

Abstract
The HslUV proteolytic machine consists of HslV, a double-ring self-compartmentalized peptidase, and one or two AAA+ HslU ring hexamers that hydrolyze ATP to power the unfolding of protein substrates and their translocation into the proteolytic chamber of HslV. Here, we use genetic tethering and disulfide bonding strategies to construct HslU pseudohexamers containing mixtures of ATPase active and inactive subunits at defined positions in the hexameric ring. Genetic tethering impairs HslV binding and degradation, even for pseudohexamers with six active subunits, but disulfide-linked pseudohexamers do not have these defects, indicating that the peptide tether interferes with HslV interactions. Importantly, pseudohexamers containing different patterns of hydrolytically active and inactive subunits retain the ability to unfold protein substrates and/or collaborate with HslV in their degradation, supporting a model in which ATP hydrolysis and linked mechanical function in the HslU ring operate by a probabilistic mechanism.
Keywords: ATP-dependent protease, ATPases associated with diverse cellular activities (AAA), crystal structure, protein engineering, protein turnover, AAA+ protease, mixed hexameric rings, protein unfolding, protein degradation, HslUV
Introduction
Enzymes of the AAA+ ATPase superfamily play roles in proteolysis, protein remodeling and disaggregation, replication, transcription, membrane fusion, vesicle transport, and other cellular processes in all organisms (1, 2). These enzymes share conserved sequence and structural motifs and typically function as homohexameric or heterohexameric rings. Fueled by the energy of ATP binding and hydrolysis, AAA+ enzymes act as molecular machines that disassemble, remodel, or denature macromolecule targets. There are three general models for how subunits in AAA+ hexamers hydrolyze ATP and generate the mechanical power strokes required for function: (i) concerted ATP hydrolysis that occurs simultaneously in all subunits (3); (ii) sequential hydrolysis by individual subunits that occurs in an invariant kinetic pattern (4); and (iii) probabilistic hydrolysis in which, following a power stroke, any ATP-bound subunit has some chance of hydrolyzing ATP to drive the next power stroke (5).
The HslUV protease consists of one or two AAA+ HslU hexamers and the dodecameric HslV peptidase (Fig. 1A) (6–12). In ATP-dependent reactions, HslU hexamers recognize protein substrates, unfold any native structure that is present, and then translocate the unfolded polypeptide into the luminal chamber of HslV for degradation. In all crystal structures of the Escherichia coli or Haemophilus influenzae HslUV complexes and some structures of HslU alone, the HslU hexamer is highly symmetric and binds six ATP or ADP molecules, as might be expected for a concerted mechanism of hydrolysis (6–12). However, in other structures of HslU alone, only three or four nucleotides are bound to the hexameric HslU ring (6), and solution experiments show detectable binding of a maximum of 3–4 nucleotides and the existence of at least two types of nucleotide-binding sites (13). These results suggest that the six subunits of HslU assume non-equivalent functional roles within the hexamer and are more consistent with sequential or probabilistic models. Here, we test different models by which ATP hydrolysis could power the mechanical functions of E. coli HslU by introducing hydrolytically inactive subunits at defined positions in its hexameric ring. Using subunit cross-linking strategies involving genetic tethering or disulfide bonding, we find that HslU pseudohexamers with mixtures of hydrolytically active and inactive subunits retain protein unfolding activity and support HslV degradation. These studies support a probabilistic mechanism in which ATP hydrolysis powers mechanical function in the HslU ring and also reveal new information about interactions between HslU and HslV.
Figure 1.

HslUV structure. A, an HslU hexamer (secondary-structure representation) bound to an HslV dodecamer (surface representation; Protein Data Bank code 1G3I). The large and small AAA+ domains of HslU and its C-terminal tails are colored blue, cyan, and red, respectively. B, tandem HslU subunits connected by a genetically encoded peptide tether. C, three W-E3 hexamers in the asymmetric unit of structure 5TXV are shown in a secondary-structure representation; the fourth hexamer is shown in a ribbon representation with electron density from a composite omit map contoured at 1 σ.
Results
HslU dimers with covalent peptide tethers
HslU homohexamers containing the Walker B E257Q mutation are defective in ATP hydrolysis and protein degradation but retain the ability to bind HslV and protein substrates (13). We engineered genes to encode tandem E. coli HslU subunits connected by a 20-residue peptide tether (Fig. 1B). One encoded dimer consisted of two wild-type subunits (W-W), another had a wild-type subunit followed by an E257Q subunit (W-E), and a third had an E257Q subunit followed by a wild-type subunit (E-W). These dimers behaved like wild-type HslU during purification, suggesting that they form W-W3, W-E3, and E-W3 pseudohexamers. Indeed, the asymmetric unit of a low-resolution W-E3 crystal structure contained four hexamers similar to wild-type HslU (Fig. 1C and Table 1), although electron density for the C-terminal 8–10 residues was missing in alternating subunits of several hexamers or was generally poor throughout a hexamer, as expected if the tether disrupts normal C-terminal contacts.
TABLE 1.
Crystallographic statistics
Values in parenthesis represent the highest resolution shell.
| Protein Data Bank code | 5TXV |
| Wavelength (Å) | 0.979 |
| Space group | P 1 21 1 |
| Unit-cell dimensions (Å) | a = 86.5; b = 420.9; c = 176.5 |
| Unit-cell angles (degrees) | α = γ = 90; β = 98.6 |
| Resolution range (Å) | 49.2–7.1 (7.3–7.1) |
| Unique reflections | 18,630 (1784) |
| Completeness (%) | 98.3 (94.6) |
| Redundancy | 4.5 (4.5) |
| Rmerge | 0.105 (0.694) |
| Rmeas | 0.119 (0.782) |
| Rpim | 0.055 (0.315) |
| Rwork | 0.274 (0.356) |
| Rfree | 0.298 (0.347) |
| MolProbity score (percentile) | 100 |
| Root mean square bonds (Å) | 0.003 |
| Root mean square angles (degrees) | 0.61 |
| Clash score | 5.8 |
| Favored rotamers (%) | 99.0 |
| Poor rotamers (%) | 0.39 |
| Ramachandran favored | 98.0 |
| Ramachandran outliers (%) | 0 |
| Bad bonds/angles | 0/1 |
| Cβ deviations | 0 |
W-W3 hydrolyzed ATP at about twice the rate of the W-E3 and E-W3 enzymes (Fig. 2A), suggesting that ATP hydrolysis is largely restricted to the W subunits in these enzymes. We constructed and expressed a W-W-W trimer, but this protein was insoluble. To assay protein unfolding, we constructed an I37AArc-cp6GFP-st11-ssrA fusion protein with a thrombin cleavage site located between β-strands 5 and 6 (14). I37AArc is a denatured variant of Arc repressor that targets the substrate to HslU, and the C-terminal st11-ssrA sequence increases the substrate turnover rate ∼2-fold (15, 16). Following thrombin cleavage, unfolding of the split substrate by wild-type HslU results in an irreversible loss of GFP fluorescence. In experiments performed at different concentrations of the split substrate, unfolding by wild-type HslU and W-W3 occurred with steady-state Vmax rates that were similar, whereas Vmax for unfolding by the W-E3 and E-W3 pseudohexamers was about half of the wild-type value (Fig. 2B). Thus, pseudohexamers with alternating ATPase active and inactive subunits retain substantial protein-unfolding activity. However, compared with wild-type HslU, W-W3 supported HslV degradation very poorly (Fig. 2C) and bound HslV ∼20-fold more weakly (Table 2), probably because the peptide tether interferes with contacts between HslU and HslV (see “Discussion”). Thus, we explored a different method of constructing covalently linked HslU hexamers.
Figure 2.

Activity of genetically tethered pseudohexamers. A, rates of hydrolysis of 5 mm ATP by the genetically tethered W-W3, W-E3, and E-W3 pseudohexamers. Values are averages (n ≥ 5) ± S.D. (error bars). B, rates of unfolding of different concentrations of thrombin-split I37AArc-cp6GFP-β5/β6-st11-ssrA by wild-type HslU or genetically tethered variants. Lines, non-linear least squares fits to the Michaelis-Menten equation. Km values for all enzymes were 1–3 μm but were not well determined because of the small number of low-concentration data points. Average Vmax values ± S.D. calculated from the highest four substrate concentrations were 0.155 ± 0.003 min−1 enz−1 (HslU), 0.143 ± 0.008 min−1 enz−1 (W-W3), 0.0802 ± 0.007 min−1 enz−1 (E-W3), and 0.0716 ± 0.006 min−1 enz−1 (W-E3). Fitted Vmax values were 10–15% higher. C, the kinetics of degradation of Arc-st11-ssrA (10 μm) by HslV (10 μm) and HslU (0.3 μm) or W-W3 (0.3 μm) at 50 °C was monitored by SDS-PAGE. Reactions contained 5 mm ATP and a regeneration system.
TABLE 2.
HslV binding
Titration assays, monitored by changes in HslV peptidase activity, were performed at 25 °C in the presence of 5 mm ATP.
| Protein | K1/2 |
|---|---|
| nm | |
| Wild-type HslU | 78 ± 11 |
| W-W3 | 1603 ± 278 |
| WSSW3 | 59 ± 9 |
| WSSWSSW2 | 56 ± 5 |
| WSSESSW2 | 21 ± 7 |
| WSSE3 | 23 ± 11 |
| WSSESSE3 | 28 ± 19 |
Construction of disulfide-cross-linked HslU pseudohexamers
HslU hexamers consist of rigid body units formed by the large and small domains of adjacent subunits (17). We used the Disulfide by Design algorithm (18) to identify Glu47/Ala349 and Gln39/Thr361 as sites for potential disulfide bonds across the rigid body interfaces of an HslU hexamer. Fig. 3A shows a model of an otherwise Cys-free HslU pseudohexamer in which red subunits contain Cys47 and blue subunits contain Cys349, potentially allowing formation of three disulfide-linked dimers. To make WSSW3 pseudohexamers, the Cys47 and Cys349 subunits both had wild-type Walker B ATPase motifs. To make WSSE3 pseudohexamers, the Cys47 subunit had a wild-type Walker B sequence, and the Cys349 subunit contained the Walker B E257Q mutation to inactivate ATP hydrolysis. Fig. 3B shows a pseudohexamer in which red subunits contain Cys349, green subunits contain Cys47 and Cys361, and blue subunits contain Cys39. In this configuration, formation of disulfide-linked trimers is possible. We designed WSSWSSW trimers, WSSESSW trimers, and WSSESSE trimers by changing which subunits had wild-type or E257Q Walker B sites.
Figure 3.

Design and purification of disulfide-cross-linked HslU variants. A, spheres show the positions of Cys47 (normally Glu) in red subunits and Cys349 (normally Ala) in blue subunits of an HslU hexamer, suggesting that Cys47-Cys349 disulfides would stabilize a pseudohexamer consisting of three linked dimers. B, spheres show the positions of Cys349 in red subunits, Cys47 and Cys361 (normally Thr) in green subunits, and Cys39 (normally Gln) in blue subunits. Disulfide bond formation in this configuration would stabilize a pseudohexamer consisting of two linked trimers. C, non-reducing SDS-PAGE of purified wild-type HslU, purified WSSW, purified WSSE, purified WSSWSSW, purified WSSESSW, and purified WSSESSE. In each case, a 0.5 μm concentration of the purified protein (in hexamer equivalents) was loaded on the gel. The first lane contains molecular weight standards.
Relatively efficient formation of disulfide-linked HslU dimers or trimers was achieved by cytosolic coexpression of appropriate variants in the oxidizing SHuffle strain of E. coli (19). For example, following purification by Ni2+-NTA5 affinity chromatography, non-reducing SDS-PAGE showed ∼50% formation of WSSW and WSSE and ∼33% formation of WSSWSSW, WSSESSW, and WSSESSE (not shown). To further purify disulfide-linked pseudohexamers, we performed ion exchange chromatography and gel filtration chromatography once in the presence and once in the absence of urea, which destabilizes unlinked HslU hexamers more than linked hexamers. Following the final chromatography step, the WSSW, WSSE, WSSWSSW, WSSESSW, and WSSESSE variants had purities of >95% (Fig. 3C). The disulfide-linked pseudohexamers bound HslV slightly more tightly than wild-type HslU (Table 2).
ATP hydrolysis by disulfide-linked pseudohexamers
Like the basal ATP hydrolysis activities of the genetically tethered pseudohexamers, those of the disulfide-linked enzymes were roughly proportional to the number of hydrolytically active W subunits in each pseudohexamer (Fig. 4, A and B). Compared with wild-type HslU, however, the WSSW3 and WSSWSSW2 pseudohexamers were ∼3-fold more hydrolytically active (Fig. 4A), possibly as a consequence of small conformational changes stabilized by the disulfide bonds. In the presence of HslV, the ATPase rate of wild-type HslU was stimulated ∼3-fold, as observed previously (13, 20, 21), and ATP hydrolysis by WSSW3 and WSSWSSW2 was stimulated ∼2-fold, but ATP hydrolysis by WSSE3, WSSESSW2, or WSSESSE2 was not markedly stimulated (Fig. 4C). Thus, the presence of E subunits suppressed normal HslV stimulation of ATP hydrolysis by HslU pseudohexamers.
Figure 4.
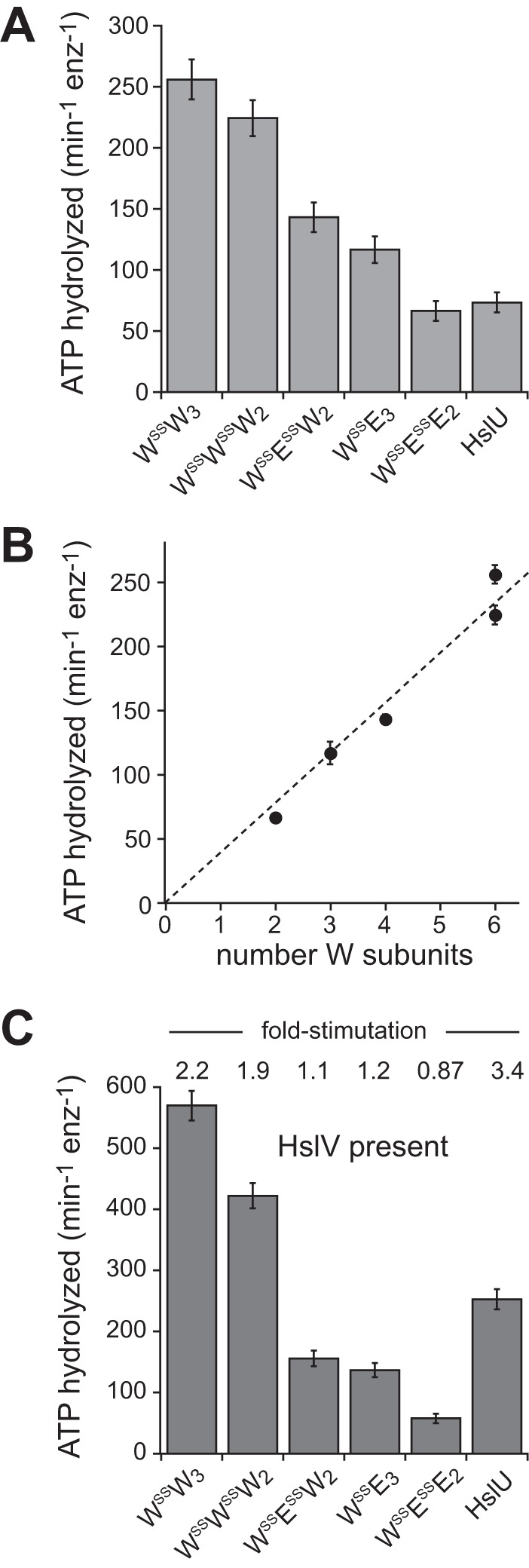
ATP hydrolysis by disulfide-cross-linked variants. A, basal rates of ATP hydrolysis by disulfide-cross-linked variants and wild-type HslU. Assays contained 0.3 μm HslU or pseudohexamers and 5 mm ATP. Values are averages of at least three replicates ± 1 S.D. (error bars). B, basal ATP hydrolysis rates for disulfide-cross-linked pseudohexamers plotted as a function of the number of W subunits. C, rates of ATP hydrolysis determined in the presence of 0.9 μm HslV dodecamer (other conditions as in A). The numbers above each bar represent the rate in the presence of HslV divided by the rate in the absence of HslV.
Degradation supported by disulfide-linked pseudohexamers
Arc repressor, a good substrate for HslUV degradation, is a metastable dimer that unfolds/dissociates with a half-life of ∼10 s but refolds in milliseconds to maintain a predominantly native structure (15, 22). We assayed the ability of different disulfide-linked pseudohexamers to support HslV degradation of Arc-CysA, where CysA designates a unique cysteine labeled with an Alexa-488 fluorophore (23), as autoquenching of the fluorophores in the native protein is relieved upon degradation. The WSSW3 and WSSWSSW2 pseudohexamers supported HslV degradation of a nearly saturating concentration of Arc-CysA at rates comparable with the wild-type HslU hexamer (Fig. 5A). Importantly, WSSESSW2 and WSSE3 also supported degradation at 35–45% of the wild-type rate, demonstrating that pseudohexamers with only three or four hydrolytically active subunits also have substantial degradation activity. Degradation supported by WSSESSE2 proceeded very slowly, at ∼3% of the wild-type rate.
Figure 5.

Degradation rates and energetic efficiencies of disulfide-cross-linked variants. A, rates of degradation of Arc-CysA (30 μm) assayed by increased fluorescence. B, rates of degradation of Arc-cp6GFP-st11-ssrA (20 μm) assayed by decreased fluorescence. C, degradation rates for Arc-CysA from A were divided by the number of wild-type subunits in the HslU variant and plotted against this number. D, degradation rates for Arc-cp6GFP-st11-ssrA from B were divided by the number of wild-type subunits in the HslU variant and plotted against this number. E, rates of ATP hydrolysis in the presence of 20 μm Arc-cp6GFP-st11-ssrA and 1 μm HslV. F, energetic efficiency determined by dividing the ATPase rates by the degradation rates. For A–D, experiments were performed using 0.3 μm HslU or variants and 0.9 μm HslV. For E and F, experiments were performed using 0.5 μm HslU or variants and 1 μm HslV. All assays contained 5 mm ATP and were performed at 37 °C. Values in A–E are averages (n ≥ 3) ± 1 S.D. (error bars). Error bars in F, propagated errors. In C and D, values for the three variants with six wild-type subunits were offset slightly on the x axis to allow visualization of error bars.
The degradation defects caused by E subunits in pseudohexamers were more severe for Arc-cp6GFP-st11-ssrA, a stable substrate that wild-type HslUV degrades ∼5-fold more slowly than Arc-CysA. WSSW3 and WSSWSSW2 supported HslV degradation of a high concentration of Arc-cp6GFP-st11-ssrA at nearly wild-type rates, but WSSESSW2 had only ∼20% activity, WSSE3 had only ∼10% activity, and WSSESSE2 was inactive (Fig. 5B). Degradation rates were normalized by dividing by the number of wild-type subunits in HslU or different variants and are plotted in Fig. 5C for the Arc-CysA substrate and Fig. 5D for the Arc-cp6GFP-st11-ssrA substrate. For Arc-CysA, there was a sharp discontinuity between two and three wild-type subunits. For Arc-cp6GFP-st11-ssrA, by contrast, the major discontinuity was between four and six subunits.
To determine the energetic efficiency of degradation of Arc-cp6GFP-st11-ssrA, we assayed the rate of ATP-hydrolysis for each disulfide-linked pseudohexamer in the presence of HslV and Arc-cp6GFP-st11-ssrA (Fig. 5E). We then divided the ATPase rate by the degradation rate to determine the average number of ATPs hydrolyzed during degradation of a single substrate (Fig. 5F). Notably, WSSW3, WSSE3, WSSWSSW2, and WSSESSW2 all had similar energetic efficiencies, hydrolyzing ∼500 ± 100 ATPs for each substrate degraded. Assuming that power strokes are tightly coupled to ATP hydrolysis, this result suggests that the WSSESSW2 and WSSE3 pseudohexamers use approximately the same number of power strokes as hexamers with six wild-type subunits to unfold and translocate Arc-cp6GFP-st11-ssrA. Thus, the slower degradation activities of the WSSESSW2 and WSSE3 enzymes compared with pseudohexamers with only wild-type subunits are principally a consequence of their slower rates of ATP hydrolysis.
Discussion
Our studies show that E. coli HslU variants containing hydrolytically active and inactive subunits at specific positions in the hexameric AAA+ ring can hydrolyze ATP, unfold proteins, and degrade substrates in collaboration with HslV. As we discuss below, these results support a probabilistic model of ATP hydrolysis and provide insights into the multivalent interactions between HslU and HslV that are required for efficient protein degradation.
Genetic tethering allowed us to express and purify HslU pseudohexamers consisting of a trimer of linked dimers. However, the W-W3 enzyme binds HslV poorly, suggesting that the tether interferes with HslV binding. Consistently, disulfide-linked pseudohexamers bind HslV well. In crystal structures of HslU hexamers alone, the C-terminal tails dock into a pocket, and the α-carboxyl group forms a salt bridge with an arginine in the sensor-2 motif of the same subunit (6, 10). These tail interactions were disrupted in several subunits in our low-resolution structure of W-E3 pseudohexamers, as expected if the attached tether prevents proper packing of these residues. In the H. influenzae HslUV complex, the C-terminal tails are detached from HslU and pack into grooves on HslV, with the HslU α-carboxylate forming a salt bridge with an HslV lysine side chain (7, 11, 12). In E. coli HslUV structures, by contrast, the tails remain docked into HslU (8, 9). Moreover, deletion of the five C-terminal residues of E. coli HslU does not prevent stimulation of HslV peptidase activity or degradation (20). Nevertheless, peptides corresponding to the C-terminal residues of HslU activate peptide cleavage by E. coli HslV, and mutations in HslV predicted to disrupt contacts with the C-terminal tails prevent HslU activation (20, 24). Our results support the importance of the C-terminal tails in high-affinity HslV binding and indicate that more than three tails of E. coli HslU must interact optimally with HslV to allow tight binding and efficient proteolysis.
The pseudohexamers that we studied have basal ATP hydrolysis rates roughly proportional to their total number of hydrolytically active W subunits. This result supports a model in which the W subunits in these pseudohexamers contribute independently to basal ATPase activity, making concerted or strictly sequential models unlikely. For example, if ATP hydrolysis in a specific subunit required prior hydrolysis in a neighboring subunit, as expected in a strictly sequential model, then a non-linear relationship between W subunits and ATP hydrolysis would be expected. HslV stimulates ATP hydrolysis by WSSW3 and WSSWSSW2 but caused little change in hydrolysis by WSSESSW2, WSSE3, or WSSESSE2. Because the E subunits in these latter enzymes also increase pseudohexamer affinity for HslV, stronger interactions between E subunits and HslV might restrict HslV-induced conformational changes required for higher ATPase activity in neighboring W subunits and thus explain the lack of ATPase stimulation. Alternatively, HslV binding might slow ATP dissociation from inactive E subunits, which becomes rate-limiting for hydrolysis in W subunits, especially if only a subset of subunits is nucleotide-bound at any given time.
Our strategies for engineering HslU rings with defined mixtures of active and inactive subunits were motivated by prior studies that applied these subunit-cross-linking methods to different hexameric AAA+ unfoldases and remodeling machines (5, 25–27). Indeed, genetic tethering experiments originally showed that ClpX also operates by a probabilistic mechanism, because ClpX pseudohexamers containing different combinations of hydrolytically active and inactive subunits unfold and degrade protein substrates in collaboration with the ClpP protease (5). Before the current study, however, it was not obvious that HslU and ClpX would operate using similar probabilistic mechanisms of ATP hydrolysis. First, HslU and ClpX contain unique family-specific auxiliary domains. The I domain of HslU emerges from the top of the AAA+ ring, between the Walker A and Walker B ATPase motifs, and regulates ATP hydrolysis, degradation, and autoinhibition (9, 20, 23). The N domain of ClpX, by contrast, serves as a docking site for some adaptors/substrates, but its deletion has little effect on ATP hydrolysis or degradation of many ClpXP substrates (28). Second, in crystal structures, the large and small AAA+ domains of each HslU subunit assume orientations that create a potential nucleotide binding site, whereas the corresponding domains of ClpX adopt structures that allow nucleotide binding in some subunits but prevent binding in other subunits (6–12, 29). Third, HslU hexamers make symmetric interactions with hexameric rings of HslV, whereas ClpX hexamers make asymmetric interactions with heptameric rings of ClpP (30).
The tethered W-E3 and E-W3 HslU pseudohexamers have ∼50% of the protein unfolding activity of the parental W-W3 enzyme, and the disulfide-linked WSSESSW2 and WSSE3 enzymes support HslV degradation of an easily degraded Arc-CysA substrate at 35–45% of the parental rates. Thus, protein unfolding and translocation by the AAA+ ring of HslU do not require ATP hydrolysis in adjacent subunits or in subunits immediately across the ring from each other. Again, these results support a model in which probabilistic hydrolysis in the AAA+ ring powers unfolding and translocation. Slower rates of ATP hydrolysis in rings with mixtures of active and inactive subunits correlate with their reduced mechanical activities, because the ATP cost of degradation of the more stable Arc-cp6GFP-st11-ssrA substrate is similar for the WSSESSW2 and WSSE3 pseudohexamers and their disulfide-linked parental enzymes containing six W subunits. Although optimal rates of ATP hydrolysis, unfolding, and degradation require six hydrolytically active HslU subunits, rings with only three or four active subunits use approximately the same number of power strokes to degrade this substrate.
We note that the WSSESSE2 pseudohexamer hydrolyzes ATP at ∼40–60% of the WSSE3 rate but supports no degradation of Arc-cp6GFP-st11-ssrA. These results suggest that some subunit-subunit communication is required for efficient unfolding and degradation by WSSESSE2 and/or that a minimum rate of ATP hydrolysis is required to unfold Arc-cp6GFP-st11-ssrA. Probabilistic models of ATP hydrolysis do not exclude coordination between ring subunits for efficient ATP hydrolysis, substrate binding, or mechanical function. In fact, the rate of ATP hydrolysis and the degree of substrate binding by HslU change in a positively cooperative fashion with ATP concentration, as expected for functional linkage between different subunits (13). In the AAA+ ring of ClpX, communication between subunits is necessary to allow staged ATP binding to drive conformational changes needed for function (31–33). Moreover, optical trapping experiments reveal that kinetic bursts of power strokes in the ClpX ring result in random patterns of shorter and longer translocation steps, supporting a probabilistic but coordinated mechanism (34, 35). Similar themes of probabilistic hydrolysis but coordinated function are seen in AAA+ unfolding rings assembled from non-identical subunits. In the Yta10/Yta12 m-AAA protease, for example, mutating the Walker A motif of either the Yta10 or the Yta12 subunits does not affect hydrolysis in the remaining wild-type subunits, but Walker B mutations in Yta12 trap ATP and prevent robust hydrolysis in adjacent Yta10 subunits (36). Finally, in the six distinct subunits of the Rpt1–6 AAA+ ring of the 26S proteasome, Walker B mutations in individual subunits have a wide range of effects on ATP hydrolysis and mechanical activity, requiring a model with some subunit-subunit coordination within the context of an inherently probabilistic mechanism of ATP hydrolysis (37).
Experimental procedures
Cloning, expression, and protein purification
Mutants were constructed and cloned by standard PCR techniques unless otherwise noted. To construct genetically linked HslU dimers, a gene encoding two HslU subunits separated by the 20-residue ASGAGGSEGGGSEGGTSGAT linker was cloned into the pet11a vector (Novagen). HslU mutants used to make disulfide-cross-linked pseudohexamers were constructed in the cysteine-free C262A/C288SHslU background with or without the E257Q mutation. Cysteine-free HslU supports robust ATP hydrolysis and substrate degradation (38–40). To make disulfide-cross-linked dimers, a gene encoding untagged E47CHslU was cloned into the first multiple cloning site (MCS1) of the pCOLADuet-1 (Novagen) vector between the NcoI and BamHI sites and a gene encoding His6-ENLYFQS-A349CHslU was cloned into MCS2 between the NdeI and XhoI sites, where His6 is the hexahistidine tag and ENLYFQS is the sequence recognized and cleaved by the tobacco etch virus protease. E47CHslU had a glycine residue inserted after the initiator methionine as a result of cloning. This pCOLADuet-1 vector was transformed into the of E. coli SHuffle T7 Express strain (New England Biolabs) for expression. To make disulfide-cross-linked trimers, a gene encoding untagged A349CHslU was cloned into MCS1 of pCOLADuet-1 between the NcoI and BamHI sites, and a gene encoding His6-ENLYFQS-E47CT361CHslU was cloned into MCS2 between the NdeI and XhoI sites. This vector was co-transformed with a pet12b (Novagen) vector encoding the untagged Q39CHslU variant into the SHuffle T7 Express strain. Both A349CHslU and Q39CHslU contained an additional glycine after the initiator methionine as a result of cloning. A gene encoding the Arc repressor from phage P22 followed by a cysteine residue and a hexahistidine tag (Arc-Cys-His6) was cloned and expressed in a pet21b vector (Novagen). Wild-type HslU and HslV were expressed from pet12b vectors. Arc-cp6GFP-st11-ssrA and I37AArc-cp6GFP-β5/β6-st11-ssrA (in which a GGTEGSLVPRGSGESGGS sequence between β-stands 5 and 6 allows thrombin cleavage and generation of a split substrate), and Arc-st11-ssrA were expressed from pet21b vectors as described (14, 15, 23).
Genetically linked HslU dimers were expressed and purified as described previously for wild-type HslU (20). Disulfide-cross-linked HslU dimers were expressed and purified as follows. E. coli SHuffle T7 Express cells carrying the pCOLA-Duet1 vector coding for the appropriate HslU mutants were grown at 30 °C until A600 of 0.6–0.8, the temperature was shifted to 18 °C, and protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 20 h. Cells were pelleted and resuspended in buffer A (50 mm Tris, pH 7.5, 300 mm NaCl, 20 mm imidazole, 0.5 mm EDTA), and 0.5 tablets of Complete Ultra EDTA-free protease inhibitor mixture (Roche Applied Science) and 1.5 μl of benzonase (250 units/μl; Sigma) per liter of the original culture were added. Cells were sonicated, the lysate was cleared by centrifugation, and 0.1% (v/v) PEI was added to the supernatant. This precipitate was cleared by centrifugation, and the supernatant was loaded onto Ni2+-NTA beads equilibrated in buffer B (50 mm Tris, pH 7.5, 300 mm NaCl, 20 mm imidazole). The Ni2+-NTA beads were washed extensively with buffer B, and protein was eluted with buffer C (50 mm Tris, pH 7.5, 300 mm NaCl, 250 mm imidazole). The eluate was diluted 3-fold with buffer D (50 mm Tris, pH 7.5, 10% (v/v) glycerol, 1 mm EDTA), loaded onto a Mono Q 10/100 GL column (GE Healthcare), and eluted with a linear gradient from 150 to 500 mm NaCl in buffer D (120 ml total). Appropriate Mono Q fractions were pooled, concentrated using an Amicon Ultra-15 centrifugal filter unit, and chromatographed on a Superdex 200 16/60 column (GE Healthcare) equilibrated in buffer E (50 mm Tris, pH 7.5, 300 mm NaCl, 10% (v/v) glycerol, 1.5 m urea, 1 mm EDTA), and 1-ml fractions were collected. Fractions with the highest purity of the cross-linked dimer as judged by non-reducing SDS-PAGE were pooled, concentrated, and run on another Superdex 200 16/60 column equilibrated in buffer F (50 mm Tris, pH 7.5, 300 mm NaCl, 10% (v/v) glycerol, 1 mm EDTA). Fractions with the highest purity of cross-linked dimer were pooled and concentrated. The concentration of cross-linked dimer was determined in hexameric equivalents by measuring the absorbance at 280 nm using 148,545 m−1 cm−1 as the molar extinction coefficient. Concentrated protein was divided into small aliquots and was flash-frozen at −80 °C.
Disulfide-cross-linked HslU trimers were expressed and purified largely as described for cross-linked dimers. His6 tags were present on Cys349 subunits for dimers and Cys47/Cys361 subunits for trimers. After precipitation with PEI, the sample was cleared by centrifugation, and the supernatant was loaded onto a 5-ml HisTrap HP column (GE Healthcare) equilibrated in buffer B. The column was washed with 75 ml of buffer B, the sample was eluted with a gradient of 20–500 mm imidazole in buffer B (100 ml total), and 2-ml fractions were collected. Fractions were pooled and dialyzed overnight against buffer G (50 mm Tris, pH 7.5, 150 mm NaCl, 10% glycerol (v/v), 1 mm EDTA) at 4 °C. The dialyzed material was loaded onto a Mono Q 10/100 GL column, and purification then proceeded by the method described for cross-linked dimers. The concentration of the cross-linked trimer was determined in hexameric equivalents by measuring absorbance at 280 nm using 147,180 m−1 cm−1 as the molar extinction coefficient. Concentrated protein was aliquoted and flash-frozen at −80 °C.
A pet21b vector carrying Arc with a C-terminal CHHHHHH tail (Arc-Cys-His6) was transformed into the E. coli X90 (λDE3) slyD::kan hslUV::tet strain, and cells were grown to an A600 of 0.6–0.8 at 37 °C. Protein expression was induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside and continued for 4 h at room temperature. Cells were resuspended and lysed, and Arc-Cys-His6 was purified by Ni2+-NTA affinity chromatography as described for purification of cross-linked HslU dimers. After Ni2+-NTA purification, protein was concentrated and injected onto a Superdex 75 16/60 column (GE Healthcare) equilibrated in buffer H (50 mm Tris, pH 7.5, 300 mm NaCl, 10% glycerol (v/v), 1 mm EDTA, 2 mm DTT). Fractions containing the Arc protein were concentrated and flash-frozen at −80 °C. Arc-Cys-His6 was labeled with the maleimide derivative of Alexa-488 (Thermo Fisher Scientific) to generate Arc-CysA (23). His6-tagged wild type HslU, His6-tagged HslV, I37AArc-cp6GFP-β5/β6-st11-ssrA, Arc-cp6GFP-st11-ssrA, and Arc-st11-ssrA were expressed and purified as described (23).
Biochemical assays
Unless noted, assays were performed at 37 °C in PD buffer (25 mm HEPES, pH 7.5, 5 mm KCl, 10% glycerol (v/v), 20 mm MgCl2, 0.032% Igepal CA-630). Hydrolysis of 5 mm ATP was measured using an NADH-coupled assay (41) by monitoring the loss of absorbance at 340 nm on a Spectramax M5 plate reader (Molecular Devices). Cleavage of I37AArc-cp6GFP-β5/β6-st11-ssrA with thrombin was performed as described (14). Rates of unfolding of different concentrations of thrombin-split I37AArc-cp6GFP-β5/β6-st11-ssrA were determined by changes in cp6GFP fluorescence (excitation, 467 nm; emission, 511 nm) in assays that contained 0.5 μm HslU or tethered variants and 5 mm ATP. Degradation of 30 μm Arc-CysA (monomer equivalents) was measured on a Spectramax M5 plate reader by monitoring the increase in fluorescence (excitation, 480 nm; emission, 520 nm). Degradation of 20 μm Arc-cp6GFP-st11-ssrA (monomer equivalents) was measured by monitoring the decrease in GFP fluorescence on a Spectramax M5 plate reader (excitation, 467 nm; emission, 511 nm). Degradation reactions contained 5 mm ATP and a regeneration system consisting of 16 mm creatine phosphate and 10 μg/ml creatine kinase. HslV activation assays were performed at 25 °C in the presence of ATP as described (13, 23), and K½ values were determined by fitting to a hyperbolic equation.
Crystallography
The W-E3 pseudohexamer was crystallized by the hanging drop method using 100 mm BisTris (pH 5.8), 26% (w/v) PEG 3350, and 260 mm ammonium sulfate as the well solution. Molecular replacement using Phaser (42) was initially used to solve the structure using a 1HQY hexamer (17) as the search model. We then replaced each 1HQY subunit with a 5JI3 subunit (23) to improve geometry and used rigid body refinement of individual domains, refinement of one B-factor and TLS group per subunit, and very tightly constrained positional refinement with torsional NCS constraints in Phenix (43). Coot (44) was used for model building, and MolProbity (45) was used to assess the geometry of the model.
Author contributions
J. C. performed experiments with HslU subunits linked by genetically encoded tethers. A. R. N. designed the split substrate for unfolding experiments. S. E. G., R. A. G., and R. T. S. performed crystallographic experiments. V. B. performed all remaining experiments. V. B. and R. T. S. wrote the manuscript. V. B., J. C., S. E. G., A. R. N., R. A. G., T. A. B, and R. T. S. contributed to the design and interpretation of experiments and approved the final manuscript.
This work was supported by National Institutes of Health Grant AI-16892. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5TXV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NTA
- nitrilotriacetic acid
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- enz
- enzyme concentration.
References
- 1. Ogura T., and Wilkinson A. J. (2001) AAA+ superfamily ATPases: common structure–diverse function. Genes Cells 6, 575–597 [DOI] [PubMed] [Google Scholar]
- 2. Snider J., Thibault G., and Houry W. A. (2008) The AAA+ superfamily of functionally diverse proteins. Genome Biol. 9, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gai D., Zhao R., Li D., Finkielstein C. V., and Chen X. S. (2004) Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 119, 47–60 [DOI] [PubMed] [Google Scholar]
- 4. Enemark E. J., and Joshua-Tor L. (2006) Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–275 [DOI] [PubMed] [Google Scholar]
- 5. Martin A., Baker T. A., and Sauer R. T. (2005) Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature 437, 1115–1120 [DOI] [PubMed] [Google Scholar]
- 6. Bochtler M., Hartmann C., Song H. K., Bourenkov G. P., Bartunik H. D., and Huber R. (2000) The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature 403, 800–805 [DOI] [PubMed] [Google Scholar]
- 7. Sousa M. C., Trame C. B., Tsuruta H., Wilbanks S. M., Reddy V. S., and McKay D. B. (2000) Crystal and solution structures of an HslUV protease-chaperone complex. Cell 103, 633–643 [DOI] [PubMed] [Google Scholar]
- 8. Wang J., Song J. J., Franklin M. C., Kamtekar S., Im Y. J., Rho S. H., Seong I. S., Lee C. S., Chung C. H., and Eom S. H. (2001) Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure 9, 177–184 [DOI] [PubMed] [Google Scholar]
- 9. Song H. K., Hartmann C., Ramachandran R., Bochtler M., Behrendt R., Moroder L., and Huber R. (2000) Mutational studies on HslU and its docking mode with HslV. Proc. Natl. Acad. Sci. U.S.A. 97, 14103–14108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trame C. B., and McKay D. B. (2001) Structure of Haemophilus influenzae HslU protein in crystals with one-dimensional disorder twinning. Acta Crystallogr. D Biol. Crystallogr. 57, 1079–1090 [DOI] [PubMed] [Google Scholar]
- 11. Sousa M. C., Kessler B. M., Overkleeft H. S., and McKay D. B. (2002) Crystal structure of HslUV complexed with a vinyl sulfone inhibitor: corroboration of a proposed mechanism of allosteric activation of HslV by HslU. J. Mol. Biol. 318, 779–785 [DOI] [PubMed] [Google Scholar]
- 12. Kwon A. R., Kessler B. M., Overkleeft H. S., and McKay D. B. (2003) Structure and reactivity of an asymmetric complex between HslV and I-domain deleted HslU, a prokaryotic homolog of the eukaryotic proteasome. J. Mol. Biol. 330, 185–195 [DOI] [PubMed] [Google Scholar]
- 13. Yakamavich J. A., Baker T. A., and Sauer R. T. (2008) Asymmetric nucleotide transactions of the HslUV protease. J. Mol. Biol. 380, 946–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nager A. R., Baker T. A., and Sauer R. T. (2011) Stepwise unfolding of a β barrel protein by the AAA+ ClpXP protease. J. Mol. Biol. 413, 4–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burton R. E., Baker T. A., and Sauer R. T. (2005) Nucleotide-dependent substrate recognition by the AAA+ HslUV protease. Nat. Struct. Mol. Biol. 12, 245–251 [DOI] [PubMed] [Google Scholar]
- 16. Sundar S., McGinness K. E., Baker T. A., and Sauer R. T. (2010) Multiple sequence signals direct recognition and degradation of protein substrates by the AAA+ protease HslUV. J. Mol. Biol. 403, 420–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J., Song J. J., Seong I. S., Franklin M. C., Kamtekar S., Eom S. H., and Chung C. H. (2001) Nucleotide-dependent conformational changes in a protease-associated ATPase HslU. Structure 9, 1107–1116 [DOI] [PubMed] [Google Scholar]
- 18. Dombkowski A. A. (2003) Disulfide by DesignTM: a computational method for the rational design of disulfide bonds in proteins. Bioinformatics 19, 1852–1853 [DOI] [PubMed] [Google Scholar]
- 19. Lobstein J., Emrich C. A., Jeans C., Faulkner M., Riggs P., and Berkmen M. (2012) SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Fact. 11, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seong I. S., Kang M. S., Choi M. K., Lee J. W., Koh O. J., Wang J., Eom S. H., and Chung C. H. (2002) The C-terminal tails of HslU ATPase act as a molecular switch for activation of HslV peptidase. J. Biol. Chem. 277, 25976–25982 [DOI] [PubMed] [Google Scholar]
- 21. Sundar S., Baker T. A., and Sauer R. T. (2012) The I domain of the AAA+ HslUV protease coordinates substrate binding, ATP hydrolysis, and protein degradation. Protein Sci. 21, 188–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Milla M. E., and Sauer R. T. (1994) P22 Arc repressor: folding kinetics of a single-domain, dimeric protein. Biochemistry 33, 1125–1133 [DOI] [PubMed] [Google Scholar]
- 23. Baytshtok V., Fei X., Grant R. A., Baker T. A., and Sauer R. T. (2016) A structurally dynamic region of the HslU intermediate domain controls protein degradation and ATP hydrolysis. Structure 24, 1766–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ramachandran R., Hartmann C., Song H. K., Huber R., and Bochtler M. (2002) Functional interactions of HslV (ClpQ) with the ATPase HslU (ClpY). Proc. Natl. Acad. Sci. U.S.A. 99, 7396–7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Glynn S. E., Nager A. R., Baker T. A., and Sauer R. T. (2012) Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat. Struct. Mol. Biol. 19, 616–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Biter A. B., Lee S., Sung N., and Tsai F. T. F. (2012) Structural basis for intersubunit signaling in a protein disaggregating machine. Proc. Natl. Acad. Sci. U.S.A. 109, 12515–12520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamasaki T., Oohata Y., Nakamura T., and Watanabe Y. H. (2015) Analysis of the cooperative ATPase cycle of the AAA+ chaperone ClpB from Thermus thermophilus by using ordered heterohexamers with an alternating subunit arrangement. J. Biol. Chem. 290, 9789–9800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baker T. A., and Sauer R. T. (2012) ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 1823, 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glynn S. E., Martin A., Nager A. R., Baker T. A., and Sauer R. T. (2009) Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 139, 744–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grimaud R., Kessel M., Beuron F., Steven A. C., and Maurizi M. R. (1998) Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. J. Biol. Chem. 273, 12476–12481 [DOI] [PubMed] [Google Scholar]
- 31. Stinson B. M., Nager A. R., Glynn S. E., Schmitz K. R., Baker T. A., and Sauer R. T. (2013) Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine. Cell 153, 628–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hersch G. L., Burton R. E., Bolon D. N., Baker T. A., and Sauer R. T. (2005) Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell 121, 1017–1027 [DOI] [PubMed] [Google Scholar]
- 33. Stinson B. M., Baytshtok V., Schmitz K. R., Baker T. A., and Sauer R. T. (2015) Subunit asymmetry and roles of conformational switching in the hexameric AAA+ ring of ClpX. Nat. Struct. Mol. Biol. 22, 411–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sen M., Maillard R. A., Nyquist K., Rodriguez-Aliaga P., Pressé S., Martin A., and Bustamante C. (2013) The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell 155, 636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cordova J. C., Olivares A. O., Shin Y., Stinson B. M., Calmat S., Schmitz K. R., Aubin-Tam M.-E. Baker T. A., Lang M. J., and Sauer R. T. (2014) Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell 158, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Augustin S., Gerdes F., Lee S., Tsai F. T., Langer T., and Tatsuta T. (2009) An intersubunit signaling network coordinates ATP hydrolysis by m-AAA proteases. Mol. Cell 35, 574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beckwith R., Estrin E., Worden E. J., and Martin A. (2013) Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat. Struct. Mol. Biol. 20, 1164–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yoo S. J., Kim H. H., Shin D. H., Lee C. S., Seong I. S., Seol J. H., Shimbara N., Tanaka K., and Chung C. H. (1998) Effects of the Cys mutations on structure and function of the ATP-dependent HslVU protease in Escherichia coli. The Cys287 to Val mutation in HslU uncouples the ATP-dependent proteolysis by HslVU from ATP hydrolysis. J. Biol. Chem. 273, 22929–22935 [DOI] [PubMed] [Google Scholar]
- 39. Seong I. S., Oh J. Y., Yoo S. J., Seol J. H., and Chung C. H. (1999) ATP-dependent degradation of SulA, a cell division inhibitor, by the HslVU protease in Escherichia coli. FEBS Lett. 456, 211–214 [DOI] [PubMed] [Google Scholar]
- 40. Yakamavich J. A. (2008) Control of HslUV Protease Function by Nucleotide Binding and Hydrolysis, Ph.D. thesis, Massachusetts Institute of Technology, Cambridge, MA [Google Scholar]
- 41. Nørby J. G. (1988) Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 156, 116–119 [DOI] [PubMed] [Google Scholar]
- 42. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]