

Abstract
Chimeric antigen receptor-modified T cells (CAR T-cells) produce pro-inflammatory cytokines that increase expression of T cell checkpoint signals such as PD-L1, which may inhibit their functionality against solid tumors. In this study, we evaluated in human tumor xenograft models the pro-inflammatory properties of an oncolytic adenovirus (Onc.Ad) with a helper-dependent Ad (HDAd) that expresses a PD-L1 blocking mini-antibody (mini-body) (HDPDL1), as a strategy to enhance CAR T-cell killing. Co-administration of these agents (CAd-VECPDL1) exhibited oncolytic effects with production of PD-L1 mini-body locally at the tumor site. On their own, HDPDL1 exhibited no anti-tumor effect and CAd-VECPDL1 alone reduced tumors only to volumes comparable to Onc.Ad treatment. However, combining CAd-VECPDL1 with HER2.CAR T-cells enhanced anti-tumor activity compared to treatment with either HER2.CAR T-cells alone, or HER2.CAR T-cells plus Onc.Ad. The benefits of locally produced PD-L1 mini-body by CAd-VECPDL1 could not be replicated by infusion of anti-PD-L1 IgG plus HER2.CAR T-cells and co-administration of Onc.Ad in a HER2+ prostate cancer xenograft model. Overall, our data document the superiority of local production of PD-L1 mini-body by CAd-VECPDL1 combined with administration of tumor-directed CAR T-cells to control the growth of solid tumors.
Introduction
Intratumoral treatment with oncolytic adenoviral vectors expressing an immunomodulatory molecule (Armed Onc.Ads) is safe and has shown some clinical benefit in patients with solid tumors (1). However, local treatment with Armed Onc.Ad has limited anti-tumor effect against metastasized tumors (1). Additionally, Onc.Ads have low transgene capacity (2, 3), limiting the potential to enhance anti-tumor immunity by adding multiple genetic modifications. We have shown that tumor cells co-infected with Onc.Ad and Helper-dependent Ads (HDAds), which have a cargo capacity of up to 34 kb and therefore can express multiple immunomodulatory molecules in a single vector, replicate both Onc.Ad and HDAd. Infection with this dual Ad gene therapy (CAd-VEC) leads to multiple cycles of production and release of both the oncolytic and the immunogenic components (4). Although CAd-VEC significantly suppressed tumor growth compared to treatment with either Onc.Ad or HDAd alone in an immunocompetent mouse model (4), it was insufficient to cure bulky or metastasized tumors.
Chimeric antigen receptors (CARs) usually combine the extracellular antigen recognition domains of a monoclonal antibody and a T-cell receptor signaling domain (CAR T-cells) (5). CAR T-cells can be systemically administered and home to both primary and metastasized tumors (5), overcoming the limited systemic anti-tumor effects of locally administered Ad-based cancer immunotherapies (1). Striking clinical successes against B-cell malignancies have been reported when CAR T-cells are directed to target antigen CD19, which is highly expressed on both malignant and normal B cells (6). Solid tumors have proven trickier, because many express a range of inhibitory cytokines (7) and immune checkpoint ligands (8) that impair the recruitment and sustained activation of effector T-cells. Thus, additional immunomodulation is likely required to increase CAR T-cell efficacy against solid tumors.
Recent clinical trials with immune-checkpoint inhibitors have improved tumor-specific T cell responses (9). PD-L1 expression on solid cancer cells is induced or increased in the presence of Th1 cytokine IFNγ (10), one of the cytokines expressed by activated CAR T-cells (11). CAR-dependent activation of CAR T-cells at the tumor site therefore may increase the expression of PD-L1 on target cancer cells, decreasing the anti-tumor effect of CAR T-cells through the PD-1:PD-L1 interaction (12).
As there are toxicities associated with systemic infusion of anti-PD-L1 antibody (13), we hypothesized that local secretion of our functional checkpoint blockade through a single combination agent, CAd-VEC, would be simpler, safer and perhaps more efficacious than combining three separate treatment modalities — oncolytic viruses, checkpoint inhibitors and CAR T-cells. We hypothesized that a CAd-VEC expressing anti-PD-L1 mini-antibody (PD-L1 mini-body) could block the PD-1:PD-L1 interaction between CAR T-cells and cancer cells locally while lysing tumor cells, and that combining these treatment modalities would yield potent anti-tumor effects in solid tumors. Here, we demonstrate that CAd-VEC expressing a PD-L1 blocking mini-antibody (CAd-VECPDL1) enhances the anti-tumor effect of CAR T-cells against human solid cancer cells in vitro and in vivo. Our “all-in-one” strategy proved more potent than the combination of anti-PD-L1 IgG and CAR T-cells with or without additional Onc.Ad pretreatment in vivo.
Materials and Methods
Adenoviral vectors (HDAds and Onc.Ads)
HDAd HDΔ28E4EGFP construct containing the EGFP transgene driven by the CMV promoter (HDAdeGFP) was produced as described elsewhere (4). HDAd without transgene (HDAd0) was produced as described elsewhere (14). To generate the PD-L1 mini-body, anti-human PD-L1 scFv encoding human IgG signal peptide and the single chain variable region of the YW243.55.S70 was fused with hinge, CH2 and CH3 regions of human IgG1 with a C-terminal HA tag. The PD-L1 mini-body complementary DNA was inserted into CMV promoter with polyA signal sequences. After confirmation of sequence and expression, this expression cassette was inserted into pHDΔ28E4 vector, and HDΔ28E4 PD-L1 mini-body (HDAdPD-L1) was rescued as described elsewhere (15). Onc.Ad5Δ24 was produced as described elsewhere (4, 16).
Cell lines
Human prostate cancer cell line PC-3, human non-small cell lung carcinoma cell line A549, human hepatocellular carcinoma cell line HepG2 and human squamous cell carcinoma line SiHa were obtained from ATCC (Manassas, VA) in 2014. Cell lines were authenticated by utilizing Short Tandem Repeat (STR) profiling by ATCC. Cells were cultured under recommended conditions.
Primary cells
Human PBMCs were isolated using Ficoll-Paque Plus according to manufacturer’s instructions (Axis-Shield). For preparation of mature dendritic cells (mDCs), PBMCs were cultured in dendritic cell medium (Cell Genix) for 2 hours at 37°C, and non-adherent cells were removed. Remaining monocytes were cultured in DC medium supplemented with 400U/mL of IL-4 and 800U/mL of GM-CSF. Fresh media with cytokines were supplemented every 3 days. The mDCs were induced by addition of TNFα, PGE-1, IL-1β and IL-6 on day 6 and cultured for 48 hours (17). CD4+ T-cells were isolated from PBMCs using MACS column according to manufacturer’s instructions (Miltenyi Biotec).
Mixed Lymphocyte Reaction
CD4+ T-cells were cultured in 96 well-round bottom plates together with allogeneic mDCs at a ratio of 10:1, using CTL medium (18). Anti-human PD-L1 IgG, Isotype IgG (Biolegend), medium of A549-infected with HDPD-L1 or HDeGFP were added, as described in Figure legends. Supernatants were collected at 5 days post co-culturing CD4+ T-cells with allogeneic mDCs, and IFNγ levels in media were measured by using the BD cytokine multiplex bead array system (BD Biosciences) according to manufacturer’s instructions. Cells were labeled with 3H-thymidine for an additional 18 hours to measure T-cell proliferation.
Co-culture experiments
Human cancer cells genetically modified to express EGFP were seeded in 12-well plates and infected with 1,000 viral particles (vp) per cell of HDAds or treated with 10 μg/mL of anti-human PD-L1 IgG, Isotype IgG (Biolegend). HER2.CAR T-cells with an effector to target ratio of 1:20 were added 48 hours post-infection and cultured for 5 additional days. Residual live EGFP+ cancer cells and T-cells were counted on the basis of EGFP and CD3 expression with Counting Beads (Life Technologies). Cell numbers were calculated per 5,000 microbeads.
PD-L1 Elisa
Immulon 2 high binding 96-well plate (VWR) was coated with 500 ng/well of recombinant human PD-L1 (BioVision). After blocking plate with PBS-T containing 3% BSA, serially diluted media of A549-infected with 1,000 vp/cell of HDegfp or HDPD-L1 mini were added and incubated at 4°C for 24 hours. Serially diluted anti-human PD-L1 antibody starting from 10 μg/well (BioLegend) was used as a positive control. After washing plate with PBS-T, HRP-labeled anti-human IgG for PD-L1 mini-body detection or HRP labeled anti-mouse IgG (BioRad) for anti-human PD-L1 and isotype antibody detection were added and incubated at room temperature for 1 hour and then we developed the washed plate. Absorbance was measured using Tecan reader (TECAN).
PD-L1 mini-body Elisa
Amounts of PD-L1 mini-body in media of cancer cells infected with 10 vp/cell of HDPD-L1 alone, Onc.Ad alone or with CAd-VECPD-L1 (Onc.Ad:HDAd=1:10) were quantified with ELISA based assay. Media of cancer cells infected with Ads were added to Immulon 2 96-well plate (VWR). Recombinant HA-tag fusion protein (Alpha Diagnostic Intl Inc.) was used as a standard. After blocking with PBS-T containing 3% BSA, anti-HA monoclonal antibody (Clone 5B1D10; Thermo Fisher Scientific) was added to the plate and incubated at 4°C for 24 hours. After washing, HRP labeled anti-mouse IgG (BioRad) was added and incubated at room temperature for 1 hour and then we developed the washed plate.
Animal experiments
After counting, 1×106 harvested cancer cells (PC-3) were re-suspended in 100 μL of PBS and subcutaneously injected into 5–6 week-old NU/J male mice. After the tumor size reached 100 mm3, 1×108 of Onc.Ad, HDPDL1 or CAd-VECPD-L1 (Onc.Ad :HDAd;1:20) were intra-tumorally injected in a volume of 20 μL. Tumor size was followed and volumes were calculated using the formula: Width2 × Length × 0.5. For PD-L1 mini-body detection in tumor and serum samples, tumors and serum were collected at dates described in Results. Tumors were homogenized with micropestles (VWR) at 4°C, and supernatants of tumor lysates were isolated via centrifugation at 2,000 rpm for 5 min. Total protein concentration of serum was measured by using Micro BCA protein assay kit (Thermo Scientific).
1×106 human cancer cells (PC-3 or SiHa) were re-suspended in a volume of 100 μL of PBS and subcutaneously injected into 5–6 week-old NSG mice (PC-3: male mice, SiHa: female mice). After the tumor size reached 100 mm3, a total of 1×107 of Onc.Ad or CAd-VECPD-L1 (Onc.Ad:HDAd;1:20) were intra-tumorally injected in a volume of 20 μL. Three days post injection of Ads, mice received 1×106 HER2.CAR T-cells intravenously. Tumor size was followed and volumes were calculated using the formula: Width2 × Length × 0.5. To track the migration and survival of HER2.CAR T-cells in vivo, T-cells were genetically modified to express eGFP.FFLuc (19). Biodistribution of HER2.CAR T-cells was assessed using the In Vivo Imaging System (Xenogen) (19).
Isolation of tumor-infiltrated HER2.CAR T-cells from tumors
After rinsing collected tumors with PBS, tumors were minced and incubated in RPMI media containing Collagenase type IV (5 mg/mL) and type I (1 mg/mL) (Thermo Fisher Scientific) at 37°C for 2 hours (20). Cells were passed 70 μm cell strainer (BD Pharmingen) and stained with antibodies described in Supplemental material.
Quantification of vector genome DNA in Ad infected cells and in Ad injected tumors
Cells were infected with 10 Vp/cell of Onc.Ad, HDPD-L1 or CAd-VECPD-L1 (Onc.Ad:HDAd; 1:10) and harvested 48 hours post-infection. Tumors were injected with a total of 1×108 Vp of Onc.Ad, HDPD-L1 or CAd-VECPD-L1 (Onc.Ad:HDAd;1:20) and harvested at indicated time points. Total DNA was extracted from infected cells or tumors, and vector copies were quantified described in Supplemental material.
Statistical analysis
Data were analyzed by Oneway ANOVA analysis of variance followed by Ranks protected least significant difference test (SigmaPlot).
Results
PD-L1 mini-body and anti-human PD-L1 IgG similarly block the PD-1:PD-L1 interaction
Cancer cells upregulate PD-L1 in the presence of IFNγ (10), which is produced by activated T-cells. We therefore co-cultured T-cells expressing 2nd generation HER2-specific CARs with CD28.ζ-signaling domains (HER2.CAR T-cells), which were recently reported to be safe in patients with sarcoma (11), with cancer cells. We evaluated the levels of PD-L1 on the HER2 positive human prostate cancer cell line PC-3 and the squamous cell carcinoma cell line SiHa, (Supplemental Fig. 1A), and the expression of PD-1 on HER2.CAR T-cells at different time points (Fig. 1A, Supplemental Fig. 1B). When co-cultured with HER2.CAR T-cells, PC-3 (Pre: 60%, Post: 100%) and SiHa cells (Pre: 20%, Post: 99%) upregulated PD-L1 expression to levels similar to those induced by recombinant IFNγ treatment (Supplemental Fig. 1A). HER2.CAR T-cells also expressed PD-1 within 24 hours of co-culture (Pre: 2%, Post: 30%), suggesting HER2.CAR T-cells express PD-1 upon activation, though PD-1 is one of multiple exhaustion markers expressed by T-cells (21).
Figure 1. HDAd-derived PD-L1 mini-body functions similarly to commercial anti-PD-L1 IgG.

(A) PC-3 and SiHa were co-cultured with HER2.CAR T-cells generated from 3 different donors (effector:target ratio of 1:20). Cells were harvested at 8, 24, 72 and 120 hours post-co-culture, and PD-1 on HER2.CAR T-cells and PD-L1 on cancer cells were analyzed by flow cytometry. Data are presented as means ± SD (n=3). (B) Schematic structure of HDAd encodes an anti-PD-L1 mini-body expression cassette (HDPD-L1). A549 cells were infected with different dosages of HDPD-L1. Media and cells were collected at 48 hours post-infection. Samples were subjected to western blotting, and PD-L1 mini-body in media was detected by anti-HA antibody. Human GAPDH in cells was detected by anti-human GAPDH antibody, HDeGFP encoding an EGFP expression cassette was used as a control. (C) A549 media infected with 1,000 vp/cell of HDPD-L1 or HDeGFP were collected at 48 hours post-infection, and binding of PD-L1 mini-body to recombinant human PD-L1 was assessed by enzyme-linked immunosorbent assay (ELISA). Anti-PD-L1 IgG or Isotype IgG were controls (10 μg/mL; highest concentration). Data are presented as means ± SD (n=3). (D) Purified CD4+ T-cells were co-cultured with irradiated allogeneic mature DCs in the presence of PD-L1 mini-body-containing medium, 10 μg/mL anti-PD-L1 IgG or isotype IgG for 5 days (T-cell:DC ratio of 10:1). IFNγ levels in the medium were measured by ELISA. Data are presented as means ± SD (n=4). *P= 0.005. The experiment was triplicated with similar results.
To test whether blocking the PD-1:PD-L1 interaction between cancer cells and HER2.CAR T-cells increases cytotoxicity locally at the tumor site, we constructed a HDAd encoding PD-L1 mini-body expression cassette (HDPDL1). We confirmed the dose-dependent expression of PD-L1 mini-body in media of non-small cell lung carcinoma A549 cells infected with HDPDL1 (Fig. 1B). We next evaluated whether the PD-L1 mini-body secreted in media of A549 cells binds to recombinant human PD-L1 protein (Fig. 1C). Although there was no binding of supernatant of A549 cells infected with control HDAd (HDeGFP), the supernatant of A549 cells infected with HDPDL1 had dose-dependent binding to rPD-L1 similar to anti-human PD-L1 IgG. To evaluate whether PD-L1 mini-body can promote T-cell responses by blocking the PD-1:PD-L1 interaction, supernatant from HDPDL1-infected A549 cells was added to an allogeneic mixed lymphocyte reaction (MLR) (18, 22) (Fig. 1D). In the presence of PD-L1 mini-body or anti-human PD-L1 IgG, IFNγ release increased 10-fold compared to an MLR in the presence of isotype IgG or supernatant of A549 cells infected with HDeGFP. The levels of IFNγ release were dependent on the dose of PD-L1 mini-body or anti-human PD-L1 IgG (Supplemental Fig. 2A). PD-L1 mini-body and anti-human PD-L1 IgG also enhanced T-cell proliferation compared to controls (Supplemental Fig. 2B). These results indicate that PD-L1 mini-body secreted from HDPDL1-infected A549 cells blocks the PD-1:PD-L1 interaction similarly to anti-human PD-L1 IgG.
PD-1:PD-L1 blockade by PD-L1 mini-body increases the killing effect of HER2.CAR T-cells in vitro
To evaluate whether PD-1:PD-L1 blockade by PD-L1 mini-body enhances HER2.CAR T-cell cancer cell killing, we co-cultured HER2.CAR T-cells with PC-3 and SiHa infected with HDPDL1 or control HDAd. We also performed co-culture experiments in the presence of 10 μg/mL anti-human PD-L1 IgG or isotype IgG as controls (Fig. 2). Live cells were counted after 5 days co-culture. Although there was no difference in live cancer cells between untreated and PC-3 cells with control HDAd or isotype IgG, PC-3 treated with anti-PD-L1 IgG or infected with HDPDL1 enhanced cancer cell killing by HER2.CAR T-cells 2–3 fold. PD-L1 blockade also increased HER2.CAR T-cell expansion 1.3–2 fold (Fig. 2A) and proliferation compared to controls (Supplemental Fig. 3). During the same co-culture experiment with SiHa cells, only those infected with HDPDL1 had 60% lower cell numbers compared to control groups. HER2.CAR T-cells in the presence of PD-L1 mini-body expanded 5-fold more than other groups (Fig. 2B).
Figure 2. Blockade of PD-L1:PD-1 interaction enhances cancer cell killing by HER2.CAR T-cells.
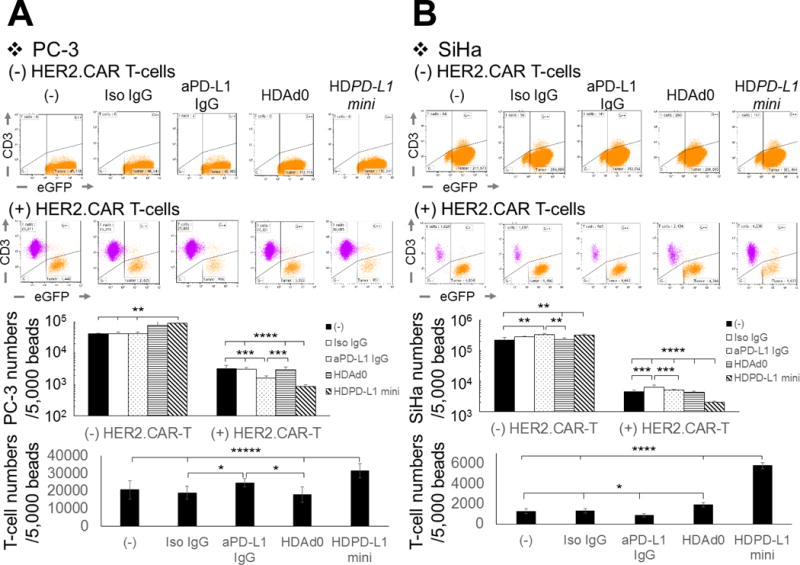
PC-3 expressing eGFP (A) and SiHa expressing eGFP (B) were infected with 1,000 vp/cell of HDAd0 or HDPD-L1. HER2.CAR T-cells were added at 24 hours post-infection (effector:target ratio of 1:20). The same co-culture experiments were performed in the presence of 10 μg/mL anti-PD-L1 IgG or isotype IgG. Cells were harvested 120 hours post-co-culture, and viable cells (7AAD-) per 5,000 counting beads were analyzed by flow cytometry. Data are presented as means ± SD (n=4). *P < 0.05, **P < 0.01, ***P=0.006, ****P=0.004, *****P < 0.001 for (A). *P < 0.03, **P < 0.01, ***P < 0.005, ****P<0.0001 for (B). The experiments were repeated with a HER2.CAR T-cells derived from a different donor with similar results.
Importantly, we observed no toxicity due to HDAd infection, PD-L1 mini-body or IgG treatments compared to untreated cells (Fig. 2). These results indicate that the cancer cell killing seen in our co-culture experiments was dependent on HER2.CAR T-cells and that PD-1:PD-L1 blockade can enhance the killing effect of these cells despite increased expression of PD-L1 on cancer cells in the presence of CAR T-cells (Fig. 1A).
CAd-VECPDL1 amplifies PD-L1 mini-body expression in human cancer cell lines in vitro and in vivo
To test whether co-infection of Onc.Ad with HDPD-L1 could amplify PD-L1 mini-body in transduced human cancer cell lines, as previously shown with HDAd transgenes (4), we co-infected HDPD-L1 with Onc.Ad (CAd-VECPDL1) into human solid cancer cell lines (non-small cell lung carcinoma A549, prostate cancer PC-3, squamous cell carcinoma SiHa, hepatocellular carcinoma HepG2), and evaluated the levels of PD-L1 mini-body in media 48 hours post-infection (Fig. 3A). A549, PC-3 and HepG2 cells infected with a CAd-VECPDL1 had 4-fold (A549), 15-fold (PC-3), 10-fold (HepG2) higher expression of PD-L1 mini-body in media compared to cells infected with HDPDL1 alone. We confirmed the amplified production of PD-L1 mini-body by Western blotting (Supplemental Fig. 4A). To test whether increased expression was dependent on the amplification of HDAd vector DNA, we quantified both HDAd and Onc.Ad vector copies using primer sets for each backbone 48 hours post-infection (Fig. 3B). Cells infected with the CAd-VECPDL1 had 1,000-fold more HDAd vector copies than cells infected with HDAd alone. To verify whether CAd-VECPDL1 induces both amplification of HDAd and the lytic effects of Onc.Ad, cellular lysis of CAd-VECPDL1 was evaluated with MTS assay 96 hours post-infection (Fig. 3C), and the LD50 of each treatment in each cell line was determined (Supplemental Table 1). Although Onc.Ad alone was more toxic than CAd-VECPDL1, CAd-VECPDL1 had dose-dependent lytic effects in infected cancer cell lines.
Figure 3. Co-infection of Onc.Ad with HDPD-L1 (CAd-VECPD-L1) amplified PD-L1 mini-body while maintaining oncolysis in vitro and in vivo.
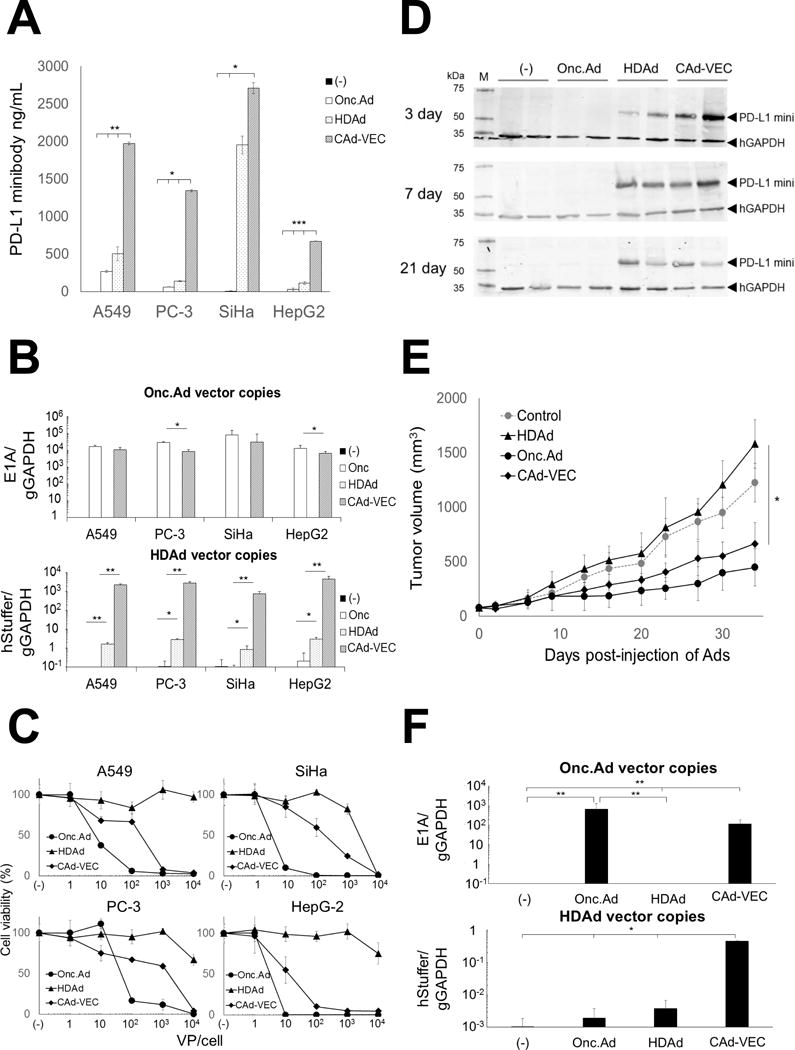
(A) A549, PC-3, SiHa and HepG2 were infected with a total 10 Vp/cell of HDPD-L1, Onc.Ad or with an CAd-VECPD-L1 (Onc.Ad:HDAd; 1:10). Medium samples were collected at 48 hours post-infection. Levels of PD-L1 mini-body in medium samples were quantified by ELISA-based assay for HA-tagged protein. Data are presented as means ± SD (n=4). *P =0.003, **P=0.002, ***P<0.001. (B) DNA samples were extracted 48 hr post-infection, and Onc.Ad and HDAd vector copy numbers were measured by quantitative PCR. Data were normalized with human genomic GAPDH. Data are presented as means ± SD (n=4). *P < 0.03, **P< 0.001. (C) Cancer cell lines were infected with increasing doses of HDPD-L1, Onc.Ad or with CAd-VECPD-L1 (Onc.Ad:HDAd= 1:10). Viable cells were analyzed at 96 hours by MTS assay. Data are presented as means ± SD (n=6). (D) PC-3 cells were transplanted into the right flanks of nude mice. A total of 1×108 Vp of Onc.Ad, HDAd or CAd-VECPD-L1 (Onc.Ad:HDAd= 1:20) were injected intratumorally. Tumors were collected and harvested at 3, 7 and 21 days post-injection. PD-L1 mini-body in tumor lysates was detected by western blotting. (E) Tumor volumes were measured at different time points. Data are presented as means ± SD (n=4). *P = 0.006. (F) Total DNA was extracted from tumors at 35 days post-injection of Ads, and the copy number of each vector determined by quantitative PCR. Data were normalized with human genomic GAPDH. Data are presented as means ± SD (n=4). *P< 0.02, **P =0.008.
To examine the PD-L1 mini-body expression and anti-tumor effect of CAd-VECPDL1 in vivo, nude mice were subcutaneously transplanted with PC-3. After the tumor volume reached 100 mm3, we injected 1×108 viral particles (Vp) of Onc.Ad, HDPDL1 or CAd-VECPDL1 intra-tumorally. Tumor samples were collected at different time points after injection, and the presence of PD-L1 mini-body in tumors was evaluated by Western Blot (Fig. 3D). PD-L1 mini-body was detected in lysates from tumors injected with HDPDL1 alone or CAd-VECPDL1 over time. PD-L1 mini-body was not detectable in the serum of mice from either group (Supplemental Fig. 4B). There was no difference in tumor growth between untreated mice and those treated with HDPDL1 alone, indicating that PD-L1 mini-body expression at the tumor site alone does not suppress tumor growth. These results are consistent with those from our in vitro experiments (Fig. 2). Mice treated with CAd-VECPDL1 demonstrated 60% lower tumor volume compared to control mice at 35 days post-injection, as did those injected with Onc.Ad alone, indicating that the Onc.Ad-dependent lytic effects are maintained in vivo (Fig. 3E). We also quantified Onc.Ad and HDPDL1 vector copies in tumors at 35 days post-injection (Fig. 3F) and found that tumors injected with CAd-VECPDL1 exhibited 100-fold more HDAd copies than those injected with HDPDL1 alone.
Combinatorial treatment with CAd-VECPDL1 and CAR T-cells prolongs survival in vivo
To test whether pre-treatment of CAd-VECPDL1 enhances the anti-tumor effects of adoptively transferred HER2.CAR T-cells in xenograft models, NSG mice were subcutaneously transplanted with PC-3, and 1×107 Vp of Onc.Ad or CAd-VECPDL1 were intratumorally injected after the tumor reached 100 mm3. Control mice were intratumorally injected with vehicle alone. 1×106 HER2.CAR T-cells genetically modified to express firefly luciferase (ffLuc) were systemically infused three days after Ad injection (19). While PD-L1 mini-body increased HER2.CAR T-cell expansion compared to HER2.CAR T-cells alone in vitro (Fig. 2A), there were no differences in ffLuc activity at tumor sites in animals treated with HER2.CAR T-cells alone or CAd-VECPDL1 with HER2.CAR T-cells (Fig. 4A). Additionally, we found that mice treated with Onc.Ad and HER2.CAR T-cells initially had 50–90% less ffLuc activity post-HER2.CAR T-cell injection than those treated with CAR T-cells alone. The decreased activity reversed at 14 days, such that mice from all three treatment groups had similar ffLuc activity. We detected PD-L1 mini-body in tumor samples 10 days after HER2.CAR T-cell injection (Supplemental Fig. 5A). While we observed both adenovirus and T-cells at tumor sites 10 days after HER2.CAR T-cell injection, it was unclear whether they co-localized there (Supplemental Fig. 5B). These results suggest that adoptive HER2.CAR T-cell treatment has a minimal impact on Onc.Ad-dependent oncolysis.
Figure 4. CAd-VECPD-L1 enhanced the anti-tumor effects of HER2.CAR T-cells in vivo.
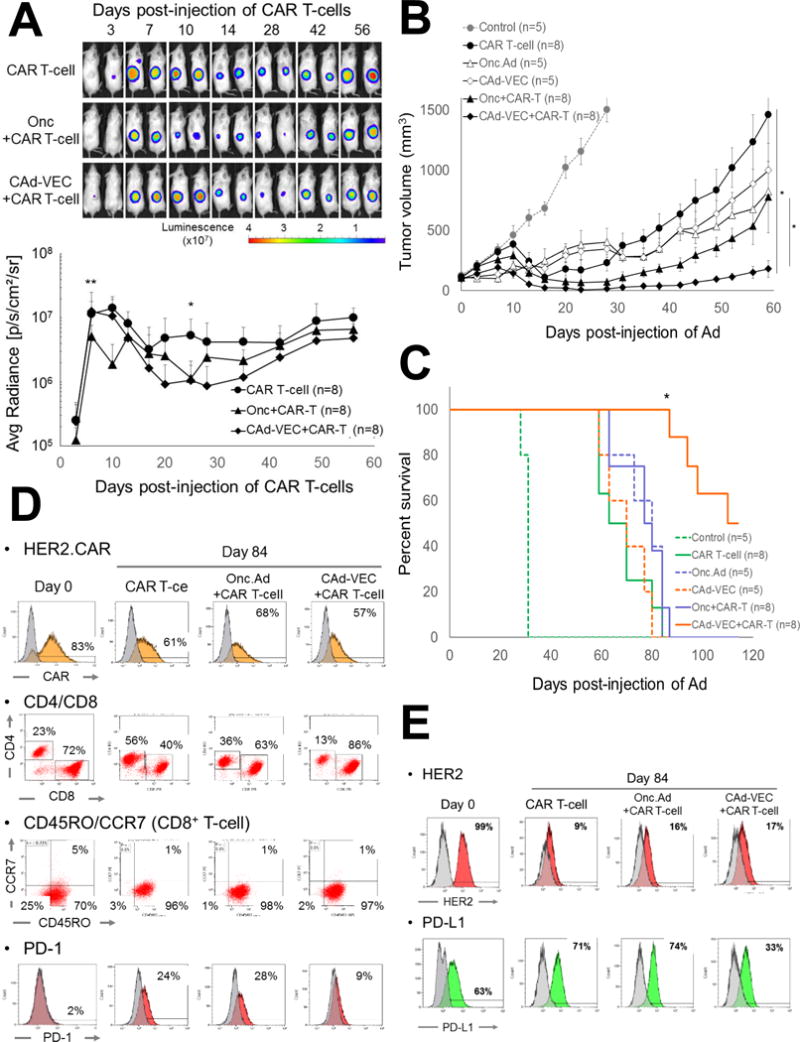
(A) PC-3 cells were transplanted into the right flanks of NSG mice. A total of 1×107 Vp of Onc.Ad or CAd-VECPD-L1 (Onc.Ad:HDAd= 1:20) were injected intra-tumorally. A total of 1×106 HER2.CAR T-cells expressing firefly luciferase (ffLuc) were systemically administered 3 days post-injection of Ads. Bioluminescence of HER2.CAR T-cells was monitored at different time points. Data are presented as means ± SD (n=8). *P =0.002, **P < 0.001. (B) Tumor volumes were measured at different time points. Data are presented as means ± SD (n=8). *P = 0.002. (C) Kaplan-Meier survival curve after administration of Ad gene therapy. The end point was established at tumor volume of > 1,500 mm3. Data are presented as means ± SD (n=8). *P < 0.001. (D) T-cells at tumor site were isolated at 84 days post-infusion, and phenotype was analyzed by flow cytometry. (E) Tumor cells were isolated at 84 days post-infusion of HER2.CAR T-cells and were cultured in vitro. Cells were harvested at 72 hours, and phenotype was analyzed by flow cytometry.
While we did not observe increased T cell expansion in mice pre-treated with CAd-VECPDL1 compared to the other groups, however mice pre-treated with Ad gene therapy (either Onc.Ad or CAd-VECPDL1) had suppressed tumor growth compared to mice treated with HER2.CAR T-cells alone (Fig. 4B). Although mice treated with CAd-VECPDL1 had similar median survival to mice treated with HER2.CAR T-cells (60 days), mice treated with CAd-VECPDL1 and HER2.CAR T-cells had 2-fold longer median survival (110 days) than mice treated with a single agent (Fig. 4C). These results indicate that PD-L1 mini-body-dependent HER2.CAR T-cell activation at the tumor site enhances the anti-tumor effect of HER2.CAR T-cells.
To evaluate how Ad gene therapy phenotypically impacts HER2.CAR T-cells and cancer cells, we collected tumors from mice 84 days post-Ad injections. We phenotyped infiltrated T-cells and residual (recurrent) cancer cells (Fig. 4D, E) and found approximately 60% of T-cells isolated from tumor samples still expressed HER2.CAR (Fig. 4D). We also phenotyped HER2.CAR T-cells at different time points to confirm the T-cells retained HER2.CAR expression (Supplemental Fig. 5C). T-cells isolated from tumors treated with Ad gene therapy had more CD8+ T-cells, but there was no difference in memory phenotype (CCR7/CD45RO) of CD8+ or CD4+ T-cells (Supplemental Fig. 5D). Although there were similar expression levels of other exhaustion markers (Tim-3 and LAG-3) (Supplemental Fig. 5D), HER2.CAR T-cells in mice treated with CAd-VECPDL1 had 30% lower PD-1 expression compared to other groups. Since PD-1+ T-cell populations are mostly CD4+ (Supplemental Fig. 1C) (23), the reduced CD4+ population in CAd-VECPDL1-treated mice may correlate with fewer PD-1+ T-cells. Since HER2.CAR T-cells still expressed the CAR, we analyzed HER2 expression on cancer cells from tumors 84 days post-Ad injection (Fig. 4E). We evaluated HER2 expression on human CD47+ cells, which is highly expressed in human cancers including prostate cancer, as tumor cells also contained mouse stroma cells (Supplemental Fig. 5E) (24, 25). Human CD47+ cells had 80–90% lower HER2 expression compared to PC-3 cells cultured in vitro, indicating that PC-3 cells downregulated HER2 expression. Interestingly, cancer cells in a mouse pre-treated with CAd-VECPDL1 showed 50% less PD-L1 expression compared to other groups (Fig. 4E). These results suggest that constitutive blockade of PD-L1 by PD-L1 mini-body leads to downregulation of PD-L1 expression on remaining cancer cells.
We performed the same experiments with NSG mice subcutaneously transplanted with SiHa cells (Fig. 5). Although 1×106 HER2.CAR T-cell alone minimally improved median survival (30 days) compared to untreated mice (24 days), mice treated with CAd-VECPDL1 and HER2.CAR T-cells had 2-fold longer median survival (42 days) than untreated mice (Fig. 5C). We collected tumors from mice treated with HER2.CAR T-cells with and without Ad treatments and phenotyped both the T-cells and tumor cells (Fig. 5D, E). Tumor cells showed reduced HER2 expression in the presence of HER2.CAR T-cells, and only the tumor sample pre-treated with CAd-VECPDL1 showed 50% less PD-L1 expression compared to other groups, similar to what we see in the PC-3 model.
Figure 5. CAd-VECPD-L1 enhanced the anti-tumor effects of HER2.CAR T-cells in SiHa xenograft model.
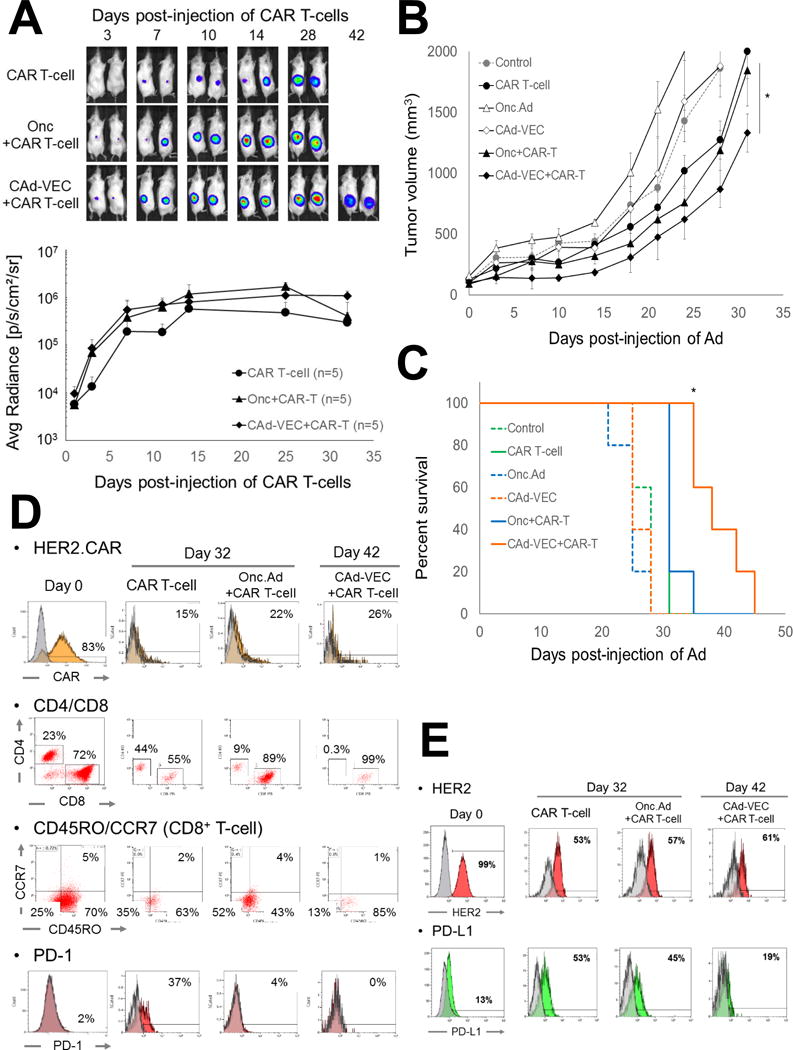
(A) SiHa cells were transplanted into the right flanks of NSG mice. A total of 1×107 Vp of Onc.Ad or CAd-VECPD-L1 (Onc.Ad:HDAd= 1:20) were injected intra-tumorally. A total of 1×106 HER2.CAR T-cells expressing firefly luciferase (ffLuc) were systemically administered 3 days post-injection of Ads. Bioluminescence of HER2.CAR T-cells was monitored at different time points. Data are presented as means ± SD (n=5). (B) Tumor volumes were measured at different time points. Data are presented as means ± SD (n=5). *P < 0.001. (C) Kaplan-Meier survival curve after administration of Ad gene therapy. The end point was established at tumor volume of > 1,500 mm3. Data are presented as means ± SD (n=5). *P = 0.003. (D) T-cells at tumor site were isolated at 32 or 42 days post-infusion, and phenotype was analyzed by flow cytometry. (E) Tumor cells were isolated at 32 or 42 days post- HER2.CAR T-cell infusion and were cultured in vitro. Cells were harvested at 72 hrs, and phenotype was analyzed by flow cytometry.
Systemic treatment of PD-L1 IgG reduces the anti-tumor effects of CAR T-cells in vivo
As anti-PD-L1 IgG is FDA approved for use in humans (9), we compared local blockade of the PD-1:PD-L1 interaction by PD-L1 mini-body to systemic anti-PD-L1 IgG treatment in NSG mice with PC-3 tumors. We systemically injected anti-PD-L1 IgG into mice treated with Onc.Ad and HER2.CAR T-cells and mice treated with HER2.CAR T-cells alone (Fig. 6A). Since pre-treatment of CAd-VECPDL1 leads to PD-L1 mini-body expression at the tumor site before HER2.CAR T-cell infiltration, we infused anti-PD-L1 IgG or isotype IgG prior to HER2.CAR T-cell injection. Intratumoral injection CAd-VECPDL1 showed significantly better anti-tumor effects than systemic anti-PD-L1 IgG in mice treated with both Onc.Ad and HER2.CAR T-cell and those treated with HER2.CAR T-cell alone (Fig. 6B). Overall, mice that received CAd-VECPDL1 had 2-fold longer median survival (110 days) compared to the IgG groups (59 days) (Fig. 6C), indicating local expression of PD-L1 mini-body is more effective than systemic anti-PD-L1 IgG treatment. Next, we examined HER2.CAR T-cell expansion at tumor sites using ffLuc (Fig. 6D). Mice pre-treated with anti-PD-L1 IgG had 30–90% lower ffLuc activity at tumor sites compared to mice injected with CAd-VECPDL1 or mice injected with isotype IgG at 1, 3 and 7 days post-HER2.CAR T-cell injection. Mice pre-treated with anti-PD-L1 IgG also had less ffLuc activity at the ventral side 1 day post-HER2.CAR T-cell injection (Supplemental Fig. 6), suggesting that systemic administration of anti-PD-L1 IgG prior to HER2.CAR T-cell infusion reduces the total number of circulating HER2.CAR T-cells. Since intratumoral CAd-VECPDL1 treatment has minimal distribution in the blood (Supplemental Fig. 4B), local PD-L1 mini-body expression may not reduce the number of HER2.CAR T-cells before they reach the tumor site, as was seen in mice systemically treated with anti-PD-L1 IgG.
Figure 6. CAd-VECPD-L1 had better anti-tumor effects than systemic PD-L1 IgG treatment.
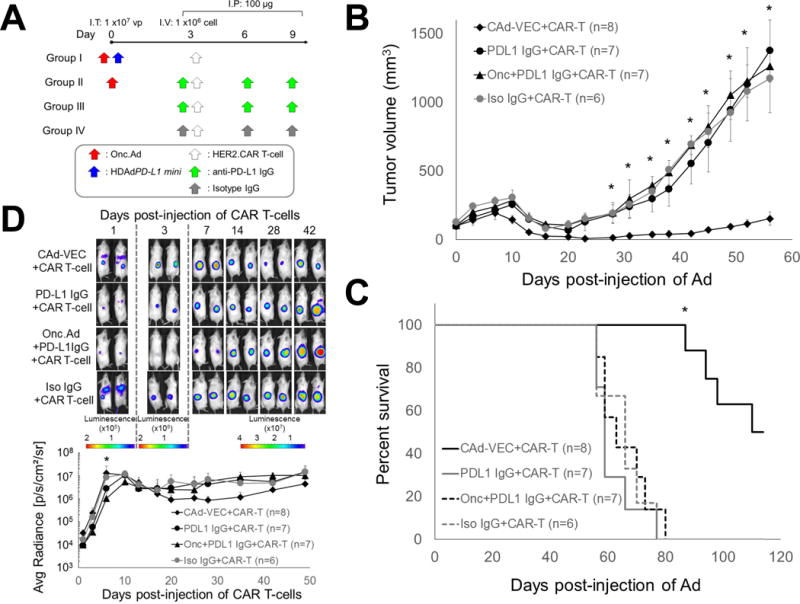
(A) PC-3 cells were transplanted subcutaneously into the right flanks of NSG mice. A total of 1×107 Vp of Onc.Ad or CAd-VEC (Onc.Ad:HDAd; 1:20) were injected intra-tumorally. A total of 1×106 HER2.CAR T-cells expressing ffLuc were systemically administered at 3 days post-injection of Ads. Mice treated with Onc.Ad with HER2.CAR T-cells or HER2.CAR T-cells alone received 3 intraperitoneal injections of 100 μg anti-PD-L1 IgG at day 0, 3 and 6 post-infusion of HER2.CAR T-cells. (B) Tumor volumes were measured at different time points. Data are presented as means ± SD (n=8 or 7). *P < 0.001. (C) Kaplan-Meier survival curve after administration of Ad gene therapy. The end point was established at tumor volume of > 1,500 mm3. Data are presented as means ± SD (n=8 or 7). *P < 0.001. (D) Bioluminescence of HER2.CAR T-cells was monitored at different time points. Data are presented as means ± SD (n=7 or 8). *P = 0.001.
Discussion
Here, we demonstrate that PD-L1 mini-body expressed by CAd-VECPDL1 can block the PD-1:PD-L1 interaction between HER2.CAR T-cells and cancer cells while maintaining cancer cell oncolysis. The combinatorial effect of Onc.Ad-mediated oncolysis and PD-L1 mini-body-mediated blockade augments the anti-tumor effect of adoptively transferred HER2.CAR T-cells. Together, CAd-VECPDL1 and HER2.CAR T-cell treatment significantly prolonged animal survival compared to treatment with HER2.CAR T-cells alone and to Onc.Ad with HER2.CAR T-cells in a xenograft mouse model of prostate cancer. Local blockade of the PD-1:PD-L1 interaction by CAd-VECPDL1 also induced superior anti-tumor effects compared to systemic administration of anti-PD-L1 IgG in combination with adoptive transfer of HER2.CAR T-cells in our xenograft mouse model.
Immune checkpoint inhibitors (e.g. PD-1, PD-L1 and CTLA-4) have been successful in treating multiple solid tumors, leading to more robust and sustained T-cell responses (9); however, many patients do not respond or subsequently relapse (26). To enhance host immune responses to cancer cells, checkpoint inhibitors have been combined with other therapeutic agents including chemotherapy, radiation therapy and oncolytic viral gene therapy (Onc.Vs) in preclinical models and clinical trials (26). Since Onc.Vs selectively lyse cancer cells and induce proinflammatory responses, combining immune checkpoint inhibitors with Onc.Vs seems to be a natural marriage (1). However, the therapeutic effect of this combination relies on the patient’s immune response, and whether this combination is safe remains unknown (1). In order to overcome these limitations, we aimed to concentrate checkpoint inhibition locally at the tumor site, thereby minimizing off-target toxicities. We tested in two murine xenograft models the combination of Onc.Ad expressing an anti-PD-L1 checkpoint inhibitor with adoptively transferred CAR-modified T-cells, as local checkpoint blockade should increase their potency. We hypothesized that this combination would create a pro-inflammatory tumor microenvironment through oncolysis by Onc.Ad and block the deleterious PD-1:PD-L1 interaction locally, enabling increased CAR T-cell activity.
We found that constitutive expression of PD-L1 mini-body at the tumor site had superior anti-tumor effects than systemic treatment of anti-PD-L1 IgG in the presence of HER2.CAR T-cells. Ten days after HER2.CAR T-cell injection, some mice that previously received systemic infusion of anti-PD-L1 IgG, but not in mice infused with isotype IgG, had transient diarrhea. Similar side effects are seen in patients with renal cell carcinoma receiving atezolizumab (anti-PD-L1 antibody) (13), suggesting that systemic treatment of anti-PD-L1 IgG with HER2.CAR T-cells leads to immune-related side effects in mice. In contrast, intratumoral administration of CAd-VECPDL1 followed by systemic HER2.CAR T-cell treatment caused no immune-related side effects (e.g. diarrhea, weight loss). Since PD-L1 mini-body expressed by CAd-VEC is localized at the tumor site with minimal circulation in the blood, our treatment may minimize toxicities related to systemic treatment of anti-PD-L1 IgG. Our results also suggest that systemic administration of anti-PD-L1 IgG prior to HER2.CAR T-cell infusion reduces the total number of circulating HER2.CAR T-cells. Stimulated naïve T-cells express both PD-L1 and PD-1, and blocking the PD-1:PD-L1 interaction leads to apoptosis through Fas:Fas ligand (FasL). HER2.CAR T-cells in mice may induce the expression of both PD-1 and PD-L1 through their TCR and/or CAR, and therefore blockade of the PD-1:PD-L1 interaction outside the tumor (e.g. in the lung) may induce unwanted Fas:FasL-dependent T-cell apoptosis (27).
Intratumoral administration of Onc.Vs, including Onc.Ads, has limited distribution to metastasized tumors (28), and investigators have been developing viral vectors to target both primary and metastasized tumors systemically. However, patients who received systemic Onc.Vs developed both Th1 and Th2 adaptive immune responses to the viruses after the first treatment, reducing the anti-tumor efficacy of the second challenge due to rejection of the virus and infected cells (29). Since CAR T-cells are generally generated from patients’ own peripheral blood and are therefore better tolerated than Onc.Vs, CAR T-cells can be infused repeatedly to cancer patients (30). Our combination of intratumoral CAd-VEC with systemic CAR T-cell therapy could overcome the inherent limitation of Onc.Vs to create enhanced anti-tumor effects.
In this study, our “all-in-one” strategy attenuated the immunosuppressive effects of the PD-1:PD-L1 interaction on adoptively transferred CAR T-cells at tumor sites, leading to superior anti-tumor effects and prolonged animal survival in a prostate cancer xenograft mouse model. This combinatorial treatment also showed significant effects in mice with subcutaneous SiHa tumors, although the potency was lower, suggesting that blocking the PD-1:PD-L1 interaction alone may be insufficient to maximize the anti-tumor effect of CAR T-cells in particularly aggressive solid tumors. Since CAd-VEC is able to deliver multiple immunomodulatory molecules (up to 34 kb) in a single HDAd vector with Onc.Ad-dependent lytic effect, in the future we can incorporate additional molecules to maximize the anti-tumor effect of adoptively transferred CAR T-cells. A previous study showed that Onc.Ad expressing chemokine can enhance the infiltration of adoptively transferred GD2.CAR T-cells at the tumor site in a neuroblastoma xenograft model, and Onc.Ad expressing cytokine enhances proliferation of those CAR T-cells, leading to superior anti-tumor effects than the combination of Onc.Ad (without transgene) with GD2.CAR T-cells (19). We will investigate whether combining PD-L1 mini-body with chemokines and/or cytokines further enhances the anti-tumor effects of adoptively transferred CAR T-cells.
Overall, we demonstrate that combined CAd-VECPDL1 and CAR T-cell treatment has significant anti-tumor effects against bulky solid tumors. Since intratumoral injection of Onc.Ads has been tested in patients with a range of solid tumors (31), our concept could readily be applied to other solid tumors and used with CARs targeting different surface molecules (e.g. GD2, PSCA) (19, 32).
Supplementary Material
One Sentence Summary.
Findings demonstrate enhanced CAR T-cell killing of tumor cells via PD-L1 blocking mini-body expressed by oncolytic adenovirus in cell lines and xenograft mouse models.
Acknowledgments
The authors would like to thank Dr. Brendan Lee in the Department of Molecular and Human Genetics at Baylor College of Medicine for his support of the project and Catherine Gillespie in the Center for Cell and Gene Therapy at Baylor College of Medicine for her editing of the paper. This work was supported by National Institutes of Health (R00HL098692) to M. Suzuki, T32HL092332 to A. Rosewell Shaw, BCM Head and Neck Seed Grant and Concern Foundation to M. Suzuki. K. Tanoue was supported by Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation, Japan Society for the Promotion of Science.
Financial Support: M. Suzuki supported by National Institutes of Health (R00HL098692), BCM Head and Neck Seed Grant and Concern Foundation. A. Rosewell Shaw supported by National Institutes of Health (T32HL092332).
Footnotes
Conflict of Interest: The authors disclose no potential conflicts of interest.
References
- 1.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nature reviews Cancer. 2014;14:559–67. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 2.Bett AJ, Prevec L, Graham FL. Packaging capacity and stability of human adenovirus type 5 vectors. Journal of virology. 1993;67:5911–21. doi: 10.1128/jvi.67.10.5911-5921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cerullo V, Koski A, Vaha-Koskela M, Hemminki A. Chapter eight–Oncolytic adenoviruses for cancer immunotherapy: data from mice, hamsters, and humans. Advances in cancer research. 2012;115:265–318. doi: 10.1016/B978-0-12-398342-8.00008-2. [DOI] [PubMed] [Google Scholar]
- 4.Farzad L, Cerullo V, Yagyu S, Bertin T, Hemminki A, Rooney C, et al. Combinatorial treatment with oncolytic adenovirus and helper-dependent adenovirus augments adenoviral cancer gene therapy. Molecular Therapy — Oncolytics. 2014;1:14008. doi: 10.1038/mto.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nature reviews Cancer. 2013;13:525–41. doi: 10.1038/nrc3565. [DOI] [PubMed] [Google Scholar]
- 6.Ramos CA, Heslop HE, Brenner MK. CAR-T Cell Therapy for Lymphoma. Annual review of medicine. 2016;67:165–83. doi: 10.1146/annurev-med-051914-021702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nature medicine. 2013;19:1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nature reviews Drug discovery. 2015;14:561–84. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 10.Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, et al. Antibody-Dependent Cellular Cytotoxicity Activity of a Novel Anti-PD-L1 Antibody Avelumab (MSB0010718C) on Human Tumor Cells. Cancer immunology research. 2015;3:1148–57. doi: 10.1158/2326-6066.CIR-15-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33:1688–96. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer research. 2015;75:2139–45. doi: 10.1158/0008-5472.CAN-15-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDermott DF, Sosman JA, Sznol M, Massard C, Gordon MS, Hamid O, et al. Atezolizumab, an Anti-Programmed Death-Ligand 1 Antibody, in Metastatic Renal Cell Carcinoma: Long-Term Safety, Clinical Activity, and Immune Correlates From a Phase Ia Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016 doi: 10.1200/JCO.2015.63.7421. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki M, Bertin TK, Rogers GL, Cela RG, Zolotukhin I, Palmer DJ, et al. Differential type I interferon-dependent transgene silencing of helper-dependent adenoviral vs. adeno-associated viral vectors in vivo. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:796–805. doi: 10.1038/mt.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki M, Cela R, Clarke C, Bertin TK, Mourino S, Lee B. Large-scale production of high-quality helper-dependent adenoviral vectors using adherent cells in cell factories. Human gene therapy. 2010;21:120–6. doi: 10.1089/hum.2009.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 17.Kaka AS, Foster AE, Weiss HL, Rooney CM, Leen AM. Using dendritic cell maturation and IL-12 producing capacity as markers of function: a cautionary tale. J Immunother. 2008;31:359–69. doi: 10.1097/CJI.0b013e318165f5d2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer immunology research. 2014;2:846–56. doi: 10.1158/2326-6066.CIR-14-0040. [DOI] [PubMed] [Google Scholar]
- 19.Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer research. 2014;74:5195–205. doi: 10.1158/0008-5472.CAN-14-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nature protocols. 2009;4:1670–80. doi: 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wherry EJ. T cell exhaustion. Nature immunology. 2011;12:492–9. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 22.Stewart R, Morrow M, Hammond SA, Mulgrew K, Marcus D, Poon E, et al. Identification and Characterization of MEDI4736, an Antagonistic Anti-PD-L1 Monoclonal Antibody. Cancer immunology research. 2015;3:1052–62. doi: 10.1158/2326-6066.CIR-14-0191. [DOI] [PubMed] [Google Scholar]
- 23.Ahrends T, Babala N, Xiao Y, Yagita H, van Eenennaam H, Borst J. CD27 agonism plus PD-1 blockade recapitulates CD4+ T cell help in therapeutic anti-cancer vaccination. Cancer research. 2016 doi: 10.1158/0008-5472.CAN-15-3130. [DOI] [PubMed] [Google Scholar]
- 24.Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nature reviews Immunology. 2015;15:73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lih CJ, Wei W, Cohen SN. Txr1: a transcriptional regulator of thrombospondin-1 that modulates cellular sensitivity to taxanes. Genes & development. 2006;20:2082–95. doi: 10.1101/gad.1441306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoos A. Development of immuno-oncology drugs - from CTLA4 to PD1 to the next generations. Nature reviews Drug discovery. 2016 doi: 10.1038/nrd.2015.35. [DOI] [PubMed] [Google Scholar]
- 27.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature medicine. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 28.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nature biotechnology. 2012;30:658–70. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nature reviews Drug discovery. 2015;14:642–62. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunological reviews. 2015;263:68–89. doi: 10.1111/imr.12243. [DOI] [PubMed] [Google Scholar]
- 31.Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I, et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:1874–84. doi: 10.1038/mt.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katari UL, Keirnan JM, Worth AC, Hodges SE, Leen AM, Fisher WE, et al. Engineered T cells for pancreatic cancer treatment. HPB : the official journal of the International Hepato Pancreato Biliary Association. 2011;13:643–50. doi: 10.1111/j.1477-2574.2011.00344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.