

Abstract
We report the first in vitro selection of DNA nanostructures that switch their conformation when triggered by change in pH. Previously, most pH-active nanostructures were designed using known pH-active motifs, such as the i-motif or the triplex structure. In contrast, we performed de novo selections starting from a random library and generated nanostructures that can sequester and release Mipomersen, a clinically approved antisense DNA drug, in response to pH change. We demonstrate extraordinary pH-selectivity, releasing up to 714-fold more Mipomersen at pH 5.2 compared to pH 7.5. Interestingly, none of our nanostructures showed significant sequence similarity to known pH-sensitive motifs, suggesting that they may operate via novel structure-switching mechanisms. We believe our selection scheme is general and could be adopted for generating DNA nanostructures for many applications including drug delivery, sensors and pH-active surfaces.
Keywords: controlled release, DNA structures, in vitro selection, nanostructures, pH-active
pH is the key
A new directed evolution method to generate DNA nanostructures which can catch and release an antisense DNA payload in response to pH change. Highly pH-selective release was achieved where up to 714-fold more payload was released at pH 5.2 compared to pH 7.5. These molecules have potential applications in drug delivery, sensors, and pH-sensitive nanomaterials.
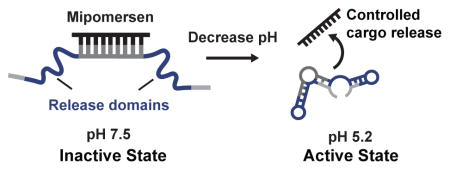
Cells often utilize changes in local pH to achieve molecular transport and processing. For example, ligands internalized by the cell are processed by the endosome for trafficking to specific cell compartments or further transported to the lysosome for degradation.[1] The pH of the late endosome (pH ~5.2) and lysosome (pH ~4.5) are significantly more acidic than the extracellular environment (pH ~7.4),[1] and there has been great interest in exploiting this pH difference to achieve controlled delivery of drugs into the cell.[2,3] In particular, there is growing interest in pH-sensitive DNA nanostructures, because they are stable, programmable, and are readily integrated with other functional nucleic acids such as antisense DNA and siRNA.[4] To date, most pH-sensitive DNA nanostructures have been constructed based on motifs that are already known to be pH-responsive. For example, a number of investigators have incorporated the “i-motif” to achieve pH-triggered structure switching.[5–8] At neutral pH, the i-motif possesses negligible secondary structure, but in acidic conditions, non-canonical base-pairs form a C-quadruplex.[9] The i-motif has been used to demonstrate rapid pH-controlled loading and release of a complementary DNA strand.[10,11] More recently, a pH-controlled “triplex switch” has been described, which switches between duplex and triplex forms.[12] Ricci and co-workers incorporated a triplex motif distal to the ligand binding site for pH-dependent allosteric control over DNA loading/release.[13] Their strategy increased the variety of DNA ligands which could be delivered by placing the pH-sensitive motif distal to the DNA binding site and the pH-sensitivity could be tuned by varying the CGC content of the triplex. [13] While demonstrably powerful, the design of nanomachines incorporating the i-motif and triplex switch is restricted by pre-determined geometry and orientation requirements.[9,12,13] Furthermore, while the triplex switch is more facilely tuned, the i-motif requires significant rational optimization to customize range of pH-sensitivity.[9, 14, 15] Moreover, strategies to tune the efficiency/rate of ligand release have not been investigated.
Here we describe a selection-based approach for the de novo generation of pH-activated DNA nanostructures (PADNAs) with optimized pH-response properties. We present a new strategy where we set selection conditions to tune the pH-sensitivity and efficiency/rate of release. Using our strategy, we discovered PADNAs that selectively release Mipomersen, an FDA-approved antisense DNA drug for the treatment of hypercholesterolemia,[16] with exceptional pH sensitivity—achieving up to a ~714-fold increase in payload release at pH 5.2 compared to 7.5.
Our starting library comprises three domains (Scheme 1A). The 17-nucleotide (nt) central domain is designed to hybridize with Mipomersen (20-nt) and thereby sequester it in an inactive state at pH 7.5. The central domain is flanked by two randomized release domains (22- and 21-nt), which are subject to selection. Finally, each PADNA molecule contains forward and reverse PCR primer-binding sites at the distal ends of the sequence (20-nt). All sequences are provided in the Supporting Information (Table S1).
Scheme 1.
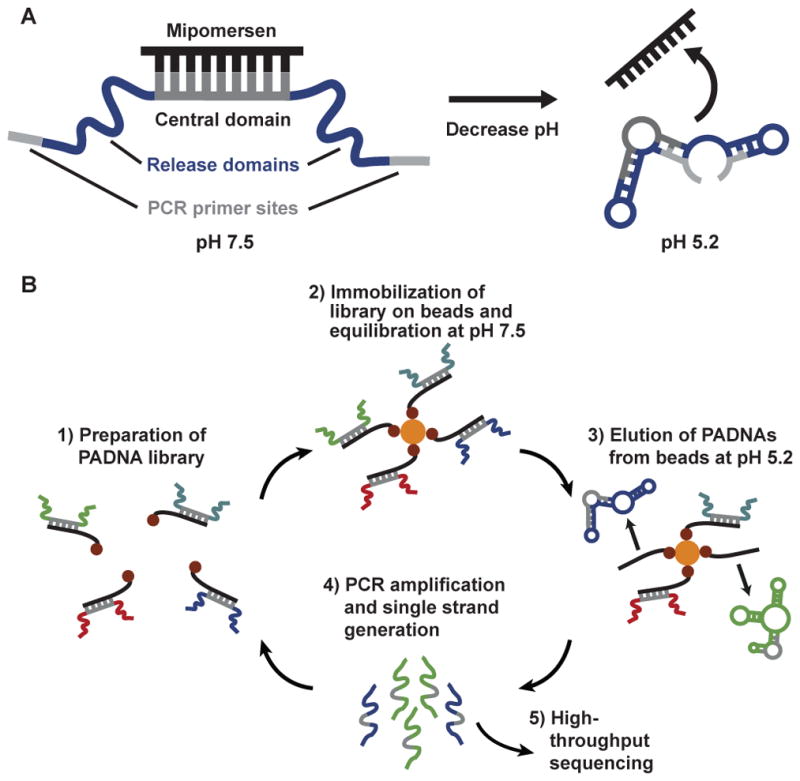
Library design and selection of pH-responsive PADNAs.
We hybridized our starting PADNA library (initially comprising 100 picomoles or 6 x 1013 molecules) with 150 pmol of biotinylated Mipomersen, and then immobilized the assembly onto streptavidin (SA)-coated magnetic beads (Scheme 1B). We incubated overnight in pH 7.5 buffer, and then discarded PADNAs that were not immobilized on the beads. Next, we challenged the assemblies with pH 5.2 buffer and collected the PADNAs that released from the beads. We PCR amplified and generated single-stranded products for the next round of selection or for high-throughput sequencing. We performed 12 rounds of selection, increasing stringency in each round by decreasing the PADNA:Mipomersen ratio (for full methods, see Supporting Information).
At the end of each round, we performed PCR amplification of samples taken from the eluents, loaded them onto a polyacrylamide gel and determined the “release ratio”—the amount of PADNAs released at pH 5.2 in comparison to pH 7.5 (Figure 1). This ratio increased over the course of selection, as seen by the differences in band intensities, reaching ~27 by R12. PADNAs showed minimal release in the presence of excess SA, confirming that they do not bind to the SA that was used during selection to conjugate Mipomersen to the beads.
Figure 1.
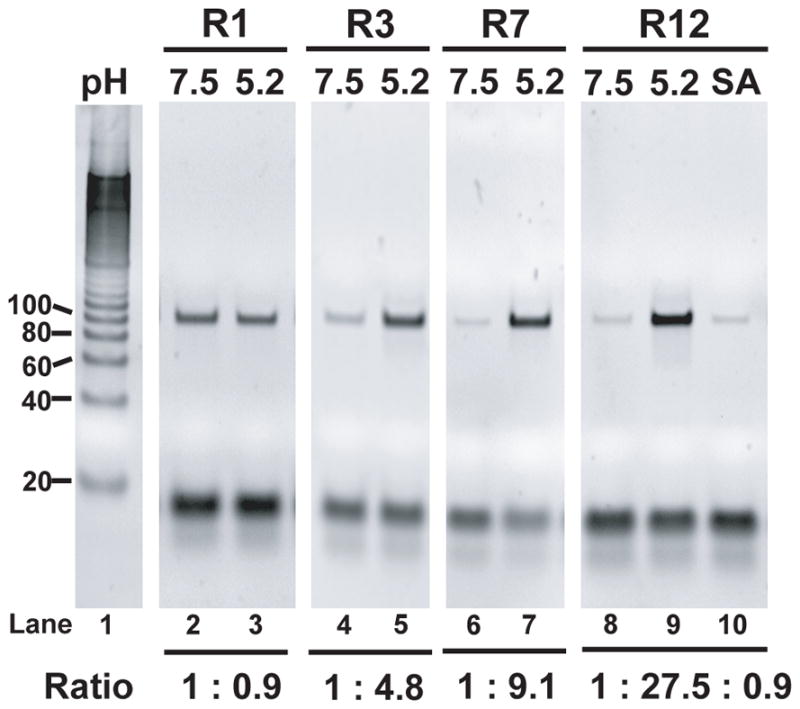
Gel images comparing the DNA eluted after 30 minutes in pH 7.5 or pH 5.2 PBSMT over multiple rounds of selection (lanes 2–9). Lane 1 contains the 20-bp ruler. Lane 10 shows elution of the R12 pool for 30 minutes in pH 7.5 buffer with 1 uM Streptavidin.
We analyzed the R12 pool with high-throughput sequencing, obtaining more than 2 million sequences. Surprisingly, the R12 pool maintained a high level of diversity, where the highest copy number of any single sequence did not exceed 91. We found that none of the 100 most abundant PADNAs contained known pH-responsive elements such as the i-motif, poly-dA repeats,[17] or the long homopyrimidine/homopurine runs that are signatures of DNA triplexes.[18] These results suggest that PADNAs are novel pH-sensitive structures that are mechanistically distinct from previously reported motifs.
We used qPCR to obtain the release ratio for the ten most abundant PADNAs. The release ratio of the top ten PADNAs ranged from 93–714 (Table S2). We focused our analysis on PADNA-1because it was the most highly represented in the R12 pool. PADNA-1 exhibits an excellent release ratio of 145, as quantified by qPCR (Figure 2A). We confirmed that the structure-switching function of PADNA-1 arises from the selected release domains by comparing our results with negative controls (Table S2, bottom two rows). We also measured release across a range of pH gradients (Figure 2B). First, we challenged the PADNA-1/Mipomersen complex with pH 9.6, and found minimal release. This indicates PADNA-1 responds specifically to acidic pH. Second, we observed that PADNA-1 shows increasing release in proportion to decreasing pH, rather than a sudden release of all Mipomersen at pH 5.2 (Figure 2B). This property is advantageous for continuous distribution of Mipomersen throughout the endosomal maturation process, rather than delivery restricted to the late endosome where the pH is ~5.2.
Figure 2.

Efficiency and specificity of Mipomersen release by PADNA-1. (A) qPCR measurements show PADNA-1 release from magnetic beads in response to shifting the pH from 7.5 to 5.2. (B) qPCR measurements of PADNA-1 release across a range of pH from alkaline (9.6) to acidic (4.5).
We monitored pH-dependent Mipomersen release via a real-time fluorescence assay. Specifically, we modified Mipomersen with 5-carboxytetramethyl-rhodamine (TAMRA) on T4 and PADNA-1 with Black Hole Quencher 2 (BHQ2) on T54 (Table S1). We chose TAMRA for these experiments because its fluorescence is known to be stable in acidic pH.[19] When Mipomersen hybridizes to PADNA-1, TAMRA fluorescence is quenched. When Mipomersen is released by acidic pH, the fluorescence is restored (Scheme S1).
First, we challenged the complex with pH 9.6 and observed no change in signal, confirming that PADNA-1 does not release in response to basic pH shift (Figure 3A). This result is consistent with our qPCR measurements (Figure 2B). When we decreased the pH to 5.2, however, we observed rapid Mipomersen release (<1 min response time). We note that the maximum release at pH 5.2 observed in this assay was 50.5% of total Mipomersen. We attribute this to the TAMRA and BHQ2 modifications, which may impede release by stabilizing the duplex.[20] Finally, we cycled the pH back to 7.5 and found that Mipomersen re-loaded onto PADNA-1, with free Mipomersen decreasing to ~15% of its maximum signal over the course of 120 minutes. Re-association appears to be a slow reaction whereas release is a fast process. This property is advantageous for drug delivery because it ensures a high rate of delivery with low rates of reabsorption during endosomal uptake. We further confirmed the reversibility of PADNA-1 loading and release by performing repeated cycling between pH 7.5 and 5.2 (Figure S2). Next, we used our fluorophore-quencher assay to monitor continuous stepwise pH titration between 7.5 and 5.2, and found that the release of Mipomersen increased proportionally with the gradual lowering of pH (Figure 3B). These results agree with pH titration experiments performed via qPCR (Figure 2B).
Figure 3.
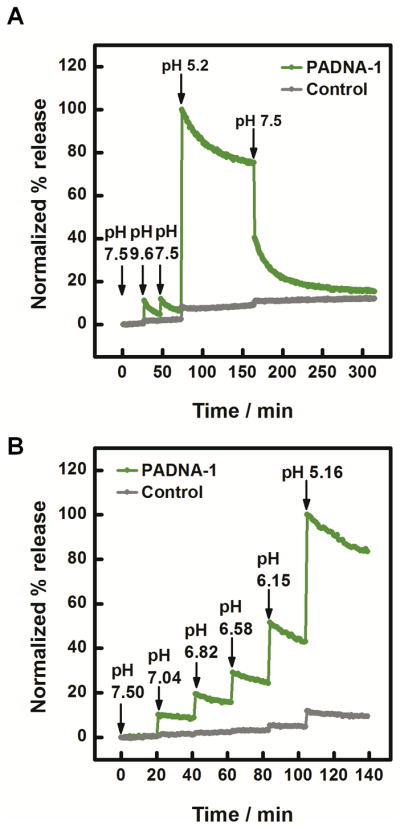
(A) Real-time measurement of fluorophore-labeled Mipomersen release from quencher-labeled PADNA-1. (B) Real-time titration experiments showed that the fraction of released Mipomersen increases monotonically with decreasing pH. The poly-T control (gray), is a negative control sequence where the release domains of PADNA-1 are replaced with polyT (Table S1). As expected, the polyT control showed minimal response under identical conditions. Data were normalized to the maximum value of release at pH 5.2. Details for this calculation are described in Figure S1. We note that the fluorescence signal measured in these experiments appears to decay due to photobleaching of TAMRA (Figure S1).
M-fold software predicted highly-ordered secondary structure for PADNA-1,[21] where the release domains and primer regions hybridize extensively with the central domain (Scheme 2).
Scheme 2.

Predicted secondary structure of PADNA-1. Red dots are high-free-energy mismatched base-pairs, orange dashes are low-free-energy mismatches, black dashes are canonical base-pairs. The central domain consists of 17 bases marked by filled black circles.
We hypothesize that part of the structure-switching mechanism arises from the reversible pH-dependent stabilization of mismatch base-pairs, including two C-A pairs at sites (1) and (2). Protonation of the adenosine N1 moiety reorganizes a C-A mismatch into a C•A+ wobble pair which has been reported to contribute up to -10 kcal/mol to the free energy.[22] We predict that at pH 5.2, PADNA-1 adopts a low-pH conformation in which the primer and release domains hybridize with the central domain, thereby displacing Mipomersen. Interestingly, the pKa of these C-A mismatches can be estimated from Figure 3B to be ~5.5, which differs from previously reported values (~7.5) measured under similar conditions.[22, 23] We believe that secondary/tertiary interactions with C-A mismatches may shift the pKa. For example, it has been shown that multiple C-A mismatches in the same duplex can lower the pKa.[24] In Nature, a lead-dependent ribozyme has been shown to contain multiple adenine bases with perturbed pKa values thought to be supported by tertiary interactions.[25]
To test our model, we substituted C-T for the C-A mismatches at either or both sites (1) and (2) in PADNA-1, and then used qPCR to measure the resulting change in release ratio (Figure 4A). The C-T basepair is a pH-insensitive analog to C-A, and would thus be expected to impede pH-triggered function. C-T substitution at site (1) reduced the release ratio to 6, and substitution at both sites knocked out the release function completely. Our results suggested that C•A+ wobble pairs play a major role in stabilizing the low pH conformation of PADNA-1 that displaces Mipomersen from the central domain.
Figure 4.

(A) MUT-1 showed decreased release at pH 5.2, yielding a reduced ΔCt value of 2.76 +/− 0.03. MUT-2 showed almost complete loss of function, yielding a ΔCt of 0.71 +/− 0.03. Data are also shown for MUT-1 release at pH 7.5 (essentially identical to data for MUT-2 and PADNA-1). Release from PADNA-1 at pH 5.2 is also shown, with the fit line reproduced from Figure 2A for comparison. (B) MUT-CC yielded a ΔCt of 1.71 +/− 0.07.
Interestingly, we also examined mutants where we replaced the C-A mismatches with G-C or A-T Watson Crick base-pairs. We found that these mutants released ~80% of loaded Mipomersen at pH 7.5 according to fluorescence monitoring (Figure S3). Since G-C/A-T basepairs would be expected to promote intramolecular folding of PADNA-1 at pH 7.5, these results support the hypothesis that stabilization of basepairs at sites (1) and (2) makes a large contribution to the structure-switching release mechanism.
Since C-C mismatches are also known to form protonated wobble pairs (C+•C),[26] we tested whether mutants with C-C substitutions at sites (1) and (2) could rescue structure-switching function (Figure 4B). However, such mutants only exhibited a release ratio of 3. Based on these observations, we believe that the structure-switching mechanism relies on specific tertiary interactions with protonated adenine residues, and this is a subject of future investigations.
In this work, we report a discovery-driven approach to identify pH-activated, structure-switching DNA nanostructures. PADNAs respond specifically to a shift from neutral to acidic pH, undergoing a conformational change that results in highly selective release of the antisense drug Mipomersen. For example, PADNA-1 releases Mipomersen with 145-fold selectivity at pH 5.2 versus 7.5. None of the pH-dependent structure-switching sequences we generated were related to known motifs, suggesting that they may operate via novel structure-switching mechanisms. We found strong evidence that the reversible formation of C•A+ wobble pairs contributes to the pH-responsive function of PADNA-1. Although our results are preliminary, PADNAs demonstrate excellent potential for tightly regulated drug delivery. In vitro selection offers exciting opportunities to discover new functional DNA motifs and we anticipate that it could be adapted to identify other sequences with distinct pH response characteristics and drug release profiles.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health, DARPA (N66001-14-2-4055), ARO (W911NF-10-2-0114), CIRM Training Grant (TG2-01151) and the Garland Initiative. We thank M. Nakamoto and M. Eisenstein for editing the manuscript.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Faye Yi Fong, Materials Department, University of California at Santa Barbara, Santa Barbara, CA 93106.
Dr. Seung Soo Oh, Materials Department, University of California at Santa Barbara, Santa Barbara, CA 93106.
Prof. Craig J. Hawker, Department of Chemistry and Biochemistry, University of California at Santa Barbara, Santa Barbara, CA 93106
Prof. H. Tom Soh, Department of Electrical Engineering and Department of Radiology, Canary Center at Stanford University, 3155 Porter Drive, Stanford, CA 94305
References
- 1.Duncan R, Richardson SCW. Mol Pharm. 2012;9:2380–2402. doi: 10.1021/mp300293n. [DOI] [PubMed] [Google Scholar]
- 2.Ganta S, Devalapally H, Shahiwala A, Amiji M. J Cont Rel. 2008;126:187–204. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 3.Schmaljohann D. Adv Drug Deliv Rev. 2006;58:1655–1670. doi: 10.1016/j.addr.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 4.Charoenphol P, Bermudez H. Acta Biomat. 2014;10:1683–1691. doi: 10.1016/j.actbio.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Modi S, Swetha MG, Goswami D, Gupta GD, Mayor S, Krishnan YA. Nat Nano. 2009;4:325–330. doi: 10.1038/nnano.2009.83. [DOI] [PubMed] [Google Scholar]
- 6.Son S, Nam J, Kim J, Kim S, Kim WJ. ACS Nano. 2014;8:5574–5584. doi: 10.1021/nn5022567. [DOI] [PubMed] [Google Scholar]
- 7.Keum JW, Bermudez H. Chem Comm. 2012;48:12118–12120. doi: 10.1039/c2cc37471d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J, Lee YM, Kang Y, Kim WJ. ACS Nano. 2014;8:9358–9367. doi: 10.1021/nn503349g. [DOI] [PubMed] [Google Scholar]
- 9.Nesterova IV, Nesterov EE. JACS. 2014;136:8843–8846. doi: 10.1021/ja501859w. [DOI] [PubMed] [Google Scholar]
- 10.Liu DS, Balasubramanian S. Angew Chem Int Ed. 2003;42:5734–5736. doi: 10.1002/anie.200352402. [DOI] [PubMed] [Google Scholar]
- 11.Liu DS, Bruckbauer A, Abell C, Balasubramanian S, Kang DJ, Klenerman D, Zhou D. JACS. 2006;128:2067–2071. doi: 10.1021/ja0568300. [DOI] [PubMed] [Google Scholar]
- 12.Idili A, Vallée-Bélisle A, Ricci F. JACS. 2014;136:5836–5839. doi: 10.1021/ja500619w. [DOI] [PubMed] [Google Scholar]
- 13.Porchetta A, Idili A, Vallee-Belisle A, Ricci F. Nano Lett. 2015;15:4467–4471. doi: 10.1021/acs.nanolett.5b00852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nesterova IV, Briscoe JR, Nesterov EE. JACS. 2015;137:11234–11237. doi: 10.1021/jacs.5b06645. [DOI] [PubMed] [Google Scholar]
- 15.Nesterova IV, Elsiddieg SO, Nesterov EE. J Phys Chem B. 2013;117:10115–10121. doi: 10.1021/jp405230g. [DOI] [PubMed] [Google Scholar]
- 16.Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, Lachmann RH, Gaudet D, Tan JL, Chasan-Taber S, Tribble DL. Lancet. 2010;375:998–1006. doi: 10.1016/S0140-6736(10)60284-X. [DOI] [PubMed] [Google Scholar]
- 17.Chakraborty S, Sharma S, Maiti PK, Krishnan Y. Nuc Acids Res. 2009;37:2810–2817. doi: 10.1093/nar/gkp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank-Kamenetskii MD, Mirkin SM. Ann Rev Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- 19.Li T, Famulok M. JACS. 2013;135:1593–1599. doi: 10.1021/ja3118224. [DOI] [PubMed] [Google Scholar]
- 20.Moreira BG, You Y, Behlke MA, Owczarzy R. Biochem Biophys Res Comm. 2005;327:473–484. doi: 10.1016/j.bbrc.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Zuker M. Nuc Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siegfried NA, O’Hare B, Bevilacqua PC. Biochem. 2010;49:3225–3236. doi: 10.1021/bi901920g. [DOI] [PubMed] [Google Scholar]
- 23.Boulard Y, cognet JAH, Gabarro-Arpa J, Le Bret M, Sowers LC, Fazakerley GV. Nuc Acids Res. 1992;20:1933–1941. doi: 10.1093/nar/20.8.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C, Gao HT, Gaffney BL, Jones RA. JACS. 1991;113:5486–5488. [Google Scholar]
- 25.Legault P, Pardi A. JACS. 1997;119:6621–6628. [Google Scholar]
- 26.Phan AT, Mergny JL. Nuc Acids Res. 2002;30:4618–4625. doi: 10.1093/nar/gkf597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.