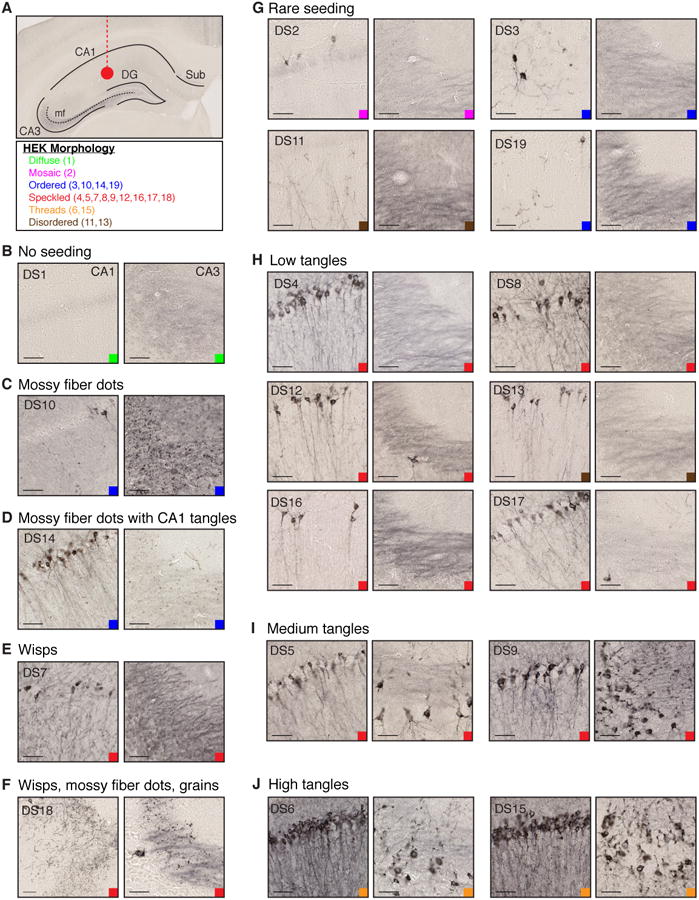
(A) Tau strains (10 μg) were injected into the left hippocampus of young PS19 mice (n=3 per condition, see Supplemental Table 1). Mouse brains were collected 8-weeks after injection. Relevant regions are indicated on a representative mouse hippocampus (DG, dentate gyrus; mf, mossy fibers; Sub, Subiculum). HEK Morphology table indicates the inclusion patterns in various strains, grouped by text color. Color-coded squares indicate these HEK cell-associated patterns in all images (B-J).
(B) DS1 injection produces no AT8 tau pathology. Representative images of CA1 and CA3 are displayed. Scale bars represent 50 μm. See Figure S3A for whole hippocampal images for DS1-19.
(C) DS10 produces AT8 positive mossy fiber “dot” pathology, with limited CA1 pathology as observed previously (Sanders et al., 2014). See Figure S3C for contralateral mf pathology.
(D) DS14 seeds mossy fiber dots similar to DS10, as well as tangle-like pathology, indicating that it is a distinct strain despite its other similar features to DS10. See Figure S3C for contralateral mossy fiber and CA1 pathology.
(E) DS7 induces “wisps” that resemble neuropil threads, but may fall within axon terminals and the dendritic tree of pyramidal neurons.
(F) DS18 pathology includes wisps and mossy fiber dots similar to DS7 and 10 respectively, as well as “grains” that are found throughout much of the hippocampus. See Figure S3D for data indicating that these phenotypes spread to distant synaptically connected locations including the entorhinal cortex.
(G-J) Several strains produce different levels of tangle-like AT8 pathology in CA1 and CA3 of the hippocampus.
(G) DS2, 3, 11, and 19 induce rare AT8 pathology in pyramidal CA1 neurons. The localization of AT8 staining varies in certain cases (cell body versus axonal pathology in DS2 and 11 respectively).
(H) DS4, 8, 12, 13, 16, and 17 induce slightly stronger tangle-like pathology in CA1 of the hippocampus (“low tangles”). CA3 shows limited or no tangle pathology at this time point.
(I) DS5 and 9 produces AT8 tangle-like tau pathology that reaches CA3 of the hippocampus as well as CA1 pyramidal cells (“medium tangles”). Tangles appear relatively consolidated within the soma of neurons. See Figure S3B for spread of tau pathology to the contralateral hippocampus and ipsilateral EC.
(J) DS6 and 15 display the highest level of tangle-like AT8 pathology (“high tangles”). Highly consolidated pathology was observed throughout cell bodies and axons of CA1 and CA3 neurons. See Figure S3B for spread of tau pathology to the contralateral hippocampus and ipsilateral EC.
Pathology varied greatly between putative strains, and was often, but not always, consistent with seeding activity observed in culture (Figure 1D-G). Strains with low seeding activity (DS2, 3, 11, 19) produced a “rare seeding” phenotype in vivo, with limited AT8 pathology localized in CA1 of the ipsilateral hippocampus (Figure 3G, S3A). These strains appear different in terms of their AT8 subcellular localization (soma versus axonal pathology in DS2 and 11), but this may reflect different levels of maturation of tau aggregates (e.g. pretangles and tangles). Several strains induced low level, yet consistent tangle-like pathology in CA1 of the hippocampus (DS4, 8, 12, 13, 16, 17) (Figure 3H, S3A). Strains with the highest seeding activity in culture (DS5, 6, 9, 15) caused widespread tangle-like tau pathology throughout several hippocampal regions (Figure 3I, J). Pathology from these robust strains spread to distant regions such as the entorhinal cortex (EC) and contralateral hippocampus (Figure S3B).
Several putative strains induced unique pathology in the hippocampus. DS10 typically produced AT8-positive “dots” in the mossy fiber tracts of the ipsilateral and contralateral hippocampus, while mostly sparing CA1 pyramidal neurons (Figure 3C, S3C) as was observed in previous work (Sanders et al., 2014). DS14, which shared the same ordered cellular phenotype as DS10, also produced mossy fiber dots (Figure 3D). However, this strain showed higher seeding activity in culture, and additionally induced CA1 tangle-like pathology in the ipsilateral and contralateral hippocampus (Figure S3C).
DS7 produced “wisps” that resemble neuropil threads (Figure 3E) while inducing weaker AT8 pathology in the main axon branches. This contrasted with several other speckled phenotype strains, which primarily induced AT8 pathology in the main axon (Figure 3H-I). DS18 produced wisps, mossy fiber dots, and “grains,” which are AT8 positive puncta found throughout the hippocampus (Figure 3F). DS18-inoculated mice also developed grain pathology in the contralateral hippocampus and wisps in the EC, indicating these specific phenotypic features can spread to distant regions (Figure S3D).
Critically, strains with matching limited proteolysis fingerprints produced similar histopathology in vivo (DS3 and 19; DS6 and 15; DS12 and 16) (Figure 3F,G,I). These pairs of similar strains (which may propagate identical tau aggregate conformations) displayed similar seeding activity and toxicity levels, and induced similar phenotypes in primary neuron culture. Importantly, DS6 and 15 derive from distinct aggregate sources (aged PS19 mice and recombinant fibrils, respectively), indicating that these strain-based phenotypes are conformation-specific rather than source-specific.
Stability of distinct tau prion strains
We previously demonstrated that DS9 and 10 propagate unique conformations, and produce identical phenotypes upon re-introduction into DS1 cells. To test whether other strains meet these same criteria for stable prion strains (Sanders et al., 2014), we transduced cell lysate from strains with distinct cellular morphology, seeding activity, and/or in vivo phenotypes into naïve DS1 cells (DS1, 4, 6, 7, 9, 10, or 11). We first performed a blinded analysis of cell morphology from a polyclonal population at 5 and 8 days after transduction. The original DS1 and secondary polyclonal DS1 cell lines contained no aggregate-positive cells (Figure S2F-H). Blinded counts of DS4, 7, and 9 demonstrated the polyclonal population maintained the nuclear speckled phenotype, while DS10 and 11 secondary lines were readily scored as ordered and disordered. DS6 threads that project from a large juxtanuclear aggregate are only readily apparent when assessing morphology on a population level rather than within individual cells. However, transduction of this cell line reliably induced overt “threads” in the vast majority of secondary cells at 5 days after transduction. By 8 days, tau aggregates in DS6 secondary cells appeared to mature, and the cellular morphology and blinded scoring results resembled that of the original DS6 cell line (Figure S2F-H).
To further examine the stability of each strain phenotype, monoclonal secondary cell lines that stably propagate aggregates were isolated by unbiased single-cell sorting at 4 days after transduction of DS4, 6, 7, 9, 10, and 11 into the LM1 cell line. Secondary strains displayed the same cellular morphology as the original cell lines (Figure S7A). We tested the seeding activity of each secondary cell line compared to the original strains by transducing cell lysate into a biosensor cell line that expresses tau RD(P301S)-CFP/YFP, and quantifying fluorescence resonance energy transfer (FRET) by flow cytometry (Holmes et al., 2014). Secondary cell lines produced seeding activity similar to their respective source strain (Figure S7C). Thus, these are stable strains that induce unique cellular phenotypes even upon serial passage in culture.
Specific strains reliably induce astrocytic pathology
Several tauopathies, including CBD and PSP, feature unique glial cell pathology (Kovacs, 2015). While the mechanisms that underlie these patterns are not known, previous work suggested that inoculation of CBD patient lysate into PS19 mice is sufficient to induce astrocytic pathology (Boluda et al., 2015). Interpretation of this result was limited by the potential for individual patient brains to contain multiple strains (Sanders et al., 2014). Further, two different paradigms were used for purification of AD versus CBD derived tau, which might also have affected the observed phenotypes (Boluda et al., 2015). We thus assessed astrocytic pathology after inoculation with isolated, individual tau strains.
Eight weeks after inoculation with DS7 and DS9, we observed tau pathology reminiscent of astrocytic plaques in multiple animals, as noted by small AT8-positive inclusions arranged in ring-like structures (Figure 4A) (Yoshida, 2014). Strains that produced higher levels of tau pathology such as DS6 did not show similar plaque pathology at this time point (Figure 4A). Co-staining with GFAP and AT8 indicated that these accumulations consist of phospho-tau within or directly adjacent to astrocytes as is typical of astrocytic plaques (Figure 4B) (Yoshida, 2014). DS12, 15, 16, and 18 induced a small degree of astrocytic plaque-like pathology at 8 weeks (Figure S4A-B). Given these findings, we retrospectively quantified the number of animals with astrocytic plaque-like pathology after completion of the time course injection experiment described below. DS7 or DS9 inoculations induced astrocytic plaque-like pathology in the majority of inoculated mice by eight weeks (Figure S4A). In contrast, DS4 and DS6 induced limited plaque pathology by 12 weeks, which was far less robust than the level observed in DS7 and 9 inoculated animals (Figure S4A, C). Thus, this phenotype is likely independent of seeding activity, and suggests specific tau conformations preferentially and predictably induce astrocytic tau pathology.
Figure 4. Specific strains induce astrocytic tau pathology.
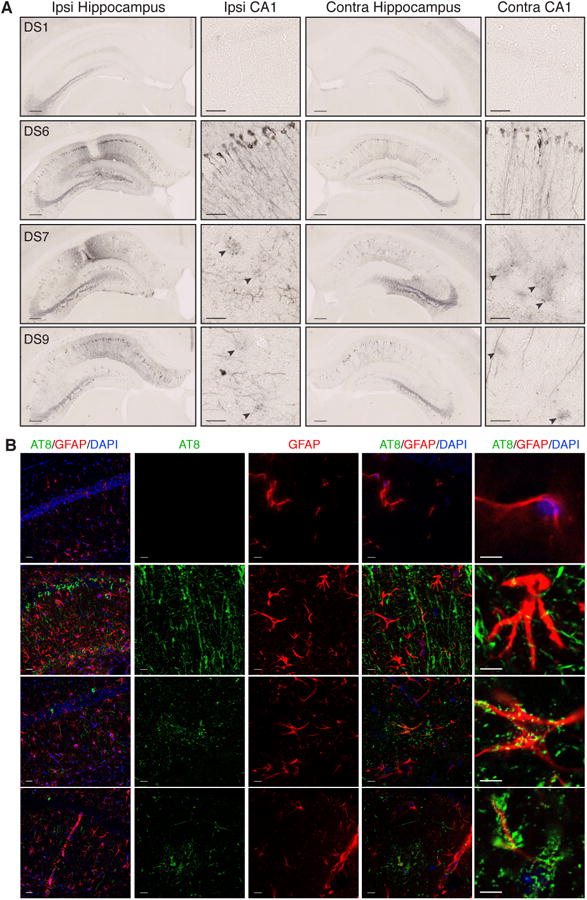
(A) AT8 tau pathology 8-weeks after injection with DS1, 6, 7, or 9. DS1 does not induce tau pathology. DS6, 7 and 9 develop strong AT8 staining in ipsilateral and contralateral hippocampi. DS7 and 9 develop diffuse, circular-shaped accumulations of AT8 staining that do not appear to localize to a neuronal cell body (black arrow heads). Scale bars represent 250 μm for the whole hippocampus, and 50 μm for CA1.
(B) Co-staining of AT8 (green) for phospho-tau, GFAP (red) for astrocytes, and DAPI (blue) for cell nuclei. DS1 shows limited GFAP staining, and no AT8 pathology. DS6 shows strong AT8 staining with limited overlap of AT8 staining. DS7 and 9 injected mice display astrocytic plaquelike pathology that either deposits within or around GFAP-positive processes of astrocytes. Scale bars represent 25 μm for left column, and 10 μm for all remaining images. For further quantification and representative images of other strains that display limited astrocytic plaque pathology, see Figure S4.
While two CBD-derived strains showed a small degree of astrocytic plaque-like pathology (DS12, 16), they were sparse at this time point (Figure S4A, B). We examined mice inoculated with DS12 and 16 at 6 months after injection, and observed increased levels of astrocytic pathology (Figure S4D). This suggests these two CBD-derived strains produce robust astrocytic plaque pathology after an extended incubation period compared to DS7 or DS9. DS11 and 13, which were also isolated from CBD patients, did not induce astroctyic plaque pathology 8 weeks after injection. Individual CBD patients likely have multiple tau strains present in their brains (Sanders et al., 2014), which may give rise to the overall pattern of histopathology observed in patients. Alternatively, DS11 and 13 may simply require more time to produce robust astrocytic plaque pathology in this mouse model, as was observed for DS12 and 16 (Figure S4B, D).
Regional vulnerability to specific strains
Tauopathies feature accumulation of tau pathology in distinct brain regions (Arnold et al., 2013), yet the mechanisms that underlie these patterns are not well understood. To test whether strains differentially induce pathology in specific brain regions, we inoculated DS1, 4, 6, 7, 9, 10 or 11 into six locations per mouse: sensory cortex (SC), caudate/putamen (CP), visual cortex (VC), hippocampus (Hip), thalamus (Thal), and inferior colliculus (IC) (Figure 5A). We chose these strains based on their unique limited proteolysis patterns, different tau pathology induced in the hippocampus, and their low (DS7, 11), medium (DS4, 10) or high (DS6, 9) seeding activity in culture. Further, DS4 and 11 derive from AD and CBD brain homogenates respectively. Patients with these diseases have different patterns of tau deposition (Arnold et al., 2013).
Figure 5. Tau strains preferentially seed pathology in specific brain regions.
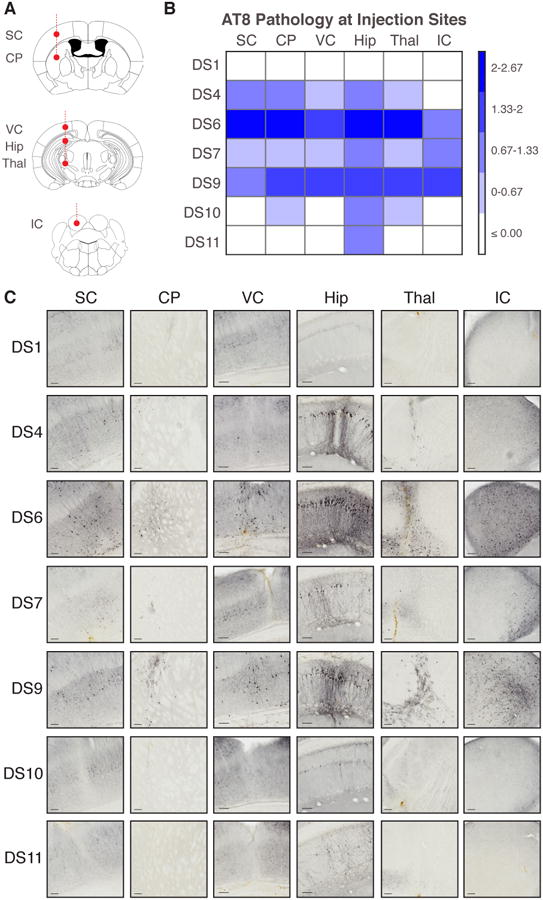
(A) Six tau strains were injected simultaneously into six brain regions: sensory cortex (SC); caudate/putamen (CP); visual cortex (VC); hippocampus (Hip); thalamus (Thal); inferior colliculus (IC) (5 μg per region). Mice that received DS1 (negative control), 4, 6, 7, 9, 10, or 11 strain injections were kept for 5-weeks post-inoculation before assessment of AT8 tau pathology (n=3 per condition).
(B) Strains preferentially induced tau pathology in specific brain regions. Slices that contained the injection sites were stained for AT8 phospho-tau. Each injection site was assessed in a blinded fashion for tau pathology on a 0-3 scale (none, low, medium, high). The level of background AT8 pathology at each injection site was accounted for by subtracting the level of pathology present in DS1 mice within each brain region. A binned heat map represents the level of pathology observed at the injection site for each strain. Note differences in regional vulnerability.
(C) Representative images are displayed for each brain region injected with the different tau strains. Scale bars represent 100 μm. DS10 mossy fiber pathology is shown in Figure S5A.
After five weeks, we quantified the level of AT8 pathology these strains induced at each injection site in a blinded fashion (Figure 5B). All produced hippocampal pathology consistent with the previous injection paradigm, illustrating the reproducibility of these phenotypes (Figure 5C). Strains with the strongest seeding activity in culture (DS6, 9) produced pathology in every injected region. DS4, a medium-seeding strain, induced moderate pathology in each region except the IC. DS11 pathology was entirely limited to the hippocampus (Figure 5B, C).
DS10 again induced pathology specific to the mossy fiber tracts of the hippocampus (Figure S5A), with limited pathology in the caudate/putamen and thalamus. Of note, it did not produce any pathology in the injected cortical regions. In contrast, DS7 produced limited AT8 pathology in each targeted brain region (Figure 5B, C). The specificity of DS10, despite its strong seeding activity, and promiscuity of DS7, despite its weak seeding activity (Figure 1D-G), were remarkable. These studies indicate tropism of certain strains for specific brain regions (mossy fiber tracts, cortical structures, IC) that is independent of simple metrics such as seeding activity.
Strains induce different rates of spread of tau pathology along neuronal networks
Even within a single clinical syndrome, tauopathy patients experience rapid or slow rates of progression (Armstrong et al., 2014; Thalhauser and Komarova, 2011). PrP strains show different lag phases and rates of neurodegeneration in animal models of prion diseases (Collinge and Clarke, 2007), suggesting this phenomenon may be linked to specific aggregate conformations. While several factors may contribute to the rate of degeneration observed in tauopathy patients, rapid spread of tau pathology likely accelerates this process. Thus, we tested the relationship between strain characteristics and rates of spread of tau pathology.
To control for differences in insoluble material, we first quantified the insoluble tau present in lysate from DS1, 4, 6, 7, 9, and 10 prepared for this time course experiment. As expected, each strain contained a large amount of insoluble tau (Figure 6A, B). We hypothesized that a strain's ability to seed aggregation of endogenously expressed monomeric tau would primarily determine the rate of spread of pathology. We predicted that strains such as DS6 and 9 with high seeding activity in culture and in vivo (Figure 1D, 5B-C) would produce rapid spread of pathology even after inoculation of reduced levels of insoluble tau. To test this hypothesis, we included DS6 and DS9 lysate diluted 1:10 as part of this time course experiment.
Figure 6. Strains induce different rates of tau pathology spread.
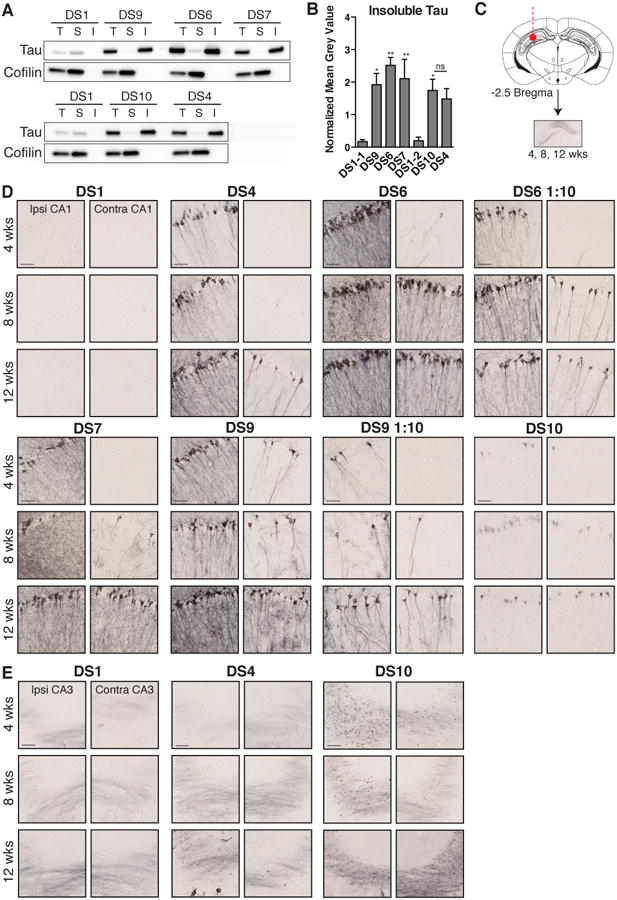
(A) Sedimentation analysis was performed on cell lysate used for the time course inoculation experiment. Each strain contains a large amount of insoluble material (T, total; S, soluble; I, insoluble). Western blot analysis of insoluble tau was performed on three biological replicates. For each experiment, the soluble fraction was loaded at 2× the concentration of the total and insoluble fractions. A cofilin loading control was performed on the blots to verify the same amount of cell lysate was added for each strain.
(B) The level of insoluble tau present in each strain was quantified by measuring the mean grey value of the insoluble tau western blot band. Samples were normalized to the mean grey value of cofilin in the total cell lysate fraction. DS1-1 and DS1-2 represent biological replicates of DS1. ANOVA shows strains have significantly more insoluble tau than DS1. A two-way t-test demonstrates DS10 and DS4 do not contain significantly different levels of insoluble tau (ns for P > 0.5; * for P ≤ 0.05; ** for P ≤ 0.01). Error bars represent S.E.M. of biological triplicates.
(C) Strains were inoculated into the hippocampus of young PS19 mice (n=5-6 per condition per time point, see Supplemental Table 1). DS6 and 9 lysate diluted 1:10 in HEK293 cell lysate were also injected (n=4-5 per condition per time point). Mice were collected at 4, 8, or 12 weeks.
(D) Representative images of ipsilateral and contralateral CA1 are displayed for each strain at 4, 8 and 12 weeks post-injection. AT8-positive tau pathology spreads to the contralateral hippocampus at different time points. Diluted DS6 and 9 lysate show faster spread than concentrated DS4, and more robust spread than DS7 and 10 at 8-weeks post-injection. Scale bars represent 50 μm. See Figure S6 for data regarding strain-specific rod microglial phenotype present at 12 weeks after inoculation.
(E) Spread of mossy fiber dot pathology occurs by eight weeks in DS10 mice. Dot pathology appears eventually to develop in DS4 mice, but spread appears delayed compared to DS10.
We injected cell lysate from each condition into the hippocampus of young PS19 mice and collected brains at 4, 8, and 12 weeks post-injection (Figure 6C). While each strain induced tau pathology in the contralateral hippocampus, this occurred at different time points (Figure 6D, E). DS6 and DS9 pathology progressed rapidly to the contralateral hippocampus, beginning as early as 4 weeks (Figure 6D). DS10 induced mossy fiber dots and limited CA1 pathology in the ipsilateral and contralateral hippocampus by 8 weeks (Figure 6D, E). DS7 wisp pathology also spread to the contralateral hippocampus by 8 weeks (Figure 6D).
While dilution of DS6 and 9 decreased the initial level of pathology induced upon inoculation, we observed tau pathology in the contralateral hippocampus at 8 weeks. In contrast, DS4 did not show tau pathology in the contralateral hippocampus until 12 weeks (Figure 6D, E). Thus, stronger strains induced more rapid spread of pathology even with a reduced amount of insoluble tau inoculum, presumably due to more efficient seeding and spread of endogenous tau aggregates.
While these inoculations induced robust neuronal tau pathology and strain-specific patterns of astrocytic tau pathology, we did not observe overt neuronal loss at these time points (data not shown). However, to begin to assess the functional effects of distinct strains, we examined the patterns of microglial pathology induced at 12 weeks after inoculation. Compared to DS1 inoculated mice, several strains displayed Iba1-positive rod microglial phenotype, with elongated projections that align along axons of CA1 pyramidal neurons (Figure S6A-C). We performed blinded quantification of ramified and rod microglia ipsilateral and contralateral to the site of inoculation. DS6 induced the largest degree of rod microgliosis, while DS7 produced very limited rod microglial pathology. DS4, 9 and 10 also produced rod microglia, but we observed this phenotype in the contralateral hippocampus primarily in DS6 and DS10 inoculated mice.
To test the stability of tau strains in vivo, we transduced brain homogenate from the hippocampus of mice at eight weeks after inoculation into the naïve DS1 cell line. DS4, 6, 9, and 10 produced morphologies consistent with the original inoculum (Figure S2F-H, Figure S7D). However, brain-derived DS7 produced a mixture of cellular morphologies. This is in contrast to DS7 cell lysate, which stably induces its cellular phenotype in culture, suggesting DS7 may imperfectly template its conformation onto full-length P301S tau in this mouse model.
We isolated monoclonal lines derived from inoculated mouse tissue by transducing brain homogenate into the DS1 line and sorting single aggregate-containing cells. Tau derived from mice inoculated with DS6 strain readily produced thread-containing inclusions in the population of converted cells (Figure S2F, S7D). However, isolation of monoclonal lines was not possible due to toxicity (0/36 individual colonies survived). In contrast, transduction of mouse-derived tissue inoculated with DS7 produced few inclusions in the population overall, with only one resultant monoclonal line. This is consistent with the low seeding activity of the original DS7 line. Multiple secondary lines were derived from brains originally inoculated with DS4, 9 and 10 (Figure S7B). FRET-based seeding activity of the secondary cell lines resembled that of the original lines (Figure S7C). Thus, several of these strains stably propagate their phenotype even upon passage through mice.
To assess AT8 pathology induced by each strain, we performed a blinded analysis of AT8 staining in slices at the level of the locus coeruleus, hippocampal injection site, and caudate/putamen. We averaged AT8 pathology rankings for each region and displayed them as a heat map to visualize the spread of tau pathology (Figure 7A). We subsequently created a limited heat-map for each strain that focuses on specific brain regions, several of which developed pathology over time (Figure 7B). DS10 once again displayed marked neuronal specificity, with strong pathology only in the mossy fiber tracts (Figure 7A, B). Blinded analysis also confirmed that DS4 exhibits slower kinetics than DS6 or 9 even when the latter strains are diluted 10-fold (Figure 7B). DS4 did not develop strong pathology outside the ipsilateral hippocampus until 12 weeks after injection, while these stronger strains showed AT8 staining in distant brain regions by 8 weeks. DS7 induced robust pathology at the injection site as observed previously (Figure 3, 5). However, the spread of DS7 pathology was relatively slow, and appeared limited to the hippocampus (Figure 7A, B).
Figure 7. Strain dictates the rate and pattern of spread of tau pathology.

(A) Slices from mice injected with each strain at each time point were stained for AT8 pathology. Tau pathology was quantified in a blinded fashion on a 0-3 scale, and averaged for each location within a given condition (n=5-6 per condition). A continuous heat map was generated. Note differential rates of spread and regional vulnerability. Regions are listed on the x-axis, and conditions/time points are on the y-axis.
(B) Limited heat maps were generated from the above data set (Figure 7A). Ipsilateral (Ip) and contralateral (Con) regions were included to assess patterns and rates of spread of pathology (retrosplenial cortex, RS; entorhinal cortex, EC; sensory cortex, SC; thalamus, Thal; CA1 of hippocampus, CA1; locus coruleus, LC; subiculum, Sub). Time points are arranged in order from earliest (4-weeks) to latest (12-weeks). Diluted DS6 and 9 lysates are also displayed (DS6 1:10 and DS9 1:10).
(C) Homogenized tissue from the hippocampus, thalamus, or sensory cortex of mice 8-weeks after inoculation with strains was applied to tau biosensor cell lines. After 48-hours, cells were collected and flow cytometry was performed to quantify the level of seeding activity in each region by integrated FRET density (IFD = percent FRET-positive cells*median fluorescent intensity of FRET positive cells) (Holmes et al., 2014). DS4 induces lower spread of seeding activity to the contralateral hippocampus at 8-weeks. DS10 induces high seeding activity despite limited AT8 pathology, while DS7 induces low seeding activity despite high AT8 pathology. DS6 and DS9 also induce seeding activity in the ipsilateral thalamus. A one-way ANOVA with Bonferroni correction for multiple comparisons was performed between ipsilateral DS1 and every other sample within a given region. (* P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001). Error bars represent S.E.M., n = 4-5. See Figure S7 for data regarding secondary cell line isolation of strains derived from inoculated mice or strain cell lysate.
DS6 and 9 spread pathology rapidly to specific brain regions after 4 weeks. DS6 strongly targeted the ipsilateral retrosplenial cortex and LC, while DS9 pathology spread most strongly to the ipsilateral entorhinal cortex and thalamus. Despite these initial differences, by 8 weeks the patterns of pathology induced by these strains largely resembled one another (Figure 7B). While diluted lysate of DS6 and 9 induced lower levels of pathology at early time points, these induced pathology that spread faster and farther than DS4, 7 or 10, and followed similar patterns to that of undiluted DS6 and 9 lysate (Figure 7B).
We next performed a seeding assay on tissue from the ipsilateral and contralateral hippocampus, thalamus, and sensory cortex at eight weeks after injection as a second metric of tau pathology. We transduced homogenized brain regions into the FRET biosensor cell line (Holmes et al., 2014), and quantified seeding after two days in culture. We have observed that all seeding activity measured after inoculation of DS9 into the hippocampus of tau knockout mice dissipates by approximately six weeks (data not shown), which suggests any signal identified at this time point likely derives from induced aggregation of endogenous tau expressed in this mouse line.
Several strains displayed robust seeding in the ipsilateral hippocampus (DS4, 6, 9, 10, and 1:10 diluted DS6 and 9). While each of these strains spread to the contralateral hippocampus, DS4 showed distinctly less contralateral seeding activity as a percentage of the ipsilateral hippocampus (Figure 7C). Further, DS6 and 9 showed variable yet significant seeding activity in the ipsilateral thalamus. The level of seeding observed in DS1 inoculated mice is consistent with the level observed in this mouse model at 4-5 months of age, suggesting this signal is due to spontaneous aggregation of endogenous tau that normally occurs within this model (Holmes et al., 2014). DS7 showed remarkably low seeding activity, while DS10 induced relatively robust seeding from the hippocampus despite only inducing mossy fiber AT8 tau pathology (Figure 6E, 7C). This is consistent with the seeding activity for the original inoculum as observed by the split-luciferase assay (Figure 1D, 2G-I). These results are consistent with the different rates of spread observed by AT8 immunohistochemistry for DS4 versus DS6 and 9, while providing a separate metric to quantify the spread of pathology for strains such as DS10.
Discussion
Overview of Findings
We have tested whether specific tau prion strains can account for critical neuropathological features that are used to discriminate tauopathies. We characterized strains by various biochemical and biological metrics, including inclusion morphology, seeding activity in dividing cells and primary neurons, detergent solubility, cellular toxicity, limited proteolysis, and re-introduction into reporter cells. We inoculated all 18 strains individually into the hippocampi of a transgenic tauopathy mouse model, causing distinct patterns of tau pathology in cell bodies, axons, and dendrites. To test for regional vulnerability, we injected 6 strains into 6 different brain regions, and waited 5 weeks to evaluate pathology. Many induced pathology in all regions (DS6, 7, and 9), while others exhibited restricted patterns in which very little or no pathology occurred despite direct exposure of neurons to a particular strain (DS4, 10, 11). We next tested whether strains exhibited unique rates of propagation through the brain. In this case we observed correlations with in vitro parameters: strains with high seeding activity tended to spread more rapidly through the brain, with one important exception—strain DS10—which only spread to the contralateral mossy fiber tracts of the hippocampus. These observations suggest that distinct tau prion strains could account for many of the features observed in human tauopathies.
Limitations of this Experimental Approach
It is impossible at this time to directly control tau strain production in vivo using transgenic mice, or to propagate strains faithfully and indefinitely in vitro without using cultured cells. This work relies on a cell model that expresses a truncated form of tau with a fluorescent protein tag, an imperfect system has nonetheless allowed us to propagate distinct tau prion strains indefinitely. Likewise, in animal models, we utilized a model that expresses full-length human tau (1N4R) that contains a disease-associated mutation (P301S) (Yoshiyama et al., 2007). While this obviously deviates from sporadic tauopathy that occurs in most individuals, it has enabled us to rapidly and reliably induce unique tau pathology based on local inoculation. We cannot exclude the possibility that inoculated tau prions themselves are moving throughout the brain and inducing pathology based on local uptake rather than true trans-neuronal propagation (Rey et al., 2013). Nonetheless, our observations suggest a fundamental disease mechanism whereby strain-specific differences govern seeding, propagation, and specific regional vulnerability.
The Utility of Studying Isolated Strains
Our prior work has indicated that human tauopathy brains, even those carefully defined by histopathology, exhibit enormously diverse strain content within and between individuals (Sanders et al., 2014). Thus syndromes that appear to be clinically and neuropathologically identical are potentially quite distinct in terms of their strain composition. This presents obvious challenges when attempting to define strains present in human disease based principally on inoculation of purified aggregates into mice. Fibril preparations created in vitro also exhibit tremendous conformational heterogeneity, as clearly illustrated by studies of α-synuclein in which one dominant fibril structure shifts to another upon serial seeding reactions (Guo et al., 2013). In this work, we have stably propagated strains with specific biochemical properties in a simple culture system based on expression of tau RD-YFP. While technical limitations have restricted our ability to define the specific structures of tau aggregates present in these strains, this work suggests a single dominant structure is faithfully propagated in each line. This has enabled us to make predictions about phenotypes in vivo based on properties observed in vitro.
Prion Strains Characterized In Vitro
Scrapie prion strains have distinct, yet reproducible patterns of incubation time, neuropathology, and behavioral phenotypes (Collinge and Clarke, 2007). Strains are presumably distinct amyloid structures that faithfully replicate in a living system, and produce well-defined pathology. Thus if a strain is identified, it is possible to predict incubation time and resultant pathology (Collinge and Clarke, 2007). In this work we characterized multiple tau strains from recombinant, mouse, and patient sources in vitro and in vivo. Detailed characterization of multiple tau prion strains in vitro using several metrics (inclusion morphology, solubility, seeding efficiency, limited proteolysis) allowed us to make predictions regarding their effects in vivo. For instance, DS6 and 9 induced robust spread of tau pathology even upon dilution, which was predicted by in vitro seeding activity. Importantly, while seeding activity correlated best with induction of local and distant pathology, this is an imperfect metric. For example DS10 inoculation produces very limited pathology in vivo despite its relatively strong seeding activity in vitro. This indicates a major role for other as yet unidentified strain-specific parameters. With further, more detailed study, we hope to link specific structural characteristics to various steps in pathogenesis, i.e. to define the “logic” that predicts biological effects. For instance, cell-type specificity (or at least preference) might be based on differential strain binding affinities to heparan sulfate proteoglycans (Holmes et al., 2013). Likewise, post-translational modifications of monomeric tau within a target cell might render it more or less vulnerable to conversion by a specific strain.
Distinct Cellular Pathologies in Vivo
Tauopathies are defined histopathologically by several criteria, especially the pattern of intracellular tau accumulation: neurofibrillary tangles, Pick bodies, threads, grains, axonal puncta, etc. (Kovacs, 2015). Although we have readily observed patterns of pathology reminiscent of those described in patients, we have not attempted to link human patterns of pathology to those in P301S mice, which express only a single isoform of mutant tau. Instead, we wish to emphasize how conformational differences in tau prion strains are sufficient to create an enormous pathological diversity: neurofibrillary tangles, soma versus axonal accumulation, grain-like structures, dendritic and axonal terminal deposits that resemble threads, and astrocytic plaques.
The presence or absence of specific glial pathology also contributes to the definition of tauopathies (Yoshida, 2014). We observed that certain strains produce AT8-postive pathology in patterns reminiscent of astrocytic plaques described in tauopathies, with localization of phospho-tau inclusions along the processes of GFAP-positive astrocytes (Yoshida, 2014). We cannot attribute these effects to tau seeding activity or toxicity, as we observed these phenomena in DS7 (low seeding, low toxicity) and DS9 (high seeding, high toxicity). Distinct strains also induced different levels of rod microglial pathology.
Given that inoculates were identical with the exception of tau structure, and the transgenic mouse model expresses only a single tau isoform, we conclude that tau prion strains themselves dictate the resultant glial pathology. Further, three strain pairs (DS3/DS19; DS12/16; and DS6/15) that displayed similar biochemical features and limited proteolysis patterns produced similar patterns of neuropathology in vivo. These strains served to internally validate the methods we developed, and highlight the close relationship of strain type to induced pathology.
Rates of Propagation in Vivo
Neurodegenerative diseases progress at different rates for unknown reasons. Similar to previous observations regarding PrP prion strains (Legname et al., 2006), our data indicate that the characteristics of an individual tau prion strain are sufficient to dictate the rate at which pathology spreads throughout the nervous system. Seeding activity correlates with this phenomenon, but cannot completely explain it. Instead, the rate of spread appears to reflect a unique interaction of specific strains with vulnerable cells. For example, strain DS10 seeds very strongly in vitro, but fails to propagate pathology outside of the hippocampal mossy fibers. Likewise, DS6 seeds very strongly in vitro, but exhibits a longer lag phase as it spreads to specific brain regions such as the entorhinal cortex versus the retrosplenial cortex. In contrast, DS9 showed more rapid spread to the entorhinal than the retrosplenial cortex. Taken together, our results suggest that the rate of propagation must be strongly influenced by raw seeding potential, i.e. the ability to convert monomeric tau upon direct introduction to the cytoplasm via lipofectamine, but also the ability of a given strain to spontaneously enter and replicate within a vulnerable cell.
Distinct Regional Vulnerabilities
Regional, or “selective” neuronal vulnerability in neurodegenerative diseases has long mystified investigators. PrP prion strains appear to account for differential regional involvement of the brain (Collinge and Clarke, 2007). Within the limits of our experimental system, our identification and characterization of distinct tau strains has allowed us to test whether aggregate structure itself defines regional vulnerability. We have found striking strain-specific regional differences, both in the pattern of spread from a single hippocampal inoculum, and also in vulnerability to a direct injection. These effects correlated with individual tau strains, independent of an inoculum dose that could have accounted for vulnerability. For example, 10-fold dilution of a potent strain, DS9, produced patterns of spreading pathology very similar to a full dose. Further, low-dose DS9 spread at a faster rate than a full dose of DS4, even though the initial “seed burden” of DS9 was less. By contrast, strain DS10 has relatively high seeding activity yet it selectively targets the mossy fibers of the hippocampus, and does not convert tau in several other brain regions even after direct inoculation.
Others previously observed that different tau fibril preparations produce unique patterns of pathology based on inoculation of tau purified from tauopathy brains or crude homogenates (Boluda et al., 2015; Clavaguera et al., 2013), and two groups have inoculated unique α-synuclein preparations (Bousset et al., 2013; Guo et al., 2013; Peelaerts et al., 2015). However, no prior studies can attribute these effects to a specific, well-characterized strain or structure, or make predictions about the behavior of a strain in vivo from the biochemical properties of that inoculum. Given that we can now link specific pathology patterns to single tau prion strains, the experiments described here should enable new approaches to define how the structural characteristics of prions dictate neuronal vulnerability.
Implications for Diagnosis and Therapy
We propose that tau prion strains will explain the diversity of human neuropathology, and will be required for mechanistic understanding of disease. Precise diagnosis of tauopathy now depends on histopathology, yet we observed different strain composition patterns in putatively identical pathological syndromes (Sanders et al., 2014). Further, diversity and evolution may confound efforts to target a specific strain, as for PrP (Giles et al., 2010; Weissmann et al., 2011). Tau prion strains as defined by specific conformations should have enormous power to help elucidate the structural determinants that underlie and predict the pathological patterns of diverse human tauopathies, and to devise appropriate therapies.
Experimental procedures
Generation of monoclonal strain library
Cells stably expressing tau RD(P301L/V337M) fused to YFP (DS1) were treated with recombinant tau fibrils or cell/brain homogenate. Resultant monclonal lines were isolated and analyzed for inclusion morphology, seeding by split luciferase assay, and protease digestion as previously described (Sanders et al., 2014). These analyses identified 18 putatively distinct strains (DS2-19).
Limited proteolysis
Cell lysates (60μg) in PBS/Triton X-100 were digested with pronase (30 μg/mL) for one hour at 37°C, then resolved by SDS-PAGE and western blot probed with primary anti-tau antibody 2B11.
Sedimentation analysis of strain library
Cell lysates were spun at 186,000 × g for 60 minutes. Pellets were washed with 1 mL lysis buffer, and spun for an additional 30 minutes. Samples were resolved by SDS-PAGE and western blot using rabbit polyclonal anti-tau antibody ab64193 (AbCam).
Toxicity and seeding assay
LM10 cells, a monoclonal cell line expressing tau RD(P301L/V337M)-CFP and tau RD(P301L/V337M)-YFP at high levels, were transduced with 20 μg of clarified cell lysate. After 72 hours, FRET-positive cells were sorted by FACS (Holmes et al., 2014), and re-plated. After 7 days the number of FRET-positive and FRET-negative cells were quantified by flow cytometry.
Animal maintenance and inoculation experiments
PS19 mice expressing 4R1N P301S human tau under the murine prion promoter (Yoshiyama et al., 2007) were used. Strains were inoculated intracerebrally via 10 μL gas-tight Hamilton syringes.
Histology and quantification of AT8 pathology
50 μm frozen sections were used. For DAB stain, biotinylated AT8 primary antibody (Thermo Scientific) was used. Images of AT8-stained slices were collected via Olympus Nanozoomer 2.0-HT (Hamamatsu). The level of tau pathology present in each region was determined by blinded analysis with a semi-quantitative 0-3 scale (no pathology, mild, moderate, and severe). Pathology was averaged among biological replicates and plotted as a heat map using MATLAB as described.
Supplementary Material
Acknowledgments
We thank Ann McKee, Bill Seeley, and Lea Grinberg for providing invaluable reagents for this study. We thank Peter Davies for providing MC1 antibody. We thank Matthew Brier, Jennifer Furman, Brandon Holmes, Suzanne Schindler and Niall Prendergast for providing guidance and critiques during the preparation of this manuscript. This work was funded by NIH/NIA grant F30AG048653 (SKK); NIH grant F31NS086251 (DWS); NIH/NIA R01AG048678, NIH/NINDS R01NS071835, the Tau Consortium, and the Cure Alzheimer's Fund (MID). This work was supported by the Hope Center Alafi Neuroimaging Laboratory at Washington University in St. Louis, Neuro-Models Facility, Whole Brain Microscopy Facility, Moody Foundation Flow Cytometry Facility, and High Throughput Screening core at University of Texas, Southwestern.
Footnotes
Supplemental information: Supplemental information includes seven figures, three tables, and detailed description of experimental procedures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Armstrong MJ, Castellani RJ, Reich SG. “Rapidly” Progressive Supranuclear Palsy. Mov Disord Clin Pract. 2014;1:70–72. doi: 10.1002/mdc3.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Toledo JB, Appleby DH, Xie SX, Wang LS, Baek Y, Wolk DA, Lee EB, Miller BL, Lee VMY, et al. Comparative survey of the topographical distribution of signature molecular lesions in major neurodegenerative diseases. J Comp Neurol. 2013;521:4339–4355. doi: 10.1002/cne.23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S, Zheng-Fischhöfer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow EM, Mandelkow E. Structure, Microtubule Interactions, and Paired Helical Filament Aggregation by Tau Mutants of Frontotemporal Dementias †. Biochemistry. 2000;39:11714–11721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68:7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boluda S, Iba M, Zhang Bin, Raible KM, Lee VMY, Trojanowski JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer's disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129:221–237. doi: 10.1007/s00401-014-1373-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, et al. Structural and functional characterization of two alpha-synuclein strains. Nature Communications. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proceedings of the National Academy of Sciences. 2013 doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nature Publishing Group. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. A General Model of Prion Strains and Their Pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, et al. Propagation of Tau Pathology in a Model of Early Alzheimer's Disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J Biol Chem. 2009a;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J Biol Chem. 2009b;284:3546–3551. doi: 10.1074/jbc.M805627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K, Glidden DV, Patel S, Korth C, Groth D, Lemus A, DeArmond SJ, Prusiner SB. Human prion strain selection in transgenic mice. Ann Neurol. 2010;68:151–161. doi: 10.1002/ana.22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VMY. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. Journal of Biological Chemistry. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Bin Zhang, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, et al. Distinct alpha-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell. 2013;154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, Belaygorod L, Cairns NJ, Holtzman DM, Diamond MI. Proteopathic tau seeding predicts tauopathy in vivo. Proceedings of the National Academy of Sciences. 2014;111:E4376–E4385. doi: 10.1073/pnas.1411649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proceedings of the National Academy of Sciences. 2013;110:E3138–E3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VMY. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci. 2013;33:1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles TPJ, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nature Publishing Group. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Kovacs GG. Invited review: Neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol. 2015;41:3–23. doi: 10.1111/nan.12208. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Legname G, Nguyen HOB, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A. 2006;103:19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. Journal of Biological Chemistry. 2015;290:14893–14903. doi: 10.1074/jbc.M115.652693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. Journal of Biological Chemistry. 2010;285:34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. 2015;522:340–344. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Kuceyeski A, Weiner M. A Network Diffusion Model of Disease Progression in Dementia. Neuron. 2012;73:1204–1215. doi: 10.1016/j.neuron.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Petit GH, Bousset L, Melki R, Brundin P. Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol. 2013;126:555–573. doi: 10.1007/s00401-013-1160-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, et al. Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron. 2014:1–18. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, Holmes BB, Diamond MI. Prions and Protein Assemblies that Convey Biological Information in Health and Disease. Neuron. 2016;89:433–448. doi: 10.1016/j.neuron.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalhauser CJ, Komarova NL. Alzheimer's disease: rapid and slow progression. Journal of the Royal Society Interface. 2011;9:119–126. doi: 10.1098/rsif.2011.0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Jucker M. Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci. 2015;38:87–103. doi: 10.1146/annurev-neuro-071714-033828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann C, Li J, Mahal SP, Browning S. Prions on the move. Nature Publishing Group. 2011;12:1109–1117. doi: 10.1038/embor.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M. Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology. 2014;34:555–570. doi: 10.1111/neup.12143. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VMY. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW. Predicting Regional Neurodegeneration from the Healthy Brain Functional Connectome. Neuron. 2012;73:1216–1227. doi: 10.1016/j.neuron.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.