

Abstract
Bacterial sepsis involves a complex interaction between the host immune response and bacterial LPS. LPS binds Toll-like receptor (TLR) 4, which leads to the release of proinflammatory cytokines that are essential for a potent innate immune response against pathogens. The innate immune system is tightly regulated, as excessive inflammation can lead to organ failure and death. MicroRNAs have recently emerged as important regulators of the innate immune system. Here we determined the function of miR-718, which is conserved across mammals and overlaps with the 5′ UTR of the interleukin 1 receptor-associated kinase (IRAK1) gene. As IRAK1 is a key component of innate immune signaling pathways that are downstream of most TLRs, we hypothesized that miR-718 helps regulate the innate immune response. Activation of TLR4, but not TLR3, induced the expression of miR-718 in macrophages. miR-718 expression was also induced in the spleens of mice upon LPS injection. miR-718 modulates PI3K/Akt signaling by directly down-regulating phosphatase and tensin homolog (PTEN), thereby promoting phosphorylation of Akt, which leads to a decrease in proinflammatory cytokine production. Phosphorylated Akt induces let-7e expression, which, in turn, down-regulates TLR4 and further diminishes TLR4-mediated proinflammatory signals. Decreased miR-718 expression is associated with bacterial burden during Neisseria gonorrhoeae infection and alters the infection dynamics of N. gonorrhoeae in vitro. Furthermore, miR-718 regulates the induction of LPS tolerance in macrophages. We propose a role for miR-718 in controlling TLR4 signaling and inflammatory cytokine signaling through a negative feedback regulation loop involving down-regulation of TLR4, IRAK1, and NF-κB.
Keywords: Akt PKB, bacterial pathogenesis, lipopolysaccharide (LPS), microRNA (miRNA), phosphatase and tensin homolog (PTEN), Toll-like receptor 4 (TLR4), miR-718
Introduction
Despite numerous clinical trials and general advances in medicine, sepsis is a major factor in at least one-third of hospital deaths (1–4). Although endotoxin was discovered well over a century ago, the fundamental role of circulating endotoxin in the blood of most patients with septic shock remains enigmatic and a subject of considerable controversy (5). The most potent microbial mediator implicated in the pathogenesis of sepsis and septic shock is bacterial LPS.
LPS is a Toll-like receptor 4 (TLR4)3 agonist that induces the immune system to generate systemic production and release of proinflammatory cytokines and chemokines. LPS engages the TLR4/MD2 receptor complex, leading to recruitment of MyD88. The MyD88-TLR4/MD2 signaling complex then recruits IRAK1, a key regulatory kinase in the TLR4/MD2 receptor pathway, which plays an essential role in TLR4 signaling (6–8). Notably, IRAK1-deficient mice are poorly responsive to LPS and have impaired TNFα and IL-6 production and a decreased ability to activate NF-κB (8–10). IRAK1 is phosphorylated during LPS activation, and the levels of IRAK1 protein subsequently decline as the protein is degraded. This chain of events triggers NF-κB activation, which results in the transcription of proinflammatory genes. The rapid induction of cytokines such as TNFα, IL-1β, and IL-6 is essential for immediate defense against infection and a long-lasting adaptive immunity against pathogens (11, 12). Although cytokine production is important for the efficient control of the dissemination of invading pathogens, overproduction of these cytokines (a condition known as septic shock syndrome) is harmful for the host and may lead to tissue damage, organ failure, and death (13). Thus, TLRs are double-edged swords, and abnormal activation of their signaling can be deleterious. Therefore, immune cells must employ a multilayered control system to avoid excessive inflammation and allow tissue repair.
Recently, microRNAs (miRNAs) have emerged as key regulators of TLR signaling, and several miRNAs are induced by TLR activation, targeting mRNAs of genes that encode components of the TLR signaling pathway (14). miRNAs are endogenous 18- to 24-nucleotide single-stranded noncoding RNA molecules that perform important regulatory functions in animals by targeting mRNAs for cleavage or translational repression. This process involves base-pairing between the miRNAs and a partially complementary sequence in the 3′ UTRs of their target mRNAs (15, 16). Many mammalian miRNAs are located within introns or exons of other protein-coding and non-coding genes (17, 18). These miRNAs are typically co-transcribed in the same direction as their “host genes” (18–22). Despite the large number of miRNAs transcribed from host genes, only a few have been studied in detail, and any functional relationship with their host genes is poorly understood.
Here we report that the evolutionarily conserved miR-718, which overlaps with the 5′ UTR of the IRAK1 gene, is induced by LPS and is a negative regulator of inflammation in macrophages. We determined the role of miR-718 in mediating innate immune responses and the mechanism by which it controls the intensity of the inflammatory response in macrophages through targeting PTEN (a protein/lipid phosphatase that acts as a negative regulator of the PI3K/Akt pathway). Understanding the role of miR-718 in LPS-activated cells may help with the design of novel interventions for sepsis and potentially expand the use of Anti-miR-718 to treat inflammatory diseases beyond sepsis.
Results
miR-718 overlaps with the 5′ UTR of the Irak1 gene
Using our genome analysis pipeline, we identified several miRNAs that are hosted by innate immunity protein-coding genes (supplemental Table S1). We prioritized miR-718 for further study, as it was within the 5′ UTR of IRAK1, which encodes interleukin 1 receptor-associated kinase 1, a key component of TLR and IL-1R signaling pathways (Fig. 1, A and B). Furthermore, pre-miR-718 is highly conserved in mammals (Fig. 1C), prompting us to further characterize its role in the regulation of inflammation. miR-718 also overlaps with the 5′ UTR of Irak1 in the mouse genome (Fig. 1D).
Figure 1.

Highly conserved miR-718 is encoded in the 5′ UTR of the IRAK1 gene. A–D, schematic of the mouse miR-718 transcript that overlaps with the 5′ UTR of Irak1.
LPS stimulation induces miR-718 expression in multiple cell types and in vivo in mice
In immortalized BMDMs (iBMDMs), miR-718 was induced by LPS (Fig. 2A) and lipo-oligosaccharide stimulation (supplemental Fig. S1A), both of which are strong stimulators of IRAK1. This is similar to miR-146, which is a classical LPS-induced miRNA (supplemental Fig. S1B). In contrast, miR-718 was not induced in iBMDMs stimulated with poly(I:C), a TLR3 ligand (supplemental Fig. S1A).
Figure 2.

miR-718 expression is induced by LPS and N. gonorrhoeae and is dependent on IRAK1. A, immortalized mouse BMDMs from WT and Irak1/miR-718-dKO were stimulated with LPS (100 ng/ml) for 2 h or cultured in medium alone. B, immortalized BMDMs from WT and Irak1/miR-718-dKO were infected with various m.o.i. of N. gonorrhoeae or LPS. C, immortalized microglia were stimulated with LPS (100 ng/ml) for 2 h or cultured in medium alone. D, mice were injected with 1 mg/kg body weight LPS or PBS (5 mice/group). After 4 h, spleens were harvested. RNA from the cells in A–D was extracted, and miR-718 levels were quantitated by TaqMan qPCR. miR-718 levels are shown relative to levels of small non-coding RNA (sno202). The data shown represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments with similar results. A.U., arbitrary units. *, p < 0.05; **, p < 0.005; and ***, p < 0.0005.
miR-718 was not induced in IRAK1-deficient cells, consistent with the large deletion in the region of the IRAK1 gene in these mice that encompasses the miR-718 locus; i.e. IRAK1 knockout mice are actually Irak1/miR-718 double knockouts (dKOs) (Fig. 2A) (8). To identify maximal induction of miR-718 expression upon bacterial infection, we infected iBMDMs with Neisseria gonorrhoeae at an increasing multiplicity of infection (m.o.i.). miR-718 was induced in a dose-dependent manner in N. gonorrhoeae-infected wild-type macrophages (Fig. 2B) but not in Irak1/miR-718-dKO macrophages. miR-718 was also induced in other cell types, such as immortalized microglia cells (Fig. 2C). To examine whether miR-718 was induced under in vivo conditions, we determined the expression of miR-718 in the spleens of mice intraperitoneally injected with LPS. miR-718 levels were significantly increased 4 h after LPS administration (Fig. 2D).
miR-718 negatively regulates proinflammatory cytokines
Because miR-718 was induced in LPS-stimulated macrophages, we investigated whether miR-718 miRNA regulates immune genes. Using lentiviral vectors, we generated a stable miR-718-expressing cell line (miR-718). This system allows the expression of miRNA precursor transcripts in their native sequence context to ensure interaction with endogenous processing machinery, leading to authentic mature miRNAs. As a control, we generated a stable cell line expressing the corresponding empty vector (Con miR). We also used shRNA-mediated miR-718 knockdown to establish a stable cell line suppressing miR-718 (Anti-miR-718) and used the corresponding empty vector to generate a stable control cell line (Con Inh).
To assess the kinetics of LPS-induced miR-718 expression, IRAK1 expression, and TNFα production, miR-718 and Anti-miR-718 macrophages were treated with LPS and harvested at various time points from 2 to 24 h. miR-718 was potently induced within 2 h of stimulation, peaked at 12 h, and was significantly decreased at 24 h, suggesting that miR-718 is an early-induced miRNA (Fig. 3, A and B). Increased expression of miR-718 led to decreased expression of Irak1 mRNA (Fig. 3C). Similarly, decreased expression of miR-718 led to increased expression of Irak1 mRNA (Fig. 3D). TNFα protein levels were decreased when miR-718 was overexpressed (Fig. 3E) and increased when miR-718 was silenced (Fig. 3F). Similar trends were observed for TNFα production at various concentrations of LPS (supplemental Fig. S2, A and B). Cells treated with LPS and nigericin also secreted less IL-1β when miR-718 was overexpressed (supplemental Fig. S2C). We confirmed that Irak1 protein levels were significantly decreased in miR-718-overexpressing macrophages, including under basal conditions when endogenous miR-718 levels are low (Fig. 2G). Infection of Anti-miR-718 cells with various m.o.i. of N. gonorrhoeae led to a significant increase in TNFα production (supplemental Fig. S2D).
Figure 3.

miR-718 represses the production of proinflammatory cytokines in macrophages. A–F, stable macrophage cell lines expressing miR-718 or the corresponding empty vector control (Con miR) (A, C, and E) and Anti-miR-718 or the corresponding empty vector control (Con Inh) (B, D, and F) were stimulated with 100 ng/ml LPS at various time points. RNA was extracted, and miR-718 levels were quantitated by TaqMan qPCR (A and B). IRAK1 levels were quantitated by SYBR Green qPCR (C and D). ELISA was performed to measure TNFα release (E and F). The data shown represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments with similar results. G, Western blotting was performed with lysates from Con miR and miR-718 macrophages stimulated with 100 ng/ml LPS for 2 h or medium (Med). ns, not significant. A.U., arbitrary units. *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
Upon recognition of specific pathogen-associated molecular patterns, most TLRs activate pathways that lead to the nuclear translocation of the transcription factor NF-κB, which regulates the transcription of proinflammatory cytokines. To investigate whether miR-718 expression affects NF-κB activity, we co-transfected an NF-κB-Luc reporter construct as well as an miR-718 construct or an empty vector into 293T cells. We detected a significant decrease in NF-κB activity in the presence of miR-718, suggesting that miR-718 negatively regulates NF-κB activity (Fig. 4A and supplemental Fig. S3A). In the canonical NF-κB pathway, the p65 (RelA) and p50 subunits form heterodimers that bind DNA and regulate transcription. In a resting state, these subunits interact with IκBα in the cytoplasm. Upon activation of this pathway, IκBα is ubiquitinated and degraded, which leads to the release of the p65 and p50 subunits and their subsequent translocation into the nucleus. In miR-718 cells, IkBα degradation is inhibited upon LPS stimulation (Fig. 4B), suggesting that it is still bound to p65 in the cytosol. To confirm these results, we stained for p65 and performed immunofluorescence studies to assess p65 translocation into the nucleus in the presence and absence of miR-718. In resting cells, p65 is predominantly located in the cytosol of macrophages (Fig. 4, C, first and third rows, and D). In control cells, LPS stimulation resulted in the rapid translocation of p65 into the nucleus in the vast majority of cells (Fig. 4, C, second row, and D), whereas, in macrophages expressing miR-718, p65 failed to translocate into the nucleus in most of the cells (Fig. 4, C, fourth row, and D), suggesting that there is a distinct intracellular distribution of p65 and, therefore, NF-κB activation in macrophages expressing miR-718.
Figure 4.

NF-κB activity is reduced in miR-718-overexpressing macrophages stimulated with LPS. A, 293T cells were transiently transfected with TLR4, NF-κB-Luc (firefly), control (Renilla) Luc, the miR-718 construct, or Control miR constructs and then stimulated with various concentrations of LPS for 2 h. After 48 h, 293T cells were lysed, and the normalized firefly luciferase activity (firefly luciferase activity/Renilla luciferase activity) was calculated. RLU, relative light units. B, whole cell extracts from Con miR and miR-718 macrophages stimulated with 100 ng/ml LPS for 2 h were analyzed for IκBα, P65, and β-actin protein expression by immunoblotting. Data are representative of three independent experiments. Med, medium. C, Con miR and miR-718 macrophages stimulated with 100 ng/ml LPS for 2 h were fixed, permeabilized, and stained with Alexa Fluor 647 anti-p65 NF-κB subunit antibody and DAPI (nucleus) and subjected to confocal microscopy. Scale bars = 20 μm. Images are representative of at least 10 fields of view and three independent experiments. D, the presence of nuclear P65 was quantified in 10–15 fields by confocal microscopy. Mean ± S.D. of triplicate determinations are shown. *, p < 0.05; **, p < 0.005.
miR-718 targets PTEN for degradation
Although we demonstrated that miR-718 controls proinflammatory cytokine production, its direct target(s) remained to be identified. Based on sequence complementarity between miR-718 and PTEN mRNA, PTEN was a predicted miR-718 target (Fig. 5A). PTEN was recently confirmed by another study to be targeted by miR-718 (23). To validate that miR-718 suppresses PTEN expression and function and to determine whether miR-718 could directly repress PTEN mRNA through a 3′ UTR interaction, we co-transfected 293T cells with a PTEN-3′ UTR-Luc reporter construct in the absence and presence of the miR-718 expression vector. We observed a significant reduction in luciferase levels in cells co-expressing PTEN-3′ UTR-Luc and miR-718 (Fig. 5B and supplemental Fig. S4A). In contrast, no change in luciferase activity was observed in the presence of miR-416, suggesting that PTEN is specifically targeted by miR-718 (Fig. 5B).
Figure 5.

miR-718 targets PTEN for degradation. A, Schematic of the seed sequence of miR-718 within the 3′ UTR of PTEN. B, the PTEN-3′ UTR-Luc reporter construct was co-transfected with miR-718 expression vector, miR-146 expression vector, or miRNA negative control (Neg. Con) into 293T cells. After 48 h, 293T cells were lysed, and the normalized firefly luciferase activity (firefly luciferase activity/Renilla luciferase activity) was calculated. Data shown represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments with similar results. C, whole cell extracts from Con miR and miR-718 macrophages stimulated with 100 ng/ml LPS were analyzed for PTEN, total Akt, and phosphorylated Akt (P-Akt-Ser473) protein expression by immunoblotting. Med, medium. D, Con miR and miR-718 macrophages stimulated with 100 ng/ml LPS for 2 h were fixed, permeabilized, and stained with PTEN antibody and DAPI (Nucleus) and subjected to confocal microscopy. E, Con miR and miR-718 macrophages stimulated with 100 ng/ml LPS for 2 h were fixed, permeabilized, and stained with phosphorylated Akt (P-Akt) antibody and DAPI (Nucleus) and subjected to confocal microscopy. In D and E, images are representative of at least 10 fields of view and three independent experiments. Scale bars = 20 μm. F, immortalized Con miR, miR-718, Irak1/miR-718-dKO, and TLR4-KO macrophages were pretreated with 10 μm Akt inhibitor IV and then stimulated with LPS for 2 h. TNFα production was measured by ELISA. The data shown represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments with similar results. *, p < 0.05.
PTEN is a PI3K-specific phosphatase that negatively regulates the Akt signaling pathway, and levels of PTEN expression inversely correlate with Akt phosphorylation (24). Thus, we examined the protein levels of PTEN and phosphorylated Akt in macrophages overexpressing miR-718. As expected, overexpression of miR-718 led to decreased expression of PTEN and increased levels of phosphorylated Akt (Fig. 5C). Immunofluorescence studies also demonstrated that PTEN was down-regulated in miR-718 macrophages (Fig. 5D, bottom panels), whereas it was highly expressed in Anti-miR-718 macrophages (supplemental Fig. S4B, bottom panel). In contrast, the phosphorylated form of Akt was highly expressed in miR-718 macrophages (Fig. 5E, bottom panels), whereas it was down-regulated in Anti-miR-718 macrophages (supplemental Fig. S4C, bottom panel). These data suggest that PTEN protein levels are decreased by overexpression of miR-718, which, in turn, enhances PI3K/Akt signaling. The Akt pathway prevents excessive innate immune responses via limiting Th1 polarization (25, 26). To assess whether the miR-718-induced suppression of proinflammatory cytokine expression occurred through the activation of the Akt pathway, we inactivated Akt using Akt inhibitor IV. miR-718 macrophages failed to suppress TNFα production in the presence of the Akt inhibitor (Fig. 5F). We confirmed these results by using two additional Akt inhibitors (API-1 and MK-2206) (supplemental Fig. S4, D and E) and Anti-miR-718 cells (supplemental Fig. S4F). Overall, these data indicated that, by targeting PTEN, miR-718 suppresses the production of proinflammatory cytokines through activation of Akt.
miR-718 represses TLR4 cell surface expression
LPS induces the release of proinflammatory cytokines through the innate immune receptor TLR4. Cell surface expression of TLR4 impacts the extent of this response and, therefore, should be tightly regulated. To test whether miR-718 expression can affect TLR4, we measured TLR4 cell surface expression by flow cytometric analysis of unstimulated and LPS-stimulated Con miR and miR-718 macrophages. LPS stimulation down-regulated TLR4 expression in Con miR cells (Fig. 6A). In contrast, TLR4 was already expressed at low levels in unstimulated miR-718 macrophages, and, therefore, LPS stimulation only caused a minor reduction in TLR4 levels. To confirm these results, we used a strategy in which cell surface proteins were biotinylated and subsequently immunoprecipitated and purified using NeutrAvidin gel. We observed reduced biotinylated TLR4 in miR-718 cells compared with Con miR cells, suggesting that miR-718 is inducing the internalization and subsequent degradation of the receptor (Fig. 6B). There was no significant change in cell surface expression of TLR4 in Irak1/miR-718-dKO macrophages, suggesting that miR-718 expression is indeed required to suppress TLR4 expression and functions as a physiological inhibitor of TLR4 expression.
Figure 6.

TLR4 cell surface expression is repressed in the presence of miR-718. A, Con miR and miR-718 macrophages were stimulated with 100 ng/ml LPS for 2 h, stained for TLR4 using TLR4-phosphatidylethanolamine (PE)-conjugated antibody, and analyzed by flow cytometry. B, cell surface protein biotinylation and isolation were performed using the Pinpoint cell surface protein isolation kit from Pierce. Con miR, miR-718, Con Inh, Anti-miR-718, and Irak1/miR-718-dKO cells were stimulated with 100 ng/ml LPS for 2 h and then labeled with EZ-Link sulfo-NHS-SS-biotin. These cells were then lysed and isolated with immobilized NeutrAvidin gel. The bound proteins were released by incubation with SDS-PAGE sample buffer containing 50 mm DTT. The flow-through and elution were kept for analysis of TLR4 protein expression by immunoblotting. C, immortalized Con miR, miR-718 and TLR4-KO macrophages were either left untreated or pretreated with 10 μm Akt inhibitor IV and then stimulated with LPS for 2 h. RNA was extracted, and TLR4 expression was quantitated by SYBR Green qPCR. Levels of TLR4 are shown relative to levels of β-actin. D, immortalized Con miR and miR-718 macrophages were either left untreated or pretreated with 10 μm of Akt inhibitor IV and then stimulated with LPS for 2 h. RNA was extracted, and let-7e expression was quantitated by TaqMan qPCR. Levels of let-7e are shown relative to levels of sno-202. E, immortalized Con miR, miR-718, and TLR4-KO macrophages were transfected with 50 nm as-let-7e or SCR-miRNA using Lipofectamine 2000. RNA was extracted 48 h after miRNA transfection, and TLR4 mRNA was quantitated by SYBR Green qPCR. F, immortalized Con Inh, Anti-miR-718, and TLR4-KO macrophages were transfected with 50 nm let-7e or SCR-miRNA using Lipofectamine 2000. RNA was extracted 48 h after miRNA transfection, and TLR4 mRNA was quantitated by SYBR Green qPCR. G, immortalized Con miR and miR-718 macrophages were transfected with 50 nm as-let-7e or SCR-miRNA using Lipofectamine 2000. Cells were then stimulated with LPS for 2 h, and TNFα production was measured by ELISA. The data shown represent the mean ± S.D. of triplicate determinations and are representative of three independent experiments with similar results. A.U., arbitrary units. *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
To test whether miR-718 suppresses TLR4 expression via activation of Akt, we pretreated unstimulated and LPS-stimulated Con miR, miR-718, and TLR4-KO macrophages with an Akt inhibitor. In the presence of Akt inhibitor IV, miR-718 failed to suppress TLR4 expression, suggesting that miR-718-mediated down-regulation of TLR4 is through the activation of Akt (Fig. 6C). These results were confirmed using an additional Akt inhibitor (MK-2206) (supplemental Fig. S5A). Phosphorylated Akt induces the production of let-7e, which targets TLR4 (27, 28). To test whether miR-718 is suppressing TLR4 through the Akt-mediated induction of let-7e, we pretreated unstimulated and LPS-stimulated Con miR and miR-718 macrophages with an Akt inhibitor. miR-718 induced let-7e production in an Akt-dependent manner, as let-7e production was markedly decreased in the presence of Akt inhibitor IV (Fig. 6D) A similar experiment using an additional Akt inhibitor (MK-2206) was performed, and similar results were obtained (supplemental Fig. S5B). Finally, to determine whether miR-718-mediated suppression of TLR4 expression is via let-7e, we transfected Con miR, miR-718, and TLR4-KO macrophages with let-7e antisense. In the absence of let-7e, miR-718 failed to repress TLR4 expression (Fig. 6E), whereas introduction of let-7e restored the ability of miR-718 to suppress TLR4 expression, as determined by qPCR (Fig. 6F). These results suggest that the Akt-mediated up-regulation of let-7e is crucial for miR-718 to suppress TLR4 expression. To determine whether the suppression of the LPS-triggered TLR4 signaling responses in miR-718 macrophages is derived from the altered expression of let-7e, we transfected Con miR and miR-718 macrophages with as-let-7e and stimulated the cells with LPS. In the absence of let-7e, miR-718 no longer repressed TNFα production (Fig. 6G), whereas the introduction of let-7e abolished TNFα production (supplemental Fig. S5C). These results suggest that let-7e mediates the miR-718 suppression of TNFα production.
miR-718 regulates the induction of LPS tolerance in macrophages
Given that miR-718 affects PTEN expression and Akt regulates endotoxin sensitivity and tolerance (27), we investigated whether miR-718 controls LPS tolerance. BMDMs were primed with a low concentration of LPS for 18 h, and then the cells were washed and restimulated with various concentrations of LPS for 5 h. TNFα was measured in the culture supernatants. miR-718 macrophages became tolerant to subsequent LPS stimulation and produced significantly lower levels of TNFα (supplemental Fig. S6A), whereas blockade of miR-718 expression abrogated the induction of LPS-induced tolerance (supplemental Fig. S6B). These results suggest that miR-718 mediates LPS tolerance in macrophages.
miR-718 facilitated N. gonorrhoeae replication in macrophages
To test directly whether miR-718 expression is involved in macrophage immune responses against N. gonorrhoeae infection, we infected Con Inh and Anti-miR-718 macrophages with N. gonorrhoeae and assessed the number of colony-forming units detected over time in cells. Anti-miR-718 macrophages exhibited the ability to control N. gonorrhoeae replication, and markedly fewer bacteria were recovered from Anti-miR-718 macrophages compared with Con Inh macrophages (supplemental Fig. S6C). In contrast, a significantly higher bacterial burden was detected in miR-718 macrophages (supplemental Fig. S6D). Overall, these results suggest that miR-718 contributes to immune responses against N. gonorrhoeae infection.
Discussion
We demonstrate that miR-718 is an anti-inflammatory miRNA that overlaps with the 5′ UTR of the Irak1 gene. Indeed, the coupled expression of miR-718 and its host transcript, Irak1, ensures that this miRNA will be available to regulate Irak1 whenever it is activated (29). There are other examples of miRNAs encoded within protein-coding genes that regulate their host gene; for example, the expression of miR-9-1, which is encoded in intron 2 of the human CROC-4 gene, highly correlates with the expression of this gene (30). Irak1 is a key component of an inflammatory pathway that is downstream of most TLRs. The transcription of miR-718 from the same genomic location provides a tightly controlled negative feedback loop that potentially protects mammals from excessive inflammation and tissue damage.
We validated PTEN as a target of miR-718. PTEN was originally identified as a tumor suppressor gene, mutated in various cancers (31, 32). Of particular interest, the PTEN mRNA has a very large 3′ UTR (about 3.3 kb) (33). There are several other miRNA target sites in the 3′ UTR of PTEN (such as miR-21, 214, 216a, 217, and 26a) (34–38), suggesting that PTEN expression is tightly regulated. Consistent with our studies, a recent study showed that, by targeting PTEN, miR-718 mediates angiogenesis through activation of Akt (23).
The Akt pathway is critical in cell proliferation, cell survival, and cancer (39, 40), suggesting that miR-718 could also function as an oncogene. The Akt pathway has been shown to suppress NF-κB and the expression of proinflammatory cytokines in the early phase of innate immune responses (25, 41–44), and Akt has also been shown recently to regulate the immune response to LPS by regulating the expression of key miRNAs (27). One of these miRNAs is let-7e, which represses TLR4, whereas the other miRNA, miR-155, represses SOCS1(27).
Through gain- and loss-of-function experiments, we showed that miR-718, by repressing PTEN, enhances PI3K/Akt signaling, which directly represses proinflammatory cytokine expression. On the other hand, phosphorylated Akt indirectly (via let-7e) represses TLR4 expression and its downstream signaling molecules, such as IRAK1 and NF-κB, suggesting that, by targeting the PTEN/Akt pathway, miR-718 is indirectly suppressing multiple proinflammatory pathways (summarized in Fig. 7A). Consistent with our findings that miR-718 regulates the surface expression of TLR4, earlier studies demonstrated that let-7e in mouse macrophages (27) and let-7i in human cholangiocytes (28) target TLR4 for degradation. Considering the regulatory role of miR-718 in LPS-stimulated macrophages, it will be important to identify additional pathways and genes that are targeted by miR-718 during an innate immune response. These target genes most likely regulate the magnitude of the host innate immune response to Gram-negative bacterial infections.
Figure 7.
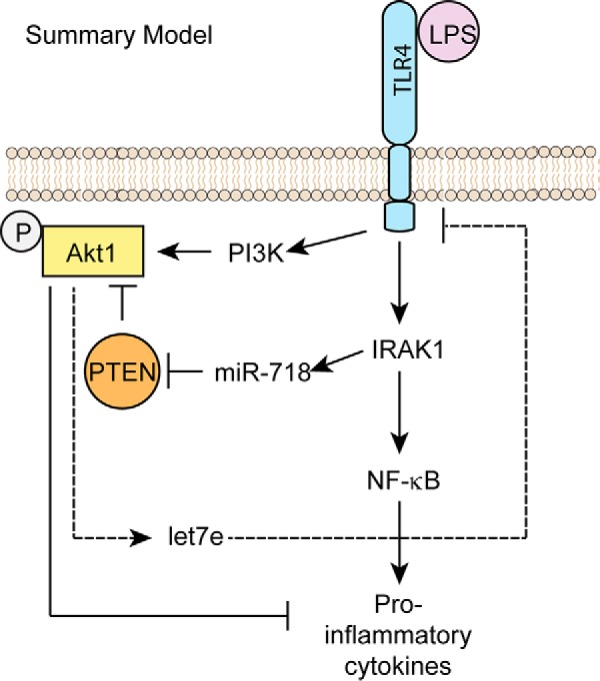
Summary model of how miR-718 suppresses proinflammatory cytokine production. A, LPS up-regulates miR-718, which targets PTEN for degradation, thereby promoting Akt activation. Phosphorylated Akt suppresses proinflammatory cytokines and also up-regulates the expression of another miRNA, let-7e, which suppresses TLR4 expression.
Notably, besides LPS, other TLR ligands, such as Pam3CSK4, also induce miR-718 transcription (data not shown) whereas poly(I:C) does not. It is important to characterize the inducers/upstream pathways of miR-718 transcription in the future by stimulating macrophages with a panel of ligands that induce the innate immune response as well as proinflammatory cytokines such as IL-1β and TNFα. To understand the function of miR-718 in vivo, it is important to generate tissue-specific and/or inducible miR-718-deficient mice. We anticipate that miR-718-null mice will be hyperresponsive to bacterial challenges and, as a result, susceptible to endotoxic shock. Future research using animal models will help us understand the exact role of miR-718 in the context of disease. It is also possible that miR-718, by establishing LPS-induced tolerance, evades recurrent bacterial infection and lowers the LPS-induced mortality rate.
Experimental procedures
Genome analysis pipeline
Version 15 of miRBase (17) was scanned for human miRNAs that overlapped with other genes. We used version 52 of the Ensembl API (Application Program Interface) (45) to retrieve gene ontology terms for all protein-coding genes that hosted a miRNA. GO terms from the biological process ontology were manually inspected to identify host genes involved in innate immunity (supplemental Table S1). miR-718 was prioritized for experimental characterization.
Reagents
Lipofectamine 2000 was from Invitrogen. DAPI was from Molecular Probes/Invitrogen. LPS, poly(I:C) and nigericin were from Invivogen (San Diego, CA). DMEM was from Cellgro (Manassas, VA), and low-endotoxin FBS was from Atlas Biologicals (Fort Collins, CO). Akt inhibitor IV was from Calbiochem. The Akt inhibitor API-1 was from Sigma-Aldrich (St. Louis, MO). The Akt inhibitor MK-2206 was from Cayman Chemical (Ann Arbor, MI). Cell surface protein biotinylation and isolation were performed using the Pinpoint cell surface protein isolation kit from Pierce.
Mice, cells, cell culture, stimulations, and ELISA
C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mouse strains were bred and maintained under specific pathogen-free conditions in the animal facilities at the University of Massachusetts Medical School in accordance with the Institutional Animal Care and Use Committee. Immortalized macrophage cell lines were generated from wild-type, TLR4−/−, and IRAK1−/− mice (from James A. Thomas, Baylor College of Medicine, described in Ref. 8). A cell line stably overexpressing miR-718 was generated using a lentiviral pMIRNA1-miR718 vector; as a control, we constructed a cell line stably expressing an empty vector pMIRNA1 (Con miR). To silence miR-718 expression, we generated a cell line stably silencing miR-718 (Anti-miR-718) using a lentiviral miRzip-miR718 vector and, as a control, a stable cell line expressing the empty vector miRzip (Con Inh). All lentiviral vectors were from System Biosciences (Palo Alto, CA). The production of viral particles in HEK293T cells was performed using the psPAX2 and pMD2.G lentiviral packaging vectors. All cells were cultured in DMEM supplemented with ciprofloxacin and 10% FBS. Anti-miR-718 cells were maintained with 5 μg/ml puromycin. 2 × 105 macrophages were plated and stimulated the following day with the indicated amounts of LPS and nigericin. All transient transfections were performed with Lipofectamine 2000 in accordance with the instructions of the manufacturer. Nigericin (10 μm) was added 1 h before supernatants were collected. Cytokine measurements were performed using ELISA kits for mouse IL-1β and TNFα (R&D Systems).
Immunoblot analysis
Immunoblot analysis was performed using antibodies directed against IRAK1, phospho-Akt-Ser473, Akt, PTEN, NF-κB, P65, and IκBα (Cell Signaling Technologies, Danvers, MA), TLR4 (Santa Cruz Biotechnology, Dallas, TX), and β-actin (Sigma).
Transfection of antisense miRNAs
For miRNA transfection studies, iBMDMs were seeded in 6-well plates and transfected with 50 nm let-7e miRNA, antisense let-7e miRNA (as-let-7e), or a scrambled (SCR) miRNA (Ambion, Foster City, CA) using Lipofectamine 2000 in accordance with the instructions of the manufacturer. Real-time PCR was performed 24 h after transfection.
RNA extraction and real-time PCR
Immortalized mouse BMDMs, immortalized microglia, and HEK293 cells (5 × 106 cells/condition) were plated and stimulated with varying concentrations of LPS. RNA was extracted with the RNeasy kit (Qiagen, Valencia, CA), cDNA was synthesized using iScript Reverse Transcription Supermix (Bio-Rad), and quantitative RT-PCR analysis was performed with qPCR primers purchased from IDT (Coralville, IA) and iQ SYBR Green Supermix (Bio-Rad). Gene expression data are presented as a ratio of gene copy number per 100 copies of actin ± S.D. For miRNA (miR-718 and miR-146) detection, small RNAs were extracted using the MirVana PARIS kit (Applied Biosystems), and TaqMan microRNA assays (Applied Biosystems) were used to quantitate miRNA expression. Expression of sno202 was used as an internal control.
Luciferase assay
The PTEN-3′ UTR-Luc reporter construct was a gift from Eric N. Olson (46), and the NF-κB-Luc reporter construct is described in Ref. 47. The above reporter constructs were transfected in iBMDMs, and luciferase activity was measured with the luciferase reporter assay system (Promega, Madison, WI).
Flow cytometry
Cells were washed with PBS containing 1% FBS and resuspended in a 1:200 dilution of phycoerythrin-conjugated anti-mouse TLR4 antibody (eBioscience, San Diego, CA) for 30 min in the dark and on ice. The cells were then washed twice with PBS containing 1% FBS and analyzed by flow cytometry.
Confocal microscopy
Confocal microscopy was performed on a Leica TCS SP8 AOBS confocal laser-scanning microscope. For P65 staining, NF-κB P65 antibody as primary antibody and anti-rabbit IgG AF647 as secondary antibody were used (both from Cell Signaling Technologies).
N. gonorrhoeae culture
N. gonorrhoeae was harvested from an overnight culture on chocolate agar plates, repassaged onto fresh chocolate agar, and grown for 6 h at 37 °C in an atmosphere containing 5% CO2.
Statistical analysis
Student's t test was used to statistically analyze differences between groups. p <0.05 was considered significant.
Author contributions
D. R. C., K. A. F., D. T. G., P. K., O. F. H., and E. A. K.-J. oversaw the whole project. O. F. H. and P. K. designed and conducted the experiments with help from S. A. and F. R. O. F. H., P. K., D. R. C., and D. T. G. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Eric Olson for the PTEN-3′ UTR-Luc reporter construct. We also thank Melanie Trombly for assistance with manuscript preparation.
This work was supported by National Institutes of Health Grant R37GM54060. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S6 and Table S1.
- TLR4
- Toll-like receptor 4
- BMDM
- bone marrow-derived macrophage
- IRAK
- interleukin 1 receptor-associated kinase
- miRNA
- microRNA
- PTEN
- phosphatase and tensin homolog
- dKO
- double knockout
- m.o.i.
- multiplicity of infection
- Luc
- luciferase
- qPCR
- quantitative PCR
- as
- antisense
- SCR
- scrambled.
References
- 1. Angus D. C., Linde-Zwirble W. T., Lidicker J., Clermont G., Carcillo J., and Pinsky M. R. (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 29, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 2. Hotchkiss R. S., and Karl I. E. (2003) The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348, 138–150 [DOI] [PubMed] [Google Scholar]
- 3. Liu V., Escobar G. J., Greene J. D., Soule J., Whippy A., Angus D. C., and Iwashyna T. J. (2014) Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA 312, 90–92 [DOI] [PubMed] [Google Scholar]
- 4. Hotchkiss R. S., and Sherwood E. R. (2015) Immunology: getting sepsis therapy right. Science 347, 1201–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Opal S. M. (2010) Endotoxins and other sepsis triggers. Contrib. Nephrol. 167, 14–24 [DOI] [PubMed] [Google Scholar]
- 6. Kawagoe T., Sato S., Matsushita K., Kato H., Matsui K., Kumagai Y., Saitoh T., Kawai T., Takeuchi O., and Akira S. (2008) Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol. [DOI] [PubMed] [Google Scholar]
- 7. Li X., Commane M., Burns C., Vithalani K., Cao Z., and Stark G. R. (1999) Mutant cells that do not respond to interleukin-1 (IL-1) reveal a novel role for IL-1 receptor-associated kinase. Mol. Cell Biol. 19, 4643–4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas J. A., Allen J. L., Tsen M., Dubnicoff T., Danao J., Liao X. C., Cao Z., and Wasserman S. A. (1999) Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J. Immunol. 163, 978–984 [PubMed] [Google Scholar]
- 9. Li S., Strelow A., Fontana E. J., and Wesche H. (2002) IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. U.S.A. 99, 5567–5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki N., Suzuki S., Duncan G. S., Millar D. G., Wada T., Mirtsos C., Takada H., Wakeham A., Itie A., Li S., Penninger J. M., Wesche H., Ohashi P. S., Mak T. W., and Yeh W. C. (2002) Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 416, 750–756 [DOI] [PubMed] [Google Scholar]
- 11. Suffredini A. F., Fantuzzi G., Badolato R., Oppenheim J. J., and O'Grady N. P. (1999) New insights into the biology of the acute phase response. J. Clin. Immunol. 19, 203–214 [DOI] [PubMed] [Google Scholar]
- 12. Siebert S., Tsoukas A., Robertson J., and McInnes I. (2015) Cytokines as therapeutic targets in rheumatoid arthritis and other inflammatory diseases. Pharmacol. Rev. 67, 280–309 [DOI] [PubMed] [Google Scholar]
- 13. Beutler B., Milsark I. W., and Cerami A. C. (1985) Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 229, 869–871 [DOI] [PubMed] [Google Scholar]
- 14. O'Neill L. A., Sheedy F. J., and McCoy C. E. (2011) MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunol. 11, 163–175 [DOI] [PubMed] [Google Scholar]
- 15. Bartel D. P. (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 [DOI] [PubMed] [Google Scholar]
- 16. Nelson P., Kiriakidou M., Sharma A., Maniataki E., and Mourelatos Z. (2003) The microRNA world: small is mighty. Trends Biochem. Sci. 28, 534–540 [DOI] [PubMed] [Google Scholar]
- 17. Griffiths-Jones S., Saini H. K., van Dongen S., and Enright A. J. (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodriguez A., Griffiths-Jones S., Ashurst J. L., and Bradley A. (2004) Identification of mammalian microRNA host genes and transcription units. Genome Res. 14, 1902–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Poliseno L., Salmena L., Riccardi L., Fornari A., Song M. S., Hobbs R. M., Sportoletti P., Varmeh S., Egia A., Fedele G., Rameh L., Loda M., and Pandolfi P. P. (2010) Identification of the miR-106b∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 3, ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baskerville S., and Bartel D. P. (2005) Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lagos-Quintana M., Rauhut R., Lendeckel W., and Tuschl T. (2001) Identification of novel genes coding for small expressed RNAs. Science 294, 853–858 [DOI] [PubMed] [Google Scholar]
- 22. Lau N. C., Lim L. P., Weinstein E. G., and Bartel D. P. (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294, 858–862 [DOI] [PubMed] [Google Scholar]
- 23. Xue M., Yao S., Hu M., Li W., Hao T., Zhou F., Zhu X., Lu H., Qin D., Yan Q., Zhu J., Gao S. J., and Lu C. (2014) HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic Acids Res. 42, 9862–9879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wan X., and Helman L. J. (2003) Levels of PTEN protein modulate Akt phosphorylation on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcomas cells. Oncogene 22, 8205–8211 [DOI] [PubMed] [Google Scholar]
- 25. Fukao T., and Koyasu S. (2003) PI3K and negative regulation of TLR signaling. Trends Immunol. 24, 358–363 [DOI] [PubMed] [Google Scholar]
- 26. Fukao T., Tanabe M., Terauchi Y., Ota T., Matsuda S., Asano T., Kadowaki T., Takeuchi T., and Koyasu S. (2002) PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 3, 875–881 [DOI] [PubMed] [Google Scholar]
- 27. Androulidaki A., Iliopoulos D., Arranz A., Doxaki C., Schworer S., Zacharioudaki V., Margioris A. N., Tsichlis P. N., and Tsatsanis C. (2009) The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 31, 220–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X. M., Splinter P. L., O'Hara S. P., and LaRusso N. F. (2007) A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J. Biol. Chem. 282, 28929–28938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barik S. (2008) An intronic microRNA silences genes that are functionally antagonistic to its host gene. Nucleic Acids Res. 36, 5232–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jeffrey P. L., Capes-Davis A., Dunn J. M., Tolhurst O., Seeto G., Hannan A. J., and Lin S. L. (2000) CROC-4: a novel brain specific transcriptional activator of c-fos expressed from proliferation through to maturation of multiple neuronal cell types. Mol. Cell Neurosci. 16, 185–196 [DOI] [PubMed] [Google Scholar]
- 31. Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S. I., Puc J., Miliaresis C., Rodgers L., McCombie R., Bigner S. H., Giovanella B. C., Ittmann M., Tycko B., Hibshoosh H., Wigler M. H., and Parsons R. (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 [DOI] [PubMed] [Google Scholar]
- 32. Steck P. A., Pershouse M. A., Jasser S. A., Yung W. K., Lin H., Ligon A. H., Langford L. A., Baumgard M. L., Hattier T., Davis T., Frye C., Hu R., Swedlund B., Teng D. H., and Tavtigian S. V. (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362 [DOI] [PubMed] [Google Scholar]
- 33. Bar N., and Dikstein R. (2010) miR-22 forms a regulatory loop in PTEN/AKT pathway and modulates signaling kinetics. PLoS ONE 5, e10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huse J. T., Brennan C., Hambardzumyan D., Wee B., Pena J., Rouhanifard S. H., Sohn-Lee C., le Sage C., Agami R., Tuschl T., and Holland E. C. (2009) The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 23, 1327–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kato M., Putta S., Wang M., Yuan H., Lanting L., Nair I., Gunn A., Nakagawa Y., Shimano H., Todorov I., Rossi J. J., and Natarajan R. (2009) TGF-β activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat. Cell Biol. 11, 881–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meng F., Henson R., Wehbe-Janek H., Ghoshal K., Jacob S. T., and Patel T. (2007) MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xiao C., Srinivasan L., Calado D. P., Patterson H. C., Zhang B., Wang J., Henderson J. M., Kutok J. L., and Rajewsky K. (2008) Lymphoproliferative disease and autoimmunity in mice with increased miR-17–92 expression in lymphocytes. Nat. Immunol. 9, 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang H., Kong W., He L., Zhao J. J., O'Donnell J. D., Wang J., Wenham R. M., Coppola D., Kruk P. A., Nicosia S. V., and Cheng J. Q. (2008) MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 68, 425–433 [DOI] [PubMed] [Google Scholar]
- 39. Montaner S. (2007) Akt/TSC/mTOR activation by the KSHV G protein-coupled receptor: emerging insights into the molecular oncogenesis and treatment of Kaposi's sarcoma. Cell Cycle 6, 438–443 [DOI] [PubMed] [Google Scholar]
- 40. Engelman J. A. (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer 9, 550–562 [DOI] [PubMed] [Google Scholar]
- 41. Lee Y. G., Lee J., Byeon S. E., Yoo D. S., Kim M. H., Lee S. Y., and Cho J. Y. (2011) Functional role of Akt in macrophage-mediated innate immunity. Front. Biosci. 16, 517–530 [DOI] [PubMed] [Google Scholar]
- 42. Luyendyk J. P., Schabbauer G. A., Tencati M., Holscher T., Pawlinski R., and Mackman N. (2008) Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J. Immunol. 180, 4218–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guha M., and Mackman N. (2002) The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J. Biol. Chem. 277, 32124–32132 [DOI] [PubMed] [Google Scholar]
- 44. Pahan K., Raymond J. R., and Singh I. (1999) Inhibition of phosphatidylinositol 3-kinase induces nitric-oxide synthase in lipopolysaccharide- or cytokine-stimulated C6 glial cells. J. Biol. Chem. 274, 7528–7536 [DOI] [PubMed] [Google Scholar]
- 45. Yates A., Akanni W., Amode M. R., Barrell D., Billis K., Carvalho-Silva D., Cummins C., Clapham P., Fitzgerald S., Gil L., Girón C. G., Gordon L., Hourlier T., Hunt S. E., Janacek S. H., et al. (2016) Ensembl 2016. Nucleic Acids Res. 44, D710–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Small E. M., O'Rourke J. R., Moresi V., Sutherland L. B., McAnally J., Gerard R. D., Richardson J. A., and Olson E. N. (2010) Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc. Natl. Acad. Sci. U.S.A. 107, 4218–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee E. S., Kalantari P., Tsutsui Section S., Klatt A., Holden J., Correll P. H., Power Section C., and Henderson A. J. (2004) RON receptor tyrosine kinase, a negative regulator of inflammation, inhibits HIV-1 transcription in monocytes/macrophages and is decreased in brain tissue from patients with AIDS. J. Immunol. 173, 6864–6872 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.