

Abstract
Activation of nuclear factor erythroid 2-related factor 2 (Nrf2, a transcription factor) and/or inhibition of mammalian target of rapamycin (mTOR) are implicated in the suppression of vascular smooth muscle cell (VSMC) proliferation. The present study has examined the likely regulatory effects of sulforaphane (SFN, an antioxidant) on Nrf2 activation and platelet-derived growth factor (PDGF)-induced mTOR signaling in VSMCs. Using human aortic VSMCs, nuclear extraction and siRNA-mediated downregulation studies were performed to determine the role of Nrf2 on SFN regulation of PDGF-induced proliferative signaling. Immunoprecipitation and/or immunoblot studies were carried out to determine how SFN regulates PDGF-induced mTOR/p70S6K/S6 versus ERK and Akt signaling. Immunohistochemical analysis was performed to determine SFN regulation of S6 phosphorylation in the injured mouse femoral artery. SFN (5 μM) inhibits PDGF-induced activation of mTOR without affecting mTOR association with raptor in VSMCs. While SFN inhibits PDGF-induced phosphorylation of p70S6K and 4E-BP1 (downstream targets of mTOR), it does not affect ERK or Akt phosphorylation. In addition, SFN diminishes exaggerated phosphorylation of S6 ribosomal protein (a downstream target of p70S6K) in VSMCs in vitro and in the neointimal layer of injured artery in vivo. Although SFN promotes Nrf2 accumulation to upregulate cytoprotective genes (e.g., heme oxygenase-1 and thioredoxin-1), downregulation of endogenous Nrf2 by target-specific siRNA reveals an Nrf2-independent effect for SFN-mediated inhibition of mTOR/p70S6K/S6 signaling and suppression of VSMC proliferation. Strategies that utilize local delivery of SFN at the lesion site may limit restenosis after angioplasty by targeting mTOR/p70S6K/S6 axis in VSMCs independent of Nrf2 activation.
Keywords: Sulforaphane, Nrf2, mTOR, PDGF, Vascular smooth muscle cells
Graphical abstract
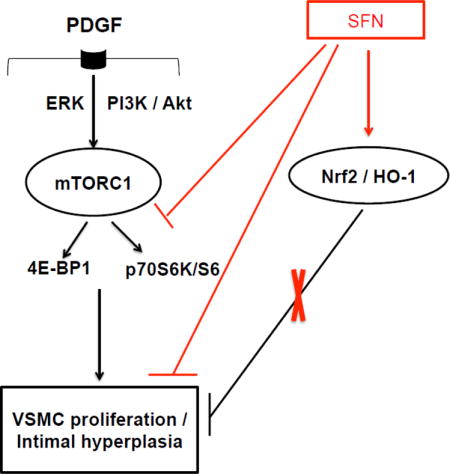
Sulforaphane (SFN) inhibits downstream targets of mTORC1, including p70S6K/S6 and 4E-BP1 phosphorylation to attenuate PDGF-induced VSMC proliferation. Although SFN activates Nrf2 transcription factor to induce HO-1 expression, Nrf2 downregulation using target-specific siRNA reveals that Nrf2/HO-1 signaling does not contribute to SFN inhibition of VSMC proliferation.
1. Introduction
Neointimal hyperplasia is one of the key biological events that underlie restenosis after angioplasty [1, 2]. Upon endothelial injury during revascularization procedures, vascular smooth muscle cells (VSMCs) undergo phenotypic transition from the contractile to proliferative state [3], thereby contributing to neointima formation. Despite the use of drug-eluting stents (e.g., rapamycin/sirolimus) to limit neointimal hyperplasia, in-stent restenosis still remains a major clinical problem [4]. It is well accepted that antioxidants play a significant role in reducing the risk of cardiovascular disease [5, 6]. Studies by several investigators including our recent findings demonstrate that sulforaphane (SFN, an antioxidant derived from cruciferous vegetables) inhibits neointima formation in vivo and VSMC proliferation in vitro [7–11]. In particular, SFN inhibits: i) the expression/activation of pro-inflammatory transcription factors such as NF-κB (p65 subunit), GATA6, and AP-1 in the injured artery or VSMCs [7, 8]; ii) platelet-derived growth factor (PDGF)-induced VSMC proliferation through upregulation of p53 and p21 that promote cell cycle arrest at G1/S phase [8, 10]; and iii) leptin-induced VSMC proliferation through diminished p70S6 kinase (p70S6K)/S6 ribosomal protein signaling [9]. It is noteworthy that PDGF, a potent mitogen released at the site of arterial injury [12], induces VSMC proliferation through the intermediary activation of signaling components such as mitogen-activated protein/ERK kinase1 (MEK1)/ extracellular signal-regulated kinase (ERK), phosphoinositide 3-kinase (PI 3-kinase)/Akt, and mammalian target of rapamycin (mTOR)/p70S6K [13–18]. However, the likely regulatory effects of SFN on PDGF-induced key proliferative signaling events have not yet been fully examined in VSMCs.
Of importance, mTOR exists as rapamycin-sensitive mTOR complex 1 (mTORC1) and rapamycin-insensitive mTOR complex 2 (mTORC2). mTORC1 signaling is activated in VSMCs upon arterial injury or in response to PDGF [13, 14, 17–21]. The downstream targets of mTORCl include p70S6K/S6 and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) [13–18, 22]. While mTOR interacts with regulatory associated protein of mTOR (raptor) in mTORC1 to regulate the phosphorylation of p70S6K and 4E-BP1 [23–25], mTORC2 complex that contains rapamycin-insensitive companion of mTOR (rictor) has been shown to phosphorylate Akt on serine 473 residue [26, 27]. Thus, it is important to examine how SFN regulates PDGF-induced mTORC1 signaling in VSMCs.
As an isothiocyanate, SFN is known to interact with specific cysteine residues of kelch-like ECH associated protein 1 (Keap1), thereby promoting the activation of nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor [28]. Due to its essential role as an Nrf2 activator, SFN is a potent inducer of Nrf2-mediated, antioxidant response element (ARE)-driven cytoprotective genes, including heme oxygenase-1 (HO-1). Studies involving Nrf2 overexpression or downregulation have documented a critical role for Nrf2 in suppressing VSMC proliferation [29] and neointima formation [30, 31]. Nevertheless, it is unclear as to whether SFN-mediated Nrf2 activation contributes to inhibition of VSMC proliferation.
The objectives of the present study are to determine the extent to which SFN regulates PDGF-induced mTOR/p70S6K signaling versus Nrf2 expression in VSMCs. We hypothesize that SFN attenuates VSMC proliferation through inhibition of PDGF-induced mTOR/p70S6K signaling and/or upregulation of Nrf2 expression. In the present study, we examined the effects of SFN on PDGF-induced activation of p70S6K versus 4E-BP1 (downstream targets of mTOR), in conjunction with Akt (a downstream target of PI 3-kinase) and ERK1/2 (a downstream target of MEK1). In addition, this study has examined SFN regulation of S6 ribosomal protein, a downstream target and a functional read-out of p70S6K [32]. Since the activation of p70S6K/S6 is determined by the complex formation between mTOR and raptor (key components of mTOR complex 1; mTORC1) [25], select studies have examined how SFN regulates the association between mTOR and raptor in VSMCs. Furthermore, studies involving target-specific downregulation of Nrf2 have examined the likely intermediary role of Nrf2 toward SFN regulation of VSMC proliferative phenotype.
2. Materials and Methods
2.1. Materials
SFN (cat# 574215) and pure proteome protein A/G mix magnetic beads (cat# LSKMAGAG02) were purchased from EMD Millipore (Billerica, MA). All surgical tools were purchased from Roboz Surgical Instrument (Gaithersburg, MD). The primary antibody for SM α-actin was purchased from Abcam (cat# ab5694; Cambridge, MA). The primary antibody for Ki-67 (cat# RM-9106-S1), Mayer’s hematoxylin (cat# TA-125-MH), and NE-PER Nuclear and Cytoplasmic Extraction kit (cat# 78835) were purchased from Thermo Scientific (Hanover, IL). The primary antibody for thioredoxin-1 (Trx-1; cat# sc-13526) was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). The primary antibodies for cyclin D1 (cat# 2978), phosphorylated retinoblastoma protein at serine 795 (pRb; cat# 9301), phosphorylated protein kinase B at serine 473 (pAktSer473; cat# 4060) and threonine 308 (pAktThr308; cat# 9275), Akt (cat# 4691), phosphorylated extracellular signal-regulated kinase1/2 at Threonine 202/204 (pERK1/2; cat# 4370), ERK1/2 (cat# 4695), phosphorylated ribosomal protein S6 at serine 235/236 (pS6; cat# 4857), S6 (cat# 2217), phosphorylated p70 S6 kinase at threonine 389 (pp70S6K; cat# 9234), p70S6K (cat# 2708), phosphorylated eukaryotic translation initiation factor 4E-binding protein 1 at serine 65 (p4E-BP1; cat# 9451), 4E-BP1 (cat# 9644), phosphorylated PDGFRβ at tyrosine 751 (pPDGFRβ; cat# 3161), PDGFRβ (cat# 3169), phosphorylated glycogen synthase kinase3β at serine 9 (pGSK3β; cat# 9336), GSK3β (cat# 9315), phosphorylated mTOR at serine 2448 (pmTOR; cat# 5536], mTOR (cat# 2972), regulatory associated protein of mTOR (Raptor; cat# 2280), Keap1 (cat# 4617S), Nrf2 (cat# 12721), heme oxygenase-1 (HO-1; cat# 5061), cAMP-response element-binding protein (CREB; cat# 9197), β-actin (cat# 4970), GAPDH (cat# 5174), anti-rabbit IgG, HRP-linked secondary antibody (cat# 7074), SignalStain antibody diluent (cat# 8112L), and SignalStain Boost IHC Detection Reagent (HRP, Rabbit) [cat# 8114S] were purchased from Cell Signaling Technology (Danvers, MA). Goat anti-mouse IgG (H+L)-HRP conjugate (cat# 1721011) was purchased from Bio Rad (Hercules, CA). Goat anti-rabbit secondary antibody (A-11037), prolong gold anti-fade mountant with DAPI (P36931), Silencer Select Negative Control #1 siRNA (cat# 4390843) and NFE2L2 (Nrf2) silencer select siRNA (cat# 4392420: s9493) were purchased from Life Technologies (Grand Island, NY). 10% neutral buffered formalin (cas# 50-00-0) was purchased from Globe Scientific Inc. (Paramus, NJ). ImmPACT DAB Peroxidase (HRP) Substrate (cat# SK-4105) was purchased from Vector Laboratories, Inc. (Burlingame, CA). All other chemicals were from Fisher Scientific (Fair Lawn, NJ) or Sigma-Aldrich (St. Louis, MO).
2.2. Methods
2.2.1. Cell culture and treatments
Human aortic VSMCs were purchased from ATCC (Manassas, VA) and maintained in vascular cell basal media with smooth muscle growth supplement (SMGS) and antibiotic-antimycotic solution in a humidified atmosphere of 95% air and 5% CO2 (passage 3 to 4), as described [22]. After the attainment of confluence (~7–10 days), VSMCs were trypsinized and seeded onto petri dishes. Subconfluent VSMCs were subjected to SMGS (serum)-deprivation for 48 h to achieve quiescence. Serum-deprived VSMCs were then exposed to 30 ng.mL−1 PDGF with or without SFN treatment, as described in the legends to respective figures. The chosen concentration of PDGF was based on our previous studies with human aortic VSMCs [18, 22]. Dimethyl sulfoxide (DMSO; 0.05%) was used as vehicle control for SFN (5 μM), as described [33]. The concentration of SFN (5 μM) used in the present study was within the biologically relevant SFN concentration (2 to 20 μM), previously reported in plasma samples of rodents or humans after ingestion of SFN or SFN-rich diet [34, 35].
To determine the acute stimulatory effects of PDGF on key proliferative signaling components, VSMCs were incubated with PDGF for 6 min, as described [18]. To determine the long-term effects of PDGF on signaling events including cyclin D1 and pRb, VSMCs were exposed to PDGF for 48 h [18, 22].
To determine whether SFN regulates mTOR signaling (e.g., pS6, a functional read-out of p70S6K) and Nrf2 expression as a function of time, VSMCs were exposed to SFN between 6 min and 48 h. These findings provided the basis for studying the effects of acute (3 h) and prolonged (48 h) SFN treatment on PDGF-induced proliferative signaling and Nrf2 nuclear translocation in VSMCs, as described in the respective figure legends.
2.2.2. Cell counts
VSMCs were equally seeded onto petri dishes (~4 × 105 cells/60 mm dish) and maintained in culture for 48 h. After the attainment of uniform subconfluency in all dishes, VSMCs were deprived of serum for 48 h. Subsequently, VSMCs were treated with SFN (5 μM) or vehicle control (DMSO, 0.05%) for 30 min followed by exposure to PDGF (30 ng.mL−1) for 96 h. At the 48 h time point, the respective cells were replenished with fresh media with and without SFN or PDGF. After 96 h, VSMCs were trypsinized and the changes in cell number were determined using Countess Counter (Life Technologies), as described [36].
2.2.3. Immunoblot analysis
VSMC lysates (15 μg protein each) were electrophoresed using precast 4–12% NuPage minigels (Life Technologies), and the resolved proteins were transferred to PVDF membranes (EMD Millipore). The membranes were blocked with 5% milk in tris-buffered saline with tween (TBST) and probed with the indicated primary antibodies. After extensive washes, the immunoreactivity was detected using horseradish peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence. The protein bands were quantified using image J software, as described [37].
2.2.4. Immunoprecipitation studies
Serum-deprived VSMCs were exposed to SFN (5 μM) or vehicle control (DMSO, 0.05%) for 3 h followed by stimulation with or without PDGF (30 ng.mL−1) for 6 min. VSMC lysates were prepared using 1× RIPA buffer with protease inhibitor cocktail (P8340, Sigma Aldrich, St. Louis, MO) and phosphatase inhibitor cocktail (524629, EMD Millipore, Billerica, MA). The lysates were then vortexed and centrifuged at 5000 × g at 4°C for 10 min. Aliquots of supernatants (200 μg protein) were incubated overnight at 4°C with either anti-raptor (cat# 2280, Cell Signaling Technology, Danvers, MA) or anti-mTOR (cat# 2972, Cell Signaling Technology, Danvers, MA) primary antibody at a dilution of 1:50, with continuous mixing. The respective antigen-antibody complexes were subjected to immunoprecipitation by mixing with magnetic beads (50 μL per sample) for 30 min at room temperature (RT). The tubes containing immunocomplexes were placed in a magnetic stand and the supernatant was discarded. The beads were washed three times with phosphate-buffered saline containing 0.05% tween 20 (PBS-T). Elution step was performed by mixing the beads with 2× Laemmli buffer containing dithiothreitol and bromophenol blue and heating at 90°C for 10 min. The eluted samples were then used for immunoblotting.
2.2.5. Nuclear and cytoplasmic protein extractions
Nuclear and cytoplasmic protein fractions were obtained using nuclear and cytoplasmic extraction kit according to manufacturer’s instructions (Thermo Scientific). Briefly, serum-deprived VSMCs were exposed to SFN (5 μM) or vehicle control (DMSO, 0.05%) for 3 h. Cells were harvested with trypsin-EDTA and then centrifuged at 500 × g for 5 min. The cell pellet was washed using ice-cold PBS. Subsequently, ice-cold cytoplasmic extraction reagents (CER I and CER II) were mixed with protease/phosphatase inhibitor cocktail and added to the cell pellet to facilitate cell membrane disruption and the release of cytoplasmic contents. After recovering the nuclear pellet from the cytoplasmic extract by centrifugation (16,000 × g for 5min), the nuclear proteins were extracted using nuclear extraction reagent (NER) plus protease/ phosphatase inhibitor cocktail.
2.2.6. Nucleofection of VSMCs with Nrf2 siRNA
Subconfluent VSMCs were transfected with 500 pmoles of Nrf2 siRNA using Amaxa nucleofector-II device U-025 program (Lonza, Germany). Scrambled (Scr.) siRNA-transfected cells served as controls. Nrf2-and Scr. siRNA-transfected VSMCs were incubated in complete medium for 48 or 72 h. Subsequently, VSMCs were serum-deprived for 45 or 24 h, respectively, and then subjected to treatments as described in the legends to the respective figures.
2.2.7. In vivo studies
2.2.7.1. Animal welfare and ethical statement
All animal experiments were performed in accordance with the Charlie Norwood Veterans Affairs Medical Center Institutional Animal Care and Use Committee (IACUC) guidelines and were approved by the committee. Male C57BL/6J mice (12 weeks old, 24–25 g body weight, Jackson Laboratories, Bar Harbor, ME) were maintained in a room at a controlled temperature of 23°C with a 12:12-h dark-light cycle. Control diet (CON; D14022802) was obtained from Research Diet (New Brunswick, NJ) and fed to mice ad libitum.
2.2.7.2. Model selection
In the chosen mouse model of arterial injury, femoral artery was subjected to guide wire injury to induce neointimal hyperplasia, as described [38].
2.2.7.3. Grouping and randomization of mice
Mice were divided into 2 treatment groups (7 mice per group), where mice were assigned randomly to the groups. In one group, SFN solution (in 0.12% DMSO vehicle) was injected (s.c.) daily at a dose of 0.5 mg.kg−1 [9]. In the other group, 0.12% DMSO was injected (s.c.) daily at a dose of 10 mL.kg−1 (equivalent to injection volume in SFN-treated group).
2.2.7.4. Experimental protocol
Left femoral artery injury was performed in all mice one day after the start of treatments according to the previously described protocol [38] and according to the IACUC-approved ACORP number 14-09-077. Under anesthesia with 2% isoflurane, the skin was shaved at the incision site and cleansed with povidone-iodine pads 3 times. After exposing the left femoral artery, a small muscular branch was isolated, ligated distally and looped proximally. Transverse arteriotomy was performed in the muscular branch and the incision was extended slightly using microsurgery forceps to allow the insertion of a straight spring wire (0.38 mm in diameter, No. C-SF-15-15, Cook, Bloomington, IN) into the femoral artery for 1 min, allowing denudation and dilatation of the artery. The proximal portion of the muscular branch was then secured by suturing to restore the blood flow in the main femoral artery. Right femoral arteries were used as sham controls as described [9]. Mice were monitored for body weight and signs of distress for 3 days after surgery. Twenty one days after femoral artery injury (23rd day of SFN treatment), mice were anesthetized with 2% isoflurane and then perfused with 0.9% NaCl solution via the left ventricle, followed by perfusion fixation with 4% paraformaldehyde (PFA) in PBS, pH 7.4. The femoral arteries were carefully excised, fixed in 4% PFA for 1–2 days before being processed for frozen sections.
2.2.7.5. Morphometric analysis of femoral artery
From every femoral artery, cross-sections (5 μm each) were stained with haematoxylin and eosin (H&E) or elastic van gieson (EVG). Images were taken by AxioCam high-resolution camera (HRc) attached to an Observer Z1 microscope (Carl Zeiss Microimaging, Inc., Thornwood, NY) using 20× magnification power. Microscopic images of the arterial sections were analyzed using image analysis software (Axiovision, release 4.8.2 SP3). The intimal area from each section was determined by subtraction of the lumen area from the internal elastic lamina (IEL) area (i.e., intimal area = IEL area − lumen area). The medial area was determined by subtraction of the IEL area from the external elastic lamina (EEL) area (i.e., medial area = EEL area − IEL area) as described [9]. Note: In the control group, the final n value was 5 since 2 out of 7 mice died during the experimental period. Cryosectioning, H&E staining and EVG staining of injured femoral arteries were performed in a blinded manner.
2.2.7.6. Immunofluorescence analysis of femoral artery
Immunofluorescence analysis was performed for smooth muscle α-actin (SM α-actin) to confirm that the neointimal layer contains smooth muscle cells. In addition, immunofluorescence analysis was carried out for Ki-67, a marker of cell proliferation. The cross sections of injured femoral arteries were placed in a humidified chamber and fixed in 4% PFA, and then blocked by incubation with 5% normal goat serum for 1 h. This was followed by exposure of the sections to the primary antibody specific for SM α-actin (1:200 dilution) or Ki-67 (1:30 dilution) for 1 h at RT or overnight at 4°C, respectively. The sections were then processed as described previously [9] and the images were captured using confocal microscope at 20× magnification power.
2.2.7.7. Immunohistochemical analysis of femoral artery
Immunohistochemical analysis was performed for pS6 to elucidate the effect of SFN on mTORC1 activation in the neointima. The cross-sections of injured femoral arteries were washed, placed in a humidified chamber, fixed in 10% neutral buffered formalin, and then blocked by incubation with 5% normal goat serum for 1 h. After decanting the blocking serum, the sections were exposed to the primary antibody specific for pS6 (1:75 dilution in signalstain antibody diluent) for 1 h at RT and washed 3 times. Hydrogen peroxide was added to the sections to block endogenous peroxidases and left for 10 min at RT. The sections were then washed 3 times, incubated with signalstain immunohistochemistry detection reagent (HRP-rabbit) for 30 min at RT, washed again 3 times and incubated with diaminobenzidine (DAB) working solution for 8 min. All previous washing steps were done using tris-buffered saline (TBS) with 0.025% triton X-100. The slides were then rinsed in running tap water for 5 min, counterstained with Mayer’s hematoxylin, and rinsed again in running tap water for 5 min. The slides were then dehydrated by immersing in 95% ethanol (2 times, 10 seconds each), 100% ethanol (2 times, 10 seconds each), cleared by immersing in xylene (2 times, 10 seconds each), mounted with mounting medium (SP15-500, Fisher Scientific), and stored at RT for subsequent microscopic visualization. The images were taken using AxioCam high resolution camera (HRc) attached to an Observer Z1 microscope (Carl Zeiss Microimaging, Inc., Thornwood, NY) at 20 × magnification power.
2.2.8. Statistical analysis
Protein bands for western blot (for each in vitro experiment) were normalized using appropriate controls as mentioned in the figure legends, and expressed as either fold mean control to limit unwanted sources of variation or as arbitrary units when protein bands were undetectable under control conditions for the most part. Data shown in the bar graphs reflect the means ± SEM. Morphometric analyses data are expressed as means ± SEM of 5–7 mice per group.
Statistical analysis was performed using unpaired student t-test for Rb phosphorylation, nuclear Nrf2, Nrf2 expression, and morphometric analyses of injured femoral arteries. For time-dependent changes in pS6, Keap1, Nrf2 and HO-1, the data were analyzed by repeated measures one-way ANOVA followed by Tukey’s multiple comparisons test. All other measured parameters were compared using regular two-way ANOVA followed by Tukey’s multiple comparisons test. For siRNA data, the effects of SFN treatment (SFN versus vehicle) and PDGF exposure (PDGF versus control) were compared using regular two-way ANOVA followed by Tukey’s multiple comparisons test for each siRNA (Nrf2 and Scr.) separately. Values of P<0.05 were considered statistically significant. Statistical analyses were carried out using GraphPad Prism software (GraphPad Software Inc. V6.0f, San Diego, CA, USA).
3. Results
3.1. SFN inhibits PDGF-induced VSMC proliferation, cyclin D1 expression, and Rb phosphorylation
Previous studies have shown that SFN at 0.5 to 10 μM concentrations inhibits PDGF– induced VSMC proliferation [8, 10] by preventing cell cycle progression from G1 to S phase [10]. In the present study, SFN treatment at 5 μM concentration led to a marked decrease in PDGF-induced VSMC proliferation by 93.5% (Figure 1A). In addition, SFN diminished PDGF-induced increase in cyclin D1 expression and Rb phosphorylation by 82.9% and 70.5%, respectively (Figure 1B).
Figure 1.
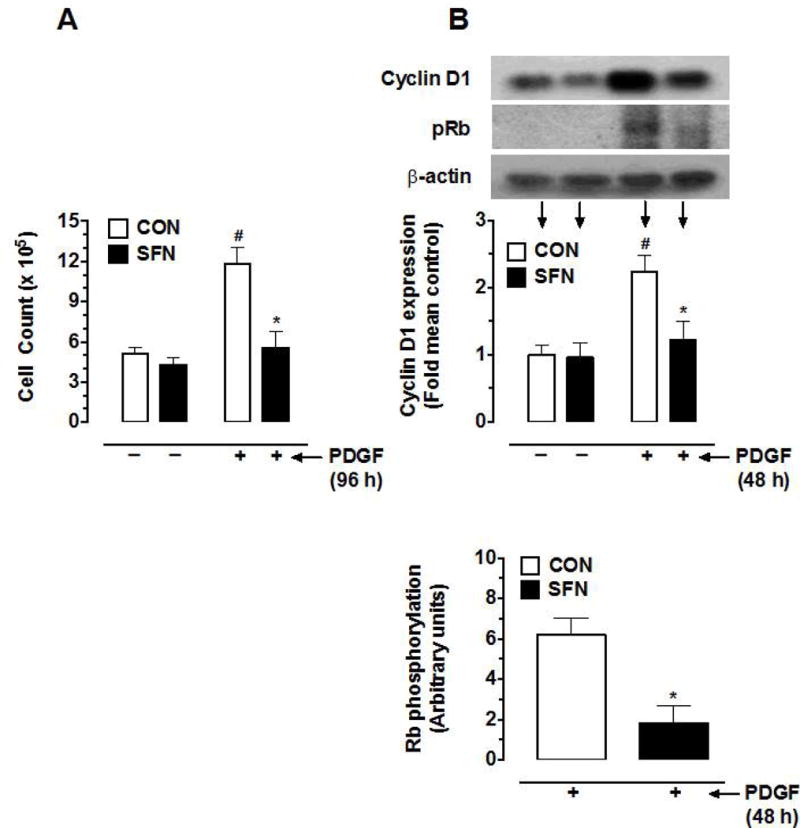
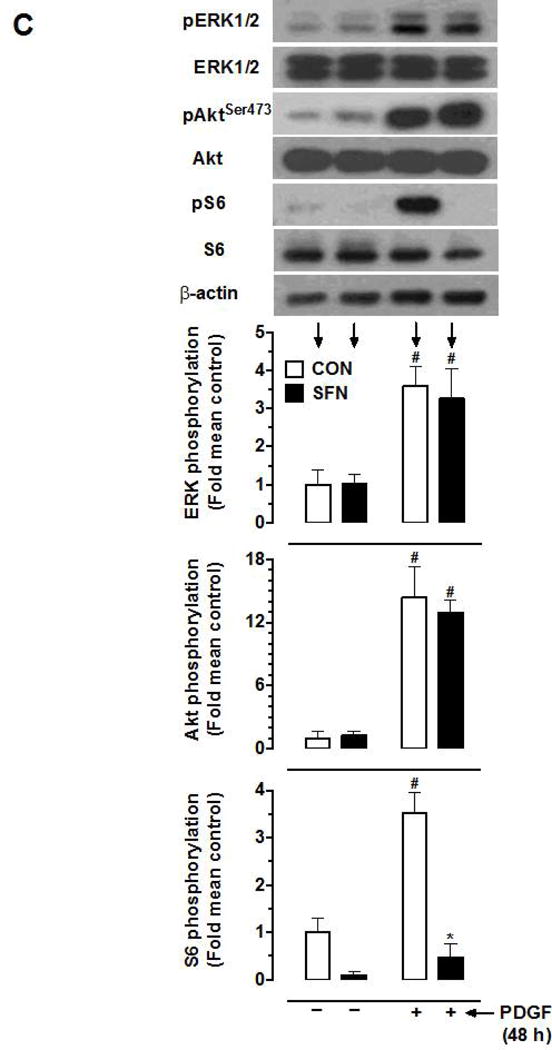
Effects of SFN on PDGF-induced key proliferative signaling in VSMCs. Serum-deprived VSMCs were pretreated with SFN (5 μM) or DMSO (vehicle) for 30 min followed by incubation with or without PDGF (30 ng.mL−1) for 96 h or 48 h. (A) At 96 h time point, the changes in cell number were determined using automated counter (n = 6). (B and C) At 48 h time point, the cell lysates were subjected to immunoblot analysis using primary antibodies specific for cyclin D1, pRb, pERK, ERK, pAktSer473, Akt, pS6 and S6, as described (n = 4–5). β-actin was used as an internal control. The data shown in the bar graphs are the means ± SEM. #, * P<0.05 compared with CON (− SFN) and PDGF (− SFN), respectively; two-way ANOVA followed by Tukey’s multiple comparisons test (for all except pRb; unpaired student t-test).
3.2. SFN suppresses basal and PDGF-induced phosphorylation of S6, but not ERK1/2 and Akt, upon prolonged exposure in VSMCs
PDGF-induced VSMC proliferation is mediated by Ras/Raf/MEK1/ERK, PI3-kinase/Akt, and mTOR/p70S6K signaling. Previously, we have reported that exposure of VSMCs to PDGF for a prolonged time interval (48 h) leads to a sustained increase in the phosphorylation of ERK1/2, Akt, and p70S6K [18]. Figure 1C shows that SFN treatment (5 μM) did not result in significant changes in basal and PDGF (48 h)-induced phosphorylation of ERK1/2 and Akt. However, it inhibited basal S6 phosphorylation by 90.2% and completely abolished PDGF-induced increase in S6 phosphorylation.
3.3. SFN inhibits the basal phosphorylation of S6 in a sustained manner at increasing time intervals
As shown in Figure 2, treatment of VSMCs with a fixed concentration of SFN (5 μM) led to a transient but insignificant increase in S6 phosphorylation at 6 and 20 min time intervals. This was followed by a decrease in S6 phosphorylation (by 58.2%) at 3 h time point that persisted up to 48 h time point (92.4%).
Figure 2.
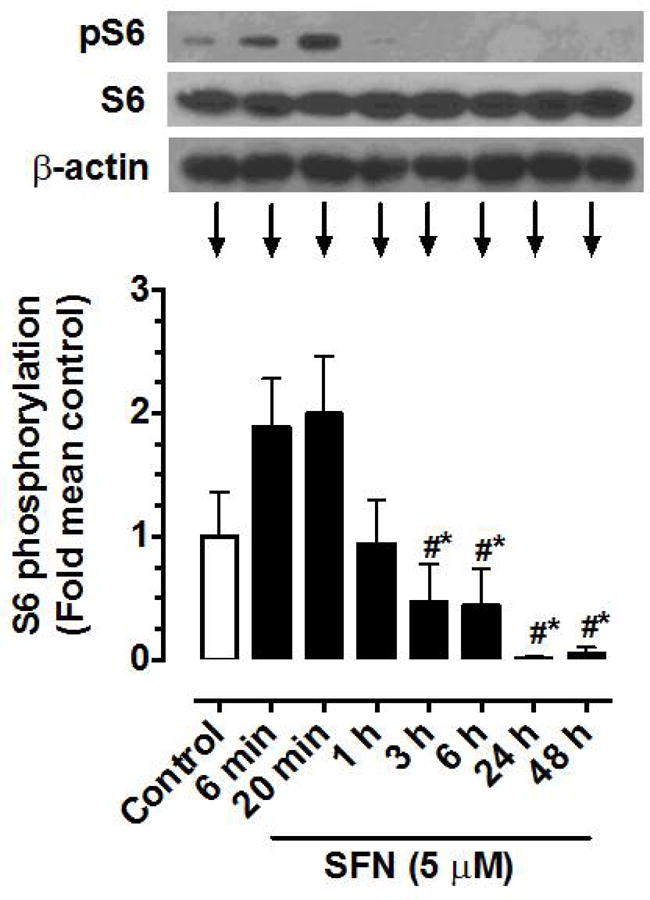
Time-dependent effects of SFN on the phosphorylation of S6 ribosomal protein in VSMCs. Serum-deprived VSMCs were treated with SFN (5 μM) at increasing time intervals (6 min to 48 h). VSMC lysates were then subjected to immunoblot analysis using primary antibody specific for pS6 and S6, as described. β-actin was used as an internal control. The data shown in the bar graph are the means ± SEM from 5 separate experiments. #, * P<0.05 compared with SFN-treated for 6 min and 20 min, respectively; repeated measures one-way ANOVA followed by Tukey’s multiple comparisons test.
SFN suppresses basal and PDGF-induced phosphorylation of p70S6K and S6, but not ERK and Akt, upon acute exposure in VSMCs
To further confirm the inhibitory effects of SFN on basal and PDGF-induced S6 phosphorylation and the lack of effects on ERK1/2 and Akt phosphorylation, VSMCs were treated with SFN (5 μM) for 3 h based on the time course study. SFN-treated VSMCs were then subjected to acute exposure to PDGF (6 min). As shown in Figure 3A, SFN did not affect the phosphorylation of PDGF receptor-β and ERK1/2. In addition, SFN did not affect PDGF-induced phosphorylation of AktSer473 and AktThr308, suggesting lack of inhibitory effects on the full activation of Akt [39]. Furthermore, SFN did not affect PDGF-induced phosphorylation of GSK-3β, a downstream target of Akt [40]. However, SFN inhibited basal and PDGF-induced increase in phosphorylation of p70S6K by 96.4% and 54.4% and completely abolished both basal and PDGF-induced increase in S6 phosphorylation (Figure 3B). With regard to 4E-BP1 (an inhibitor of translation initiation), SFN treatment showed a modest inhibition of basal phosphorylation (18.5%), but led to a significant inhibition of PDGF-induced increase in phosphorylation by 71.6%.
Figure 3.
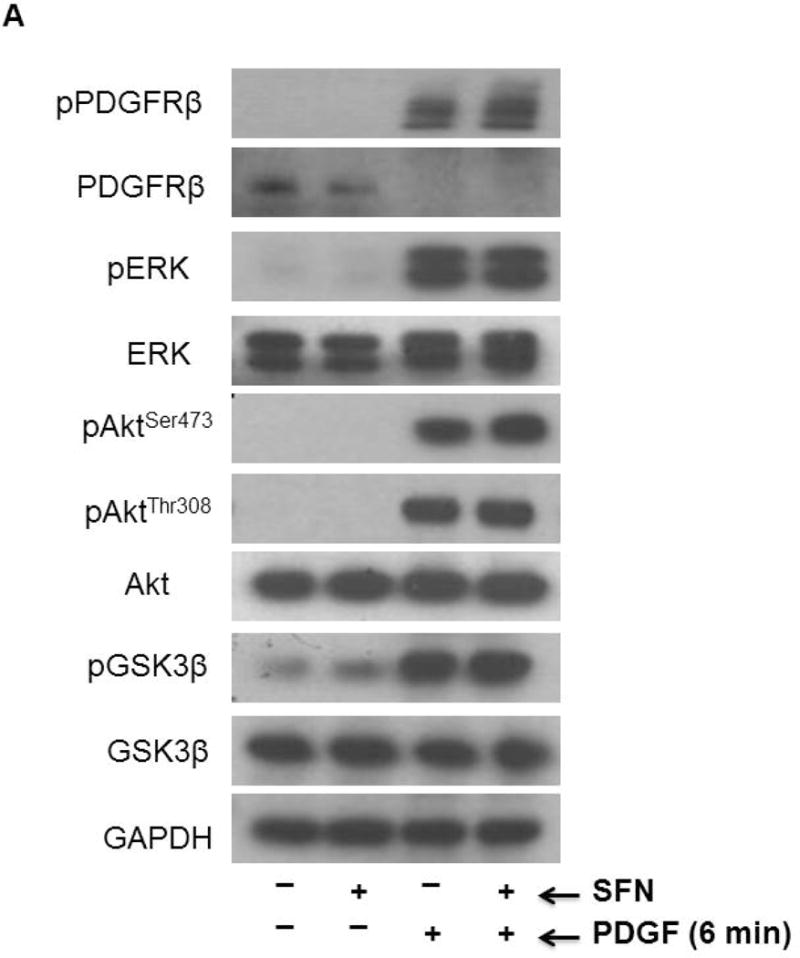
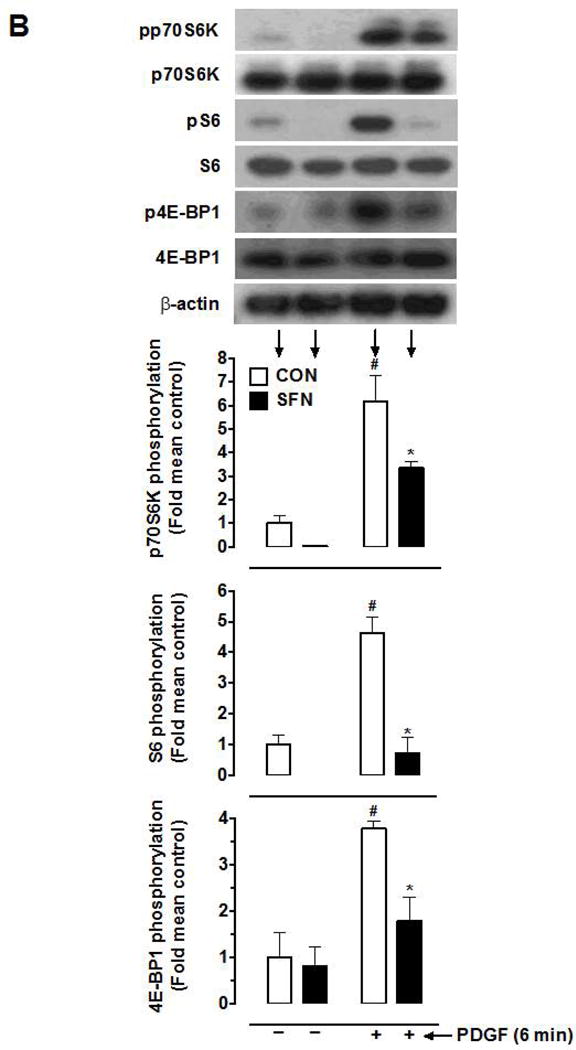
Effects of SFN on key proliferative signaling events induced by acute PDGF exposure in VSMCs. Serum-deprived VSMCs were pretreated with SFN (5 μM) or DMSO (vehicle) for 3 h followed by stimulation with or without PDGF (30 ng.mL−1) for 6 min. (A and B) VSMC lysates were then subjected to immunoblot analysis using primary antibodies specific for pPDGFRβ, PDGFRβ, pERK1/2, ERK1/2, pAktSer473, pAktThr308, Akt, pGSK3β, GSK3β, pp70S6K, p70S6K, pS6, S6, p4E-BP1 and 4E-BP1, as described. GAPDH or β-actin was used as internal control. The data shown in the bar graphs are the means ± SEM from 3 separate experiments. #, * P<0.05 compared with CON (− SFN) and PDGF (− SFN), respectively; two-way ANOVA followed by Tukey’s multiple comparisons test.
3.4. SFN attenuates mTOR phosphorylation without affecting mTOR-raptor association
Since mTOR (an upstream kinase) regulates the phosphorylation state of p70S6K/S6 and 4E-BP1 [41, 42], the likely regulatory effect of SFN (5 μM, 3 h) on basal and PDGF (6 min)-induced mTOR activation was determined. Of note, mTOR has four phosphorylation sites (Thr2446 [43], Ser2448, Ser2481 [44] and Ser1261 [45]). In addition, a decrease in mTOR phosphorylation at Ser2448 residue correlates with a decrease in mTORC1 activity [32]. As shown in Figure 4A, SFN showed a modest inhibition of basal mTORSer2448 phosphorylation (30.3%), but significantly suppressed PDGF-induced increase in mTORSer2448 phosphorylation by 96.4%.
Figure 4.
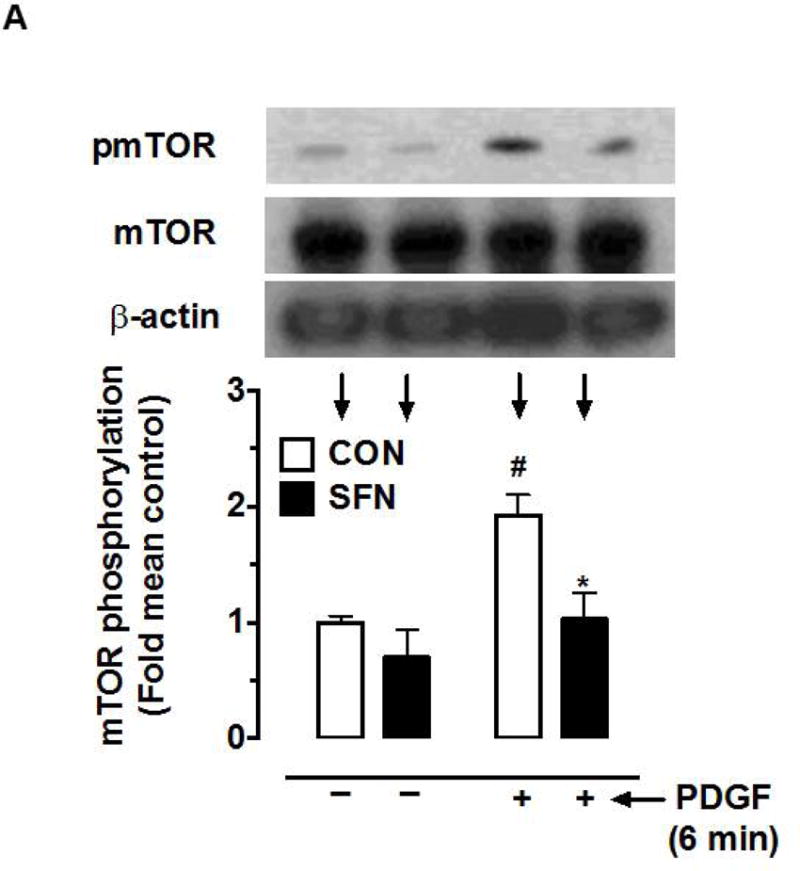
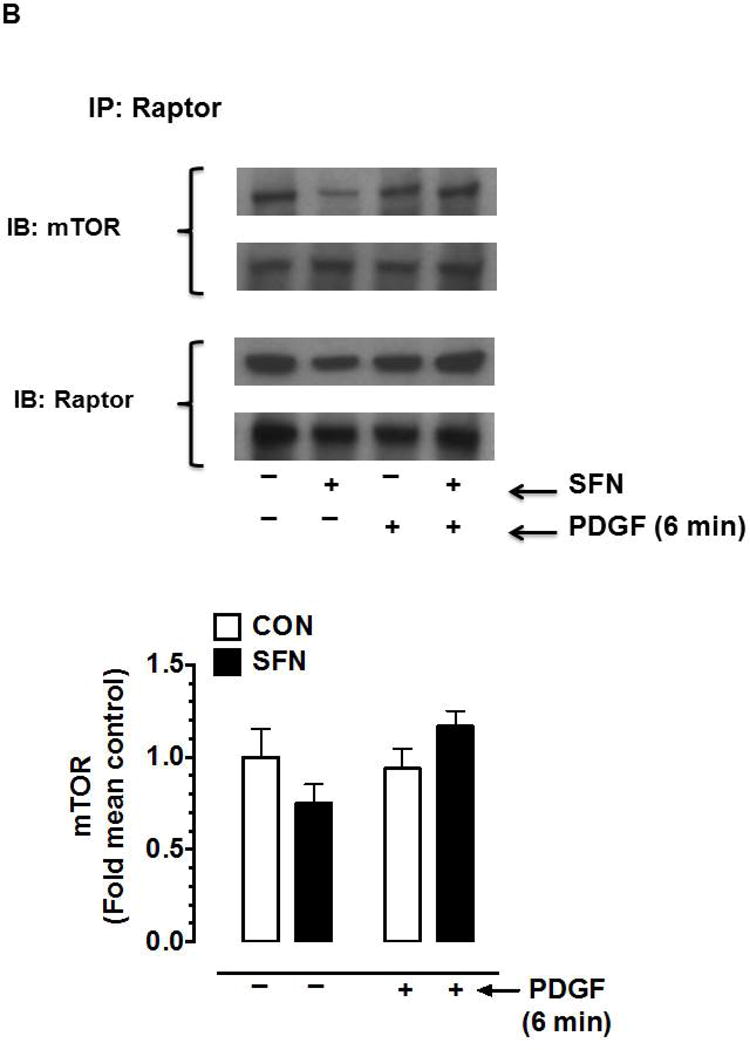
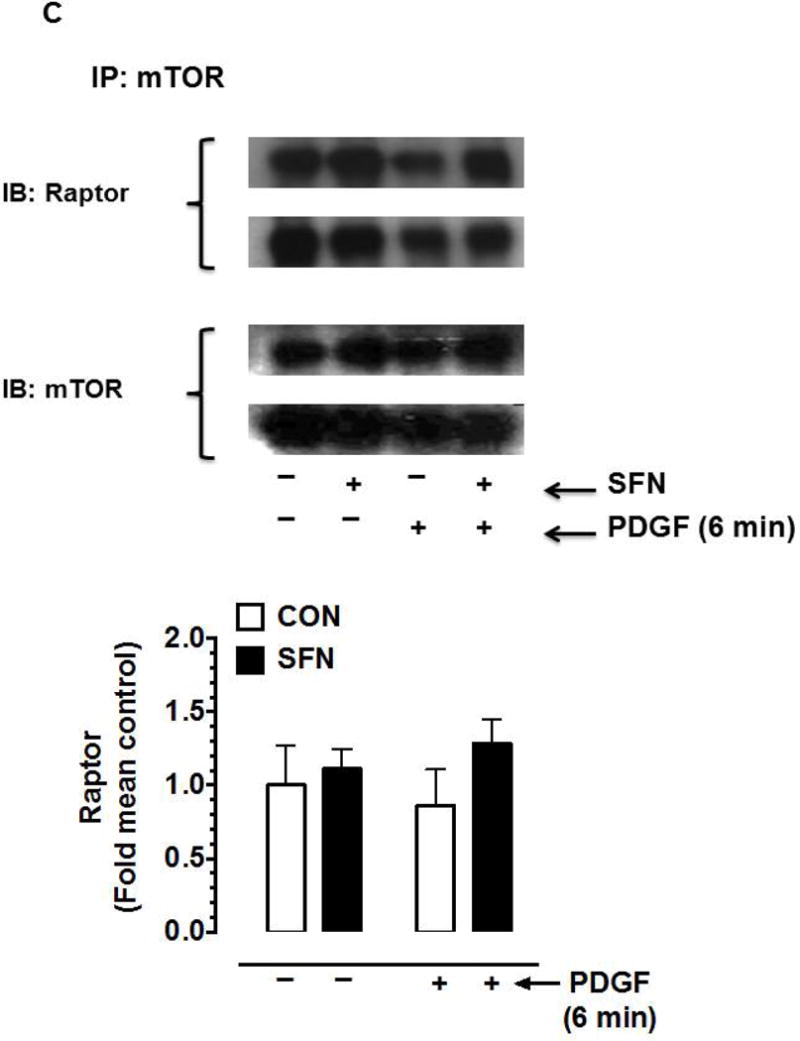
Effects of SFN on mTOR phosphorylation and mTOR-raptor association in VSMCs. (A) Serum-deprived VSMCs were pretreated with SFN (5 μM) or DMSO (vehicle) for 3 h followed by stimulation with or without PDGF (30 ng.mL−1) for 6 min. VSMC lysates were then subjected to immunoblot analysis using primary antibodies specific for pmTOR and mTOR. The data shown in the bar graph are the means ± SEM from 3 separate experiments. #, * P<0.05 compared with CON (− SFN) and PDGF (− SFN), respectively; two-way ANOVA followed by Tukey’s multiple comparisons test. (B and C) Serum-deprived VSMCs were treated with SFN (5 μM) or DMSO (vehicle) for 3 h followed by stimulation with or without PDGF (30 ng.mL−1) for 6 min. The cell lysates were then subjected to immunoprecipitation (IP) using primary antibody specific for raptor (B) or mTOR (C). The respective immunocomplexes were used for immunoblot (IB) analysis using the indicated primary antibodies. Blots representative of 2 individual n values are shown for each. The data shown in the bar graph are the means ± SEM from 3 separate experiments.
Although it has been shown that mTOR interacts with raptor in mTORC1 to regulate the phosphorylation of p70S6K and 4E-BP1 [23], a decrease in mTORC1 activity may occur independent of a change in the association between mTOR and raptor [32]. Hence, select studies were performed to determine the interaction between mTOR and raptor in SFN-treated VSMCs. Immunoprecipitation studies using raptor (Figure 4B) and mTOR (Figure 4C) primary antibodies did not show significant differences in the association with mTOR and raptor, respectively.
3.5. SFN enhances the expression of Nrf2 and HO-1 proteins in VSMCs and promotes Nrf2 accumulation in the nuclear fraction
Nuclear translocation of Nrf2 is required for Nrf2-mediated induction of antioxidant and phase 2 genes [46]. SFN has been shown to upregulate Nrf2 [47], promote its nuclear translocation [48] and increase the expression of antioxidant genes including HO-1 in rat VSMCs [47–49]. In the present study using human aortic VSMCs, SFN (5 μM) treatment enhanced the expression levels of Nrf2 and HO-1 proteins in a time-dependent manner (Figure 5). In particular, a significant increase in Nrf2 expression was observed at 1 to 48 h time points. In addition, a significant increase in HO-1 occurred at 6 to 48 h time points. SFN treatment did not affect Keap1 protein expression up to 24 h. However, it led to significant (~2.9 fold) increase in Keap1 expression at 48 h time point.
Figure 5.
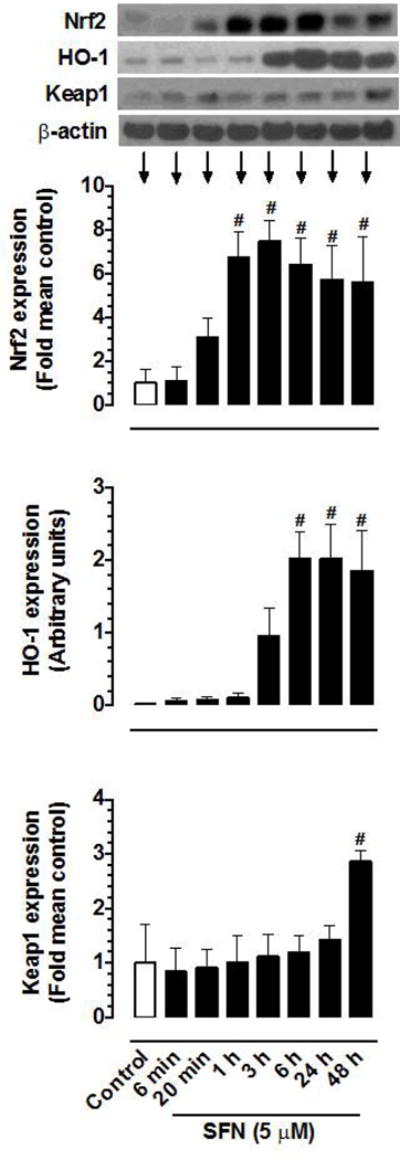
Time-dependent effects of SFN on the expression of Nrf2, HO-1, and Keap1 proteins in VSMCs. Serum-deprived VSMCs were treated with SFN (5 μM) at increasing time intervals (6 min to 48 h). VSMC lysates were then subjected to immunoblot analysis using primary antibodies specific for Nrf2, HO-1, and Keap1, as described. β-actin was used as an internal control. The data shown in the bar graphs are the means ± SEM from 3 (Keap1) or 4 (Nrf2 and HO-1) separate experiments. # P<0.05 compared with control (vehicle-treated cells); repeated measures one-way ANOVA followed by Tukey’s multiple comparisons test.
Under control conditions, Nrf2 expression was relatively higher in the nuclear fraction compared with cytoplasmic fraction (Figure 6A). SFN treatment (5 μM, 3 h) led to marked increases in Nrf2 expression in the nuclear and cytoplasmic fractions. In particular, SFN enhanced Nrf2 expression in the nuclear fraction by ~2.7 fold (Figure 6B).
Figure 6.
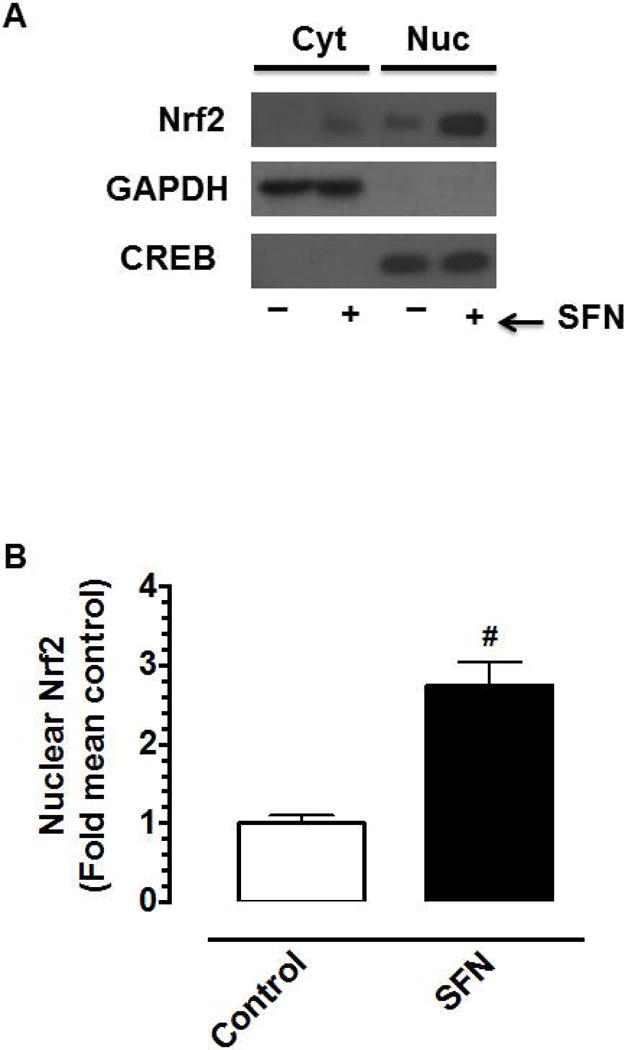
Effects of SFN on Nrf2 protein expression in the nuclear fraction of VSMCs. (A–B) Serum-deprived VSMCs were treated with SFN (5 μM) or DMSO (vehicle) for 3 h. Subsequently, cytoplasmic (Cyt) and nuclear (Nuc) proteins were extracted for immunoblot analysis using primary antibody specific for Nrf2. GAPDH and CREB were used as cytoplasmic and nuclear markers, respectively. Nuclear Nrf2 bands were normalized to their respective CREB bands. The data shown in the bar graph are the means ± SEM from 4 separate experiments. # P<0.05 compared with control (vehicle-treated cells); unpaired student t-test.
3.6. SFN attenuates mTOR signaling independent of Nrf2 activation
Nrf2 activators have been shown to exhibit anti-proliferative effects in vitro and in vivo, in part, through Nrf2-mediated upregulation of HO-1 [50–54]. To determine whether SFN-induced Nrf2/HO-1 and/or mTOR signaling contribute to inhibition of PDGF receptor signaling, VSMCs were subjected to target-specific downregulation of Nrf2 using specific siRNA. Figure 7 shows that transfection of VSMCs with Nrf2 siRNA resulted in the downregulation of Nrf2 protein (by ~89%), which prevented SFN-mediated upregulation of HO-1 and Trx1. However, Nrf2 downregulation did not affect SFN-mediated inhibition of PDGF receptor signaling including phosphorylation of p70S6K, 4E-BP1 and S6. In addition, SFN-mediated inhibition of PDGF-induced cyclin D1 expression remained essentially the same under Nrf2 downregulated conditions.
Figure 7.
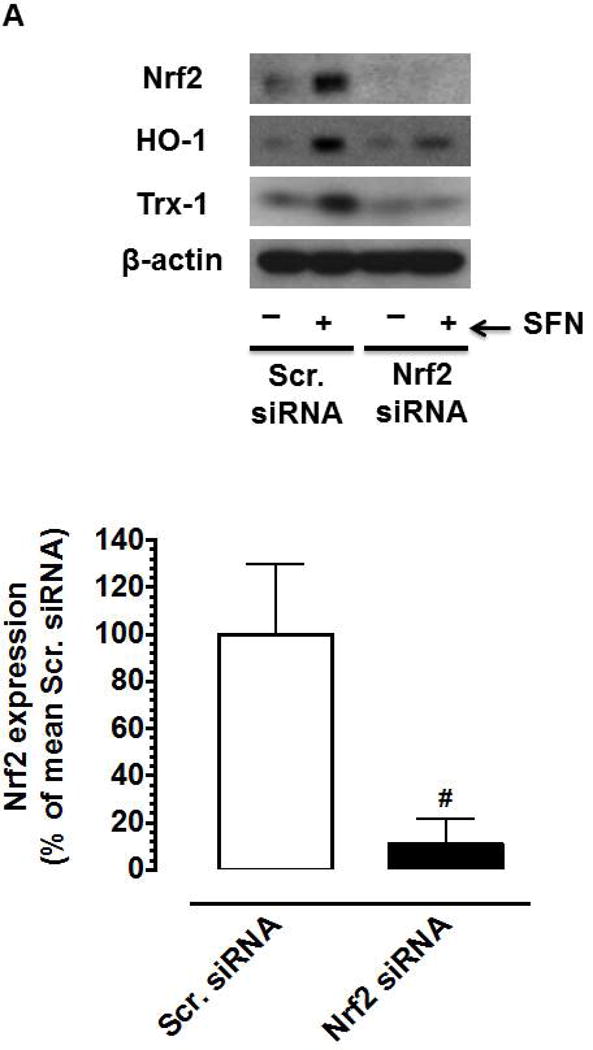
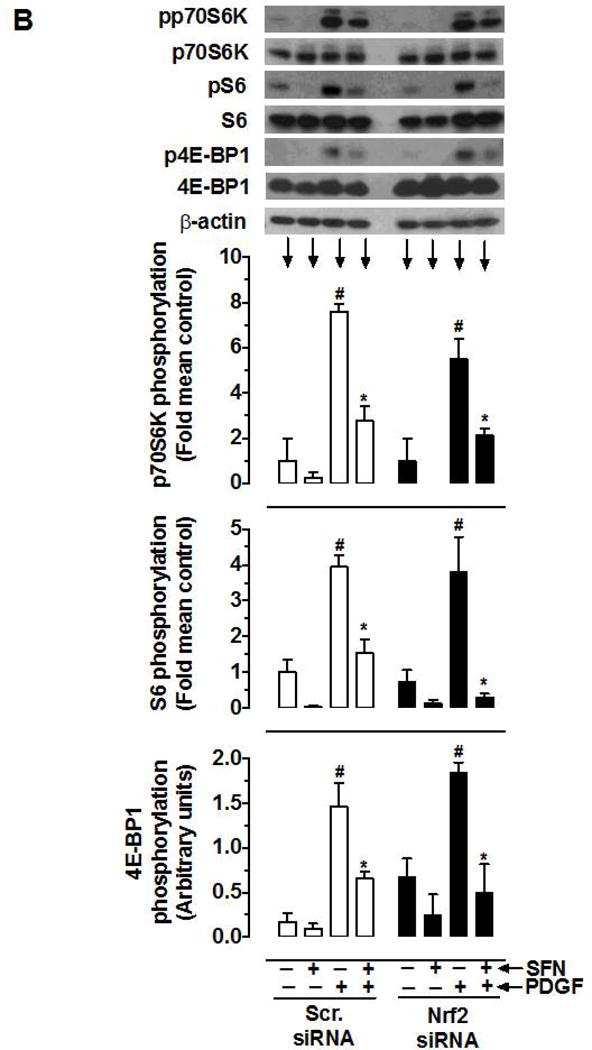
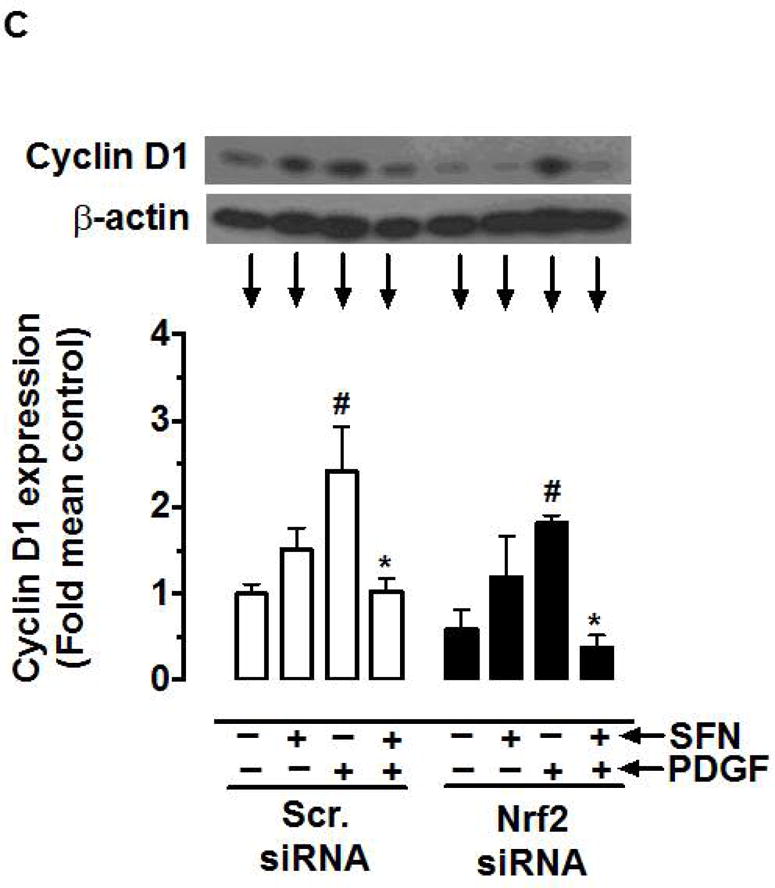
Effects of Nrf2 downregulation on HO-1/Trx-1 expression and PDGF-induced key proliferative signaling in VSMCs. VSMCs were transfected with scrambled (Scr.) or Nrf2 siRNA followed by maintenance in culture for 72 h or 48 h, and subsequent serum deprivation for 45 h or 24 h, respectively. (A) Representative immunoblots showing Nrf2 downregulation and its effects on SFN (3 h and 48 h)-induced HO-1 and Trx-1 expression, respectively, using specific primary antibodies. The data shown in the bar graph are the means ± SEM (n = 3). # P<0.05 compared with Scr siRNA; unpaired student t-test. (B) After 45 h serum deprivation, VSMCs were treated with SFN (5 μM) or DMSO (vehicle) for 3 h followed by exposure to PDGF (30 ng.mL−1) for 6 min. VSMC lysates were then subjected to immunoblot analysis using primary antibodies specific for pp70S6K, p70S6K, pS6, S6, p4E-BP1 and 4E-BP1 (n = 3). (C) After 24 h serum deprivation, VSMCs were pretreated with SFN (5 μM) or DMSO (vehicle) for 30 min followed by exposure to PDGF (30 ng.mL−1) for 48 h. VSMC lysates were then subjected to immunoblot analysis using primary antibody specific for cyclin D1 (n = 3). β-actin was used as an internal control. The data shown in the bar graphs are the means ± SEM. #, * P<0.05 compared with CON (− SFN) and PDGF (− SFN) of the same siRNA group, respectively; two-way ANOVA followed by Tukey’s multiple comparisons test.
3.7. SFN attenuates injury-induced neointima formation and Ki-67 expression
Previously, SFN has been shown to attenuate neointima formation after arterial injury in rats with normal metabolic milieu [8, 10, 11] and in mice with diet-induced obesity and metabolic aberrations [9]. In accordance with these earlier studies, SFN treatment in mice (subcutaneous injection, 23 days) significantly attenuated neointima formation in the injured femoral artery, as evidenced by decreases in neointima/media ratio (by 31.4%) and neointimal area (by 36%) compared with control group (Figure 8A–B). SFN treatment did not result in significant changes in lumen or medial areas (data not shown). Immunofluorescence analysis of injured femoral arteries showed that the neointimal layer is mainly composed of smooth muscle cells (Figure 8C). In addition, immunofluorescence analysis of the injured femoral arteries also showed a marked decrease in Ki-67 immunoreactivity (marker of cellular proliferation) in the neointimal layer in SFN-treated group (Figure 8D).
Figure 8.
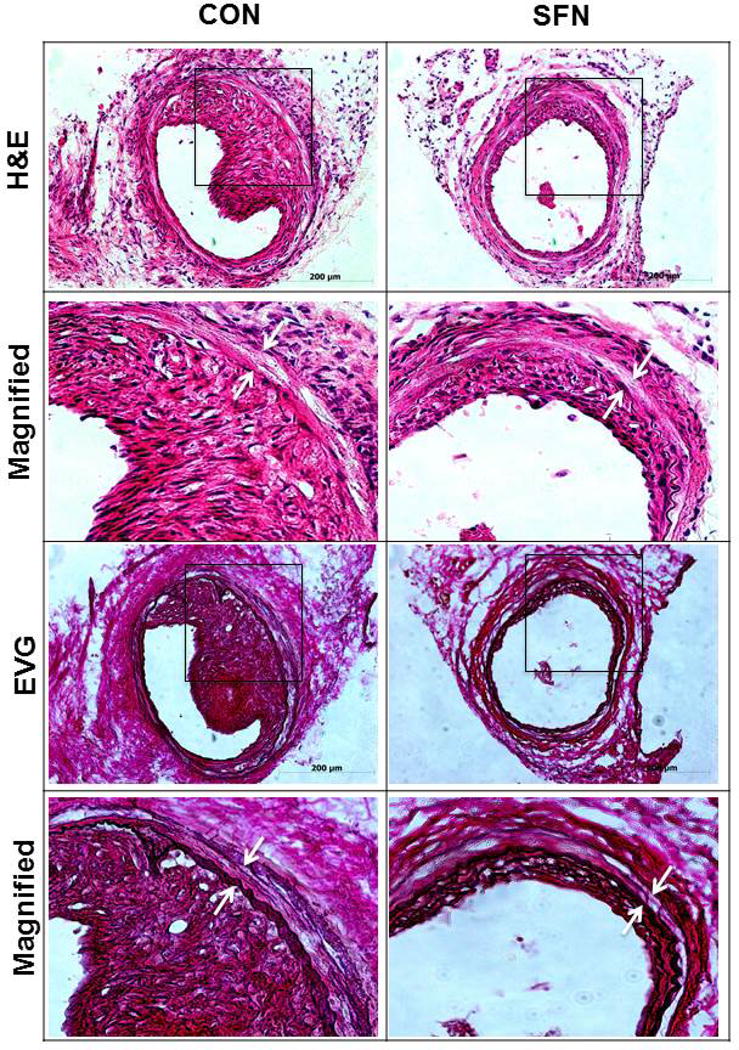
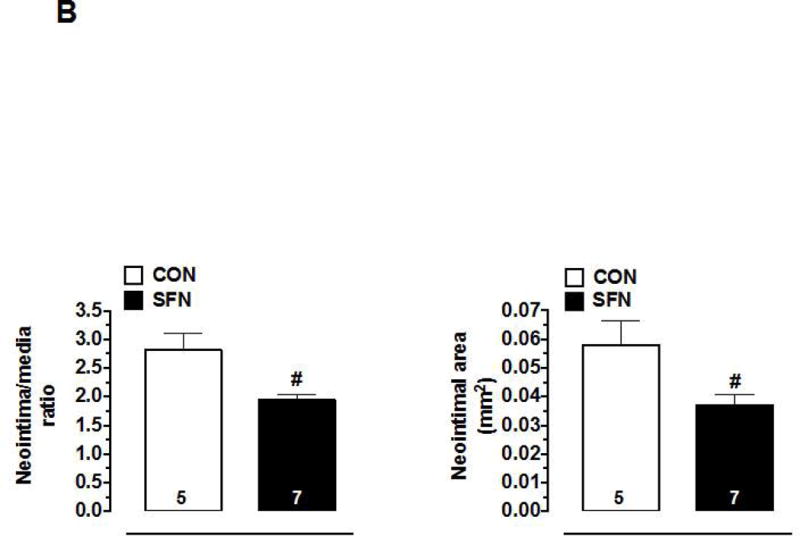
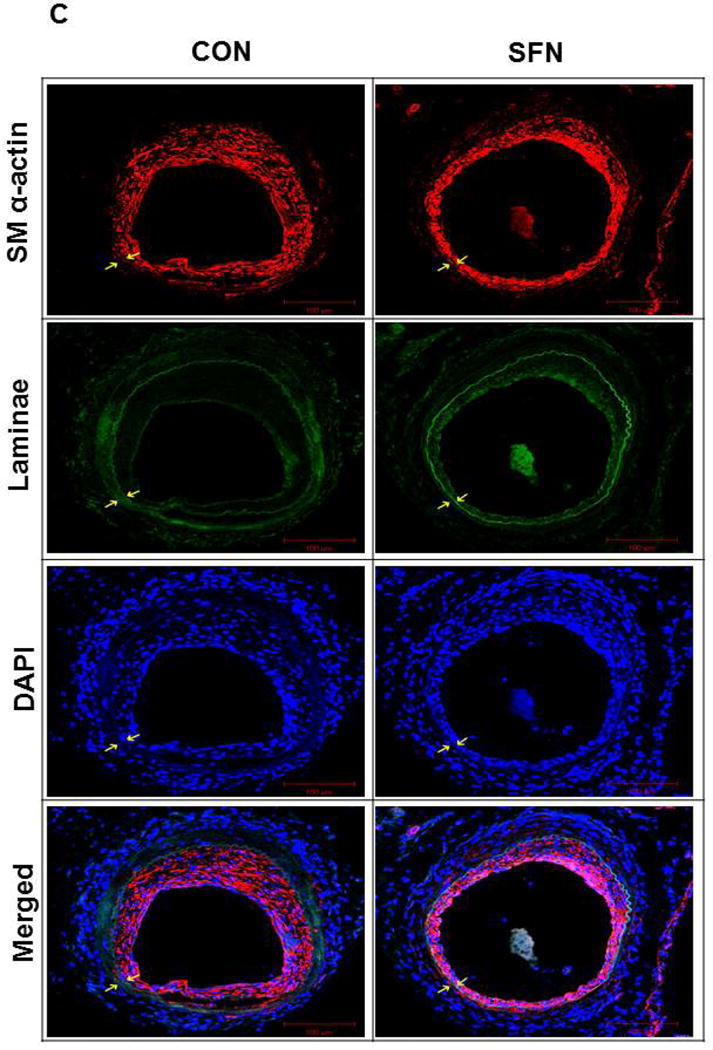
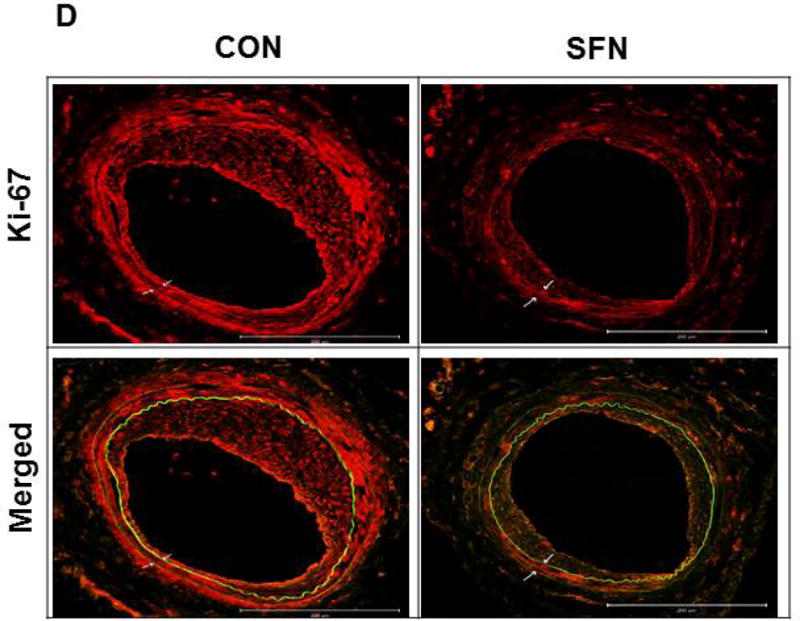
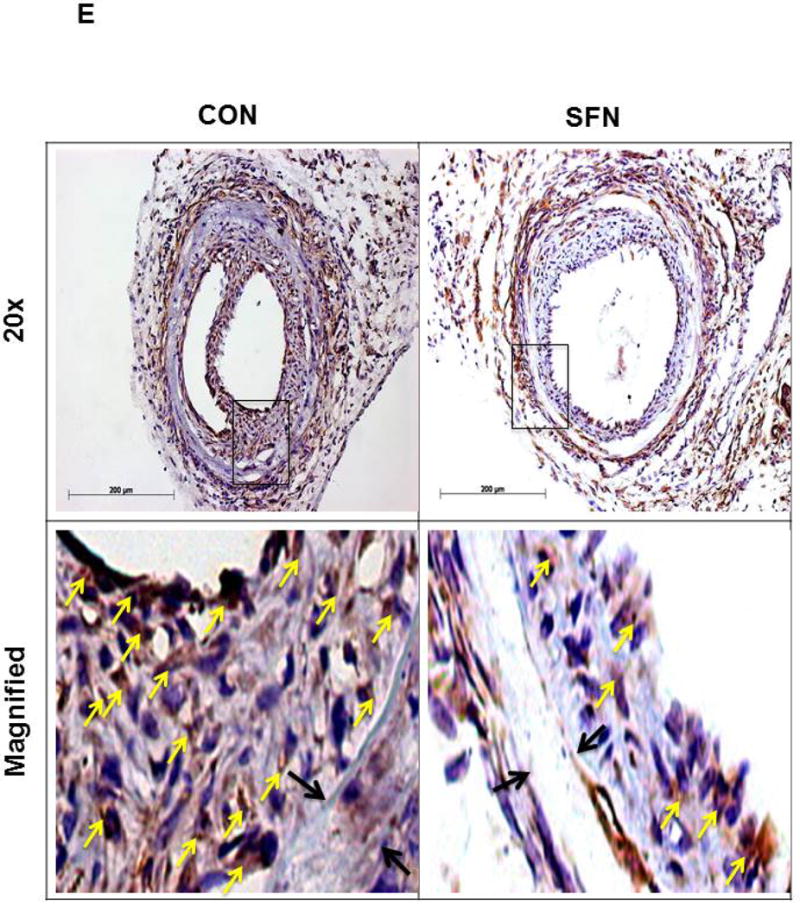
Effects of SFN on neointima formation and S6 phosphorylation in mouse femoral artery after guide wire injury. SFN was injected daily (0.5 mg.kg−1, s.c.) starting 1 day before femoral artery injury and for the next 21 days until sacrifice. (A) H&E and EVG staining of injured femoral arteries from SFN or vehicle-treated mice (B) Morphometric analyses of injured femoral arteries that include neointima/media ratio and neointimal area. The data shown are the means ± SEM. Number of mice in each group is shown at the bases of the bars. # P<0.05 compared with control (vehicle-treated mice); unpaired student t-test. (C) Representative images from confocal immunofluorescence analysis of smooth muscle (SM) α-actin (red), nuclei (DAPI, blue), elastin autofluorescence (laminae, green) and merged images in the injured femoral arteries from SFN or vehicle-treated mice. (D) Representative images from confocal immunofluorescence analysis of Ki-67 (red) and the merged [Ki-67 (red) merged with elastin autofluorescence (green)] in the injured femoral arteries from SFN or vehicle-treated mice. Magnification 20×, and scale bars represent 100–200 μm. Arrows indicate internal and external elastic laminae. (E) Representative images from immunohistochemical analysis of pS6 in the injured femoral arteries from SFN or vehicle-treated mice. Positive cells were visualized using diaminobenzidine (DAB) staining. Magnification 20×; scale bars represent 200 μm (upper panel). Black arrows indicate internal and external elastic laminae, while yellow arrows indicate phosphorylated S6 (lower panel).
3.8. SFN attenuates S6 phosphorylation in the neointimal layer of the injured femoral arteries
As shown in Figure 8E, immunohistochemical analysis revealed a decrease in the phosphorylation state of S6 ribosomal protein in the neointimal layer of SFN-treated group compared with control. These data suggest that the diminution in S6 phosphorylation, an index of decreased mTORC1 activity, may contribute in part to SFN-mediated attenuation of neointima formation at the lesion site.
4. Discussion
Previous studies have shown that activation of Nrf2 transcription factor [29–31] or inhibition of mTOR/p70S6K signaling [13–22] plays a critical role toward attenuating VSMC proliferation/neointima formation after arterial injury. Despite the fact that SFN is an Nrf2 activator [28], the likely intermediary role of activated Nrf2 toward SFN inhibition of VSMC proliferation has not yet been investigated. In addition, there are no reports that document the potential inhibitory effects of SFN on mTOR/p70S6K signaling induced by PDGF, a potent mitogen released at the site of arterial injury. The present study demonstrates that SFN inhibits PDGF-induced activation of mTOR/p70S6K/S6 axis in a sustained manner thereby abrogating enhanced VSMC proliferation, independent of its role in Nrf2 activation. In particular, SFN at 5 μM concentration inhibits PDGF-induced activation of mTOR without affecting mTOR association with raptor in VSMCs. In addition, SFN inhibits PDGF-induced phosphorylation of p70S6K and 4E-BP1, downstream targets of mTOR. Furthermore, SFN abolishes exaggerated phosphorylation of S6 ribosomal protein (a downstream target of p70S6K), which is induced upon acute or prolonged PDGF exposure. Notably, SFN-induced decrease in neointima formation is accompanied by a decrease in S6 phosphorylation in the mouse model of femoral artery injury. While SFN inhibits mTOR signaling in VSMCs, there are no detectable changes in PDGF-induced activation of ERK and Akt. Of importance, downregulation of endogenous Nrf2 by target-specific siRNA reveals an Nrf2-independent effect for SFN inhibition of mTOR/p70S6K/S6 axis, which may contribute to diminished cyclin D1 expression and suppression of VSMC proliferation. Strategies that utilize local delivery of SFN at the lesion site may limit restenosis after angioplasty by targeting mTOR/p70S6K/S6 axis in VSMCs independent of Nrf2 activation.
From a mechanistic standpoint, SFN-mediated increase in Nrf2 accumulation and nuclear translocation observed in the present study with VSMCs may be attributed to its well-characterized role as an electrophile [55]. In this regard, SFN has been shown to modify distinct cysteine residues in Keap1 by interacting with sulfhydryl groups. The consequent disruption of cytoplasmic Keap1-Nrf2 complex would prevent Nrf2 from undergoing proteasomal degradation, thereby facilitating its accumulation and nuclear translocation [56–58]. Nrf2 has been shown to heterodimerize with small Maf protein to promote the transcription of ARE-driven cytoprotective genes, including HO-1, Trx-1, and rate-limiting enzymes of glutathione synthesis [30, 59, 60]. Accordingly, adenoviral overexpression of Nrf2 induces the expression of HO-1 and key enzymes of glutathione synthesis in VSMCs, as described [29]. The present findings reveal that activation of endogenous Nrf2 by SFN results in the upregulation of HO-1 and Trx-1 expression in VSMCs.
Of importance, Nrf2 downregulation using target-specific siRNA suppresses SFN-mediated upregulation of HO-1 and Trx-1 in the present study, suggesting a critical relationship between Nrf2 activation and cytoprotective gene expression in VSMCs. However, under these conditions, SFN-mediated inhibition of PDGF-induced cyclin D1 expression and key proliferative signaling remains essentially the same, suggesting that SFN-mediated Nrf2 activation does not play an intermediary role toward suppressing PDGF-induced VSMC proliferation. At this juncture, it is important to note that pharmacological agents such as curcumin, trans-resveratrol, sulfasalazine (anti-inflammatory/immune-modulatory agent), and dimethylfumarate (an anti-psoriasis drug) inhibit VSMC proliferation in vitro and in vivo presumably through Nrf2-mediated upregulation of HO-1, as reported in recent studies [50–54]. However, none of these studies has utilized Nrf2 gene-silencing strategy to support an obligatory role of Nrf2 activation toward inhibition of VSMC proliferation. Notably, Ashino et al. have shown that Nrf2 downregulation by target-specific siRNA does not affect PDGF-induced VSMC proliferation [30]. However, a study by Levonen et al. has demonstrated that adenoviral delivery of Nrf2 gene results in anti-proliferative effects that are counterbalanced by its anti-apoptotic effects in vivo, thereby contributing to no significant changes in neointimal hyperplasia [29]. Together, although the role of Nrf2 toward inhibition of VSMC proliferation/neointima formation remains controversial, the present findings support the notion that Nrf2 activation is not required at least for SFN-mediated inhibitory effects on PDGF-induced VSMC proliferation. Future studies should employ Nrf2-null mice to confirm the intermediary role of Nrf2 transcription factor toward SFN-mediated inhibition of neointima formation after arterial injury.
In addition to targeting Keap1, SFN has been shown to interact with cysteine residues in several other proteins, including NF-κB [61], AP-1 [62], tubulin [63], and TLR4 [64], thereby accounting for its anti-inflammatory and/or anti-proliferative effects. As a thiol-reactive agent [65], SFN may thus have the potential to directly regulate mTORC1 activity in VSMCs. Previously, a redox-sensitive mechanism has been reported for the control of mTORC1 activity, where phenylarsine oxide is shown to react with the sulfhydryl groups of two closely spaced cysteine residues in mTOR to enhance mTORC1 activity. This is evidenced by increased p70S6K phosphorylation and destabilization of mTOR-raptor complex in HEK293 cells [25]. In addition, conditions that create a reducing environment could inhibit mTORC1 activity possibly through reduction of disulfide bonds in the FATC domain of mTOR [66]. However, the present study with VSMCs reveals that SFN does not affect the association between mTOR and raptor. It diminishes the phosphorylation of mTORC1 downstream targets, suggesting decreased mTORC1 activity by SFN. Thus, the likely interaction of SFN with cysteine residues in mTOR does not provide a plausible explanation for its inhibitory effect on mTORC1 activation in VSMCs. As an alternative mechanism, SFN may dysregulate tuberous sclerosis complex (TSC) and/or Rheb, upstream regulators of mTORC1 [67–69]. Future studies are clearly warranted to determine whether SFN affects TSC and/or Rheb signaling to inhibit mTOR/p70S6K/S6 axis in VSMCs.
Of importance, SFN-mediated inhibition of mTORC1 activation is clearly different from classical mTORC1 inhibitors (e.g., rapamycin and ATP-competitive inhibitors) with regard to mechanistic and functional considerations. For instance, SFN inhibits PDGF-induced phosphorylation of mTOR and its downstream signaling components, p70S6K/S6 and 4E-BP1, thereby suppressing VSMC proliferation. Rapamycin (sirolimus) is known to interact with FK506-binding protein 12 (FKBP12) to form a complex, which inhibits mTORC1 activity in an allosteric manner by binding to FKBP12-Rapamycin-Binding (FRB) domain of mTOR [67, 70]. Its role in mTORC1 inhibition is reflected by diminished phosphorylation of p70S6K and S6, whereas 4E-BP1 phosphorylation remains refractory with prolonged rapamycin treatment [70]. This increase in 4E-BP1 phosphorylation by rapamycin may allow protein synthesis to occur sufficient enough to promote cell proliferation [70]. However, at least in VSMCs, SFN treatment is not associated with an increase in 4E-BP1 phosphorylation. Furthermore, ATP-competitive mTOR inhibitors that compete with ATP in the catalytic domain of mTOR are currently in clinical trials, and these inhibitors have been shown to inhibit kinase-dependent functions of mTORC1 and mTORC2 [70, 71]. While SFN inhibits mTORC1 signaling, it does not appear to diminish mTORC2 activation (as reflected by its lack of effect on the phosphorylation AktSer473, a downstream target of mTORC2). Further studies are clearly warranted that should determine whether SFN regulates mTORC2 (rictor-mTOR) kinase activity in VSMCs [26, 27]. From a therapeutic standpoint, rapamycin is used clinically for the treatment of restenosis after angioplasty. While it is beneficial in inhibiting exaggerated VSMC proliferation, its use is also associated with diminished endothelial cell proliferation. This would prevent reendothelialization at the lesion site, thereby increasing the risk of thrombosis with rapamycin treatment [72]. Future studies should determine the likely beneficial effects of SFN toward promoting reendothelialization in the injured vessel wall.
In conjunction with the observed inhibitory effects of SFN on VSMC proliferation/neointima formation (present study), it is pertinent to note that SFN administration to wild-type rodents does not result in changes in body weight, food intake, lipid profile, blood pressure, and heart rate, as reported in previous studies [9, 73, 74]. However, in high fat diet- or western diet-fed obese mice, SFN attenuates weight gain with no significant effects on food intake [9, 74]. In addition, SFN treatment improves systemic lipid profile. For instance, it decreases the circulating levels of total cholesterol [74] and triglycerides [9] in diet-induced obese mice. Furthermore, SFN decreases systolic blood pressure in spontaneously hypertensive stroke-prone rats and obese mice [9, 73], with no effect on heart rate [9].
In conclusion, SFN administration would provide a realistic treatment option to attenuate the development and/or progression of restenosis after angioplasty through its ability to target PDGF-induced mTOR, p70S6K, and 4E-BP1 signaling in VSMCs independent of Nrf2 activation. Furthermore, in view of the potential link between HO-1 induction and plaque stabilization [75], Nrf2-mediated upregulation of HO-1 by SFN may prevent plaque destabilization/rupture in vulnerable vessels thereby offering protection from acute coronary events.
Figure 9.
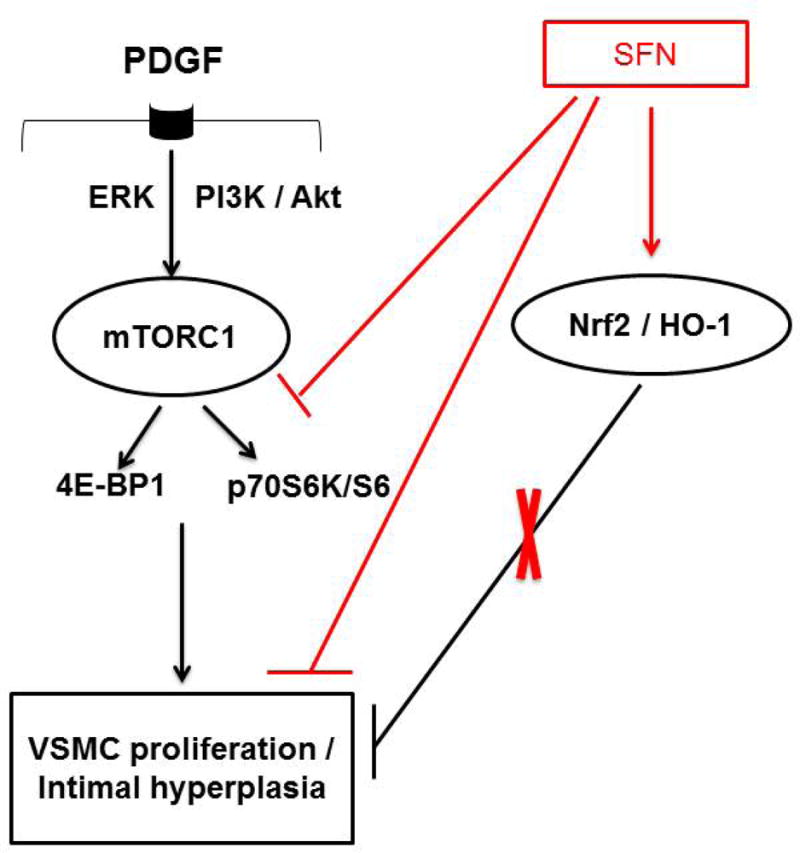
Sulforaphane (SFN) inhibits downstream targets of mTORC1, including p70S6K/S6 and 4E-BP1 phosphorylation to attenuate PDGF-induced VSMC proliferation. Although SFN activates Nrf2 transcription factor to induce HO-1 expression, Nrf2 downregulation using target-specific siRNA reveals that Nrf2/HO-1 signaling does not contribute to SFN inhibition of VSMC proliferation.
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute/National Institutes of Health Grant (R01-HL-097090), University of Georgia Research Foundation Fund, and Egyptian Government Scholarship Fund from the Egyptian Cultural and Educational Bureau.
Abbreviations
- ARE
antioxidant response element
- CREB
cAMP-response element-binding protein
- DAB
diaminobenzidine
- DMSO
dimethyl sulfoxide
- 4E-BP1
eukaryotic translation initiation factor 4E-binding protein 1
- EEL
external elastic lamina
- ERK1/2
extracellular signal-regulated kinase1/2
- EVG
elastic van gieson
- FKBP12
FK506-binding protein 12
- FRB domain
FKBP12-Rapamycin-Binding domain
- GSK3β
glycogen synthase kinase3β
- H&E
haematoxylin and eosin
- HO-1
heme oxygenase-1
- IEL
internal elastic lamina
- Keap1
kelch-like ECH associated protein 1
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- MEK1
mitogen-activated protein/ERK kinase1
- Nrf2
nuclear factor erythroid 2-related factor 2
- PBS
phosphate-buffered saline
- PDGF
platelet-derived growth factor
- PFA
paraformaldehyde
- PI 3-kinase
phosphoinositide 3-kinase
- pRb
phosphorylated retinoblastoma protein
- p70S6K
p70S6 kinase
- raptor
regulatory associated protein of mTOR
- Rheb
Ras homolog enriched in brain
- rictor
rapamycin-insensitive companion of mTOR
- RT
room temperature
- SFN
sulforaphane
- SM α-actin
smooth muscle α-actin
- SMGS
smooth muscle growth supplement
- TBST
tris-buffered saline with tween
- Trx-1
thioredoxin-1
- TSC
tuberous sclerosis complex
- VSMCs
vascular smooth muscle cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Chemical compounds studied in this article: Sulforaphane (PubChem CID: 5350); Dimethyl Sulfoxide (PubChem CID: 679)
Author Contributions
N.M.S. and L.S. conceptualized and designed experiments. N.M.S. performed experimental work. N.M.S. and L.S. analyzed and interpreted data. N.M.S. and L.S. contributed to the writing of the manuscript. The experiments were conducted at the Charlie Norwood VA Medical Center, Augusta, Georgia, USA.
Conflict of Interest
The authors declare no conflicts of interest.
References
- 1.Clowes AW, Reidy MA, Clowes MM. Mechanisms of stenosis after arterial injury. Lab Invest. 1983;49:208–15. [PubMed] [Google Scholar]
- 2.Schwartz SM, deBlois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–65. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- 3.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 4.Alfonso F, Byrne RA, Rivero F, Kastrati A. Current treatment of in-stent restenosis. J Am Coll Cardiol. 2014;63:2659–73. doi: 10.1016/j.jacc.2014.02.545. [DOI] [PubMed] [Google Scholar]
- 5.Levonen AL, Vahakangas E, Koponen JK, Yla-Herttuala S. Antioxidant gene therapy for cardiovascular disease: current status and future perspectives. Circulation. 2008;117:2142–50. doi: 10.1161/CIRCULATIONAHA.107.718585. [DOI] [PubMed] [Google Scholar]
- 6.Leopold JA. Antioxidants and coronary artery disease: from pathophysiology to preventive therapy. Coron Artery Dis. 2015;26:176–83. doi: 10.1097/MCA.0000000000000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim JY, Park HJ, Um SH, Sohn EH, Kim BO, Moon EY, et al. Sulforaphane suppresses vascular adhesion molecule-1 expression in TNF-alpha-stimulated mouse vascular smooth muscle cells: involvement of the MAPK, NF-kappaB and AP-1 signaling pathways. Vascul Pharmacol. 2012;56:131–41. doi: 10.1016/j.vph.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Kwon JS, Joung H, Kim YS, Shim YS, Ahn Y, Jeong MH, et al. Sulforaphane inhibits restenosis by suppressing inflammation and the proliferation of vascular smooth muscle cells. Atherosclerosis. 2012;225:41–9. doi: 10.1016/j.atherosclerosis.2012.07.040. [DOI] [PubMed] [Google Scholar]
- 9.Shawky NM, Pichavaram P, Shehatou GSG, Suddek GM, Gameil NM, Jun JY, et al. Sulforaphane improves dysregulated metabolic profile and inhibits leptin-induced VSMC proliferation: implications toward suppression of neointima formation after arterial injury in western diet-fed obese mice. J Nutr Biochem. 2016;32:73–84. doi: 10.1016/j.jnutbio.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoo SH, Lim Y, Kim SJ, Yoo KD, Yoo HS, Hong JT, et al. Sulforaphane inhibits PDGF-induced proliferation of rat aortic vascular smooth muscle cell by up-regulation of p53 leading to G1/S cell cycle arrest. Vascul Pharmacol. 2013;59:44–51. doi: 10.1016/j.vph.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Orozco-Sevilla V, Naftalovich R, Hoffmann T, London D, Czernizer E, Yang C, et al. Epigallocatechin-3-gallate is a potent phytochemical inhibitor of intimal hyperplasia in the wire-injured carotid artery. J Vasc Surg. 2013;58:1360–5. doi: 10.1016/j.jvs.2012.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 13.Marx SO, Jayaraman T, Go LO, Marks AR. Rapamycin-FKBP inhibits cell cycle regulators of proliferation in vascular smooth muscle cells. Circ Res. 1995;76:412–7. doi: 10.1161/01.res.76.3.412. [DOI] [PubMed] [Google Scholar]
- 14.Braun-Dullaeus RC, Mann MJ, Seay U, Zhang L, von Der Leyen HE, Morris RE, et al. Cell cycle protein expression in vascular smooth muscle cells in vitro and in vivo is regulated through phosphatidylinositol 3-kinase and mammalian target of rapamycin. Arterioscler Thromb Vasc Biol. 2001;21:1152–8. doi: 10.1161/hq0701.092104. [DOI] [PubMed] [Google Scholar]
- 15.Tang Z, Wang Y, Fan Y, Zhu Y, Chien S, Wang N. Suppression of c-Cbl tyrosine phosphorylation inhibits neointimal formation in balloon-injured rat arteries. Circulation. 2008;118:764–72. doi: 10.1161/CIRCULATIONAHA.107.761932. [DOI] [PubMed] [Google Scholar]
- 16.Lo IC, Lin TM, Chou LH, Liu SL, Wu LW, Shi GY, et al. Ets-1 mediates platelet-derived growth factor-BB-induced thrombomodulin expression in human vascular smooth muscle cells. Cardiovasc Res. 2009;81:771–9. doi: 10.1093/cvr/cvn351. [DOI] [PubMed] [Google Scholar]
- 17.Kida T, Chuma H, Murata T, Yamawaki H, Matsumoto S, Hori M, et al. Chronic treatment with PDGF-BB and endothelin-1 synergistically induces vascular hyperplasia and loss of contractility in organ-cultured rat tail artery. Atherosclerosis. 2011;214:288–94. doi: 10.1016/j.atherosclerosis.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Biswas SK, McNulty PH, Kozak M, Jun JY, Segar L. PDGF-induced vascular smooth muscle cell proliferation is associated with dysregulation of insulin receptor substrates. Am J Physiol Cell Physiol. 2011;300:C1375–C85. doi: 10.1152/ajpcell.00670.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mourani PM, Garl PJ, Wenzlau JM, Carpenter TC, Stenmark KR, Weiser-Evans MC. Unique, highly proliferative growth phenotype expressed by embryonic and neointimal smooth muscle cells is driven by constitutive Akt, mTOR, and p70S6K signaling and is actively repressed by PTEN. Circulation. 2004;109:1299–306. doi: 10.1161/01.CIR.0000118462.22970.BE. [DOI] [PubMed] [Google Scholar]
- 20.Ha JM, Yun SJ, Kim YW, Jin SY, Lee HS, Song SH, et al. Platelet-derived growth factor regulates vascular smooth muscle phenotype via mammalian target of rapamycin complex 1. Biochem Biophys Res Commun. 2015;464:57–62. doi: 10.1016/j.bbrc.2015.05.097. [DOI] [PubMed] [Google Scholar]
- 21.Xie Y, Jin Y, Merenick BL, Ding M, Fetalvero KM, Wagner RJ, et al. Phosphorylation of GATA-6 is required for vascular smooth muscle cell differentiation after mTORC1 inhibition. Sci Signal. 2015;8:ra44. doi: 10.1126/scisignal.2005482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osman I, Segar L. Pioglitazone, a PPARgamma agonist, attenuates PDGF-induced vascular smooth muscle cell proliferation through AMPK-dependent and AMPK-independent inhibition of mTOR/70S6K and ERK signaling. Biochem Pharmacol. 2016 doi: 10.1016/j.bcp.2015.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–89. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 24.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–94. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarbassov DD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem. 2005;280:39505–9. doi: 10.1074/jbc.M506096200. [DOI] [PubMed] [Google Scholar]
- 26.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 27.Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406–16. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 28.Dinkova-Kostova AT, Kostov RV. Glucosinolates and isothiocyanates in health and disease. Trends Mol Med. 2012;18:337–47. doi: 10.1016/j.molmed.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 29.Levonen AL, Inkala M, Heikura T, Jauhiainen S, Jyrkkanen HK, Kansanen E, et al. Nrf2 gene transfer induces antioxidant enzymes and suppresses smooth muscle cell growth in vitro and reduces oxidative stress in rabbit aorta in vivo. Arterioscler Thromb Vasc Biol. 2007;27:741–7. doi: 10.1161/01.ATV.0000258868.80079.4d. [DOI] [PubMed] [Google Scholar]
- 30.Ashino T, Yamamoto M, Yoshida T, Numazawa S. Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia. Arterioscler Thromb Vasc Biol. 2013;33:760–8. doi: 10.1161/ATVBAHA.112.300614. [DOI] [PubMed] [Google Scholar]
- 31.Ashino T, Yamamoto M, Numazawa S. Nrf2/Keap1 system regulates vascular smooth muscle cell apoptosis for vascular homeostasis: role in neointimal formation after vascular injury. Sci Rep. 2016;6:26291. doi: 10.1038/srep26291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosner M, Siegel N, Valli A, Fuchs C, Hengstschlager M. mTOR phosphorylated at S2448 binds to raptor and rictor. Amino acids. 2010;38:223–8. doi: 10.1007/s00726-008-0230-7. [DOI] [PubMed] [Google Scholar]
- 33.Shawky NM, Pichavaram P, Shehatou GS, Suddek GM, Gameil NM, Jun JY, et al. Sulforaphane improves dysregulated metabolic profile and inhibits leptin-induced VSMC proliferation: Implications toward suppression of neointima formation after arterial injury in western diet-fed obese mice. J Nutr Biochem. 2016;32:73–84. doi: 10.1016/j.jnutbio.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu R, Hebbar V, Kim B-R, Chen C, Winnik B, Buckley B, et al. In Vivo Pharmacokinetics and Regulation of Gene Expression Profiles by Isothiocyanate Sulforaphane in the Rat. J Pharmacol Exp Ther. 2004;310:263–71. doi: 10.1124/jpet.103.064261. [DOI] [PubMed] [Google Scholar]
- 35.Ye L, Dinkova-Kostova AT, Wade KL, Zhang Y, Shapiro TA, Talalay P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: pharmacokinetics of broccoli sprout isothiocyanates in humans. Clinica Chimica Acta. 2002;316:43–53. doi: 10.1016/s0009-8981(01)00727-6. [DOI] [PubMed] [Google Scholar]
- 36.Pyla R, Poulose N, Jun JY, Segar L. Expression of conventional and novel glucose transporters, GLUT1, -9, -10, and -12, in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2013;304:C574–89. doi: 10.1152/ajpcell.00275.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pyla R, Pichavaram P, Fairaq A, Park MA, Kozak M, Kamath V, et al. Altered energy state reversibly controls smooth muscle contractile function in human saphenous vein during acute hypoxia-reoxygenation: Role of glycogen, AMP-activated protein kinase, and insulin-independent glucose uptake. Biochem Pharmacol. 2015;97:77–88. doi: 10.1016/j.bcp.2015.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sata M, Maejima Y, Adachi F, Fukino K, Saiura A, Sugiura S, et al. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32:2097–104. doi: 10.1006/jmcc.2000.1238. [DOI] [PubMed] [Google Scholar]
- 39.Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335(Pt 1):1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 41.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes & Development. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 42.Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172–6. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng SW, Fryer LG, Carling D, Shepherd PR. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem. 2004;279:15719–22. doi: 10.1074/jbc.C300534200. [DOI] [PubMed] [Google Scholar]
- 44.Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem. 2000;275:7416–23. doi: 10.1074/jbc.275.10.7416. [DOI] [PubMed] [Google Scholar]
- 45.Acosta-Jaquez HA, Keller JA, Foster KG, Ekim B, Soliman GA, Feener EP, et al. Site-specific mTOR phosphorylation promotes mTORC1-mediated signaling and cell growth. Mol Cell Biol. 2009;29:4308–24. doi: 10.1128/MCB.01665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes & Development. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao XD, Zhou YT, Lu XJ. Sulforaphane enhances the activity of the Nrf2-ARE pathway and attenuates inflammation in OxyHb-induced rat vascular smooth muscle cells. Inflammation Res. 2013;62:857–63. doi: 10.1007/s00011-013-0641-0. [DOI] [PubMed] [Google Scholar]
- 48.Lopes RA, Neves KB, Tostes RC, Montezano AC, Touyz RM. Downregulation of Nuclear Factor Erythroid 2-Related Factor and Associated Antioxidant Genes Contributes to Redox-Sensitive Vascular Dysfunction in Hypertension. Hypertension. 2015;66:1240–50. doi: 10.1161/HYPERTENSIONAHA.115.06163. [DOI] [PubMed] [Google Scholar]
- 49.Wu L, Juurlink BH. The impaired glutathione system and its up-regulation by sulforaphane in vascular smooth muscle cells from spontaneously hypertensive rats. J Hypertens. 2001;19:1819–25. doi: 10.1097/00004872-200110000-00016. [DOI] [PubMed] [Google Scholar]
- 50.Oh CJ, Park S, Kim JY, Kim HJ, Jeoung NH, Choi YK, et al. Dimethylfumarate attenuates restenosis after acute vascular injury by cell-specific and Nrf2-dependent mechanisms. Redox Biol. 2014;2:855–64. doi: 10.1016/j.redox.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim JY, Cho HJ, Sir JJ, Kim BK, Hur J, Youn SW, et al. Sulfasalazine induces haem oxygenase-1 via ROS-dependent Nrf2 signalling, leading to control of neointimal hyperplasia. Cardiovasc Res. 2009;82:550–60. doi: 10.1093/cvr/cvp072. [DOI] [PubMed] [Google Scholar]
- 52.Kim N, Hwangbo C, Lee S, Lee JH. Eupatolide inhibits PDGF-induced proliferation and migration of aortic smooth muscle cells through ROS-dependent heme oxygenase-1 induction. Phytother Res. 2013;27:1700–7. doi: 10.1002/ptr.4924. [DOI] [PubMed] [Google Scholar]
- 53.Pae HO, Jeong GS, Jeong SO, Kim HS, Kim SA, Kim YC, et al. Roles of heme oxygenase-1 in curcumin-induced growth inhibition in rat smooth muscle cells. Exp Mol Med. 2007;39:267–77. doi: 10.1038/emm.2007.30. [DOI] [PubMed] [Google Scholar]
- 54.Kim JW, Lim SC, Lee MY, Lee JW, Oh WK, Kim SK, et al. Inhibition of neointimal formation by trans-resveratrol: role of phosphatidyl inositol 3-kinase-dependent Nrf2 activation in heme oxygenase-1 induction. Mol Nutr Food Res. 2010;54:1497–505. doi: 10.1002/mnfr.201000016. [DOI] [PubMed] [Google Scholar]
- 55.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes to cells: devoted to molecular & cellular mechanisms. 2003;8:379–91. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 56.Ahn Y-H, Hwang Y, Liu H, Wang XJ, Zhang Y, Stephenson KK, et al. Electrophilic tuning of the chemoprotective natural product sulforaphane. Proc Natl Acad Sci USA. 2010;107:9590–5. doi: 10.1073/pnas.1004104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu C, Eggler AL, Mesecar AD, van Breemen RB. Modification of keap1 cysteine residues by sulforaphane. Chemical Research in Toxicology. 2011;24:515–21. doi: 10.1021/tx100389r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Ann Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 60.Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M. Genetic Evidence that Small Maf Proteins Are Essential for the Activation of Antioxidant Response Element-Dependent Genes. Mol Cell Biol. 2005;25:8044–51. doi: 10.1128/MCB.25.18.8044-8051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhauser C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem. 2001;276:32008–15. doi: 10.1074/jbc.M104794200. [DOI] [PubMed] [Google Scholar]
- 62.Dickinson SE, Melton TF, Olson ER, Zhang J, Saboda K, Bowden GT. Inhibition of activator protein-1 by sulforaphane involves interaction with cysteine in the cFos DNA-binding domain: implications for chemoprevention of UVB-induced skin cancer. Cancer Res. 2009;69:7103–10. doi: 10.1158/0008-5472.CAN-09-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mi L, Xiao Z, Hood BL, Dakshanamurthy S, Wang X, Govind S, et al. Covalent binding to tubulin by isothiocyanates. A mechanism of cell growth arrest and apoptosis. J Biol Chem. 2008;283:22136–46. doi: 10.1074/jbc.M802330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Youn HS, Kim YS, Park ZY, Kim SY, Choi NY, Joung SM, et al. Sulforaphane suppresses oligomerization of TLR4 in a thiol-dependent manner. J Immunol. 2010;184:411–9. doi: 10.4049/jimmunol.0803988. [DOI] [PubMed] [Google Scholar]
- 65.Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci USA. 2001;98:3404–9. doi: 10.1073/pnas.051632198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dames SA, Mulet JM, Rathgeb-Szabo K, Hall MN, Grzesiek S. The solution structure of the FATC domain of the protein kinase target of rapamycin suggests a role for redox-dependent structural and cellular stability. J Biol Chem. 2005;280:20558–64. doi: 10.1074/jbc.M501116200. [DOI] [PubMed] [Google Scholar]
- 67.Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nature Genetics. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 68.Yoshida S, Hong S, Suzuki T, Nada S, Mannan AM, Wang J, et al. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J Biol Chem. 2011;286:32651–60. doi: 10.1074/jbc.M111.238014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cavell BE, Syed Alwi SS, Donlevy AM, Proud CG, Packham G. Natural product-derived antitumor compound phenethyl isothiocyanate inhibits mTORC1 activity via TSC2. Journal of Natural Products. 2012;75:1051–7. doi: 10.1021/np300049b. [DOI] [PubMed] [Google Scholar]
- 70.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10:868–80. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 71.Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1:27–36. doi: 10.1007/s12154-008-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luscher TF, Steffel J, Eberli FR, Joner M, Nakazawa G, Tanner FC, et al. Drug-eluting stent and coronary thrombosis: biological mechanisms and clinical implications. Circulation. 2007;115:1051–8. doi: 10.1161/CIRCULATIONAHA.106.675934. [DOI] [PubMed] [Google Scholar]
- 73.Wu L, Noyan Ashraf MH, Facci M, Wang R, Paterson PG, Ferrie A, et al. Dietary approach to attenuate oxidative stress, hypertension, and inflammation in the cardiovascular system. Proc Natl Acad Sci USA. 2004;101:7094–9. doi: 10.1073/pnas.0402004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choi K-M, Lee Y-S, Kim W, Kim SJ, Shin K-O, Yu J-Y, et al. Sulforaphane attenuates obesity by inhibiting adipogenesis and activating the AMPK pathway in obese mice. J Nutr Biochem. 2014;25:201–7. doi: 10.1016/j.jnutbio.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 75.Larsen K, Cheng C, Duckers HJ. Regulation of vulnerable plaque development by the heme oxygenase/carbon monoxide system. Trends Cardiovasc Med. 2010;20:58–65. doi: 10.1016/j.tcm.2010.04.001. [DOI] [PubMed] [Google Scholar]