

Abstract
Oxygen‐dependent regulation of the erythropoietin gene is mediated by the hypoxia‐inducible factor (HIF) family of transcription factors. When oxygen is plentiful, HIF undergoes hydroxylation by a family of oxygen‐dependent prolyl hydroxylase domain (PHD) proteins, promoting its association with the von Hippel‐Lindau (VHL) ubiquitin E3 ligase and subsequent proteosomal degradation. When oxygen is scarce, the PHD enzymes are inactivated, leading to HIF accumulation and upregulation not only of erythropoietin expression, but also the expression of hundreds of other genes, including those coordinating cardiovascular and ventilatory adaptation to hypoxia. Nevertheless, despite the identification of over 50 mutations in the PHD‐HIF‐VHL pathway in patients with previously unexplained congenital erythrocytosis, there are very few reports of associated cardiopulmonary abnormalities. We now report exaggerated pulmonary vascular and ventilatory responses to acute hypoxia in a 35‐year‐old man with erythrocytosis secondary to heterozygous mutation in PHD2, the most abundant of the PHD isoforms. We compare this phenotype with that reported in patients with the archetypal disorder of cellular oxygen sensing, Chuvash polycythemia, and discuss the possible clinical implications of our findings, particularly in the light of the emerging role for small molecule PHD inhibitors in clinical practice.
Keywords: Hypoxia, hypoxia‐inducible factor, prolyl hydroxylase domain protein, pulmonary circulation, ventilation
Introduction
Oxygen‐dependent gene expression is exemplified by erythropoietin (EPO) production, which is mediated by the hypoxia‐inducible factor (HIF) family of transcription factors (Wang and Semenza 1993; Wang et al. 1995). The HIF transcriptional complex is a heterodimer comprising α and β subunits, the former of which is regulated by oxygen availability mainly through the action of a family of 2‐oxoglutarate‐dependent prolyl hydroxylase domain (PHD) proteins (Epstein et al. 2001). When oxygen is plentiful, hydroxylation of two specific proline residues in the HIFα protein promotes its association with the von Hippel‐Lindau (VHL) ubiquitin E3 ligase, leading to its degradation in the ubiquitin‐proteosomal pathway. When oxygen is scarce, the PHD enzymes are inactivated, leading to HIFα accumulation and formation of the transcriptional complex through binding to nuclear HIFβ (Ivan et al. 2001; Jaakkola et al. 2001). Human HIFα exists as three isoforms (HIF1α, 2α and 3α), which regulate overlapping sets of genes (Schodel et al. 2011). Similarly, three PHD isoforms have been described (PHD1, 2 and 3), of which PHD2 is the most abundant (Appelhoff et al. 2004).
Since the elucidation of the PHD‐HIF‐VHL pathway, mutations in its constituent proteins have been identified in previously unexplained cases of congenital erythrocytosis. Chuvash polycythemia, for example, is endemic in the Upper Volga region of Russia, with sporadic cases reported elsewhere, and is characterized by severe erythrocytosis. It is now known to be due to a homozygous loss‐of‐function mutation (598C>T) in the VHL gene, leading to inappropriate HIFα stabilization, activation of HIF‐mediated gene transcription and EPO upregulation (Ang et al. 2002). Importantly, this condition highlights the importance of HIF signaling not only for EPO regulation, but also in the regulation of systemic oxygen delivery more broadly. Patients with Chuvash polycythemia display elevated minute ventilation and pulmonary artery pressures at baseline, and profoundly enhanced pulmonary vascular, ventilatory and cardiac responses to acute hypoxia (Bushuev et al. 2006; Smith et al. 2006, 2008b), a phenotype reminiscent of that seen in lowlanders acclimatized to the hypoxia of high altitude.
To date, over 50 oxygen sensing mutations have been identified in patients with congenital erythrocytosis (Bento et al. 2014; Camps et al. 2016; McMullin 2016). The majority are loss‐of‐function mutations in VHL or EGLN1 (PHD2), but gain‐of‐function mutations in the EPAS1 (HIF2α) gene have also been described. Cardiopulmonary abnormalities have been reported in Chuvash polycythemia, in two infants with distinct VHL mutations (Bond et al. 2011; Sarangi et al. 2014), and in a small number of patients with HIF2α mutations (Gale et al. 2008; Formenti et al. 2011). Abnormal cardiopulmonary physiology associated with PHD2 deficiency has also been demonstrated in mice (Bishop et al. 2013; Dai et al. 2016; Hodson et al. 2016; Kapitsinou et al. 2016), but to the best of our knowledge there are no reports of an analogous cardiopulmonary phenotype in PHD‐deficient humans. We now report exaggerated ventilatory and pulmonary vascular responses to hypoxia in a patient with heterozygous PHD2 mutation.
Case Report
We assessed the cardiopulmonary phenotype of a 35‐year‐old man who had been diagnosed with polycythemia 6 years earlier, with peak hemoglobin and hematocrit of 191 g/L and 0.54, respectively. As reported previously (Percy et al. 2006), serum EPO was normal despite the elevated hemoglobin, suggesting dysregulation of erythropoietin secretion. Following extensive investigation, this was attributed to heterozygosity for a 950C>G mutation in the PHD2 gene, corresponding to a proline‐arginine substitution. In vitro, the mutated PHD protein was less effective at binding and hydroxylating HIF1α and HIF2α, and in cell culture overexpression of mutant PHD2 was less effective at suppressing HIF reporter gene activity, compared with wild type protein (Percy et al. 2006).
The patient was recruited through his consultant physician, and provided written, informed consent for the study, which was approved by the Oxfordshire Clinical Research Ethics Committee. After baseline measurements including spirometry, serum iron indices and arterial blood gas sampling (Table 1), the patient was exposed to a 10‐min period of mild eucapnic hypoxia (end‐tidal PO2 70 mmHg), followed by a 10‐min period of moderate eucapnic hypoxia (end‐tidal PO2 50 mmHg), using a purpose‐built end‐tidal forcing gas control system (Robbins et al. 1982). During hypoxia, minute ventilation was measured continuously, and systolic pulmonary artery pressure (SPAP) and cardiac output were estimated beat‐by‐beat using non‐invasive Doppler echocardiography. This methodology has previously been used to assess a group of three patients with Chuvash polycythemia, plus six healthy control subjects. These results have been reported elsewhere (Smith et al. 2006), but are reproduced in Figure 1 and Table 1, for comparison with the current case.
Table 1.
Physical characteristics and blood indices in a patient with erythrocytosis secondary to PHD2 mutation, compared with values from previously published studies on patients with Chuvash polycythemia and healthy control participants (Smith et al. 2006)
| Healthy control participants Mean ± SD (n = 6) | Chuvash polycythemia Mean ± SD (n = 3) | PHD2‐deficient patient (n = 1) | |
|---|---|---|---|
| Physical characteristics | |||
| Age (year) | 24 ± 5 | 22 ± 5 | 35 |
| Height (cm) | 173 ± 10 | 174 ± 5 | 175 |
| Weight (kg) | 73 ± 11 | 59 ± 10 | 80 |
| Body mass index (kg/m2) | 24 ± 2 | 20 ± 5 | 26 |
| Full blood count | |||
| Hemoglobin (g/L) | 137 ± 17 | 141 ± 21 | 178 |
| Hematocrit | 0.42 ± 0.05 | 0.48 ± 0.08 | 0.52 |
| Mean cell volume (fL) | 89 ± 3 | 64 ± 6a | 85 |
| Serum iron studies | |||
| Iron (μmol/L) | 17 ± 4 | 4 ± 1a | 16 |
| Ferritin (ng/ml) | 49 ± 37 | 2 ± 1 | 193 |
| Transferrin (g/L) | 2.6 ± 0.5 | 3.8 ± 0.3a | 2.2 |
| Transferrin saturation (%) | 30 ± 9 | 4 ± 1 | 32 |
| Arterial blood gases | |||
| PO2 (mmHg) | 99 ± 11 | 102 ± 3 | 107 |
| PCO2 (mmHg) | 40 ± 4 | 34 ± 2a | 36 |
| pH (mmHg) | 7.38 ± 0.02 | 7.41 ± 0.02a | 7.45 |
Significant difference compared with control participants (P < 0.05, Student's t‐test). Mean ± SD.
Figure 1.
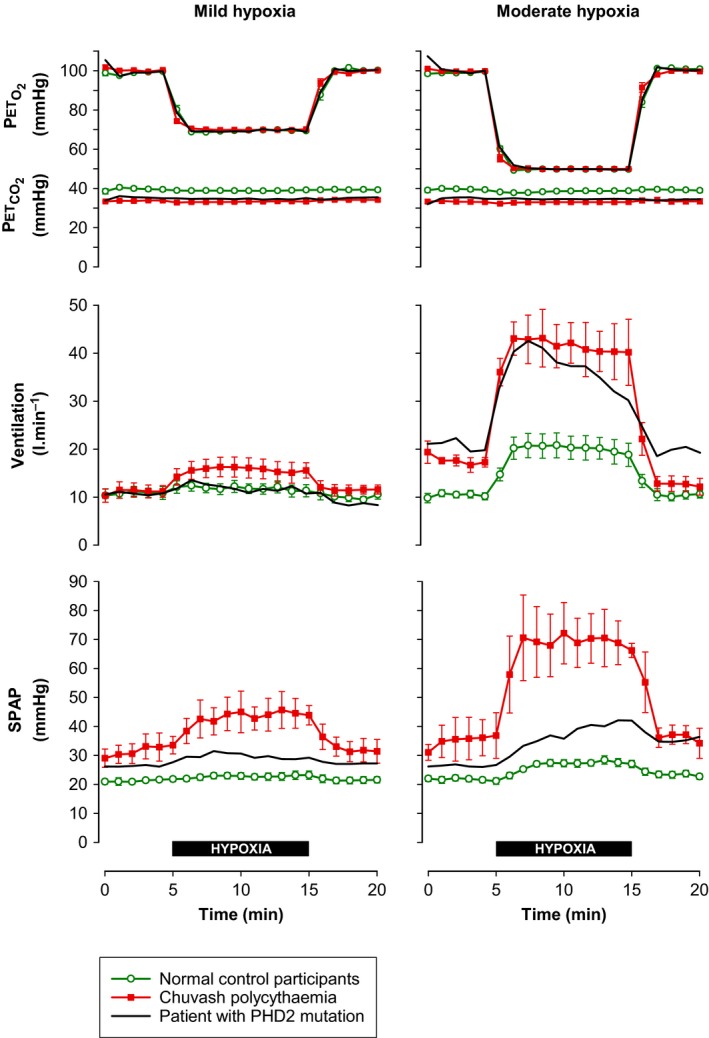
Changes in minute ventilation and systolic pulmonary artery pressure (SPAP) during 10 min of mild and moderate hypoxia in a patient with PHD2 mutation (current report), compared with previously published (Smith et al. 2006) responses in normal control participants (n = 6) and patients with Chuvash polycythemia (n = 3). End‐tidal partial pressures of oxygen (PETO2) and carbon dioxide (PETCO2) were controlled using dynamic end‐tidal forcing. Symbols show mean ± SEM.
Spirometry revealed slightly reduced forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) in the PHD2‐deficient patient, compared with age, gender and height adjusted normal values (FEV 74% and FVC 78% predicted, respectively). Similar results were seen previously in patients with Chuvash polycythemia (Smith et al. 2008b). As shown in Table 1, hemoglobin and hematocrit were elevated but mean corpuscular volume and iron studies were normal. Arterial blood gas analysis revealed a PCO2 of 36.3 mmHg with borderline alkalosis (pH 7.45), compatible with a mild elevation of basal pulmonary ventilation, relative to metabolism.
Figure 1 shows the ventilatory and pulmonary vascular responses to hypoxia in the PHD2‐deficient patient, compared with previously reported responses in patients with Chuvash polycythemia and in normal volunteers (Smith et al. 2006). With regard to minute ventilation, the patient had a normal response to mild hypoxia, but a substantially enhanced ventilatory response to moderate hypoxia, similar to that seen in the Chuvash patients. With regard to pulmonary hemodynamics, the patient had mildly elevated baseline SPAP, intermediate between the normal and Chuvash groups. During hypoxia, the magnitude of the rise in SPAP in the PHD2‐deficient patient again appeared to be intermediate between the normal response and the exaggerated response seen in Chuvash patients.
Discussion
From a cardiopulmonary perspective, the best‐characterized oxygen sensing mutations are those associated with Chuvash polycythemia, in whom we have previously described elevated basal ventilation and pulmonary artery pressure, and dramatically enhanced ventilatory, pulmonary vascular and cardiac sensitivity to acute hypoxia (Smith et al. 2006, 2008b). We have also described a small number of patients with HIF2α gain‐of‐function mutations, who also have elevated basal ventilation and pulmonary artery pressure, a moderately enhanced pulmonary vascular sensitivity to hypoxia, but a normal ventilatory sensitivity to hypoxia (Formenti et al. 2011). Using similar methodology, we now describe an intermediate phenotype associated with PHD2 mutation, with mildly elevated basal ventilation and pulmonary artery pressure, a marked increase in the ventilatory sensitivity to hypoxia, but only a relatively modest increase in the pulmonary vascular sensitivity to hypoxia.
We cannot exclude some contribution from the moderately raised blood viscosity to the elevation of pulmonary artery pressure in this case. However, pulmonary hypertension is reported in PHD2‐deficient mice in the absence of polycythemia (Dai et al. 2016; Kapitsinou et al. 2016), and in a previous patient with elevated pulmonary artery pressure and erythrocytosis secondary to HIF2α mutation, therapeutic venesection was not associated with any change in pulmonary artery pressure or the magnitude of the pulmonary vascular response to hypoxia (Formenti et al. 2011).
Several factors could contribute to the apparently milder pulmonary vascular phenotype associated with PHD2 mutation, compared with the Chuvash phenotype. First, PHD2 is reported in cell culture to be of greater importance in the regulation of HIF1α, compared with HIF2α (Appelhoff et al. 2004), while HIF2α appears to be primarily responsible for the pulmonary hypertension associated with Chuvash polycythemia, at least in mice (Hickey et al. 2010). However, despite its apparent preference for HIF1α in cell culture, PHD2 appears nonetheless to be an important regulator of HIF2α in vivo. Conditional PHD2 inactivation was recently shown in mice to result in pulmonary hypertension and enhanced ventilatory sensitivity to hypoxia (Hodson et al. 2016; Kapitsinou et al. 2016). The effects of PHD2 inactivation were dependent upon the presence of HIF2α, but were largely unaffected by concomitant loss of HIF1α.
Second, it has also been reported in mouse models of Chuvash polycythemia that abnormal erythrocytosis results not only from HIF upregulation, but also from VHL‐dependent, HIF‐independent activation of the JAK‐STAT signaling pathway, leading to enhanced sensitivity of erythroid precursor cells to erythropoietin (Russell et al. 2011). If this pathway also contributes to the pulmonary vascular phenotype in human Chuvash polycythemia, it may not be recapitulated in patients with PHD2 mutations.
Third, a notable difference between the previously reported Chuvash patients and the current PHD2‐deficient patient is the presence of severe iron deficiency in the former group. This is likely to reflect the greater severity of erythropoietin dysregulation in patients with Chuvash polycythemia, in whom frequent venesection is required, with consequent iron deficiency. In contrast, the current PHD2‐deficient patient required less frequent venesection, and remained iron replete. Iron deficiency may mimic hypoxia at a cellular level by limiting the intracellular availability of iron, which is required as a co‐factor for the catalytic activity of the PHD proteins. Accordingly, it has recently been reported that iron deficiency in otherwise healthy individuals is associated with enhanced pulmonary vascular responses to hypoxia (Frise et al. 2016) and it has been suggested before that iron deficiency may contribute to the Chuvash pulmonary vascular phenotype (Sable et al. 2012). In this context, Figure 2 shows the striking similarity between the pulmonary vascular response to hypoxia in the current PHD2‐deficient patient and the corresponding response in healthy individuals exposed either to 8 h of hypoxia, or to an 8‐h infusion of the iron chelator desferrioxamine (Smith et al. 2008a).
Figure 2.
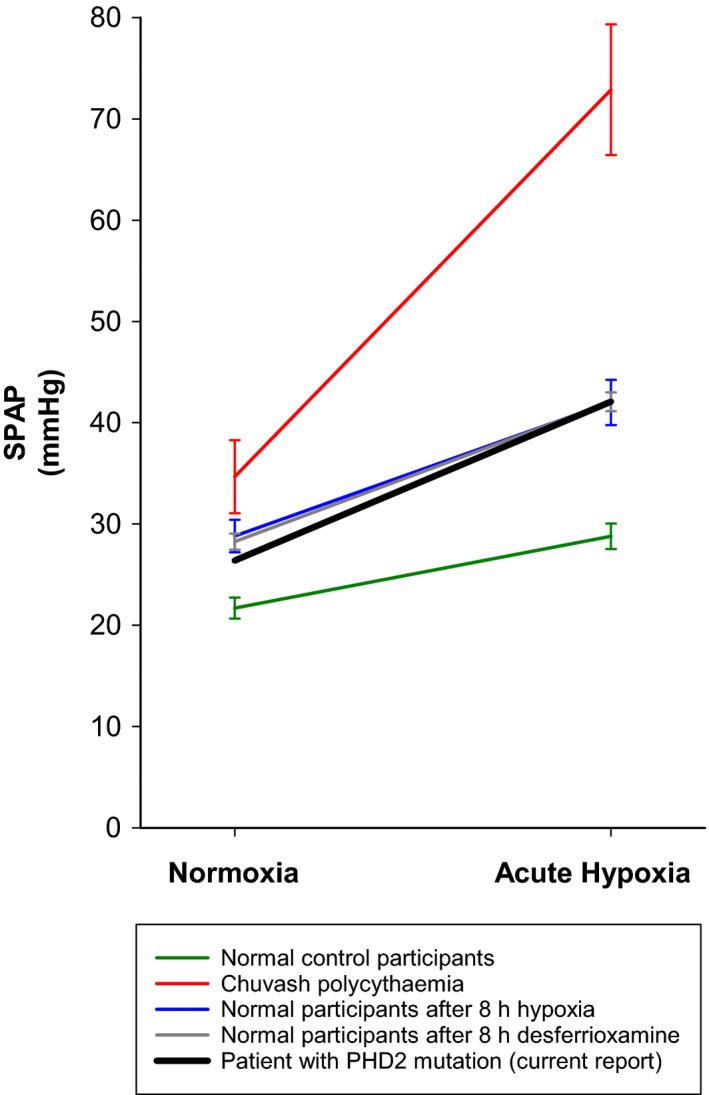
Systolic pulmonary artery pressure (SPAP) breathing room air (Normoxia) and after 10 min of hypoxia (Acute Hypoxia; end tidal PO2 50 mmHg) in a patient with PHD2 mutation, compared with previously published (Smith et al. 2006, 2008a) responses in normal control participants (n = 8), patients with Chuvash polycythemia (n = 3), healthy volunteers immediately after prolonged hypoxia (end tidal PO2 55 mmHg for 8 h; n = 8), and healthy volunteers immediately after infusion of the iron chelator desferrioxamine mesylate (4 g per 70 kg over 8 h; n = 8). Symbols show mean ± SEM.
Finally, our results may have important clinical implications. In combination with previous reports of pulmonary hypertension in patients with VHL and HIF2 mutations (Bushuev et al. 2006; Smith et al. 2006; Gale et al. 2008; Bond et al. 2011; Formenti et al. 2011; Sarangi et al. 2014), our findings support the idea that patients presenting with erythrocytosis secondary to oxygen sensing mutations should be monitored for cardiopulmonary abnormalities (Bento et al. 2014; McMullin 2016). This might be particularly important for those in whom venesection is being considered, or in patients likely to be exposed to hypoxia, such as through ascent to high altitude or during air travel. In Chuvash polycythemia, for example, flight‐induced pulmonary hypertension can develop even when baseline pulmonary artery pressures are not elevated (Smith et al. 2013; Turner et al. 2015). Our results are also significant in the light of the emerging role for small molecule PHD inhibitors in clinical practice, for example to upregulate EPO production in the treatment of patients with renal anemia (Brigandi et al. 2016; Provenzano et al. 2016). Early phase clinical trials investigating these inhibitors have now been completed. Our results raise the possibility that these agents may produce significant alterations in cardiorespiratory control that have not yet been widely considered.
Conflict of Interest
PHM is a founder, shareholder and director of ReOx Ltd.
Acknowledgments
We are grateful to the patient for his participation.
Talbot N. P. , Smith T. G. , Balanos G. M. , Dorrington K. L. , Maxwell P. H. , Robbins P. A.. Cardiopulmonary phenotype associated with human PHD2 mutation. Physiol Rep, 5 (7), 2017, e13224, doi: 10.14814/phy2.13224
Funding Information
No funding information provided.
References
- Ang, S. O. , Chen H., Hirota K., Gordeuk V. R., Jelinek J., Guan Y., et al. 2002. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 32:614–621. [DOI] [PubMed] [Google Scholar]
- Appelhoff, R. J. , Tian Y. M., Raval R. R., Turley H., Harris A. L., Pugh C. W., et al. 2004. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia‐inducible factor. J. Biol. Chem. 279:38458–38465. [DOI] [PubMed] [Google Scholar]
- Bento, C. , Percy M. J., Gardie B., Maia T. M., van Wijk R., Perrotta S., et al. 2014. Genetic basis of congenital erythrocytosis: mutation update and online databases. Hum. Mutat. 35:15–26. [DOI] [PubMed] [Google Scholar]
- Bishop, T. , Talbot N. P., Turner P. J., Nicholls L. G., Pascual A., Hodson E. J., et al. 2013. Carotid body hyperplasia and enhanced ventilatory responses to hypoxia in mice with heterozygous deficiency of PHD2. J. Physiol. 591:3565–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond, J. , Gale D. P., Connor T., Adams S., de Boer J., Gascoyne D. M., et al. 2011. Dysregulation of the HIF pathway due to VHL mutation causing severe erythrocytosis and pulmonary arterial hypertension. Blood 117:3699–3701. [DOI] [PubMed] [Google Scholar]
- Brigandi, R. A. , Johnson B., Oei C., Westerman M., Olbina G., De Zoysa. J., et al. 2016. A novel hypoxia‐inducible factor‐prolyl hydroxylase inhibitor (GSK1278863) for Anemia in CKD: a 28‐day, phase 2A randomized trial. Am. J. Kidney Dis. 67:861–871. [DOI] [PubMed] [Google Scholar]
- Bushuev, V. I. , Miasnikova G. Y., Sergueeva A. I., Polyakova L. A., Okhotin D., Gaskin P. R., et al. 2006. Endothelin‐1, vascular endothelial growth factor and systolic pulmonary artery pressure in patients with Chuvash polycythemia. Haematologica 91:744–749. [PubMed] [Google Scholar]
- Camps, C. , Petousi N., Bento C., Cario H., Copley R. R., McMullin M. F., et al. 2016. Gene panel sequencing improves the diagnostic work‐up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica 101:1306–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Z. , Li M., Wharton J., Zhu M. M., and Zhao Y. Y.. 2016. Prolyl‐4 Hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through Hypoxia‐Inducible Factor‐2alpha. Circulation 133:2447–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, A. C. , Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., et al. 2001. C. elegans EGL‐9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107:43–54. [DOI] [PubMed] [Google Scholar]
- Formenti, F. , Beer P. A., Croft Q. P., Dorrington K. L., Gale D. P., Lappin T. R., et al. 2011. Cardiopulmonary function in two human disorders of the hypoxia‐inducible factor (HIF) pathway: von Hippel‐Lindau disease and HIF‐2alpha gain‐of‐function mutation. FASEB J. 25:2001–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frise, M. C. , Cheng H. Y., Nickol A. H., Curtis M. K., Pollard K. A., Roberts D. J., et al. 2016. Clinical iron deficiency disturbs normal human responses to hypoxia. J. Clin. Invest. 126:2139–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale, D. P. , Harten S. K., Reid C. D., Tuddenham E. G., and Maxwell P. H.. 2008. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 112:919–921. [DOI] [PubMed] [Google Scholar]
- Hickey, M. M. , Richardson T., Wang T., Mosqueira M., Arguiri E., Yu H., et al. 2010. The von hippel‐lindau chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J. Clin. Invest. 120:827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson, E. J. , Nicholls L. G., Turner P. J., Llyr R., Fielding J. W., Douglas G., et al. 2016. Regulation of ventilatory sensitivity and carotid body proliferation in hypoxia by the PHD2/HIF‐2 pathway. J. Physiol. 594:1179–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan, M. , Kondo K., Yang H., Kim W., Valiando J., Ohh M., et al. 2001. HIFalpha targeted for VHL‐mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468. [DOI] [PubMed] [Google Scholar]
- Jaakkola, P. , Mole D. R., Tian Y. M., Wilson M. I., Gielbert J., Gaskell S. J., et al. 2001. Targeting of HIF‐alpha to the von Hippel‐Lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science 292:468–472. [DOI] [PubMed] [Google Scholar]
- Kapitsinou, P. P. , Rajendran G., Astleford L., Michael M., Schonfeld M. P., Fields T., et al. 2016. The endothelial Prolyl‐4‐Hydroxylase Domain 2/Hypoxia‐Inducible Factor 2 axis regulates pulmonary artery pressure in mice. Mol. Cell. Biol. 36:1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullin, M. F. 2016. Congenital erythrocytosis. Int. J. Lab. Hematol. 38(Suppl 1):59–65. [DOI] [PubMed] [Google Scholar]
- Percy, M. J. , Zhao Q., Flores A., Harrison C., Lappin T. R., Maxwell P. H., et al. 2006. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc. Natl Acad. Sci. USA 103:654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano, R. , Besarab A., Wright S., Dua S., Zeig S., Nguyen P., et al. 2016. Roxadustat (FG‐4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: a phase 2, randomized, 6‐ to 19‐week, open‐label, active‐comparator, dose‐ranging, safety and exploratory efficacy study. Am. J. Kidney Dis. 67:912–924. [DOI] [PubMed] [Google Scholar]
- Robbins, P. A. , Swanson G. D., and Howson M. G.. 1982. A prediction‐correction scheme for forcing alveolar gases along certain time courses. J. Appl. Physiol. 52:1353–1357. [DOI] [PubMed] [Google Scholar]
- Russell, R. C. , Sufan R. I., Zhou B., Heir P., Bunda S., Sybingco S. S., et al. 2011. Loss of JAK2 regulation via a heterodimeric VHL‐SOCS1 E3 ubiquitin ligase underlies Chuvash polycythemia. Nat. Med. 17:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sable, C. A. , Aliyu Z. Y., Dham N., Nouraie M., Sachdev V., Sidenko S., et al. 2012. Pulmonary artery pressure and iron deficiency in patients with upregulation of hypoxia sensing due to homozygous VHL(R200W) mutation (Chuvash polycythemia). Haematologica 97:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarangi, S. , Lanikova L., Kapralova K., Acharya S., Swierczek S., Lipton J. M., et al. 2014. The homozygous VHL(D126N) missense mutation is associated with dramatically elevated erythropoietin levels, consequent polycythemia, and early onset severe pulmonary hypertension. Pediatr. Blood Cancer 61:2104–2106. [DOI] [PubMed] [Google Scholar]
- Schodel, J. , Oikonomopoulos S., Ragoussis J., Pugh C. W., Ratcliffe P. J., and Mole D. R.. 2011. High‐resolution genome‐wide mapping of HIF‐binding sites by ChIP‐seq. Blood 117:e207–e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T. G. , Brooks J. T., Balanos G. M., Lappin T. R., Layton D. M., Leedham D. L., et al. 2006. Mutation of von Hippel‐Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 3:e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T. G. , Balanos G. M., Croft Q. P., Talbot N. P., Dorrington K. L., Ratcliffe P. J., et al. 2008a. The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J. Physiol. 586:5999–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T. G. , Brooks J. T., Balanos G. M., Lappin T. R., Layton D. M., Leedham D. L., et al. 2008b. Mutation of the von Hippel‐Lindau gene alters human cardiopulmonary physiology. Adv. Exp. Med. Biol. 605:51–56. [DOI] [PubMed] [Google Scholar]
- Smith, T. G. , Chang R. W., Robbins P. A., and Dorrington K. L.. 2013. Commercial air travel and in‐flight pulmonary hypertension. Aviat. Space Environ. Med. 84:65–67. [DOI] [PubMed] [Google Scholar]
- Turner, B. E. , Hodkinson P. D., Timperley A. C., and Smith T. G.. 2015. Pulmonary artery pressure response to simulated air travel in a hypobaric chamber. Aerosp. Med. Hum. Perform. 86:529–534. [DOI] [PubMed] [Google Scholar]
- Wang, G. L. , and Semenza G. L.. 1993. Desferrioxamine induces erythropoietin gene expression and hypoxia‐inducible factor 1 DNA‐binding activity: implications for models of hypoxia signal transduction. Blood 82:3610–3615. [PubMed] [Google Scholar]
- Wang, G. L. , Jiang B. H., Rue E. A., and Semenza G. L.. 1995. Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc. Natl Acad. Sci. USA 92:5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]