

Abstract
Aim: There is an unmet need in Japan for more optimal lipid-lowering therapy (LLT) for patients with homozygous familial hypercholesterolemia (HoFH) who respond inadequately to available drug therapies and/or apheresis, to achieve goals of low-density lipoprotein cholesterol (LDL-C) reduction by 50% or to < 100 mg/dL.
Methods: In this study, Japanese patients with HoFH on stable LLT and diet were treated with lomitapide, initiated at 5 mg/day and escalated to maximum tolerated dose (up to 60 mg/day) over 14 weeks. The primary efficacy endpoint was mean percentage change from baseline to Week 26 in LDL-C. Secondary endpoints included changes in other lipid parameters and safety throughout the 56-week study (including follow-up).
Results: Nine patients entered the efficacy phase of the study and, of these, eight completed 56 weeks. Mean LDL-C was reduced by 42% (p < 0.0001) at 26 weeks, from 199 mg/dL (95% CI: 149–250) at baseline to 118 mg/dL (95% CI: 70–166). A 50% reduction in LDL-C and LDL-C < 100 mg/dL was achieved by five and six of nine patients, respectively, at 26 weeks. After 56 weeks, LDL-C was reduced by 38% (p = 0.0032) from baseline. Significant reductions in non-HDL-C, VLDL-C, triglycerides, and apolipoprotein B were also reported at Week 26. There were no new safety signals and, similar to previous studies, gastrointestinal adverse events were the most common adverse events.
Conclusion: Lomitapide, added to ongoing treatment with other LLTs, was effective in rapidly and significantly reducing the levels of LDL-C and other atherogenic apolipoprotein B-containing lipoproteins in adult Japanese patients with HoFH.
Keywords: Homozygous familial hypercholesterolemia, Lomitapide, Hypercholesterolemia, LDL-C
See editorial vol. 24: 390–392
Introduction
Homozygous familial hypercholesterolemia (HoFH) is a rare, autosomal dominant genetic disorder characterized by substantially elevated low-density lipoprotein cholesterol (LDL-C) levels. In untreated patients, total plasma cholesterol and LDL-C levels are generally ≥ 500 mg/dL.1) and markedly premature atherosclerotic cardiovascular disease (ASCVD) occurs.2–4). If left untreated, most patients will develop atherosclerosis before the age of 20 years and generally do not survive past the age of 30 years.1). The primary goal of therapy is the prevention of ASCVD including coronary artery disease by controlling hypercholesterolemia. The European Atherosclerotic Society has defined the current LDL-C target for adults with HoFH as < 100 mg/dL, or < 70 mg/dL in the presence of clinical ASCVD.1). The American Heart Association recommends similar goals with the caveat that these targets may be difficult to achieve, therefore maximal LDL-C reduction that can be well tolerated is a pragmatic target.5). The Japan Atherosclerosis Society recommends that treatment for adults with HoFH should aim for LDL-C < 100 mg/dL; however, because this target may be challenging, they have suggested a secondary target of a 50% reduction in pretreatment levels of LDL-C.6). Individuals unable to achieve these LDL-C target levels may remain at high risk for ASCVD.1, 7). A recent review of familial hypercholesterolemia in Asia found that awareness of the disease, and its diagnostic criteria, varied between countries; concluding that more resources are required to raise awareness, improve care, and increase research in Asian populations.8). Ongoing observational studies will help elucidate geographic/epidemiological differences that may impact optimal management of HoFH, including screening, in different regions.9, 10).
HoFH is commonly caused by loss of function mutations in the gene encoding the LDL-receptor (LDLR).3), but can also be caused by mutations in several other genes resulting in impairment of the LDLR pathway.11–13). Consequently, treatments with statins and PCSK9 inhibitors, which rely on upregulating hepatic LDLR, have reduced effects in most patients with HoFH, whose LDLRs are absent or defective. Since patients with HoFH usually do not achieve the recommended LDL-C target by using these treatments, adjunctive lipoprotein apheresis is also recommended. Apheresis can transiently reduce LDL-C by more than 50%.17, 18). However, the rapid re-accumulation of circulating LDL-C requires that apheresis be repeated frequently, every 1–2 weeks, and means that LDL-C targets are unlikely to be maintained. Even with regular lipoprotein apheresis, many patients with HoFH still develop coronary artery disease and/or aortic valve stenosis.19). More effective treatments for HoFH are required to prevent atherosclerosis in these patients.
Lomitapide (Juxtapid [AEGR-733], Aegerion Pharmaceuticals Inc., Cambridge, MA, USA; Lojuxta, Aegerion Pharmaceuticals Ltd, Uxbridge, UK) is an inhibitor of the microsomal triglyceride transfer protein that transfers triglycerides within the liver onto apolipoprotein B during the assembly of very-low-density lipoproteins (VLDLs), which are the precursors of circulating LDLs.20, 21). Lomitapide is indicated as an adjunctive therapy for adults with HoFH in a number of countries, including the USA, Canada, countries in the EU, Taiwan, and Korea.21–23).
Aim
In Japan, there is an unmet need for more optimal lipid-lowering therapy (LLT) for patients with HoFH who respond inadequately to available drug therapies and/or apheresis. The primary aim of this study was to evaluate, in Japanese adult patients with HoFH, the efficacy of lomitapide in reducing LDL-C in combination with other LLTs. Secondary aims were to evaluate the safety and tolerability of lomitapide and to evaluate changes in other lipid parameters, hepatic fat, and xanthomas.
Methods
Study Design
In this phase 3, single-arm, open-label study, adult patients with HoFH added lomitapide to their maximally tolerated, stable, LLT. After initial screening, including informed consent (during Weeks −12 to −6), the study progressed in three phases (Fig. 1): −6–0 weeks, pre-treatment run-in during which both diet and other LLTs were stabilized; 0–26 weeks, dose-escalation and efficacy measures; and > 26–56 weeks, safety measures. At the conclusion of this trial, patients had the option of continuing in a long-term extension trial.
Fig. 1.
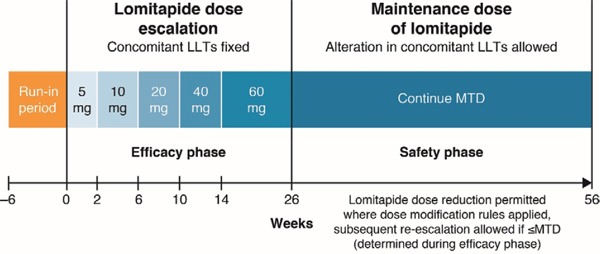
Study design
LLT, lipid-lowering therapy; MTD, maximum tolerated dose.
During the run-in phase, the diet stabilization target required patients to obtain < 20% of food energy as fat and to take daily supplements of vitamin E (400 IU) and fatty acids (≥ 200 mg linoleic acid, 210 mg alpha-linolenic acid, 110 mg eicosapentaenoic acid, and 80 mg docosahexaenoic acid). Supplements of vitamin E, which is largely transported by LDL-C, were given in anticipation of LDL-C reductions. The stabilization target for other LLTs (including lipoprotein apheresis or plasmapheresis if applicable) was the maximum tolerated dose (MTD). The run-in phase started with a screening visit between ȡ12 weeks and −6 weeks. Adherence to the diet stabilization target was assessed by reviewing patients' self-completed dietary records (supplied by the sponsor along with detailed instructions) at Week −2 ± 1 and at all subsequent study visits.
During the efficacy phase, oral lomitapide was initiated at 5 mg/day and escalated to each patient's MTD (up to a maximum of 60 mg/day). This dose is supported by previously reported pharmacokinetic results in Japanese patients.24). MTD was defined as the highest dose during Weeks 0–26 that did not result in unacceptable adverse events (AEs), including elevated liver function tests (LFTs). Patients attended study visits at Weeks 0, 2, 6, 10, 14, 18, 22, and 26. The last permitted dose increase was at Week 22.
In the safety phase, lomitapide was continued at the patient's MTD for an additional 30 weeks. Lomitapide dose could be decreased if dose modification rules were applied, and subsequent re-escalation was allowed up to the MTD that was determined during the efficacy phase. Adjustments to background LLTs were also allowed at the investigator's discretion. In addition to the study visits during the efficacy phase, patients were assessed at Weeks 31, 36, 41, 46, 51, and 56.
The study was conducted in accordance with the International Council for Harmonisation (ICH) Guidance for Industry E6, Guideline for Good Clinical Practice, which is consistent with the ethical principles that have their origins in the Declaration of Helsinki. The protocol and patient informed consent form were reviewed and approved by an Institutional Review Board and/or Independent Ethics Committee covering each participating facility before the study began.
Patients
Patients were recruited from six centers in Japan. Japanese men and women ≥ 18 years of age were eligible if diagnosed with functional HoFH. Diagnosis had to be based on at least one of the following criteria: documented functional mutation(s) in two LDL receptor alleles or alleles known to affect LDL receptor functionality; skin fibroblast or lymphocyte LDL receptor activity < 20% normal; fasting LDL-C ≥ 300 mg/dL on maximally tolerated LLT, and both parents having documented untreated total cholesterol (TC) > 250 mg/dL; or untreated TC ≥ 500 mg/dL and triglycerides < 300 mg/dL, and both parents having documented untreated TC > 250 mg/dL.
Patients were excluded if they had uncontrolled hypertension, history of chronic renal insufficiency, or significant liver disease. Patients who required use of potentially hepatotoxic medications, especially those that could induce microvesicular or macrovesicular steatosis, were also excluded, as were patients who required use of strong or moderate CYP3A4 inhibitors or simvastatin > 10 mg/day, and patients who were unable to limit their alcohol consumption to no more than one alcoholic drink per day.
Outcomes
The primary efficacy endpoint was the mean percentage change from baseline to Week 26 in directly measured LDL-C at the MTD of lomitapide. Key secondary efficacy endpoints were the mean percentage change in other lipid parameters: TC; non-high-density lipoprotein cholesterol (non-HDL-C); VLDL cholesterol (VLDL-C); triglycerides; apolipoprotein B; lipoprotein(a); HDL-C; and apolipoprotein A1. TC, LDL-C, HDL-C, and triglycerides were measured enzymatically; non-HDL-C and VLDL-C were calculated. Lipoprotein(a) and apolipoprotein B were measured by immunonephelometry. Apolipoprotein A1 was measured by Latex Agglutination. All lipid parameters were assessed at each efficacy phase visit and at Weeks 36, 46, and 56 during the safety phase. Lipid panels were obtained following a fasting period (except for water and medications) of at least 12 hours and at a consistent time point immediately prior to lipoprotein apheresis, when relevant. The timing of blood draws relative to lipoprotein apheresis was critical because the procedure causes a sharp drop in LDL-C followed by a more gradual rebound, thus consistency ensured that measures were always made at the same point on the rebound curve. A further secondary endpoint was xanthoma frequency and severity (monitored at regular clinical visits).
Long-term safety and tolerability of lomitapide was evaluated by changes in laboratory parameters, electrocardiogram results, vital signs, physical examinations, and weight. Hepatic fat percentage was assessed at baseline, Week 26, and Week 56 using magnetic resonance imaging (MRI) scans, or computed tomography/ultrasound scans if MRI was contraindicated. Scans were processed by a central reader, VirtualScopics (Rochester, NY). Because the clinical relevance of change in hepatic fat is not clearly understood and there is minimal understanding of what constitutes an unhealthy level of hepatic fat, liver safety was primarily monitored by LFTs. AEs were classified according to their severity and their relationship to the study drug. LFTs were performed at all visits during both the efficacy and safety phases: tests were for alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and total bilirubin. Other laboratory assessments, physical examination, electrocardiogram, and vital sign measurements were performed at all visits during the efficacy phase and at Weeks 36, 46, and 56 during the safety phase. Blood and urine samples were centrally analyzed by PPD.® Central Labs at their locations in Singapore, Highland Heights (KY, USA), and Zaventem, Belgium.
Statistical Analysis
For all efficacy endpoints, baseline values were taken to be the mean of data from the run-in compliance (Week −2 ± 1) visit and the Week 0 visit. Differences between baseline and Week 26 were analyzed using a mixed-model repeated measures analysis of variance, with factors for dose group (final escalated dose at Week 26), baseline LDL-C, and week of lipid assessment. The variance–covariance structure was data driven and was assessed during the data analysis. The differences between baseline and Week 26 were also analyzed using paired t-tests. In anticipation of patients with missing data, an imputation method was planned for evaluating LDL-C at Week 26; here, last observation carried forward meant that the last non-missing result prior to Week 26 was to be used if data were missing at Week 26.
Results
Subjects
Nine patients were screened and enrolled in the run-in phase, all were subsequently entered into the efficacy phase. All patients had documented LDLR defects consistent with a diagnosis of HoFH: four LDLR true homozygotes, two LDLR compound heterozygotes, and three LDLR/PCSK-9 double heterozygotes. All patients were on concomitant LLT, including six on lipoprotein apheresis. Baseline demographic and concomitant LLT data are shown in Table 1. A total of eight patients completed the 0–26-week efficacy phase, one patient discontinued at 22 weeks owing to an AE of LFT (persistent elevations of AST and ALT). Eight patients continued lomitapide during the 26–56-week safety phase and all nine patients were included in the 56-week safety analysis.
Table 1. Baseline patient characteristics and concomitant lipid-lowering treatment/medications.
| Patient # | Age, years | Gender, M/F | BMI, kg/m2 | Hepatic fat, % | LDL-C, mg/dL* | Concomitant LLT/apheresis |
|---|---|---|---|---|---|---|
| 1 | 40 | F | 19.2 | 0.1 | 199 | Rosuvastatin, ezetimibe, lipoprotein apheresis |
| 2 | 46 | M | 30.6 | 1.5 | 183 | Rosuvastatin, ezetimibe, colestilan |
| 3 | 33 | M | 24.5 | 15.7 | 331 | Ethyl eicosapentaenoic acid, lipoprotein apheresis |
| 4 | 52 | F | 18.8 | 0.2 | 259 | Ezetimibe, ethyl eicosapentaenoic acid, lipoprotein apheresis |
| 5 | 43 | M | 19.6 | 0.6 | 200 | Atorvastatin, ezetimibe, lipoprotein apheresis |
| 6 | 35 | F | 18.5 | 3.4 | 221 | Atorvastatin, ezetimibe, probucol, lipoprotein apheresis |
| 7 | 75 | M | 23.8 | 4.5 | 121 | Atorvastatin |
| 8 | 63 | F | 19.0 | 0.3 | 134 | Atorvastatin, ezetimibe |
| 9 | 66 | M | 25.3 | 2.5 | 147 | Rosuvastatin, ezetimibe, colestilan, lipoprotein apheresis |
Baseline LDL-C is the mean of the run-in visit at -2 weeks and the baseline visit at Week 0.
BMI, body mass index; LDL-C, low-density lipoprotein cholesterol; LLT, lipid-lowering therapy
Dose Escalation and Efficacy
Most patients (5/9) progressed to an MTD of 20 mg lomitapide. Of the remaining three patients who completed the efficacy phase, the MTDs were 5 mg, 10 mg, and 40 mg lomitapide.
There was a highly significant reduction in LDL-C from baseline to Week 26 (Fig. 2, Table 2). Mean LDL-C reduced by 42% from 199 mg/dL (95% CI: 149–250) at baseline to 118 mg/dL (95% CI: 70–166) at Week 26. A decrease in mean LDL-C (19%) occurred as early as the first 2 weeks on treatment, with continued reduction occurring steadily over time thereafter (Fig. 3). The reduction in LDL-C was significant across patients with and without apheresis (Fig. 2). At Week 26, a ≥ 50% reduction in LDL-C from baseline was achieved by five out of nine patients which was maintained through to Week 56. During the efficacy phase, six of the nine patients achieved the target LDL-C level of < 100 mg/dL recommended for patients with HoFH; of these six patients, three achieved levels < 70 mg/dL by Week 26 and an additional patient achieved this by Week 56.
Fig. 2.
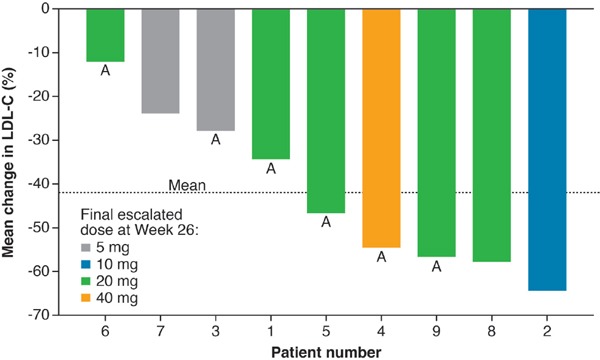
Waterfall plot of individual patient response and dose at primary endpoint (26 weeks)
A, patient receiving apheresis. LDL-C, low-density lipoprotein cholesterol
Table 2. Primary and secondary efficacy endpoints.
| Lipid parameters* | Baseline, mg/dL | Week 26,† mg/dL (n = 9) | Change from baseline, % | P-value | Week 56, mg/dL (n = 8) | Change from baseline, % | P-value |
|---|---|---|---|---|---|---|---|
| LDL-C (primary endpoint) | 199 (66) | 118 (62) | −42 (−56, −28) | 0.0001 | 115 (56) | −38 (−58, −17) | 0.003 |
| Total cholesterol | 279 (80) | 190 (66) | −32 (−42, −22) | < 0.0001 | 190 (62) | −(−41, −10) | 0.006 |
| Non-HDL-C | 228 (78) | 140 (71) | −40 (−53, −28) | < 0.0001 | 138 (63) | −35 (−53, −16) | 0.003 |
| VLDL-C | 25 (13) | 15 (10) | −42 (−52, −32) | < 0.0001 | 12 (5) | −45 (−55, −35) | < 0.0001 |
| Triglycerides‡ | 104 (61–273) | 57 (36–203) | −46 (−54, −21) | < 0.0001 | 54 (31–111) | −42 (−60, −24) | < 0.0001 |
| Apolipoprotein B | 148 (40) | 85 (45) | −45 (−59, −32) | < 0.0001 | 83 (42) | −41 (−60, −23) | 0.001 |
| Lipoprotein(a)§ | 59 (21–325) | 49 (21–179) | −19 (−45, 22) | NS | 40 (14–180) | −33 (−61, 9) | 0.014 |
| HDL-C | 50 (10) | 50 (12) | 1 (−15, 17) | NS | 53 (11) | 6 (−11, 22) | NS |
| Apolipoprotein A1 | 135 (12) | 127 (20) | −5 (−17, 7) | NS | 131 (18) | −3 (−13, 7) | NS |
Data are presented as mean (standard deviation) and mean percent change (Δ) from baseline (95% CI) except triglycerides and lipoprotein(a) that are presented as median (range) and median percent change (range)
Last observation carried forward; includes one patient who stopped lomitapide at Week 22
The mean (95% CI) percent changes from baseline in triglycerides for Weeks 26 and 56 were −42 (−51, −32) and −44 (−55, −34), respectively (both p < 0.001)
Lipoprotein(a) levels are reported as nmol/L. The mean (95% CI) percent changes from baseline in lipoprotein(a) for Weeks 26 and 56 were −14 (−31, 3) and −27 (−47, −8), respectively (both not significant).
CI, confidence internal; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; NS, not significant; SD, standard deviation; VLDL-C, very-low-density lipoprotein cholesterol
Fig. 3.
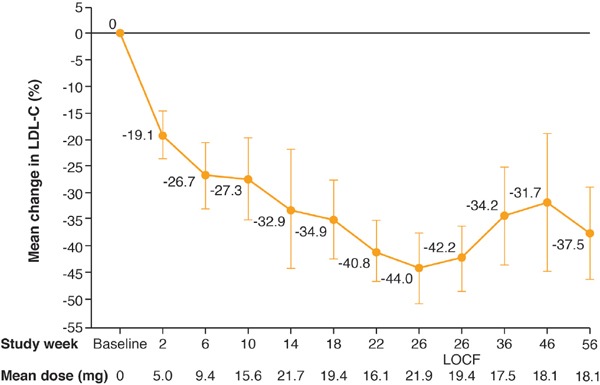
Percentage change in LDL-C during treatment (full analysis set)
LDL-C, low-density lipoprotein cholesterol; LOCF, last observation carried forward
During the safety phase from Week 26 through to Week 56, when patients were permitted to modify their background LLTs, including apheresis, there was a slight increase towards baseline in LDL-C levels between Weeks 26 and 36 with a plateau thereafter (Fig. 3). Note that the mean dose of lomitapide administered during the safety phase decreased slightly from 21.9 mg at Week 26 to 18.1 mg at Week 56. At the end of the safety phase, after 56 weeks of treatment, the mean percent change from baseline in LDL-C was clinically meaningful and statistically significant at 38% (p = 0.0032).
Three of the six patients (50%) receiving apheresis were able to increase the interval between apheresis treatments during the safety phase and maintain low LDL-C levels through to Week 56. Patient #1, who was on apheresis once per week at baseline, switched to a mostly once every 2 weeks schedule. Patient #4 increased the interval between apheresis treatments from 2 weeks at baseline to 3 weeks. Patient #5 had weekly apheresis at baseline but steadily increased the interval between treatments, reaching a monthly interval by the end of the safety phase.
Secondary endpoints included highly significant, clinically meaningful reductions in key lipid parameters: TC, non-HDL-C, VLDL-C, triglycerides, and apolipoprotein B (Table 2). The reduction in secondary lipid parameters at Week 26 was maintained through Week 56 (Table 2). At Week 26 there were smaller, non-significant, decreases in apolipoprotein A1 and lipoprotein(a), and there was no change in HDL-C (Table 2). By Week 56 the median reduction in lipoprotein(a) had improved to 27% (p = 0.0135).
Two patients showed improvement or resolution in xanthomas by Week 56. One patient (patient #2) with xanthomas on the knees, buttocks, Achilles tendon, heel, and toe at baseline was assessed by the investigator as having an improvement in xanthomas at Weeks 10, 22, and 46, and xanthomas on the knees and medial canthus were considered resolved for one patient (patient #5) at Week 56.
Dietary Compliance
During the efficacy phase, mean percentage energy from dietary fat was 19%, which was below the study requirement of < 20%. Most patients were compliant with the fat intake at most study visits. Mean percentage energy from fat over the entire efficacy phase was > 25% for only one patient (patient #3, 30%), who was noncompliant with the recommended < 20% fat at most assessments. Mean values of vitamin E, which was given as a dietary supplement, were above normal limits at baseline then decreased during treatment with lomitapide, remaining within normal limits. The dietary supplementation with fatty acid complex kept fatty acid levels within normal limits, although mean decreases from baseline were observed during treatment with lomitapide.
Safety and Tolerability
During the dose-escalation phase, all patients experienced at least one treatment-emergent AE (Table 3), which were ameliorated by dose reduction/interruption for all but one patient (patient #3), who discontinued lomitapide treatment at Week 22 as the result of persistent LFT elevations. During the safety phase, seven of the eight patients experienced at least one treatment-emergent AE (Table 3). Gastrointestinal symptoms were the most common AEs and were reported in eight of the nine patients. There was one serious AE during the safety phase (chest pain) which was considered unrelated to study treatment. There were no deaths during the study.
Table 3. Summary of treatment-emergent AEs.
| Dose of lomitapide at AE onset |
||||||
|---|---|---|---|---|---|---|
| Number of patients, n (%) | 5 mg | 10 mg | 20 mg | 40 mg | 60 mg | All patients |
| Efficacy phase | ||||||
| Patients who received the dose, n | 9 | 9 | 7 | 4 | 1 | 9 |
| Adverse events | 6 (66.7) | 6 (66.7) | 6 (85.7) | 4 (100.0) | 1 (100.0) | 9 (100.0) |
| Drug-related AEs | 5 (55.6) | 5 (55.6) | 5 (71.4) | 3 (75.0) | 1 (100.0) | 9 (100.0) |
| Severe AEs | 1 (11.1) | 1 (11.1) | 1 (14.3) | 1 (25.0) | 0 | 3 (33.3)* |
| Serious AEs | 0 | 0 | 0 | 0 | 0 | 0 |
| AEs leading to study discontinuation | 1 (11.1)† | 0 | 0 | 0 | 0 | 1 (11.1)† |
| Dose held or reduced due to an AE | 2 (22.2) | 1 (11.1) | 4 (57.1) | 3 (75.0) | 1 (100.0) | 8 (88.9) |
| Safety phase | ||||||
| Patients who received the dose, n | 2 | 2 | 5 | 1 | 0 | 8 |
| Adverse events | 2 (100.0) | 2 (100.0) | 5 (100.0) | 0 | 0 | 7 (87.5) |
| Drug-related AEs | 1 (50.0) | 1 (50.0) | 4 (80.0) | 0 | 0 | 6 (75.0) |
| Severe AEs | 0 | 0 | 0 | 0 | 0 | 0 |
| Serious AEs | 0 | 0 | 1 (20.0) | 0 | 0 | 1 (12.5) |
| AEs leading to study discontinuation | 0 | 0 | 0 | 0 | 0 | 0 |
| Dose held or reduced due to an AE | 1 (50.0) | 1 (50.0) | 1 (20.0) | 0 | 0 | 3 (37.5) |
Included abnormal liver function test (two patients) and diarrhea (one patient)
Elevated transaminases.
AE, adverse event
Liver Safety and Hepatic Fat
Treatment-emergent ALT and AST elevations to ≥ 3 × ULN were found in three of the nine patients. In two of these patients (patients #2 and #7), maximum ALT reached ≥ 3 × ULN while AST remained < 3 × ULN. In both cases, transaminase elevations were effectively managed by reduction in the lomitapide dose or temporary dose interruption with both patients completing the study through Week 56. In the third patient (patient #3), ALT levels reached ≥ 5 × ULN with AST ≥ 3 × ULN, and the patient discontinued lomitapide due to these LFT elevations. The patient had transaminases within normal ranges at Week 2, and the dose of lomitapide was increased to 10 mg. Subsequently, on Day 43, the patient developed elevated ALT (168 U/L: 4.2 × ULN) and AST (84 U/L: 2.0 × ULN). The AE was considered moderate and probably related to the study drug, thus the dose of lomitapide was reduced to 5 mg at Day 50; after elevated transaminases persisted, the dose of lomitapide was interrupted 7 days later. Maximal elevations in ALT (267 U/L: 6.7×ULN) and AST (128 U/L: 3×ULN) were noted at Week 9. Treatment with 5 mg lomitapide resumed at Week 18 (Day 127) when the patient's ALT level had improved to 92 U/L (2.3 × ULN) and their AST level had returned to within normal limits (40 U/L). The patient again experienced elevations in ALT, and treatment with lomitapide was discontinued at Week 22 (Day 155); the patient had subsequent peak elevations in ALT of 268 U/L (6.7 × ULN) and in AST of 130 U/L (3.0 × ULN). At the 26-week visit (Day 183), the patient's ALT and AST levels were 197 U/L (4.9×ULN) and 90 U/L (2.1 × ULN), respectively.
Bilirubin levels were within the normal range at all time points during the treatment period for all patients with ALT and/or AST elevations ≥ 3 × ULN; mean levels of total bilirubin and alkaline phosphatase decreased from baseline to Week 26/last observation carried forward, with mean changes of −2.3 µmol/L and −2.3 U/L, respectively. There were no reported cases of Hy's law. There were no clinically meaningful changes in hematology/coagulation parameters from baseline to Week 56.
Mean (range) percent hepatic fat, based on imaging data, increased from 3.2% (0.1–15.7%) at baseline to 15.6% (2.1–38.8%, last observation carried forward) at Week 26 (n = 9) and 12.7% (3.6–40.2%) at Week 56 (n = 8). In five patients, measures of hepatic fat remained ≤ 10% at Weeks 26 and 56. One patient (patient #6) had hepatic fat levels between 10% and 20% at both time points. The remaining three patients (patients #2, #3, and #7), all of whom had elevations in AST and/or ALT ≥ 3 × ULN, had hepatic fat levels of > 20%. In three patients who completed the study and discontinued treatment at study conclusion (patients #6, #7, and #8), hepatic fat assessments were conducted 4 weeks after discontinuation of lomitapide; all patients showed a rapid return toward baseline hepatic fat levels.
Discussion
Current LLTs for patients with HoFH do not enable all patients to reach target goals for LDL-C. The limited efficacy of statins and the transient nature of apheresis leave many patients at a high risk of developing atherosclerosis, with many developing coronary artery disease and/or aortic valve stenosis.19). Despite the development of various LLTs in recent decades, there remains a need for improved treatments for patients with HoFH. The current study demonstrated that adding lomitapide to existing LLTs (including apheresis) reduced LDL-C levels by 42% in patients with HoFH, which was similar to the result of the prior phase 3 study in non-Japanese patients.21). This reduction was sufficient to bring LDL-C levels to < 100 mg/dL for six of the nine patients, and to < 70 mg/dL for four of those six patients at least once during the study. Although it is not possible to predict the clinical consequences of these reductions with certainty, it has been reported that even sub-optimal reductions of circulating LDL-C reduces mortality in patients with HoFH.25). It should be noted that, currently, no morbidity or mortality outcome data exist for lomitapide.
This study showed that treatment with lomitapide enabled three of the six patients receiving apheresis at baseline to decrease the frequency of treatments and still maintain low LDL-C levels: this is comparable to the results of the prior phase 3 trial, in which six out of 13 patients successfully reduced or eliminated apheresis.26). In addition to LDL-C reductions, there were also substantial, highly significant, mean reductions in key secondary lipid parameters, including TC (32%), triglycerides (42%), non-HDL-C (40%), apolipoprotein B (45%), and VLDL-C (42%). These reductions in lipid parameters were maintained during the safety phase to 56 weeks. In addition, two patients had improvements in xanthoma over the 56 weeks of study, which was an encouraging result.
Because lomitapide acts in the liver, preventing the transport of lipids, hepatic AEs are directly associated with its mechanism of action; hepatic fat accumulation and LFT anomalies were considered carefully. Changes in transaminases and hepatic fat were variable across the nine patients. ALT levels ≥ 3 × ULN were seen in three patients; however, these were not associated with elevated bilirubin or alkaline phosphatase, and they resolved with dose reduction in two of the three cases. There were no indications for Hy's law, and bilirubin levels declined during treatment and did not go outside the normal limits. Mean hepatic fat percentage increased between baseline and Week 26, but it did not increase further between Weeks 26 and 56. In most cases (5/9) hepatic fat levels remained below 10%. Three patients had hepatic fat levels > 20%; these were the same patients who experienced treatment-emergent increases in ALT. In previous studies of lomitapide, the greatest elevations in transaminases and liver fat were observed in patients who reported consuming larger quantities of alcohol than were permitted in the protocol, which may have predisposed them to greater increases in these parameters.21, 27). In the current study, there was no report of protocol deviation with respect to alcohol and there were no other obvious factors that might have predisposed any patients to greater increases in hepatic fat. Consistent with results from a previous phase 2 study.27), measures of hepatic fat returned to baseline levels within 4 weeks after discontinuation of lomitapide in three patients who discontinued drug after completing the study. Although lomitapide-associated increases in hepatic fat appear to be stable and reversible, the long-term clinical implications remain unknown and warrant further investigation.
Overall, the efficacy and safety of 26 weeks of lomitapide in Japanese patients were consistent with results in the global phase 3 study and with studies of lomitapide in other patient popluations.17, 22–24). The results of the current study were also in line with the real-world results reported from a global observational registry prospectively assessing the long-term safety and effectiveness of lomitapide in real-life clinical practice (the LOWER study).28). There were some demographic differences between the current study and the prior global phase 3 study.21): mean baseline age was around 50 years in the present study versus around 30 years in the prior study; three of the nine patients in the present study had double heterozygous mutations (containing PCSK9 gain of function mutation) versus none of the 29 patients in the prior study; and baseline LDL-C was lower in the present study (199 mg/dL) versus the prior study (336 mg/dL).21). It is possible that differences in these baseline parameters outline different HoFH patient profiles in Japanese versus Caucasian populations. A recent study of four patients with HoFH found that differences in the MTP gene were associated with a strong or weak response to lomitapide.29).
There were no new safety signals and, similar to previous studies, gastrointestinal AEs were the most common AEs. In the current study, there were no further reports of gastrointestinal AEs following dose reductions or interruptions in five patients.
Conclusion
In conclusion, lomitapide, added to ongoing treatment with other LLTs, was effective in rapidly and significantly reducing the levels of LDL-C and other atherogenic apolipoprotein B-containing lipoproteins in adult Japanese patients with HoFH. Six patients achieved LDL-C < 100 mg/dL at least once over the course of the study, and half of the patients receiving apheresis were able to increase the interval between apheresis treatments and maintain low LDL-C levels.
Acknowledgments
The authors would like to thank all patients, their families, and the investigators who participated in this trial. Medical writing support for the development of this manuscript was provided by Esther Race of Choice Healthcare Solutions, and was funded by Aegerion Pharmaceuticals, Inc.
Conflicts of Interest
The authors report the following disclosures: M. Harada-Shiba has received honoraria from Astellas, Kaneka Medics, Kowa, MSD, and Pfizer, and clinical research funding from Kaneka Medics; A. Nohara has received clinical research funding from Aegerion Pharmaceuticals, Astellas Pharma, Keiai-Kai Medical, Kowa, MSD, Sanofi, Shionogi, and Synageva Biopharma; M. Yoshida has received honoraria from Daiichi Sankyo, Mitsubishi Tanabe Pharma, and MSD, and scholarship grants from: Astellas, AstraZeneca, Kowa, Nippon Boehringer Ingelheim, and Takeda Pharmaceutical; P. Foulds and Q. Chang own equity in, and are employees of, Aegerion Pharmaceuticals; K. Ikewaki, Y. Otsubo, and K. Yanagi have no disclosures to report.
References
- 1). Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen-Thiessen E, Tybjaerg-Hansen A, Watts GF, Averna M, Boileau C, Boren J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AF, Stroes E, Taskinen MR, Wiegman A, Wiklund O, Chapman MJ: Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J, 2014; 35: 2146-2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). Beigel R, Beigel Y: Homozygous familial hypercholesterolemia: long term clinical course and plasma exchange therapy for two individual patients and review of the literature. J Clin Apher, 2009; 24: 219-224 [DOI] [PubMed] [Google Scholar]
- 3). Hobbs HH, Brown MS, Goldstein JL: Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat, 1992; 1: 445-466 [DOI] [PubMed] [Google Scholar]
- 4). Kolansky DM, Cuchel M, Clark BJ, Paridon S, McCrindle BW, Wiegers SE, Araujo L, Vohra Y, Defesche JC, Wilson JM, Rader DJ: Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol, 2008; 102: 1438-1443 [DOI] [PubMed] [Google Scholar]
- 5). Gidding SS, Ann Champagne M, de Ferranti SD, Defesche J, Ito MK, Knowles JW, McCrindle B, Raal F, Rader D, Santos RD, Lopes-Virella M, Watts GF, Wierzbicki AS, on behalf of the American Heart Association Atherosclerosis, Hypertension, and Obesity in the Young Committee of the Council on Cardiovascular Disease in the Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and Council on Lifestyle and Cardiometabolic Health : The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association. Circulation, 2015; 132: 2167-2192 [DOI] [PubMed] [Google Scholar]
- 6). Teramoto T, Sasaki J, Ishibashi S, Birou S, Daida H, Dohi S, Egusa G, Hiro T, Hirobe K, Iida M, Kihara S, Kinoshita M, Maruyama C, Ohta T, Okamura T, Yamashita S, Yokode M, Yokote K, Harada-Shiba M, Arai H, Bujo H, Nohara A, Ohta T, Oikawa S, Okada T, Wakatsuki A: Familial hypercholesterolemia. J Atheroscler Thromb, 2014; 21: 6-10 [DOI] [PubMed] [Google Scholar]
- 7). Harada-Shiba M, Arai H, Oikawa S, Ohta T, Okada T, Okamura T, Nohara A, Bujo H, Yokote K, Wakatsuki A, Ishibashi S, Yamashita S: Guidelines for the management of familial hypercholesterolemia. J Atheroscler Thromb, 2012; 19: 1043-1060 [DOI] [PubMed] [Google Scholar]
- 8). Zhou M, Zhao D: Familial hypercholesterolemia in Asian populations. Journal of atherosclerosis and thrombosis, 2016; 23: 539-549 [DOI] [PubMed] [Google Scholar]
- 9). Watts GF, Ding PY, George P, Hagger MS, Hu M, Lin J, Khoo KL, Marais AD, Miida T, Nawawi HM, Pang J, Park JE, Gonzalez-Santos LB, Su TC, Truong TH, Santos RD, Soran H, Yamashita S, Tomlinson B: Translational research for improving the care of familial hypercholesterolemia: the “ten countries study” and beyond. J Atheroscler Thromb, 2016; 23: 891-900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10). Pang J, Lansberg PJ, Watts GF: International developments in the care of familial hypercholesterolemia: where now and where to next. J Atheroscler Thromb, 2016; 23: 505-519 [DOI] [PubMed] [Google Scholar]
- 11). Hegele RA: Monogenic dyslipidemias: window on determinants of plasma lipoprotein metabolism. Am J Hum Genet, 2001; 69: 1161-1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Marais AD: Familial hypercholesterolaemia. Clin Biochem Rev, 2004; 25: 49-68 [PMC free article] [PubMed] [Google Scholar]
- 13). Harada-Shiba M, Takagi A, Miyamoto Y, Tsushima M, Ikeda Y, Yokoyama S, Yamamoto A: Clinical features and genetic analysis of autosomal recessive hypercholesterolemia. J Clin Endocrinol Metab, 2003; 88: 2541-2547 [DOI] [PubMed] [Google Scholar]
- 14). Gagne C, Gaudet D, Bruckert E, Ezetimibe Study Group : Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. Circulation, 2002; 105: 2469-2475 [DOI] [PubMed] [Google Scholar]
- 15). Yamamoto A, Harada-Shiba M, Endo M, Kusakabe N, Tanioka T, Kato H, Shoji T: The effect of ezetimibe on serum lipids and lipoproteins in patients with homozygous familial hypercholesterolemia undergoing LDL-apheresis therapy. Atherosclerosis, 2006; 186: 126-131 [DOI] [PubMed] [Google Scholar]
- 16). Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, Wasserman SM, Stein EA, TESLA Investigators : Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet, 2015; 385: 341-350 [DOI] [PubMed] [Google Scholar]
- 17). Thompson GR: LDL apheresis. Atherosclerosis, 2003; 167: 1-13 [DOI] [PubMed] [Google Scholar]
- 18). Vella A, Pineda AA, O'Brien T: Low-density lipoprotein apheresis for the treatment of refractory hyperlipidemia. Mayo Clin Proc, 2001; 76: 1039-1046 [DOI] [PubMed] [Google Scholar]
- 19). Makino H, Harada-Shiba M: Long-term effect of low-density lipoprotein apheresis in patients with homozygous familial hypercholesterolemia. Ther Apher Dial, 2003; 7: 397-401 [DOI] [PubMed] [Google Scholar]
- 20). Wetterau JR, Lin MC, Jamil H: Microsomal triglyceride transfer protein. Biochim Biophys Acta, 1997; 1345: 136-150 [DOI] [PubMed] [Google Scholar]
- 21). Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Du Plessis AM, Propert KJ, Sasiela WJ, Bloedon LT, Rader DJ: Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet, 2013; 381: 40-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22). LOJUXTA: (lomitapide) summary of product characteristics Aegerion Pharmaceuticals Ltd, 2015; April [Google Scholar]
- 23). JUXTAPID: (lomitapide) prescribing information Aegerion Pharmaceuticals, Cambridge, MA, 2014; August [Google Scholar]
- 24). Taubel J, Sumeray M, Lorch U, McLean A: Pharmacokinetics and pharmacodynamics of lomitapide in Japanese subjects. J Atheroscler Thromb, 2016; 23: 606-620 [DOI] [PubMed] [Google Scholar]
- 25). Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ, Marais AD: Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation, 2011; 124: 2202-2207 [DOI] [PubMed] [Google Scholar]
- 26). Stefanutti C, Blom DJ, Averna MR, Meagher EA, Theron H, Marais AD, Hegele RA, Sirtori CR, Shah PK, Gaudet D, Vigna GB, Sachais BS, Di Giacomo S, du Plessis AM, Bloedon LT, Balser J, Rader DJ, Cuchel M, for Phase 3 HoFH Lomitapide Study Investigators : The lipid-lowering effects of lomitapide are unaffected by adjunctive apheresis in patients with homozygous familial hypercholesterolaemia - a post-hoc analysis of a Phase 3, single-arm, open-label trial. Atherosclerosis, 2015; 240: 408-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, Millar JS, Ikewaki K, Siegelman ES, Gregg RE, Rader DJ: Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. The N Engl J Med, 2007; 356: 148-156 [DOI] [PubMed] [Google Scholar]
- 28). Blom DJ, Kastelein JJ, Larrey D, Makris L, Phillips H, Bloeden L, Underberg J: Lomitapide Observational Worldwide Evaluation Registry (LOWER): one-year data. American Heart Association Scientific Sessions, 2015; Abstract 2075 [Google Scholar]
- 29). Kolovou GD, Kolovou V, Papadopoulou A, Watts GF: MTP gene variants and response to lomitapide in patients with homozygous familial hypercholesterolemia. J Atheroscler Thromb, 2016; 23: 878-883 [DOI] [PMC free article] [PubMed] [Google Scholar]