

Abstract
Repressor activator protein 1 (Rap1) performs multiple vital cellular functions in the budding yeast Saccharomyces cerevisiae. These include regulation of telomere length, transcriptional repression of both telomere-proximal genes and the silent mating type loci, and transcriptional activation of hundreds of mRNA-encoding genes, including the highly transcribed ribosomal protein- and glycolytic enzyme-encoding genes. Studies of the contributions of Rap1 to telomere length regulation and transcriptional repression have yielded significant mechanistic insights. However, the mechanism of Rap1 transcriptional activation remains poorly understood because Rap1 is encoded by a single copy essential gene and is involved in many disparate and essential cellular functions, preventing easy interpretation of attempts to directly dissect Rap1 structure-function relationships. Moreover, conflicting reports on the ability of Rap1-heterologous DNA-binding domain fusion proteins to serve as chimeric transcriptional activators challenge use of this approach to study Rap1. Described here is the development of an altered DNA-binding specificity variant of Rap1 (Rap1AS). We used Rap1AS to map and characterize a 41-amino acid activation domain (AD) within the Rap1 C terminus. We found that this AD is required for transcription of both chimeric reporter genes and authentic chromosomal Rap1 enhancer-containing target genes. Finally, as predicted for a bona fide AD, mutation of this newly identified AD reduced the efficiency of Rap1 binding to a known transcriptional coactivator TFIID-binding target, Taf5. In summary, we show here that Rap1 contains an AD required for Rap1-dependent gene transcription. The Rap1AS variant will likely also be useful for studies of the functions of Rap1 in other biological pathways.
Keywords: gene regulation, protein engineering, Saccharomyces cerevisiae, transcription regulation, yeast, altered DNA binding specificity, transcription activation, transcription activator
Introduction
Transcription activation is the first step in the carefully orchestrated process of eukaryotic protein-coding gene expression (1). This complex and evolutionarily conserved (2) process is essential for life as it controls the precise location, timing, and level of production of specific mRNA transcripts and hence the cognate encoded protein products (3, 4). Eukaryotic cells employ multiple distinct proteins, and protein complexes, to catalyze mRNA gene transcription (5). Collectively termed the transcription machinery, these proteins include the 12-subunit DNA-dependent RNA polymerase II (RNAPII)3 and six general transcription factors (GTFs) TFIIA, TFIIB, TFIID, TFIIE, TFIIF, and TFIIH. Upon interaction with accessible promoters, the GTFs seed the formation of a functional pre-initiation complex (PIC) by RNAPII. Under appropriate conditions the PIC is able to initiate mRNA gene transcription (6) and transition into early elongation complexes (ECs). PIC formation occurs upon nucleosomal DNA, and both PIC formation and PIC/EC function are controlled by a cadre of transcriptional coregulators (coactivators and corepressors), some of which act at the level of chromatin to alter the accessibility of the underlying DNA, covalently modify histones, and/or modulate the activity of the transcription machinery (7, 8).
Although together the transcription machinery and coregulators clearly play essential roles in mRNA biosynthesis, it is the enhancer DNA-binding transcriptional activator proteins that confer gene specificity to transcriptional control by selectively responding to discrete cell-external and/or cell-internal molecular cues (9). Activator proteins typically carry out this function through the use of (minimally) two modular, functionally distinct domains, a DNA-binding domain (DBD) and an activation domain (AD) (10). Upon receiving the signal for gene activation, a transcriptional activator protein binds to its specific and accessible enhancer target recognition site in chromatin through its DBD. These enhancer DNA-bound proteins then transmit a signal for activation, via their AD, which either directly or indirectly results in the activation of the mRNA gene transcription machinery on the promoter of the cis-linked target gene (10).
Activation signal transmission usually occurs through direct AD-coactivator interaction. Characterized coactivator targets are quite diverse. Some coactivators, like TFIIA, TFIIB, and TFIID, are obligate critical components of the transcription machinery (11–15), whereas others such as SAGA, Swi/Snf, p300, and OCA-B target chromatin and/or other coregulators (16–19). Identifying AD-coregulator interactions and, more importantly, understanding the mechanistic outcomes of these interactions is an active yet incompletely understood area of research (2, 4, 20). Studies that define the structure and function of transactivator ADs, and their coactivator targets, will provide critical tools for developing deeper insight into the molecular mechanisms that regulate specific target gene transcription.
Saccharomyces cerevisiae repressor activator protein 1 (Rap1) is an evolutionarily conserved protein whose cognate enhancer binding site (upstream activating sequence; UASRap1), DBD structure, and biological activities have been studied for over 30 years (21). In budding yeast, Rap1 is encoded by a single copy essential gene (22). As its name suggests, the 827-amino acid Rap1 protein performs key repression and activation functions in the cell. In addition to transcriptional regulation, Rap1 also modulates critical aspects of telomere biology, boundary element formation, and meiotic DNA recombination (23). Early sequence alignments, gross deletion analyses, and functional studies indicated that Rap1 is tripartite. The three major domains of S. cerevisiae Rap1 include a dispensable N terminus, part of which encodes a breast cancer 1 C-terminal (BRCT) homology domain (amino acids (aa) 1–360) (24–26), a central DNA-binding domain (DBD) (aa 361–599 (27)), and a C-terminal domain (aa 600–827) that is important for multiple telomere functions as well as HML/HMR mating type locus silencing (28, 29). Bioinformatic analyses have failed to identify any clear consensus AD element(s) within the protein.
Of the three domains of Rap1, only the DBD is essential, although yeast expressing just the DBD are extremely slow growing. Removal of Rap1 sequences C-terminal to the DBD produce yeast that display a slow growth phenotype (30, 31). Several analyses have shown that aa within the very C terminus of Rap1 function in silencing through direct binding of the silent information regulator (Sir) and Rap1 interacting factor (Rif) proteins (28, 29, 32–34). These proteins serve to modulate telomere length and repress telomere proximal gene transcription. Out of the many cellular functions that Rap1 performs, the telomere-specific functions of Rap1 are the best understood in terms of molecular mechanism (25, 34, 35).
Rap1 was first identified as an enhancer DNA-binding factor for ribosomal protein (RP)-encoding genes (21). Subsequent studies have extensively characterized RP gene enhancer elements (i.e. the UASRap1). This work defined the following: (a) a 13-base pair (bp) UASRap1 consensus sequence 5′-1(A/G)(C/A)A(C/T)CC(A/G)(C/A)NCA(C/T)(C/T)13-3′ (30, 36–38); (b) the contribution of each nucleotide (nt) within the enhancer to both Rap1- UASRap1 DNA binding (36) and Rap1-dependent reporter gene activity (39); and (c) there appear to be biologically important functional differences in the sequences of Rap1 DNA-binding sites involved in transcriptional activation, transcriptional repression, and telomere function(s) (40–46). The structure of the Rap1 DBD (27) in complex with multiple Rap1 DNA-binding elements has been solved by X-ray crystallography. The DBD is composed of two very similar homeodomain motifs that bind Rap1 DNA sites in tandem. Multiple specific protein-DNA contacts form throughout the protein-DNA complex. These contacts do not markedly change when Rap1 binds different DNA target sequences (47–49).
Compared with the amount of knowledge describing how Rap1 binds DNA and silences telomeres, almost nothing is known about how Rap1 performs its transcription activation function. Indeed, the existence of a putative Rap1 AD(s) has remained ambiguous. One study mapped a potential AD to Rap1 aa 630–695 through the use of Gal4 DBD-Rap1 fusions (33). However, these investigators neither assessed the putative AD function of these aa in the context of the intact protein nor precisely mapped and assessed the contributions of individual aa within the implicated domain sequence toward transcriptional activation of the UASGAL-lacZ reporter gene they utilized. Soon after, others tested this idea by direct internal deletion of putative AD sequences. Although removal of Rap1 aa 630–695 induces slow growth, such cells exhibit a 2-fold increase over wild type (WT) of the two Rap1-dependent ribosomal protein gene mRNA transcripts scored (RPL19 and RPL45), and a variable decrease in the two Rap1-dependent GE gene mRNAs scored (PYK1 mRNA ∼70% WT; PGK1 mRNA ∼10% WT) (30). These data challenge the idea that Rap1 C-terminal aa 630–695 compose an AD. Two additional sets of observations further complicate this issue. First, Morse and co-workers (50–52) have extensively documented the nucleosome remodeling activity of Rap1 and its impact on gene transcription activation. These workers found that Rap1 appears to drive transcription via a chromatin opening mechanism. In this context both Rap1 N- and C-terminal sequences are dispensable for HIS4 gene transcription activation, suggesting that the Rap1 DBD alone drives activation through a chromatin-based mechanism (50–52). Consistent with these observations, Rap1 can efficiently bind nucleosomal UASRap1 sequences, in vitro and in vivo (53, 54). Second, several other groups have reported that Rap1-heterologous DBD fusions fail to robustly drive reporter gene expression in vivo in yeast (55–57). Collectively, these data leave open the questions of which Rap1 domain(s) and via what molecular mechanism(s) Rap1 turns on target gene transcription.
Although the data noted above leaves unanswered the question of what domain(s) of Rap1 might serve as AD to stimulate target gene transcription, there is no doubt that Rap1 activates many genes in yeast, including the RP- and GE-encoding genes. Numerous studies have shown that mutation of the Rap1 UASRap1-binding sites located upstream of RP- and GE-encoding genes reduces mRNA levels by 50–90% depending on the gene in question (57–60). Other studies show that RP gene transcription absolutely depends on Rap1 (31, 61, 62), despite the fact that multiple transcription regulators, including Fhl1, Ifh1/Crf1, Sfp1, Esa1, and Hmo1, have all been shown to contribute to RP gene regulation (61–67). Confirming these studies, chromatin immunoprecipitation (ChIP)-Seq and ChIP-Exo studies document the presence of Rap1 on genes carrying UASRap1 elements (37, 45, 46, 58, 68), whereas acute depletion of nuclear Rap1 using the “anchor-away” technique (69) shows that ongoing RP gene transcription requires Rap1 (58). Finally, Rap1 exhibits another characteristic of bona fide transcription activators; it directly binds to coactivator complexes (2, 5, 70). Rap1 binds the purified coregulators Swi/Snf, a chromatin remodeler (19), and the coactivator TFIID (31). Collectively, these studies argue that Rap1 has all the predicted properties of a typical transactivator and thus likely contains an AD.
Our previous analyses of Rap1-TFIID interaction have shown that Rap1-TFIID TBP-associated factor (Taf) interaction occurs through the Taf4, Taf5, and Taf12 subunits of the complex. Furthermore, we have mapped the relevant Rap1-binding domains (RBDs) of each protein (31) and demonstrated that deletion of the cognate RBD-encoding sequences of TAF4 or TAF5 causes lethality (31, 71), results consistent with the idea that these RBDs play important roles in cell physiology. Point mutagenesis of the two RBDs induces temperature-conditional growth, decreases the affinity of Rap1-Taf4/12 and Rap1-Taf5 interaction, and concomitantly drastically and preferentially reduces cellular RP gene transcript levels (71). These data have led us to conclude that TFIID serves as a key Rap1 coactivator on the RP genes, one that transduces activating signals from Rap1 directly to the transcription machinery. Collectively, these data motivated us to redouble our efforts to identify, characterize, and ultimately generate mutant variants of an AD within Rap1 to facilitate dissection of the molecular mechanism(s) by which this potent activator turns on transcription of its target genes.
As an alternative approach toward identifying an AD within budding yeast Rap1, we devised a strategy to attempt to generate an altered DNA-binding specificity Rap1 variant (Rap1AS). Our goal was to identify a form of Rap1 that exhibited true altered specificity (AS) of DNA binding. Such a variant form of the protein, Rap1AS, would not efficiently bind at “WT” UASRap1 elements but could bind and drive the expression of an integrated selectable reporter gene from a distinct mutated form of the UASRap1 enhancer. Thus, Rap1AS would obviate the complications that arise as a result of the myriad Rap1 activities required for the proper expression of hundreds of essential Rap1-dependent genes as well as regulation of telomere function.
Generation of a Rap1AS variant would allow for the straightforward molecular genetic dissection of Rap1 structure-function relationships by scoring function/expression of the Rap1AS-dependent reporter gene in cells containing engineered deletion or point-mutated variant forms of Rap1AS. Breakthroughs in the understanding of transcription mechanisms have been made through the generation and utilization of altered DNA-binding specificity mutants of other essential transcription factors, in both prokaryotes and eukaryotes. Examples include AS variants of bacteriophage λ cro protein (72); Escherichia coli Trp repressor (73) and sigma factor σ70 (74); yeast TBP (75) and transcription factor Gcn4 (76); mammalian estrogen receptor (77, 78); and Drosophila transcription factor Engrailed (79). These altered DNA-binding variant proteins proved key in unlocking the molecular mechanisms by which these disparate DNA-binding proteins operate.
This report describes structure-based, site-directed mutagenesis of UASRap1 DNA and the gene encoding the Rap1 protein itself, coupled with a sensitive yeast screening strategy, to identify an altered DNA-binding specificity variant of Rap1, termed Rap1AS. The gene encoding Rap1AS was subjected to systematic deletion and point mutagenesis to identify a 41-amino acid-long AD within the C-terminal portion of the protein that is essential for driving high level reporter gene expression. This AD has the features of acidic activation domains described in other eukaryotes as it contains seven evolutionarily conserved hydrophobic residues within the element that contribute critically to the activation potential of Rap1AS. Mutation of all seven of these conserved residues to alanine within WT Rap1 (Rap1WT) induces a dramatic slow growth phenotype while simultaneously reducing transcription of RP- and GE-encoding genes.
Our work definitively establishes the fact that Rap1 contains an AD that is required for Rap1-dependent chromosomal gene transcription. Access to WT and multiple distinct variant forms of Rap1 will prove invaluable for further dissection of the molecular mechanisms through which this protein drives high level transcription of the vigorously transcribed RP- and GE-encoding genes. Indeed, we present the results of protein-protein interaction experiments that show that mutation of key Rap1 AD residues significantly reduces the binding of the variant Rap1 to Taf5, one of the known TFIID coactivator subunit targets of Rap1 (31, 71). Finally, we note that the Rap1AS variant should also prove useful for investigators who study the complex biological functions of Rap1 in transcriptional repression, boundary element establishment, telomere length regulation, chromatin opening, and meiotic recombination (23, 80, 81).
Results
Generation of an altered DNA-binding specificity variant Rap1AS
Generation of Rap1AS was approached in two steps. First, specific subsequences of the UASRap1 were systematically mutagenized to identify variants that significantly decreased binding of Rap1WT to UASRap1 DNA. These experiments identified a specific “inactivated” UASRap1. Second, the RAP1 sequences encoding the Rap1 amino acid residues predicted by X-ray crystallography to contact the key mutationally sensitive UASRap1 DNA bps were subjected to site-directed mutagenesis. Plasmids carrying these mutant RAP1 variants were then introduced into yeast carrying an integrated reporter gene whose expression was driven by the inactivated mutant UASRap1 enhancer. Screening many such yeast transformant colonies, each expressing a different variant form of mutated Rap1, was expected to result in the identification of rare mutant variants that had gained the ability to bind the mutant UASRap1 and thereby activate reporter gene expression driven by the inactivated UASRap1 identified in the first step of this strategy.
The structure of the Rap1 DBD (27) bound to various target DNAs guided our Rap1AS generation strategy. These Rap1-DNA structures show that the Rap1 DBD binds its recognition motif primarily via two homeodomains and an unstructured tail (47–49). Multiple studies have shown that within a homeodomain, the third, or recognition helix of the three helix bundle, mediates sequence-specific DNA recognition (82, 83). The apposition of the homeodomain recognition helix with the upstream half of the Rap1-binding sequence is shown in Fig. 1A. DNA bp2, bp3, and bp4 are contacted by the recognition helix of Rap1 DBD homeodomain 1 (Fig. 1A (bp labeled bp2, bp3, and bp4, and the homeodomain is dark blue)). These base pairs were specifically chosen for mutational analyses to avoid the 3′-half of the Rap1 DNA recognition motif because others have proposed that UASRap1 3′ sequences determine whether Rap1 exerts activator or repressor activity when bound to a particular genomic locus (41, 45).
Figure 1.
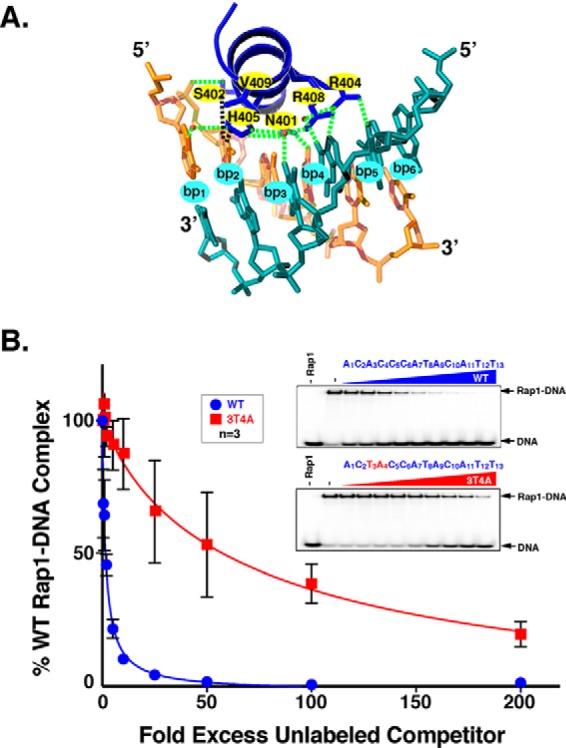
Mutation of WT UASRap1 nucleotides 3A and 4T to 3T and 4A significantly decreases binding of WT Rap1 to 3T4A UASRap1 DNA. A, Rap1 DBD homeodomain-1 recognition helix (dark blue, top) is shown in complex with its DNA recognition site labeled bp1, bp2, bp3, bp4, bp5, bp6. Top strand, tan hues (5′-1ACACCC6-3′), and bottom strand, teal hues. Hydrogen bonds between protein and DNA are indicated by dashed green lines; H-bonds mediated by H2O molecules are indicated by green dashed lines and red spheres; hydrophobic interactions are indicated by black dashed lines. The DNA base pairs mutated to create the 3T4A mutant-binding site are bp3 and bp4. Rap1 DBD recognition helix amino acid residues targeted for codon randomization mutagenesis are indicated by yellow ovals at amino acid positions N401, S402, H405, R408, and V409. Image was generated using PyMOL (130) from Protein Data Bank code 1IGN (47). B, gel shift competition DNA-binding analyses with WT UASRap1 or 3T4A UASRap1 DNAs. Gel shift binding reactions were performed by incubating 100 fmol of purified recombinant Rap1WT with 50 fmol (700 cpm/fmol) of duplex 32P-labeled WT UASRap1 DNA (5′-1ACACCCATACATT13-3′) alone (No Rap1, −Rap1), or with Rap1 and either no competitor (−), or the indicated fold molar-excess of either cold WT UASRap1 (top gel scan) or cold 3T4A UASRap1 (bottom gel scan); 0.5×, 1×, 2×, 5×, 10×, 25×, 50×, 100×, or 200× left to right (WT, blue circles; 3T4A, red squares) in a final volume of 20 μl. Reactions were fractionated on non-denaturing polyacrylamide gels, vacuum-dried, and imaged using a Bio-Rad Pharos FX imager. The amount of bound complex from each reaction was quantified using Bio-Rad Quantity One software. Data were analyzed using GraphPad Prism 7 software and are expressed as % Rap1WT-[32P]DNA complex when no competitor is present (i.e. + Rap1 and − competitor). Plot curves were generated using an [inhibitor] versus response non-linear fit. Error bars represent S.D. A representative image for each competition was chosen from among three independent replicates.
All possible single and double point mutant variants of UASRap1 bp2, bp3, and bp4 were tested for the ability to bind purified Rap1WT via gel shift competition DNA binding analyses. The assays used Rap1WT protein and 32P-labeled duplex WT UASRap1 probe and increasing concentrations of unlabeled WT or mutated duplex competitor DNAs. These experiments showed that no single base pair mutation had a significant effect on the affinity of Rap1-DNA binding (data not shown), consistent with prior mutational analysis of UASRap1 (36). However, several of the double base pair mutant UASRap1-binding sites caused a significant decrease in Rap1WT-DNA binding. The 3T4A UASRap1 mutant reduced Rap1WT binding by over 20-fold relative to the WT UASRap1 (compare loss of Rap1-DNA complex in the presence of WT and 3T4A competitor DNAs at 2-, 5-, 10-, and 25-fold mole excesses; Fig. 1B). This reduction in affinity (competition strength) was the largest of the double mutants tested (data not shown). Consequently, the 3T4A UASRap1 was selected to serve as the inactivatedUASRap1 sequence in screens for selection of Rap1AS variants.
Rap1 mutagenesis and screening strategy
To generate a Rap1AS variant capable of binding the 3T4A UASRap1 site, we utilized a mutagenesis approach recently employed to create altered DNA-binding specificity variants of the Drosophila engrailed homeodomain (79). The six amino acids of the Rap1 DBD homeodomain-1 DNA recognition helix that face DNA (Fig. 1A) were chosen as targets for codon-directed randomization mutagenesis (Fig. 2A; Rap1 aa Asn-401, Ser-402, Arg-404, His-405, Arg-408, and Val-409) (84). To identify a Rap1AS from the library, a direct yeast screening strategy was devised that could rapidly test millions of Rap1 mutant variants for the ability to functionally interact with 3T4A UASRap1in vivo. The functional interaction chosen for this screen utilized a HIS3 reporter gene. In the reporter used for Rap1AS screening, the two WT UASRap1 DNA-binding sites present in a previously characterized UASRap1-HIS3 reporter (31, 57) were replaced with two copies of the 3T4A variant UASRap1 (see Figs. 1B and 2B). HIS3 reporter gene expression can be selected by the addition of 3-aminotriazole (3-AT) to the growth media. Because aminotriazole is a competitive inhibitor of the HIS3 gene product, the enzyme imidazoleglycerol-phosphate dehydratase (85), simple growth tests can be used to select for increasing levels of HIS3 gene expression by varying the concentration of 3-AT in the growth media. Because Rap1WT does not bind 3T4A UASRap1 efficiently (cf. Fig. 1B), yeast expressing only Rap1WT have no means to drive 3T4A UASRap1-HIS3 expression, and hence they fail to grow on media containing 3-AT. By contrast, a cell expressing a Rap1AS variant that can bind and drive efficient 3T4A UASRap1-HIS3 expression will confer resistance to 3-AT (3-ATR) and thus grow in the presence of the inhibitor.
Figure 2.
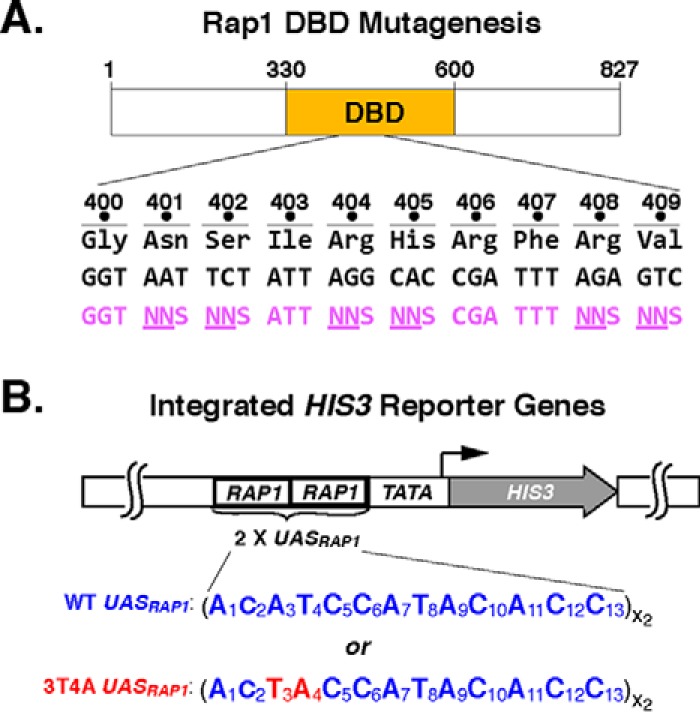
Rap1 DBD mutagenesis strategy. A, schematic of Rap1 showing the location of the DBD within the 827-aa-long protein. Amino acid sequence of DBD region aa 400–409 (black, 3-letter code) is shown along with the corresponding nt codon sequence (black) and a portion of one of the primers used for codon randomization (purple) indicating the targeted codons (NNS, N = any nt, S = G or C). B, schematic of integrated UASRap1-driven TATA-HIS3 reporter genes used in the selection of the altered DNA-binding specificity variant of Rap1. Two versions of the reporter are shown, WT (top), where HIS3 is driven by tandem copies of the WT UASRap1 enhancer sequence 1ACATCCATACACC13, or 3T4A variant UASRap1 enhancer, 1ACTACCATACACC13. Mutated nts are shown in red in FIGURE.
Rap1AS screen
The Rap1AS screen was performed using yeast strain YAM23 (relevant genotype: MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rap1Δ::HPHMX4 (pRS415 UASRap1-MYC5-Rap1) trp1Δ::KANMX4 HA3-TAF1 UASRap1(3T4A)-TATA-HIS3::TRP1::DED1, see Table 1) that was transformed to Ura+ with mutagenized Rap1 DBD library plasmids carried on the URA3-marked plasmid pRS416. An estimated 1 × 106 independent Ura+ colonies (∼1% of the total library) were plated onto SC − His + 5 mm 3-AT selective media plates. After 4 days of incubation at 30 °C, 158 His+, 3-ATR colonies were isolated. The growth phenotype of these colonies was re-tested by patching onto SC-His + 3-AT plates; 136 colonies screened true. To determine whether the His+, 3-ATR phenotype displayed by these colonies was plasmid-borne, Rap1 mutant expression plasmids were isolated from the 30 colonies that had appeared the earliest on the 3-AT-containing selection plates (ranked in order of appearance with colony 1 appearing 1st and colony 30 appearing 30th). Plasmids were recovered from these 30 strains and used to transform the original selection yeast strain, YAM23. The resulting transformants were retested for growth on SC-His + 5 mm 3-AT selective media plates. Fourteen of the 30 recovered Rap1 mutant expression plasmids conferred 3-ATR, and thus they represented putative altered DNA-binding specificity variants of Rap1.
TABLE 1.
Yeast strains used in this study
| Name | Strain | Genotype | Refs. |
|---|---|---|---|
| BY4741 | BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | 123 |
| rap1Δ | BY4741 rap1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rap1Δ::HPHMX4 (pRS416 UASADH-RAP1) | 71 |
| 3T4A-HIS3 #1 | YAM23 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rap1Δ::HPHMX4 (pRS415 UASRap1-MYC5-RAP1) trp1Δ::KANMX4 HA3-TAF1 UASRap1(3T4A)-TATA-HIS3::TRP1::DED1 | This study |
| WT-HIS3 | YAM30 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rap1Δ::HPHMX4 (pRS416 UASRap1-FLAG3-RAP1) trp1Δ::KANMX4 HA3-TAF1 UASRap1(WT)-TATA-HIS3::TRP1::DED1 | This study |
| 3T4A-HIS3 #2 | YAM31 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rap1Δ::HPHMX4 (pRS416 UASRap1-FLAG3-RAP1) trp1Δ::KANMX4 HA3-TAF1 UASRap1(3T4A)-TATA-HIS3::TRP1::DED1 | This study |
| ΔUAS-HIS3 | YAM32 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rap1Δ::HPHMX4 (pRS416 UASRap1-FLAG3-RAP1) trp1Δ::KANMX4 HA3-TAF1 UASRap1(UASRap1Δ)-TATA-HIS3::TRP1::DED1 | This study |
| 3G5G-HIS3 | YAM33 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rap1Δ::HPHMX4 (pRS416 UASRap1-FLAG3-RAP1) trp1Δ::KANMX4 HA3-TAF1 UASRap1(3G5G)-TATA-HIS3::TRP1::DED1 | This study |
Analyses of putative altered DNA-binding specificity Rap1AS variants
The 14 Rap1 mutants identified in the screen were characterized to allow selection of one variant for use as a Rap1AS in Rap1 AD mapping studies. The complete ORFs of the 14 plasmid-borne rap1AS genes were sequenced. All mutations identified were within the targeted Rap1 aa (Fig. 3A, top). Only two of the mutants (10 and 11) possessed identical sequences, indicating that the screen was likely not saturated. Sequence analyses were performed to identify any patterns present in the types of aa changes within the Rap1 variants identified in the screen (MEME motif; Fig. 3A, lower). Rap1 aa Asn-401 was frequently found mutated to a Gly or a Pro, whereas residue His-405 was frequently mutated to Trp or Ser. There were no clear aa substitution patterns for Arg-408 and Arg-409. Despite being targeted for mutagenesis, Ser-402 and Arg-404 were mutated only rarely (Ser-402) or not at all (Arg-404). These data suggest either a failure to efficiently mutagenize Rap1 aa residue 404 or a strong/absolute requirement of Arg-404 for Rap1 DNA binding.
Figure 3.

Rap1 mutagenesis screen identifies a Rap1 variant with altered DNA-binding specificity. A, amino acid sequence of mutagenized Rap1 DBD amino acids 400–409 (yellow highlighting) and 14 variant forms (#2–18) of Rap1 identified in the Rap1AS screen. Amino acid changes in these variants are indicated (green). Lower, a motif of putative Rap1AS hit homeodomain-1 recognition helix sequences generated using MEME. The size of each letter is proportional to its frequency of appearance among the Rap1 variant sequences #2–18. B, yeast growth test to assess the ability of various forms of Rap1 (WT) or variant (#2–18) to confer resistance to 5 mm 3-aminotriazole via expression of either the WT UASRap1-HIS3 reporter variant (blue) or the 3T4A UASRap1-HIS3 (red) variant and either a second copy of Rap1WT or the indicated Rap1AS screen hit. Yeast were serially diluted 1:4 (left to right) and spotted using a pinning tool onto non-selective media (+ His) and media that selected from expression of the UASRap1-HIS3 reporter (+ 3-AT). Plates were photographed after growth at 30 °C for 2 days. Images are representative of three independent biological replicates. C, gel shift competition analysis performed to compare the binding affinity of WT and Rap1AS screen hit variant 2 for binding to either WT UASRap1 or 3T4A UASRap1 DNAs. Assays were performed (as in Fig. 1B) by incubating purified Rap1WT or Rap1 variant 2 with its cognate binding site 32P-WT UASRap1 (blue) or 32P-3T4A UASRap1 (red). Binding reactions also included either no competitor (−) or 50-fold mole-excess of either cold WT UASRap1 (W, blue) or cold 3T4A UASRap1 (M, red) as shown. A representative image from two independent replicates is presented. D, plasmid shuffle analysis of altered DNA-binding specificity Rap1 variant 2 performed to test its ability to complement the rap1Δ null allele. Yeast carrying a chromosomal null RAP1 allele (rap1Δ) and a URA3-marked RAP1 covering plasmid were transformed with a test variant of RAP1, labeled rap1*: (i) a second plasmid-borne copy of RAP1 (labeled WT, blue); (ii) empty plasmid vector (labeled −, black); or (iii) the same plasmid vector expressing Rap1 variant 2 (labeled AS#2, red). Yeast were serially diluted 1:4 (left to right), and growth was scored on media lacking 5-FOA (“Unshuffled”; relevant genotype: RAP1, rap1Δ, rap1*) or containing 5-FOA (“Shuffled” relevant genotype: −, rap1Δ, rap1*). Plates were incubated at 30 °C for 2 days and then photographed; a representative image from three independent replicates is shown.
As predicted from the competition DNA binding data of Fig. 1, Rap1WT cannot utilize the 3T4A UASRap1 site to drive HIS3 expression (compare top two growth tests, Fig. 3B). By contrast, all of the putative Rap1AS variants have the ability to promote efficient expression of the 3T4A UASRap1-HIS3 reporter gene to confer 3-ATR growth (mutants #2–18; Fig. 3B). Interestingly, those mutant variants recovered from colonies that appeared the earliest during the screen grow slightly faster than those recovered from colonies that had appeared later (i.e. variants 2–14 earlier than variants 17 and 18; Fig. 3, A and B). Overall, the 3-ATR growth properties of variants 2–14 are comparable with yeast containing Rap1WT and the WT UASRap1-HIS3 reporter, although variants 17 and 18 grow somewhat slower.
To allow unambiguous Rap1 AD mapping, a true altered DNA-binding specificity mutant that fails to bind WT UASRap1 (and thereby interfere with Rap1WT essential function) was desired. To find such a Rap1 mutant whose improved 3T4A UASRap1 binding is accompanied by a reduced WT UASRap1 binding, 6 of the 14 preliminarily characterized Rap1 screen hits (see below) were selected for purification and gel shift DNA binding competition assays. Because there was no way to predict a priori, which if any of the Rap1 screen hits possessed true altered DNA-binding specificity, the six screen hits were chosen to cover the sequence variation present in the collection of Rap1 mutants whose plasmid-borne expression supported 3-ATR. Variants 5 and 10 were chosen because their sequences were highly similar to the logo that was generated by motif analysis. Rap1 variants 2, 5, 13, 17, and 18 were selected because of deviations from the sequence logo, in either the presence of additional mutations (i.e. variant 5), the absence of a mutation that was present in other variants (17 and 18), or an uncommon mutation (i.e. 2) such as a basic Arg residue at position 401 instead of an uncharged Gly, Pro, or Ala. All six of these Rap1 mutant variants bound 32P-3T4A UASRap1 in a gel shift assay (Fig. 3C, and data not shown). Out of the six, Rap1 mutants 5, 17, and 18 displayed expanded DNA-binding specificity, and they bound both WT and 3T4A UASRap1 sites with similar affinity. By contrast, Rap1 variants 2, 10, and 13 showed reduced affinity for the WT UASRap1 site. In particular, mutant 2 concomitantly displayed the largest reduction in WT UASRap1 affinity (Fig. 3C and data not shown). Consistent with its significantly reduced affinity for WT UASRap1, Rap1 mutant 2 also failed to complement the null rap1 allele (rap1Δ) in a plasmid shuffle assay (Fig. 3D), indicating that it could not efficiently bind the WT UASRap1 of (at least one) essential Rap1-dependent genes in vivo. Based upon all these data, Rap1 variant 2 showed true altered DNA-binding specificity in vitro and in vivo, and henceforth is referred to as Rap1AS (or AS).
Molecular genetic characterization of Rap1AS
Rap1AS was subjected to further characterization to ensure that it would serve as an appropriate reagent for attempted AD identification and mapping studies. The AS form of Rap1 was tested to document the following: (a) Rap1AS-driven reporter expression actually requires an enhancer and is specific to the 3T4A UASRap1 site; (b) Rap1AS efficiently drives HIS3 reporter expression as scored at the mRNAHIS3 level; (c) Rap1AS is stably expressed at levels similar to Rap1WT; and (d) when coexpressed with Rap1WT, Rap1AS does not compete with the WT protein and cause a dominant slow growth phenotype.
To demonstrate that Rap1AS drives reporter gene expression efficiently and specifically from 3T4A UASRap1, two new HIS3 reporter variants were constructed. The first, termed ΔUASRap1, completely lacks UASRap1 sites upstream of HIS3. This construct tests the hypothesis that Rap1AS might bypass the requirement for an enhancer, while simultaneously defining background expression of the UASRap1-TATA-HIS3 reporter. The second construct substituted a 3G5G UASRap1 enhancer in place of either the WT or 3T4A enhancer. This variant enhancer tested the functional DNA-binding specificity of Rap1AS, which was selected to bind the 3T4A site. Previous work has shown that both mutations (i.e. UASRap1 deletion or 3G5G substitution) dramatically reduce Rap1-driven transcription in vivo (31, 57). HIS3 expression was scored in appropriate yeast strains by 3-ATR growth and qRT-PCR-measured mRNAHIS3 assays. As shown in both growth (Fig. 4A) and HIS3 mRNA analyses (Fig. 4B), Rap1WT and Rap1AS were only able to robustly drive expression of the reporter containing their cognate DNA recognition site (WT UASRap1 and 3T4A UASRap1, respectively). It is important to note that as detailed under “Experimental Procedures,” HIS3 mRNA levels are scored by qRT-PCR using total RNA extracted from cells grown in SC + histidine to prevent any selection for reporter gene expression that occurs when 3-AT is added to growth media (i.e. mRNA analyses of Figs. 4–7). Interestingly, Rap1AS drives expression of the 3T4A UASRap1-HIS3 reporter gene mRNAHIS3 to a level greater than that observed with the WT UASRap1-HIS3 reporter (Fig. 4B). Deletion of the enhancer abolished expression of the HIS3 reporter regardless of the Rap1 form present (WT or AS; Fig. 4, A and B). Immunoblot analysis of yeast whole cell extracts shows that Rap1AS was stably expressed at levels comparable with Rap1WT (Fig. 4C; note that all constructs contain an N-terminal Myc5 tag and an NLS; see “Experimental Procedures”). Furthermore, growth curves with pseudodiploid cells (Fig. 4D; Rap1 WT/WT; Rap1 WT/AS) demonstrate that Rap1AS does not induce a dominant slow growth phenotype. Based on these data, we concluded that Rap1AS possessed all of the characteristics desired for use as a tool to search for an AD within Rap1.
Figure 4.

Functional characterization of Rap1AS. A, Rap1AS specifically drives the mutant 3T4A UASRap1-HIS3 reporter. Upper, test of the ability of WT and AS forms of Rap1 to bind different enhancers to confer aminotriazole-resistant growth using the assay detailed in Fig. 3B. Yeast strains carrying the indicated UASRap1-HIS3 reporter variant (WT, 3G5G, ΔUAS, or 3T4A, as shown) and either a second copy of Rap1WT (blue) or Rap1AS (red) were grown on non-selective (+ His) or HIS3-reporter gene selection (+ 3-AT) media. Images shown are representative of three independent replicates. Lower, detailed structures of the UASRap1-HIS3 reporters used in these growth tests. B, qRT-PCR analyses of the steady-state levels of HIS3 reporter mRNA, relative to ACT1 in the yeast strains tested in A. Data were obtained by testing three biological replicates (each indicated by a white circle) analyzed in duplicate and plotted as a percentage of the HIS3 mRNA levels present in WT Rap1 + WT UASRap1 yeast. Mean ± S.D. is depicted. *, p < 0.01; **, p < 0.001. C, steady-state protein expression levels of Rap1WT and Rap1AS. MYC-tagged Rap1 forms (top, WT, AS; Test Rap1 (Myc) IB) were scored using immunoblotting (IB) with anti-Myc IgG. A strain carrying a plasmid expressing only untagged Rap1 was used as a specificity control for the Myc antibody (labeled no tag). Prior to incubation with antibodies, blots were stained with Ponceau S to monitor total protein loading (bottom, Ponceau S). Equal protein loading was also monitored via immunoblotting with anti-actin IgG (middle, Actin IB). Images are representative of three independent replicates. D, growth curves of yeast expressing WT or AS forms of Rap1 from a plasmid carrying either a second copy of Rap1WT or Rap1AS (labeled WT/WT or WT/AS). Overnight-grown yeast starter cultures were diluted to a starting OD600 of 0.5, and the optical density of the cultures was monitored over the course of 12 h. Data shown represents the average of three biological replicates. Error bars represent S.D.
Figure 5.

Mapping the activation domain of Rap1 to amino acids 630–671. A, schematic of the Rap1 protein. Shown are the well characterized Rap1 DNA DBD (aa 330–600) and silencing domain (SD) as well as the region suspected to contain the AD (AD? 600–671). B, growth analysis of yeast strains carrying the 3T4A version of the HIS3 reporter (left column, UASRap1-red; labeled 3T4A) and various forms of Rap1 (labeled Rap1, 2nd column) either Rap1AS (AS, red), Rap1WT (WT, blue), or the indicated Rap1AS deletion mutant (ΔAD?, Δ600–610, Δ611–620, Δ621–630, Δ631–640, Δ641–650, Δ651–660, Δ660–671; red). Serial dilutions of cells were plated and grown on non-selective (+ His), or reporter gene-selective (+ 3-AT) media as in Figs. 3B and 4A. Images are representative of three independent replicates. C, qRT-PCR analysis to score reporter HIS3 mRNA expression levels in the various yeast strains tested in B. Results of these analyses are plotted as in Fig. 4B, except that data are expressed as a percentage of the relative HIS3 mRNA levels in yeast carrying Rap1AS instead of Rap1WT. Data represent three biological replicates (white circles) measured in duplicate. Mean ± S.D. is depicted. *, p < 0.05; **, p < 0.01; ***, p < 0.005. D, immunoblot analysis of Rap1AS, Rap1WT, and Rap1AS deletion variant protein expression levels. This analysis was performed as detailed in Fig. 4C. Images are representative of three independent replicates. E, schematic indicating the location of the Rap1 activation domain (AD; aa 630–671) mapped through the deletion mutagenesis experiments shown here.
Figure 6.

Activation function of the Rap1 AD depends upon evolutionarily conserved hydrophobic amino acids. A, multiple sequence alignment of S. cerevisiae (S. cer) Rap1 activation domain amino acids 630–671 with the corresponding region of Rap1 proteins from the sensu lato yeast S. castelli (S. cas.) and S. kluyveri (S. klu). Amino acids that are identical in all three yeast species are boxed in yellow. These conserved amino acids were targeted for site-directed mutagenesis. B, growth analysis of yeast strains carrying the 3T4A UASRap1-HIS3 reporter and either positive (AS, Rap1AS) and negative (WT, Rap1WT) control forms of Rap1 on the three sets of plates shown (i.e. AS, WT to N645A; AS, WT to Y665A; or AS, WT to E671E) or the indicated Rap1AS mutant variant (shown, I633A to E671A). Growth tests were performed as in Figs. 3–5 by plating serial dilutions of cells on either non-selective (+ His) or reporter gene-selective (+ 3-AT) media. Rap1AS AD point mutants that display a large decrease in growth relative to Rap1AS are boxed in blue. Images are representative of five independent replicates. C, qRT-PCR analysis performed on total RNA prepared from yeast carrying the 3T4A UASRap1-HIS3 reporter and either Rap1AS (AS), Rap1WT (WT), or the indicated Rap1AS AD point mutant variant. Analyses were performed as detailed for Fig. 5C. Data are representative of three biological replicates, each measured in duplicate. Mean ± S.D. is depicted. *, p < 0.005; **, p = 0.0001.
Figure 7.

Seven hydrophobic Rap1 AD amino acids confer Rap1 activation function. A, schematic of Rap1 illustrating the structures of two AD knock-out alleles. Middle, schematic of intact Rap1 illustrating the location of known functional domains (DBD, AD, and SD). Top, Rap1 AD sequences 630–671 that were deleted from Rap1. Bottom, location and sequence of the seven hydrophobic amino acids identified by site-directed mutagenesis and functional assays (Fig. 6, above) that were all mutated to alanine (L639A, F646A, L650A, L654A, F663A, Y665A, and I669A) to create the Rap1AS 7Ala variant. B, steady-state protein levels of Rap1 variants tested in A (AS, WT, Δ630–671, 7Ala; specificity control = no tag, as labeled; blots performed as detailed in Figs. 4C and 5D). Images are representative of two independent replicates. C, qRT-PCR analysis performed on total RNA prepared from yeast carrying the 3T4A UASRap1-HIS3 reporter and either Rap1AS (AS), Rap1WT (WT), or Rap1AS AD knock-out variants (Δ630–671 or 7Ala). Analyses were performed as detailed for Figs. 5C and 6C. Data are representative of three biological replicates, each measured in triplicate. Mean ± S.D. is depicted. *, p = 0.0002; **, p = 0.0001.
Systematic deletion mutagenesis of Rap1 identifies a potential activation domain
As described in the Introduction, prior analyses of Rap1 led to the hypothesis that an AD was located within a C-terminal portion of the protein (Fig. 5A; aa 600–671, labeled AD? (32, 46)) distinct from the sequences implicated in silencing of telomere-proximal genes whose deletion has little effect on cell viability (28, 29, 32, 34). To directly test this hypothesis, a variant of Rap1AS was generated wherein the RAP1AS ORF sequences encoding aa 600–671 were deleted. This deletion generated a protein termed ΔAD?-Rap1AS. Yeast carrying an integrated 3T4A UASRap1-HIS3 reporter were then used to score the ability of intact Rap1WT, Rap1AS, and the internally deleted ΔAD?-Rap1AS forms of the protein to promote 3-ATR growth (top three rows of yeast cell growth, labeled AS, WT, ΔAD? respectively; Fig. 5B). As shown above in Fig. 4A, positive control cells that express intact Rap1AS grew well in the presence of 3-AT due to efficient expression of HIS3 (Fig. 5, B and C). Negative control cells expressing only Rap1WT (which is unable to bind the 3T4A UASRap1 enhancer) fail to drive high level HIS3 transcription (Fig. 5C) or exhibit ATR (Fig. 5B) as expected (see Figs. 1, 3, and 4). The test strain, which expresses the ΔAD?-form of Rap1AS, was similarly unable to confer ATR or drive significant levels of HIS3 transcription (Fig. 5, B and C). All three forms of Rap1 (Rap1AS, Rap1WT, and ΔAD?-Rap1AS) were stably expressed at equivalent levels (Fig. 5D; lanes labeled AD, WT, ΔAD?). These data indicate that an activation domain indeed resides within the sequences between aa 600 and 671.
To identify the aa within the 600–671-aa interval that comprise the AD, a series of internal, N-terminal, and C-terminal deletion variants of the 600–671-aa region of Rap1AS were generated. Only data for the series of seven internal 10-aa deletions moving through Rap1 aa 600–671 are shown here (Fig. 5, B–D, labeled Δ600–610, Δ611–620, Δ621–630, Δ631–640, Δ641–650, Δ651–660, and Δ660–671). These Rap1AS variants were tested for 3T4A UASRap1-HIS3 expression by scoring growth on 3-AT-containing plates and mRNAHIS3 levels by qRT-PCR. To ensure that all constructs were stably expressed, steady-state protein expression levels were also monitored (Fig. 5D). Deletion of the first 30 aa of the 600–671 region (i.e. constructs Δ600–610, Δ611–620, and Δ621–630; Fig. 5, B and C) had no significant effect upon either growth in the presence of 3-AT or HIS3 reporter mRNA transcript levels. By contrast, removal of aa within the distal 40 aa of Rap1 AD? sequences (Δ631–640, Δ641–650, Δ651–660, and Δ660–671; Fig. 5, B and C) dramatically reduced HIS3 reporter expression as scored by ATR growth (Fig. 5B) and mRNAHIS3 levels (Fig. 5C). All Rap1AS deletion constructs were as stable as Rap1WT, results excluding any contribution of Rap1AS protein abundance to reporter gene expression (Fig. 5D). These data, which are consistent with the data obtained from the N- and C-terminal AD? deletion variants (data not shown), indicate that the Rap1 AD resides within amino acids 630–671 (domain labeled AD; Fig. 5E).
Identification of key Rap1 AD amino acids
The deletion mutagenesis-defined AD of Rap1AS (i.e. aa 630–671) was subjected to site-directed mutagenesis to identify the specific amino acid residues that contribute essential AD function. Based on the idea that evolutionary conservation implies function, Rap1 AD-like sequences from S. cerevisiae and the distantly related sensu lato yeast strains Saccharomyces castelli and Saccharomyces kluyveri were subjected to a multiple sequence alignment to identify amino acids conserved in all three yeast species (labeled S. cer, S. cas, and S. klu, respectively, Fig. 6A). Alignment shows that 24 of the 42 aa within the Rap1 AD were identical among S. cerevisiae and sensu lato yeast (Fig. 6A). Based on this sequence conservation, single point mutant variants were generated within the AD of Rap1AS by changing each of the 24 identical aa to alanine (except for Ala-670, which was mutated to arginine). These Rap1AS point mutant variants were transformed into the 3T4A UASRap1-HIS3 reporter yeast strain and tested for the ability to confer 3-ATR. Growth assays performed with very high levels of 3-AT (300 mm) revealed that eight aa, seven of which were hydrophobic, were required for robust 3-ATR growth (Fig. 6B; hydrophobic aa Leu-639, Phe-646, Leu-650, Leu-654, Phe-663, Tyr-665, and Ile-669, as well as A670R; all boxed). Steady-state protein levels of all variant proteins were essentially identical to WT (data not shown). Parallel qRT-PCR analyses of mRNAHIS3 levels in these same strains showed some correlation with ATR patterns (Fig. 6C). Importantly, the L650A mutant, which displayed the largest growth defect, also showed a significant reduction in mRNAHIS3 levels (Fig. 6C). These data suggest that Rap1 AD activity depends on key hydrophobic aa.
To test the hypothesis that the majority, if not all, of the AD activity of Rap1 is conferred by aa 630–671, particularly the seven hydrophobic aa identified by alanine mutagenesis, two additional Rap1AS AD variants were prepared. Each was predicted to ablate, or knock out (KO) AD activity. The first “AD-KO” variant of Rap1AS lacked the entire mapped Rap1 AD region aa 630–671 (Fig. 7A, top, labeled Δ630–671). The second Rap1AS AD-KO variant was created by simultaneously mutating all seven of the key (cf. Fig. 6B) hydrophobic aa to alanine (Fig. 7A, labeled 7Ala, bottom). Plasmid-borne genes encoding Rap1AS, Rap1WT, Rap1AS Δ630–671 and Rap1AS 7Ala were transformed into the 3T4A-UASRap1-HIS3 test strain, and protein and mRNAHIS3 levels were scored by immunoblot and qRT-PCR (Fig. 7, B and C, respectively). All proteins were equivalently expressed. Notably however, the two AD-KO strains both displayed drastically reduced HIS3 mRNA levels relative to the WT version of Rap1AS. Thus, 3T4A UASRap1-HIS3 reporter gene expression depends predominantly on the mapped Rap1 AD, particularly the seven key hydrophobic amino acids implicated in AD function by alanine scanning mutagenesis.
Rap1 AD Is required for normal cell growth and transcription
We reasoned that if the Rap1 AD (and key AD amino acids mapped therein using Rap1AS) truly represents the bona fide activation domain of Rap1, then introducing the 7Ala mutation into the context of the Rap1WT should cause a decrease in the transcription of chromosomal Rap1-dependent genes and a concomitant decrease in growth rate. Introducing these mutations into the WT version of the protein also controls for potential synthetic genetic interactions between the altered DBD sequences of Rap1AS and the altered sequences that were introduced into the mapped AD. To probe the question of essentiality of the Rap1 AD for growth, the ability of the 7Ala mutation in the otherwise Rap1WT protein was scored for complementation of a rap1Δ null mutant strain using the plasmid shuffle method. Rap1WT and empty plasmid vector, respectively, served as positive and negative controls for this test. Although Rap1WT efficiently and specifically complements rap1Δ, the Rap17Ala variant only weakly complemented (compare growth of WT versus 7Ala strains, row 1 versus row 3; Fig. 8A) despite the fact that the two proteins were equally expressed (Fig. 8B). The dramatic growth deficiency of the Rap17Ala-expressing strain was quantified via growth curves. The WT strain doubling time is 94 min, whereas the Rap17Ala expressing strain requires nearly twice as long (155 min) to double (Fig. 8C). Thus, consistent with expectations, the Rap1 AD is required for normal rates of cell growth.
Figure 8.

Rap1 AD is required both for normal growth and transcription of authentic chromosomal Rap1 target genes. A, ability of the Rap17Ala variant to support cell viability was assessed by plasmid shuffle analyses. Yeast carrying a chromosomal null RAP1 allele (rap1Δ) and a URA3-marked RAP1 covering plasmid were transformed with a test variant of RAP1, labeled rap1*: (i) a second plasmid-borne copy of RAP1 (labeled WT); (ii) empty plasmid vector (labeled −); or (iii) the same plasmid vector expressing the Rap17Ala variant (labeled 7Ala). Yeast were serially diluted 1:4 (left to right), and growth was scored on media either lacking 5-FOA (Unshuffled; relevant genotype, RAP1, rap1Δ, rap1*) or containing 5-FOA (Shuffled relevant genotype: −, rap1Δ, rap1*) as detailed in Fig. 3D. Images are representative of five independent replicates. B, immunoblot (IB) analysis of the in vivo steady-state levels of Rap1WT and Rap17Ala proteins. Analysis was performed as described in Figs. 5D and 7B. Images are representative of four independent replicates. C, growth curve analysis of three biological replicates of shuffled yeast strains expressing either only Rap1WT or Rap17Ala and performed and plotted as described in Fig. 4D. D, analysis of nascent RNA levels for several chromosomal yeast genes (integrated UASRap1-HIS3 reporter, ENO1, PYK1, RPS3, RPL26B, and RPL3) in cells solely expressing WT RAP1 or the 7Ala variant of RAP1. Cellular RNAs were pulse-labeled with 4sU for 2.5 min, extracted, and purified. RNA thiol groups present on 4sU-labeled RNA were chemically biotinylated, and affinity-purified. Affinity-purified RNA was analyzed via qRT-PCR to quantify the amount of nascent transcripts produced from each of the six genes noted above. qRT-PCR analyses were performed as described in Fig. 5C except that data were normalized to S. pombe β-tubulin mRNA present due to the addition of 4sU pulse-labeled S. pombe cells into each sample prior to RNA extraction. Data are representative of three biological replicates, each measured in triplicate. Mean ± S.D. is depicted. * = p < 0.002; ** = p < 0.0001. See “Experimental Procedures” for details.
Assuming that the Rap1 activation domain mapped here actually functions specifically in the activation of authentic chromosomal target genes, it is reasonable to predict that Rap1 AD loss-of-function mutants would exhibit reduced expression of both RP- and GE-encoding genes. Indeed, extensive cis-element mapping, use of conditional alleles, and ChIP-based in vivo occupancy studies all support the idea that Rap1 contributes importantly to the expression of these two gene classes (37, 45, 58, 68, 86, 87). However, two considerations preclude straightforward interpretation of the effects of mutations in Rap1 (or other transcription proteins) on cellular steady-state mRNA levels. First, unlike the synthetic chimeric UASRap1-HIS3 reporter gene studied in the experiments of Figs. 2–7, expression of both RP- and GE-encoding genes is modulated by multiple transactivator proteins in addition to Rap1 (57–59, 61–67). As a consequence, mutations in the AD of Rap1 will likely not result in as large a decrement in authentic chromosomal target gene transcription as observed with the HIS3 reporter gene (cf. Figs. 5 and 6). Second, another, and perhaps more important factor complicating interpretation of such experiments that monitor steady-state mRNA levels, is the fact that transcription and mRNA degradation are linked. Both prokaryotic and eukaryotic cells can maintain steady-state transcript levels despite loss of key proteins involved in either transcription/transcription activation or mRNA degradation by modulating these dynamic processes (88–93).
To circumvent these caveats, the 4-thiouracil nascent RNA pulse-labeling method of RNA analysis (88–90, 93, 94) was employed to monitor nascent transcript levels in cells expressing either WT Rap1 or the 7Ala AD Rap1 variant as the sole source of Rap1 (i.e. the shuffled strains analyzed in Fig. 8, A–C). (Note that both strains carry the integrated WT UASRap1-HIS3 reporter gene.) Nascent RNA levels for five actively transcribed chromosomal genes and the integrated HIS3 reporter gene were scored by qRT-PCR (see 'Experimental Procedures' for details). Four of the actively transcribed chromosomal genes are Rap1-dependent (two glycolytic, ENO1 and PYK1; two RP-encoding genes, RPS3 and RPL26B), whereas one chromosomal Rap1-independent RP-encoding gene was scored (RPL3). Alteration of the Rap1 AD by the 7Ala mutation dramatically, and specifically, reduced transcription of the known Rap1-dependent genes (UASRap1-HIS3, ENO1, PYK1, RPS3, and RPL26B) but not the Rap1-independent RPL3 gene (Fig. 8D). The specific reductions in transcription of the known Rap1-dependent genes ranged from ∼50% for the synthetic reporter gene (UASRap1-HIS3; Fig. 8D) to ∼80% for two of the Rap1-dependent chromosomal genes (ENO1 and RPL26B; Fig. 8D). Meanwhile, unlike the Rap1ΔSD, Rap1 7Ala failed to relieve transcription repression in an HMRA silencing reporter strain (data not shown) (29, 95), indicating, as expected, that the Rap1 AD plays no significant role in transcriptional repression. Collectively our data demonstrate that the Rap1 AD, mapped and characterized here through the use of the altered DNA-binding specificity variant of Rap1, is a bona fide activation domain.
Mutation of the Rap1 activation domain reduces binding to the RBD of the TFIID coactivator subunit Taf5
We have previously shown that Rap1 binds specifically, and with high affinity, to distinct RBDs within the Taf4, Taf5, and Taf12 subunits of TFIID (31, 71). Rap1-Taf RBD binding is sensitive to RBD mutation, and we found that yeast expressing such mutations in either Taf4 or Taf5 display reduced ribosomal protein mRNA gene expression (71, 96). These data argue that TFIID serves as a coactivator for Rap1. However, we did not know which domain(s) within Rap1 was responsible for Rap1-Taf RBD interactions. We theorized that if the Rap1 AD mapped herein plays a key role in Rap1-TFIID Taf-RBD interaction, then mutation of the AD should reduce Rap1-Taf binding. Because we had previously documented synthetic lethal interactions between Taf5 RBD mutants and Rap1 deletion variants (71), we decided to perform GST-Rap1/Taf5 pulldown binding experiments as a first test of our Rap1 AD-Taf RBD direct interaction hypothesis. These studies used an N-terminal RBD-containing fragment of Taf5 (aa 1–337) and either GST− alone (GST−), GST-fused to WT Rap1 (GST-Rap1 WT), GST-fused to the 7Ala variant of Rap1 (GST-Rap1 7Ala), and GST-fused to Rap1 ΔC (GST Rap1 ΔC). We observed that both the 7Ala- and the ΔC-Rap1 variants reproducibly displayed an ∼50% reduction in binding to the Taf5 RBD (Fig. 9). These results are consistent with the hypothesis that the Rap1 AD we have mapped is central to transcriptional activation.
Figure 9.
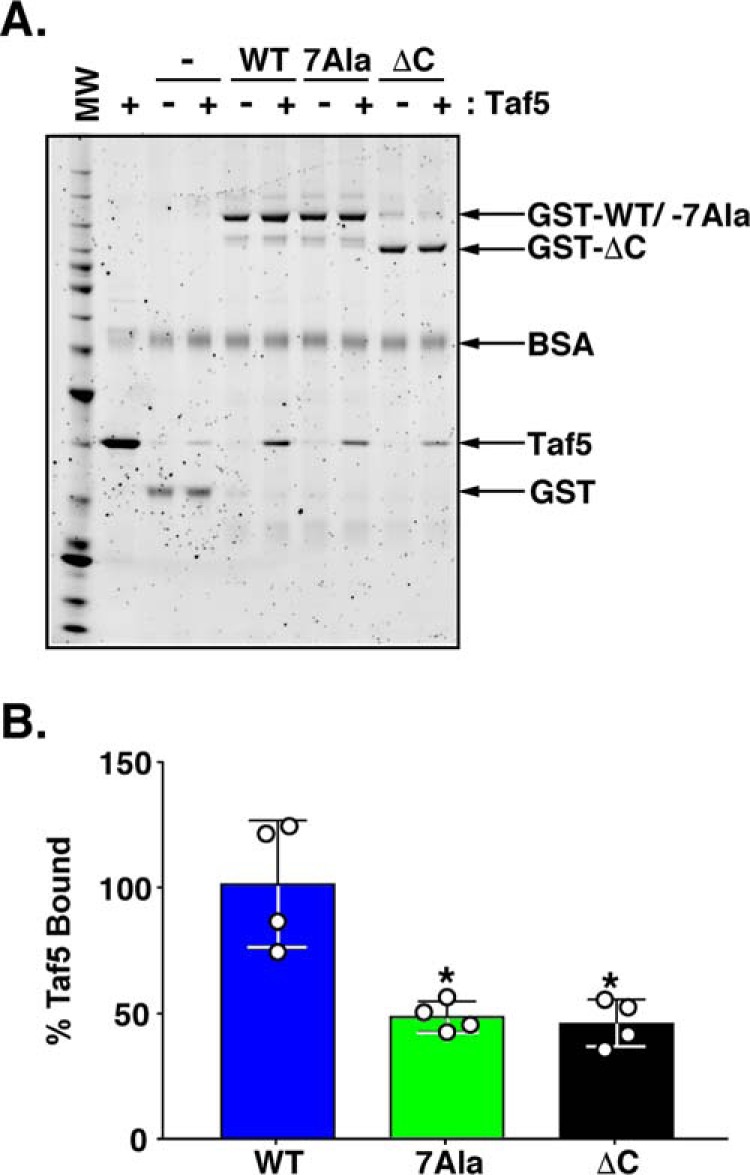
Mutation of the Rap1 AD reduces binding of Rap1 to the RBD of the TFIID coactivator subunit Taf5. A, Sypro-stained NuPAGE gel of Taf5/GST-Rap1 pulldown protein-protein binding reactions. Pulldown experiments were conducted using negative control GST−, positive control GST-WT Rap1, test GST-7Ala Rap1, and test GST-ΔC Rap1-loaded GST-agarose beads and the N-terminal RBD-containing fragment of Taf5 (aa 1–337). Glutathione-Sepharose beads loaded with either 12 pmol of GST or 6 pmol of GST-Rap1 variant (GST−, GST-WT Rap1, GST-7Ala Rap1, GST-ΔC Rap1; labels, arrows) were incubated with either 0 or 100 pmol of purified Taf5 fragment under the conditions detailed under “Experimental Procedures.” All incubations contained bovine serum albumin (BSA; label, arrow) to minimize nonspecific protein binding to the beads. Bead-bound proteins were eluted with SDS-sample buffer, heat-denatured, and fractionated on an SDS-polyacrylamide gel in parallel with molecular weight standards (MW), and 25 pmol of purified Taf5 (Taf5). Gels were stained with Sypro Ruby, and images were obtained using a Pharos FX imager. Image is representative of four independent replicates. B, quantification of the data shown in A. Quantity One software was used to score the intensity of GST−, GST-Rap1 variant, and Taf5 bands. The intensity of each Taf5 band was normalized to the intensity of each GST− or GST-Rap1 variant band, and then Taf5 binding to GST beads alone was subtracted from the Taf5 binding of all GST-Rap1 variants. Taf5 binding data obtained from four independent replicates are plotted using Graph Pad Prism 7 software as a percentage of the Taf5 binding to GST-Rap1 WT. Mean ± S.D. is depicted. *, p = 0.002.
Discussion
Transcription factor AD mapping represents a critical step in the path toward determining the mechanism by which a gene, or group of co-regulated genes, is activated. We were particularly interested in the Rap1 AD because Rap1 in budding yeast drives the transcription of 128 of the 137 genes encoding ribosomal proteins as well as many of the genes encoding the enzymes of glycolysis (37, 58, 81, 97). These two classes of genes are among the most vigorously transcribed families of genes in most organisms. Understanding the mechanisms controlling the expression of such highly transcribed classes of genes will provide valuable and potentially generalizable mechanistic insights into how highly expressed eukaryotic genes are regulated.
In budding yeast, transcription of the genes encoding RPs is controlled through the action of multiple DNA binding transcription proteins. These include Rap1, Ifh1, Fhl1, Sfp1, and Hmo1 (61–67). RP gene transcription is responsive to a variety of regulatory inputs, several of which respond to nutrient availability and/or cell size (61–64, 81, 98). These regulatory pathways appear to work through Ifh1/Fhl1 and Sfp1 (61–64) and thus appear distinct from Rap1 function(s). Meanwhile, Rap1 is the only activator that is absolutely required for transcription of the RP encoding genes, as deletion of the genes encoding these other factors (or their cognate DNA binding elements) reduces but does not eliminate RP gene expression (31, 57).
Our laboratory has had a long-standing interest in Rap1- and RP-encoding genes because transcription of this gene family requires TFIID (31, 57, 71, 96, 99–109). We have previously shown that Rap1 directly interacts with the RP gene coactivator TFIID (31) and that this binding is specific, high affinity, and mediated through distinct RBDs present in TFIID subunits Taf4, Taf5, and Taf12 (31, 71). Deletion of the RBDs from either Taf4 or Taf5 is lethal, whereas the amino acids within these ∼100–300 amino acid domains are mutable, and certain point mutations within either RBD leads to temperature-conditional growth and reduced affinity of binding to Rap1. Moreover, certain combinations of distinct, non-lethal rap1 deletion mutants with taf5ts mutants induce synthetic lethality, whereas shifting strains carrying either taf4ts or taf5ts mutant alleles to non-permissive temperatures induce a large and selective decrease in expression of essentially the entire complement of ribosomal protein encoding genes (71).
We have also previously documented and characterized important interactions between Rap1 and TFIIA. Mutations that compromise these interactions decrease TFIIA-TFIID and TFIIA-Taf4 interaction/binding, RP gene expression in vivo, and Rap1-driven reporter gene transcription in vitro (96). Finally, we have previously described unique Rap1-induced TFIIA- and TFIID-dependent DNA looping and Rap1-dependent protein structural transitions of TFIIA within a quaternary Rap1·TFIIA·TFIID·UASRap1TATA DNA complex (the enhancer-promoter reporter DNA used here) (105). Collectively, these data lead us to propose that Rap1 directly and specifically binds subunits of both TFIID and TFIIA to activate transcription of the RP encoding genes (31, 71, 96, 105, 110). As a prerequisite to advance these studies, and to more fully dissect the molecular mechanisms utilized by Rap1 to activate transcription, it is essential to have a collection of AD variants of Rap1 that are defective in RP gene transcription activation.
Unfortunately, although much is known regarding the TFIID coactivator subunit targets of Rap1, prior to the work described in this report, there was no unambiguous data regarding whether Rap1 actually contains a functional AD. As a consequence, we decided to undertake an alternative approach to identify an AD within budding yeast Rap1. Despite the challenges involved, we undertook the generation of an altered DNA-binding specificity variant of Rap1, termed Rap1AS. We planned to use Rap1AS as a novel tool to facilitate the identification and high resolution characterization of an activation domain within Rap1.
With the exception of proteins that bind DNA via zinc finger motifs, true altered DNA-binding specificity variants are fairly difficult to generate. This is underscored by the fact that few such proteins have been described in the literature. That said, successful generation of altered DNA-binding specificity variant proteins has yielded significant insights into deciphering the determinants of protein-DNA recognition while simultaneously providing novel tools to probe and dissect the activity of DNA-binding proteins. Recognition of a mutant enhancer or UAS site requires that a DBD gain the ability to bind the mutant DNA sequence via alteration in the ability of a protein to bind novel DNA sequences and/or shapes. True-altered DNA-binding specificity variants (rather than expanded DNA-binding specificity variants) display significantly reduced WT DNA sequence binding in addition to gaining mutant DNA recognition. The AS Rap1 variant identified in this report, termed Rap1AS, exhibits exactly these properties and hence was deemed ideal for initiating efforts to discover and pinpoint the AD of Rap1 absent the many complications imposed by the multiple essential cellular functions of the protein (see Introduction).
Analysis of the aa sequences of the Rap1 AS mutants recovered in our screen provides insights into the DNA recognition determinants of WT and mutant UASRap1 DNA base pairs 3 and 4. The fact that consistent aa sequence patterns were found among the mutations present at Rap1 aa 401 and 405 (see Fig. 3A) in all of the Rap1 variants suggests that these aa changes are required to enable Rap1 binding to 3T4A UASRap1. Furthermore, comparison of the sequences of altered DNA-binding specificity Rap1 variants 2, 10, and 13, with the sequences of expanded DNA-binding Rap1 variants 5, 17, and 18, suggests that the true altered DNA-binding specificity variants contained both a mutation at aa 408 and a bulky hydrophobic amino acid (i.e. Trp or Phe) at Rap1 aa 405 (cf. Fig. 3). These mutations may decrease Rap1 mutant binding to WT UASRap1by breaking a favorable water-mediated contact between Rap1 Arg-408 and WT UASRap1 base pair 3 (47) or by using a bulky hydrophobic aa at Rap1 position 405 to actively exclude binding to the WT UASRap1 element (see Fig. 1A).
Our unambiguous AD mapping data also show that the best of our altered DNA-binding specificity variants, Rap1AS, represents a powerful tool to study Rap1 structure-function relationships. Activator proteins, such as Rap1, can contain one or more ADs, usually 30–100 aa in size. ADs have been classified based on the kinds of aa that dominate the activation domain: acidic, proline-rich, serine-rich, and glutamine-rich (111, 112). Using Rap1AS, a single AD of the protein, was successfully mapped to Rap1 aa 630–671. This 41-aa region contains nine glutamic and aspartic aa, 20% of the total AD residues, and it has a predicted pI of 3.84. Thus, the Rap1 AD should be considered to be an acidic activation domain.
Although this acidic AD is located within the AD mapped using the Gal4 DBD-Rap1 fusion approach (aa 630–692) (33), our analyses provide important additional insights. Studies of acidic ADs have shown that typically they contain small clusters of key hydrophobic aa that actually confer AD activity (113–116) rather than their acidic aa (116, 117). Consistent with this principle, our mutational analyses of individual Rap1 AD amino acids showed, for the first time, that specific individual hydrophobic amino acids (i.e. Leu-639, Phe-646, Leu-650, Leu-654, Phe-663, Tyr-665, and Ile-669) contribute critically to Rap1 AD function. Our Rap1 AD analysis, however, did reveal a few interesting differences between the Rap1 AD and other well studied activator ADs. First, in contrast to other activators such as VP16, Zta, Gcn4, p53, and glucocorticoid receptor, where at least two AD mutations must be made to noticeably decrease transcriptional activity of an intact AD in reporter assays (113–116, 118), several single point Rap1 AD mutant variants (i.e. L639A, F646A, L650A, L654A, F663A, Y665A, and I669A; Fig. 6B) displayed defects in activity as scored by growth. It is also surprising that it was the non-polar L650A mutant that showed a significant decrease in mRNAHIS3 levels in qRT-PCR (Fig. 6C), instead of a bulky aromatic hydrophobic amino acid (i.e. Phe or Tyr), which have been shown to be the most critical contributors to AD function in other well characterized ADs (113–116, 118). Finally, the Rap1 AD activity is spread over a large number of contiguous aa (i.e. 7 amino acids; see Figs. 6–9) compared with other ADs whose activity is concentrated on only 2–3 aa (113, 114, 116, 118, 119). It is tempting to speculate that these seven key hydrophobic aa may represent separate/overlapping binding domains for the known distinct coactivator targets of Rap1 (19, 31, 71, 96). Alternatively, these seven critical AD aa may collectively enable Rap1 to bind with unusually high affinity to its coactivator targets and thus drive the high levels of transcription observed at the RP- and GE-encoding genes. Indeed, it was recently shown that the addition of hydrophobic aa to a small nine-residue AD sub-motif of yeast Gcn4 could increase coactivator binding affinity and transcription activity (119). We believe that the collection of the Rap1 AD point mutants generated here will prove to be powerful reagents for investigating these possibilities.
Importantly, our study also demonstrates, for the first time, that transcription of authentic chromosomal Rap1-dependent target genes and Rap1-TFIID Taf5 coactivator target binding depend on a discrete AD within Rap1. The results of the Rap1WT and Rap17Ala variant pulse-labeling studies reported here provide compelling evidence that the Rap1 AD functions to specifically modulate transcription activation of chromosomal Rap1 target genes (Fig. 8). These results are all the more notable given the number of recent studies that document the robust ability of yeast to maintain normal steady-state transcript levels in the face of mutations in genes encoding key mRNA synthesis and mRNA degradation proteins. It is highly likely that our findings would have been difficult to observe without the use of the nascent RNA labeling technique. Meanwhile, our GST-Rap1/Taf5 pulldown binding experiments (Fig. 9) show that the Rap1 AD binds coactivator targets as expected and provides a starting point for dissecting the biochemical mechanism of transcription activation by the Rap1 AD.
Because Rap1 can bind nucleosomal UASRap1 sites (54) and reposition nucleosomes (50), the Rap1 AD may target chromatin remodeling factors, in addition to TFIID and TFIIA subunits, to modulate target gene transcription. Indeed, several recent studies have suggested that the high level of RP and GE gene transcription requires altered nucleosome stabilities and/or positioning (58, 68, 120). Consistent with these observations, the ATP-dependent nucleosome remodeling complex Swi/Snf has been shown to interact with Rap1 (19). The Rap1 AD may also directly contact the chromatin remodeling RSC complex (120) and/or the histone acetyltransferase NuA4 complex, both of which have been identified as important regulators of Rap1-dependent gene transcription (65, 121, 122). Future studies will address these important questions.
In conclusion, the Rap1 AD mapping and the Rap1 AD mutants generated through this study represent an important advance in Rap1 biology. This information and the reagents generated will enable future work aimed at molecularly dissecting the mechanisms responsible for regulating the robustly transcribed RP- and GE-encoding genes. These advances were made possible by the generation of a true-altered DNA-binding specificity Rap1AS. In addition to providing new knowledge to advance protein engineering efforts, we suspect that use of the novel Rap1AS reagent will also markedly help studies of Rap1 biology move forward.
Experimental procedures
Yeast strains
All of the yeast strains for this study are listed in Table 1. These strains were derived from a rap1Δ null strain created in the BY4741 background (123). These strains carry either a pRS416 FLAG3-RAP1 or pRS415 MYC5-RAP1 covering plasmid expressed under the control of the native RAP1 promoter (RAP1 nt position −433 to +1) (30) and terminator (RAP1 nt position +2485 to +2533). These strains also carry an HA3-tagged TAF1 allele and various integrated UASRap1-HIS3 reporter genes derived from a similar reporter characterized in previous studies (31, 57).
Test Rap1 yeast expression vectors
Test RAP1 yeast expression vectors are the same as the pRS415-MYC5 RAP1 expression vector except that the SV40 nuclear localization signal (PKKKRKV (124)) was inserted via PCR between the MYC5 tag and the RAP1 open-reading frame (ORF). Mutant RAP1 test expression vectors were created either by replacing the BlpI to SphI fragment of RAP1 in plasmid pRS415-MYC5-NLS-RAP1 with the BlpI to SphI fragment of a mutant RAP1 generated via library construction (see below) or splicing by overlap extension PCR (125).
Rap1 expression and purification
To prepare Rap1 for DNA binding assays, Rap1 ORF sequences were excised from pRS416-Rap1 expression plasmids with EcoRI and XhoI and ligated into similarly digested pET28a expression vector, in-frame with the vector encoded N-terminal His6 tag. Plasmids were transformed and propagated at 37 °C in the E. coli Rosetta II (DE3) expression strain (Novagen) in Luria-Bertani media (126) supplemented with kanamycin (10 μg/ml) and chloramphenicol (34 μg/ml). His6-Rap1 expression was induced for 4 h following addition of isopropylthiogalactoside to 1 mm. Cell pellets from 500 ml of culture were resuspended in 20 ml of Lysis/Wash buffer (25 mm HEPES-NaOH (pH 7.6), 10% v/v glycerol, 300 mm NaCl, 0.01% v/v Nonidet P-40, 1 mm benzamidine, 0.2 mm PMSF). Cells were lysed by treatment with lysozyme (final concentration of 1 mg/ml) and sonication. After centrifugation, each cleared lysate was incubated with 2.5 ml of Ni-NTA-agarose (Qiagen) pre-equilibrated with an equal volume of Lysis/Wash buffer. Proteins were bound for 3 h at 4 °C, washed three times with Lysis/Wash buffer, and transferred to a disposable column (Bio-Rad), and bound proteins were eluted using Lysis/Wash buffer containing 200 mm imidazole.
To generate Rap1 for GST pulldown assays, Rap1 ORFs encoded on the pRS415 expression plasmid were subjected to PCR to generate Rap1 coding sequences with EcoRI and XhoI ends that could be cloned in-frame with the His6-GST tag in the pBG101 expression vector. His6-GST-Rap1 variants were expressed and subjected to Ni-NTA purification as described for His6-Rap1 scaled up for 2 liters of E. coli culture. Ni-NTA-purified His6-GST-Rap1 variants were diluted to reduce the concentration of NaCl in the Lysis/Wash buffer to 150 mm and permit binding to 2.5 ml of Sepharose SP Fast Flow (Pharmacia) pre-equilibrated in a disposable column (Bio-Rad) with 4 column volumes of Buffer B150 (25 mm HEPES-NaOH (pH 7.6), 10% v/v glycerol, 150 mm NaCl, 0.01% v/v Nonidet P-40, 1 mm benzamidine, 0.2 mm PMSF). After binding, Sepharose SP was washed with 4 more column volumes of B150. Bound proteins were eluted using 3 column volumes of Buffer B1000 (25 mm HEPES NaOH (pH 7.6), 10% v/v glycerol, 1 m NaCl, 0.01% v/v Nonidet P-40, 1 mm benzamidine, 0.2 mm PMSF).
Taf5 aa 1–337 expression and purification
His6 Taf5 aa 1–337 (containing the RBD) was expressed from a previously generated pET33b expression vector (71), and purified using Ni-NTA-agarose as described for His6 Rap1 scaled up for 2 liters of E. coli culture.
Rap1 DNA binding assays
10 ng (∼100 fmol) of purified His6-Rap1 WT/variant proteins was incubated with 0.6 ng (∼50 fmol, ∼35,000 dpm) of 19-bp 32P-labeled duplex DNA containing a Rap1-binding site (either WT, 5′-ATATACACCCATACATTGA-3′, or 3T4A, 5′-ATATACTACCATACATTGA-3′; UASRap1 sequences underlined; mutated residues boldface) in the presence or absence of variable amounts of unlabeled WT or 3T4A UASRap1DNA (see figure legends) for 20 min at room temperature in Binding buffer (20 mm HEPES KOH (pH 7.6), 10% v/v glycerol, 100 mm KCl, 0.1 mm EDTA, 1 mm DTT, 25 μg/ml BSA, 2.5 μg/ml poly(dG-dC) (double-stranded, alternating copolymer, Sigma)) in a final volume of 20 μl. Reactions were loaded on 0.5× TBE-buffered (44.5 mm Tris, 44.5 mm boric acid, 1 mm EDTA (pH 8.0)) 6% polyacrylamide gels and electrophoresed for 1 h at 200 V at room temperature, and the gels were vacuum-dried. [32P]DNA signals were detected via K-screen imaging using a Pharos FX imager (Bio-Rad). The intensity of the bands representing Rap1-[32P]DNA complexes were quantified using Quantity One software (Bio-Rad).
Mutagenesis of Rap1 DBD and construction of Rap1 variant expression library
Codon randomization of Asn-401, Ser-402, Arg-404, His-405, Arg-408, and Val-409 of the RAP1 DBD was achieved by overlap extension PCR (125) using the mutagenic oligonucleotide (see also Fig. 2A): 5′-CATTATGTGCCTAACCACACGGGTNNSNNSATTNNSNNSCGATTTNNSNNSTATCTTTCCAAAAGACTAGAGTACG-3′ its reverse complement, and two flanking/outside primers containing BlpI and XhoI restriction sites. Mutagenic PCR products were digested with BlpI and XhoI restriction enzymes and ligated into similarly digested pRS416-RAP1, a plasmid that drives RAP1 expression using the native RAP1 enhancer-promoter (see under “Yeast Strains”). Ligation products were electroporated into ElectroMAX DH510B cells (genotype F−mcrAΔ (mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ-rpsL nupG, Life Technologies, Inc.). Multiple electroporation reactions were used to create a library of ∼1 × 108 independent bacterial clones, a number that exceeds the 8.6 × 107 variations theoretically generated by the codon randomization of six amino acids. Sanger sequencing of library expression plasmid isolated from 10 individual clones was used to confirm that the mutagenesis targeted the expected amino acids.
Screen for Rap1AS
To screen for a mutant RAP1 variant that could drive expression of a reporter gene containing a mutant Rap1-binding site (i.e. UASRap1 3T4A), the library was introduced into yeast strain 3T4A-HIS3 #1 (see Table 1) and plated onto SC media (127) containing 5 mm 3-AT (Sigma); plates were incubated for 4 days at 30 °C. Colonies were then replica plated to SC media plates containing 5 mm 3-AT and 0.1% 5-FOA to determine whether growth of the colony on 5 mm 3-AT required Rap1 produced from the URA3-marked pRS416 RAP1 library plasmid. RAP1 expression plasmids were recovered from colonies that grew on 5 mm 3-AT and failed to grow on 5 mm 3-AT + 0.1% 5-FOA. These purified plasmid preparations were treated with the restriction endonuclease BsRG1 (New England Biolabs) that cuts the pRS415-MYC5-RAP1 covering plasmid, but not pRS416-RAP1 library plasmid. The digest was used to transform E. coli to Ampr, and the resulting plasmids were isolated to generate putative Rap1AS-encoding pRS416-RAP1 mutant plasmids. The recovered pRS416-RAP1 plasmids were retransformed into yeast strain 3T4A-HIS3 #1 and retested for growth on 5 mm 3-AT to confirm that 3-AT-resistant growth was conferred by the mutant pRS416-RAP1 plasmid.
Yeast cell growth assays
Yeast strains were grown overnight to saturation, serially diluted 1:4 in sterile water in 96-well plates, spotted using a pinning tool (Sigma) onto non-selective media plates (SC + His) and selective media plates (SC-His + 3-AT), and grown overnight at 30 °C for 2–4 days. Plate images were acquired using a ChemiDoc MP imager (Bio-Rad) and processed using ImageLab software (Bio-Rad). Images were saved at a resolution of 300 dpi.
Immunoblotting
Total protein was extracted from yeast cell pellets containing ∼1 × 107 cells (1 OD600 unit) via mild alkali treatment and heating in standard electrophoresis buffer. In brief, cell pellets were resuspended in 0.1 m NaOH (128) and incubated for 10 min at room temperature. Cells were re-pelleted; the 0.1 m NaOH was removed; and pellets were incubated for 10 min at 75 °C in SDS-PAGE sample buffer (1× LDS Sample Buffer (Life Technologies, Inc.) with DTT added to 62.5 mm). Proteins were fractionated on denaturing 4–12% BisTris polyacrylamide gels (Life Technologies, Inc.); gels were equilibrated in transfer buffer (30 mm Bicine, 25 mm BisTris, 1 mm EDTA, 60 μm chlorobutanol), and then electrotransfered to PVDF membranes pre-equilibrated in transfer buffer (GE Healthcare, Immobilon-P, 0.45 μm). Loading and transfer efficiency was monitored after transfer by staining with 0.5% w/v Ponceau S in 1% v/v acetic acid. After imaging, stain was removed using multiple changes of H2O. Blots were blocked with 5% w/v nonfat milk in 1× Tris-buffered saline (TBS) (100 mm Tris-Cl (pH 7.5), 150 mm NaCl). Myc5-Rap1 was detected using an anti-Myc antibody conjugated to HRP (Roche Applied Science catalog no. 11667203001) at a 1:2000 dilution. Endogenous actin was used as an additional loading control. Actin was detected using a 1:5000 dilution of anti-β-actin antibody (Abcam catalog no. ab8224) followed by incubation with a 1:2500 dilution of HRP-conjugated horse anti-mouse IgG antibody (Cell Signaling catalog no. 7076S). Both anti-Myc and anti-β-actin antibody incubations were performed in 1% w/v nonfat milk blocking in 1× TBS. HRP-generated immune complexes were detected by exposing blots to enhanced chemiluminescence reagent (GE Healthcare) followed by exposure to X-ray film. X-ray films were scanned at 600 dpi and saved as TIFF images.
Steady-state RNA analysis
Total RNA was extracted and purified from equal numbers of yeast cells using the hot phenol method (129). RNA concentration was measured by monitoring absorbance at 260 and 280 nm using a NanoDrop 2000c spectrophotometer. RNA integrity was assessed by agarose gel electrophoresis, ethidium bromide staining, and scanning using a Pharos FX imager (Bio-Rad). The intensity of the bands representing large (26S) and small (18S) yeast rRNAs were quantified using Quantity One software (Bio-Rad). Reverse transcription was performed using 2.5 μg of RNA from each strain using Superscript III (Life Technologies, Inc.) per the manufacturer's instructions. Priming for cDNA synthesis was achieved using oligo(dT)16 (Life Technologies, Inc.). Quantitative real time PCRs (qRT-PCR) were performed using equal amounts of RNAs (−/+ RT (reverse transcription) controls) and cDNAs using IQ real time SYBR Green PCR Supermix (Bio-Rad), an iCycler (Bio-Rad), and the gene-specific primers listed in Table 2. Relative transcript levels for each gene of interest were determined by comparing the cycle threshold (Ct) values generated from cDNA-containing reactions to standard curves obtained in parallel by measuring serial 1:10 dilutions of yeast genomic DNA and normalizing to the reference gene ACT1. Scatter plots generated using Prism 7 (GraphPad) and representing data obtained from both biological (individual points) and technical replicates of each sample are expressed relative to the normalized amount of a target gene of interest in the indicated positive control sample.
TABLE 2.
qRT-PCR primers used in this study
| Gene | Primer name | Primer sequence (5′ → 3′) |
|---|---|---|
| HIS3 | HIS3_F2 | ATGACAGAGCAGAAAGCCC |
| HIS3_R2 | GCACTCAACGATTAGCGAC | |
| ACT1 | ACT1_F2 | TGGTCGGTATGGGTCAAAAA |
| ACT1_R2 | AAGGACAAAACGGCTTGGAT | |
| ENO1 | ENO1_F | CGTCAACGATGTCATTGCTC |
| ENO1_R | CCAAAGAAACACCCAAGATAGC | |
| PYK1 | PYK1_F | CCAACCTCCACCACCGAAAC |
| PYK1_R | GGGCTTCAACATCATCAGTCCA | |
| RPS3 | RPS3_F | TACGGTGTCGTCAGATACG |
| RPS3_R | GACCAGAGTGAATCAAGAAACC | |
| RPL26B | RPL26B_F | TTTCACCGCCCCATCCTCCC |
| RPL26B_R | GGAACGGAAGCACCGTTGAC | |
| RPL3 | RPL3_F | TGGACAGACCAGGTTCTAAG |
| RPL3_R | AAATGTTCAGCCCAGACGG | |
| Tubulin | Tubulin_F | CCGCTGGTGGAAAGTATGTT |
| Tubulin_R | GCCAATTCAGCACCTTCAGT |
Pulse labeling of nascent RNA
Nascent RNA levels in yeast strains of interest were assessed using efficient and reversible RNA labeling and purification (94). 200 ml of each yeast culture grown in YPD to a cell density of ∼1.5 × 107 cells/ml (2.5 OD600/ml) were pulse-labeled for 2.5 min with 5 mm 4-thiouracil (Sigma), immediately harvested via vacuum filtration, and immersed in a 1:1 ratio of phenol/RNA Buffer A (50 mm NaAc (pH 5.5), 10 mm EDTA (pH 8), 0.5% SDS) pre-warmed to 65 °C. ∼4.8 × 108 cells (80 OD600 units) of 6-min thiouracil labeled Schizosaccharomyces pombe cells were added into each sample prior to RNA extraction to provide a reference gene for later qRT-PCR analysis. Total RNA from the resulting S. cerevisiae/S. pombe mixtures was extracted using the hot phenol method (129). RNA concentration and integrity were assessed as described above. To prepare nascent thiolated RNA for separation from total RNA, 400 μg of each RNA in 1 ml of Biotinylation Buffer (10 mm Tris-HCl (pH 7.5), 1 mm EDTA (pH 8)) was heat-denatured at 65 °C for 10 min and then chilled for 2 min on ice. RNA was biotinylated for 30 min at room temperature in the dark on a tilt board following addition of 20 μl of 1 mg/ml 2-((biotinoyl)amino)ethyl methanethiosulfonate (Biotium) dissolved in dimethylformamide. Unincorporated biotin was removed by two chloroform extractions (once with 1 ml and then with 0.75 ml), the second of which was performed using a Heavy Phase Lock Gel (5 Prime Inc.). RNA was precipitated using an equal volume (0.75 ml) of isopropyl alcohol and dissolved in 100 μl of 0.1% diethyl pyrocarbonate (DEPC)-treated H2O (Fisher). RNA was again heat-denatured at 65 °C for 10 min and then chilled on ice for 5 min. Biotinylated nascent RNAs were purified using 200 μl of a colloidal suspension of μMACs streptavidin beads/reaction (μMACs streptavidin kit, Miltenyi Biotec). Samples were placed in a light-tight container on a tilt board at room temperature for 15 min. Each RNA/bead mixture was added to a separate μMACs streptavidin kit column that had been placed in the μMACs separator and washed 2× with 0.2 ml of Nucleic Acid Wash Buffer (μMACs streptavidin kit, Miltenyi Biotec). The flow-through was collected and re-applied to the column. Columns were washed two times with 0.5 ml of the biotinylated RNA Wash Buffer (100 mm Tris-HCl (pH 7.5), 10 mm EDTA (pH 8), 1 m NaCl, 0.1% Tween 20), and biotinylated RNA was eluted with two 200-μl washes of 0.1 m DTT. Biotinylated RNA was ethanol-precipitated following addition of 40 μg of molecular biology grade glycogen (Roche Applied Science), 0.1× volume of 3 m NaOAc (pH 5.5), and 3× volumes of 100% ethanol. Precipitated RNAs were dissolved in Tris 0.1 EDTA (T.1E: 10 mm Tris-Cl (pH 7.5), 0.1 mm EDTA). cDNA synthesis was performed as described above except that gene-specific primers for genes of interest lacking a poly(A) tail were included in addition to oligo(dT)16; qRT-PCR was performed, and relative transcript levels were determined as described above, except that S. pombe β-tubulin present in the samples due to the addition of S. pombe cells was used as a reference gene (93) (see “Steady-state RNA Analysis” above for details).
GST pulldown assays
GST pulldown assays were conducted as 200-μl reactions in pulldown buffer (20 mm HEPES-NaOH (pH 7.5), 150 mm NaAc, 0.05% Triton X-100, 10% glycerol, 1 mm DTT, 25 ng/μl BSA) using 5 μl of glutathione-Sepharose 4 Fast Flow (GE Healthcare), 12 pmol of His6-GST, or 6 pmol of His6-GST-Rap1 variant and either 0 or 100 pmol of His6-Taf5. Binding reactions were incubated for 1 h at room temperature on the tilt board. Following incubation, glutathione-Sepharose beads were pelleted and washed once with pulldown buffer. Beads were pelleted again, and bead-bound material was eluted with 1× NuPAGE sample buffer (1× LDS Sample Buffer (Life Technologies, Inc.) with DTT added to 62.5 mm). Bead-bound material was fractionated on 4–12% NuPAGE gels (Life Technologies, Inc.), stained with Sypro Ruby (Life Technologies, Inc.), and imaged with Pharos FX (Bio-Rad). Binding data were quantified using Quantity One software (Bio-Rad) and plotted using Prism 7 (GraphPad).
Author contributions
P. A. W. conceived of the study and assisted with the design and interpretation of the experiments. A. N. J. performed, designed, and interpreted all experiments. Both authors wrote the manuscript.
Acknowledgments
We thank our laboratory colleagues Drs. Jordan Feigerle, Justin H. Layer, Iulia M. Gilchuk, and Chiranthani Sumanasekera for reagents, advice, and thoughtful discussions throughout the course of this work. A. N. J. would also like to thank the members of her Ph.D. thesis advisory committee, Drs. Roger Colbran, Roland Stein, William Tansey, and Bryan Venters, for their helpful guidance and suggestions. We are grateful to Dr. Lazlo Tora and Tiago Baptista for their assistance in helping to set up the nascent RNA labeling technique in our laboratory.
This work was supported in part by National Institutes of Health Grant R01GM115892 (to P. A. W.) and National Institutes of Health Cancer Center Core Grant 5P30CA06848. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- RNAPII
- DNA-dependent RNA polymerase II
- GTF
- general transcription factors
- PIC
- pre-initiation complex
- EC
- RNA polymerase II elongation complex
- DBD
- DNA-binding domain
- AD
- activation domain
- RP
- ribosomal protein
- GE
- glycolytic enzyme
- UAS
- upstream activating sequence
- aa
- amino acid(s)
- nt
- nucleotide
- Rif
- Rap1 interacting factor
- Taf
- TBP associated factor
- Sir
- Silent information regulator
- AS
- altered specificity
- 3-AT
- 3-amino-1,2,4-triazole
- SC media
- synthetic complete media
- NLS
- nuclear localization signal
- MTSEA
- (2-((biotinoyl)amino)ethyl methanethiosulfonate
- nt
- nucleotide
- RBD
- Rap1-binding domain
- ChIP-Seq/-Exo
- chromatin immunoprecipitation-sequencing/chromatin immunoprecipitation-exonuclease
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- TBP
- TATA-binding protein
- qRT
- quantitative RT
- Ni-NTA
- nickel-nitrilotriacetic acid
- 5-FOA
- 5-fluoroorotic acid
- Bicine
- N,N-bis(2-hydroxyethyl)glycine.
References
- 1. Taatjes D. J., Marr M. T., and Tjian R. (2004) Regulatory diversity among metazoan co-activator complexes. Nat. Rev. Mol. Cell Biol. 5, 403–410 [DOI] [PubMed] [Google Scholar]
- 2. Hahn S., and Young E. T. (2011) Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189, 705–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shandilya J., and Roberts S. G. (2012) The transcription cycle in eukaryotes: from productive initiation to RNA polymerase II recycling. Biochim. Biophys. Acta 1819, 391–400 [DOI] [PubMed] [Google Scholar]
- 4. Weake V. M., and Workman J. L. (2010) Inducible gene expression: diverse regulatory mechanisms. Nat. Rev. Genet. 11, 426–437 [DOI] [PubMed] [Google Scholar]
- 5. Ranish J. A., and Hahn S. (1996) Transcription: basal factors and activation. Curr. Opin. Genet. Dev. 6, 151–158 [DOI] [PubMed] [Google Scholar]
- 6. Thomas M. C., and Chiang C.-M. (2006) The general transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol. 41, 105–178 [DOI] [PubMed] [Google Scholar]
- 7. Roeder R. G. (2005) Transcriptional regulation and the role of diverse coactivators in animal cells. FEBS Lett. 579, 909–915 [DOI] [PubMed] [Google Scholar]
- 8. Jonkers I., and Lis J. T. (2015) Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 16, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Struhl K. (1995) Yeast transcriptional regulatory mechanisms. Annu. Rev. Genet. 29, 651–674 [DOI] [PubMed] [Google Scholar]
- 10. Ptashne M. (1988) How eukaryotic transcriptional activators work. Nature 335, 683–689 [DOI] [PubMed] [Google Scholar]
- 11. MA D., Olave I., Merino A., and Reinberg D. (1996) Separation of the transcriptional coactivator and antirepression functions of transcription factor IIA. Proc. Natl. Acad. Sci. U.S.A. 93, 6583–6588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ozer J., Bolden A. H., and Lieberman P. M. (1996) Transcription factor IIA mutations show activator-specific defects and reveal a IIA function distinct from stimulation of TBP-DNA binding. J. Biol. Chem. 271, 11182–11190 [DOI] [PubMed] [Google Scholar]
- 13. Kim T. K., Kim T. H., and Maniatis T. (1998) Efficient recruitment of TFIIB and CBP-RNA polymerase II holoenzyme by an interferon-β enhanceosome in vitro. Proc. Natl. Acad. Sci. U.S.A. 95, 12191–12196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanese N., Pugh B. F., and Tjian R. (1991) Coactivators for a proline-rich activator purified from the multisubunit human TFIID complex. Genes Dev. 5, 2212–2224 [DOI] [PubMed] [Google Scholar]
- 15. Chen J.-L., Attardi L. D., Verrijzer C. P., Yokomori K., and Tjian R. (1994) Assembly of recombinant TFIID reveals differential coactivator requirements for distinct transcriptional activators. Cell 79, 93–105 [DOI] [PubMed] [Google Scholar]
- 16. Brownell J. E., Zhou J., Ranalli T., Kobayashi R., Edmondson D. G., Roth S. Y., and Allis C. D. (1996) Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84, 843–851 [DOI] [PubMed] [Google Scholar]
- 17. Wallberg A. E., Pedersen K., Lendahl U., and Roeder R. G. (2002) p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by notch intracellular domains in vitro. Mol. Cell. Biol. 22, 7812–7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ren X., Siegel R., Kim U., and Roeder R. G. (2011) Direct interactions of OCA-B and TFII-I regulate immunoglobulin heavy-chain gene transcription by facilitating enhancer-promoter communication. Mol. Cell 42, 342–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tomar R. S., Zheng S., Brunke-Reese D., Wolcott H. N., and Reese J. C. (2008) Yeast Rap1 contributes to genomic integrity by activating DNA damage repair genes. EMBO J. 27, 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Green M. R. (2005) Eukaryotic transcription activation: right on target. Mol. Cell 18, 399–402 [DOI] [PubMed] [Google Scholar]
- 21. Huet J., Cottrelle P., Cool M., Vignais M. L., Thiele D., Marck C., Buhler J. M., Sentenac A., and Fromageot P. (1985) A general upstream binding factor for genes of the yeast translational apparatus. EMBO J. 4, 3539–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shore D., and Nasmyth K. (1987) Purification and cloning of a DNA binding protein from yeast that binds to both silencer and activator elements. Cell 51, 721–732 [DOI] [PubMed] [Google Scholar]
- 23. Morse R. H. (2000) RAP, RAP, open up! New wrinkles for RAP1 in yeast. Trends Genet. 16, 51–53 [DOI] [PubMed] [Google Scholar]
- 24. Callebaut I., and Mornon J.-P. (1997) From BRCA1 to RAP1: a widespread BRCT module closely associated with DNA repair. FEBS Lett. 400, 25–30 [DOI] [PubMed] [Google Scholar]
- 25. Zhang W., Zhang J., Zhang X., Xu C., and Tu X. (2011) Solution structure of Rap1 BRCT domain from Saccharomyces cerevisiae reveals a novel fold. Biochem. Biophys. Res. Commun. 404, 1055–1059 [DOI] [PubMed] [Google Scholar]
- 26. Miyake T., Hu Y.-F., David S. Y., and Li R. (2000) A functional comparison of BRCA1 C-terminal domains in transcription activation and chromatin remodeling. J. Biol. Chem. 275, 40169–40173 [DOI] [PubMed] [Google Scholar]
- 27. Henry Y. A., Chambers A., Tsang J. S., Kingsman A. J., and Kingsman S. M. (1990) Characterisation of the DNA binding domain of the yeast RAP1 protein. Nucleic Acids Res. 18, 2617–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kyrion G., Boakye K. A., and Lustig A. J. (1992) C-terminal truncation of RAP1 results in the deregulation of telomere size, stability, and function in Saccharomyces cerevisiae. Mol. Cell. Biol. 12, 5159–5173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feeser E. A., and Wolberger C. (2008) Structural and functional studies of the Rap1 C-terminus reveal novel separation-of-function mutants. J. Mol. Biol. 380, 520–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Graham I. R., Haw R. A., Spink K. G., Halden K. A., and Chambers A. (1999) In vivo analysis of functional regions within yeast Rap1p. Mol. Cell. Biol. 19, 7481–7490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garbett K. A., Tripathi M. K., Cencki B., Layer J. H., and Weil P. A. (2007) Yeast TFIID serves as a coactivator for Rap1p by direct protein-protein interaction. Mol. Cell. Biol. 27, 297–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Y., Rai R., Zhou Z.-R., Kanoh J., Ribeyre C., Yang Y., Zheng H., Damay P., Wang F., Tsujii H., Hiraoka Y., Shore D., Hu H.-Y., Chang S., and Lei M. (2011) A conserved motif within RAP1 has diversified roles in telomere protection and regulation in different organisms. Nat. Struct. Mol. Biol. 18, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hardy C. F., Balderes D., and Shore D. (1992) Dissection of a carboxy-terminal region of the yeast regulatory protein RAP1 with effects on both transcriptional activation and silencing. Mol. Cell. Biol. 12, 1209–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moretti P., Freeman K., Coodly L., and Shore D. (1994) Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev. 8, 2257–2269 [DOI] [PubMed] [Google Scholar]
- 35. Hardy C. F., Sussel L., and Shore D. (1992) A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 6, 801–814 [DOI] [PubMed] [Google Scholar]
- 36. Vignais M. L., Huet J., Buhler J. M., and Sentenac A. (1990) Contacts between the factor TUF and RPG sequences. J. Biol. Chem. 265, 14669–14674 [PubMed] [Google Scholar]
- 37. Lieb J. D., Liu X., Botstein D., and Brown P. O. (2001) Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 28, 327–334 [DOI] [PubMed] [Google Scholar]
- 38. Lascaris R. F., Mager W. H., and Planta Rudi J. (1999) DNA-binding requirements of the yeast protein Rap1p as selected in silico from ribosomal protein gene promoter sequences. Bioinformatics 15, 267–277 [DOI] [PubMed] [Google Scholar]
- 39. Nieuwint R. T., Mager W. H., Maurer K. C., and Planta R. J. (1989) Mutational analysis of the upstream activation site of yeast ribosomal protein genes. Curr. Genet. 15, 247–251 [DOI] [PubMed] [Google Scholar]
- 40. Vignais M. L., Woudt L. P., Wassenaar G. M., Mager W. H., Sentenac A., and Planta R. J. (1987) Specific binding of TUF factor to upstream activation sites of yeast ribosomal protein genes. EMBO J. 6, 1451–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Idrissi F.-Z., Fernández-Larrea J. B., and Piña B. (1998) Structural and functional heterogeneity of Rap1p complexes with telomeric and UASrpg-like DNA sequences. J. Mol. Biol. 284, 925–935 [DOI] [PubMed] [Google Scholar]
- 42. Idrissi F.-Z., Garcia-Reyero N., Fernandez-Larrea J. B., and Piña B. (2001) Alternative mechanisms of transcriptional activation by Rap1p. J. Biol. Chem. 276, 26090–26098 [DOI] [PubMed] [Google Scholar]
- 43. Li B., Nierras C. R., and Warner J. R. (1999) Transcriptional elements involved in the repression of ribosomal protein synthesis. Mol. Cell. Biol. 19, 5393–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kasahara K., Ohtsuki K., Ki S., Aoyama K., Takahashi H., Kobayashi T., Shirahige K., and Kokubo T. (2007) Assembly of regulatory factors on rRNA and ribosomal protein genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 27, 6686–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lickwar C. R., Mueller F., Hanlon S. E., McNally J. G., and Lieb J. D. (2012) Genome-wide protein–DNA binding dynamics suggest a molecular clutch for transcription factor function. Nature 484, 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rhee H. S., and Pugh B. F. (2011) Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147, 1408–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Konig P., Giraldo R., Chapman L., and Rhodes D. (1996) The crystal structure of the DNA-binding domain of yeast RAP1 in complex with telomeric DNA. Cell 85, 125–136 [DOI] [PubMed] [Google Scholar]
- 48. Taylor H. O., O'Reilly M., Leslie A. G., and Rhodes D. (2000) How the multifunctional yeast Rap1p discriminates between DNA target sites: a crystallographic analysis. J. Mol. Biol. 303, 693–707 [DOI] [PubMed] [Google Scholar]
- 49. Matot B., Le Bihan Y.-V., Lescasse R., Pérez J., Miron S., David G., Castaing B., Weber P., Raynal B., Zinn-Justin S., Gasparini S., and Le Du M.-H. (2012) The orientation of the C-terminal domain of the Saccharomyces cerevisiae Rap1 protein is determined by its binding to DNA. Nucleic Acids Res. 40, 3197–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yu L., and Morse R. H. (1999) Chromatin opening and transactivator potentiation by RAP1 in Saccharomyces cerevisiae. Mol. Cell. Biol. 19, 5279–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ganapathi M., Palumbo M. J., Ansari S. A., He Q., Tsui K., Nislow C., and Morse R. H. (2011) Extensive role of the general regulatory factors, Abf1 and Rap1, in determining genome-wide chromatin structure in budding yeast. Nucleic Acids Res. 39, 2032–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu L., Sabet N., Chambers A., and Morse R. H. (2001) The N-terminal and C-terminal domains of RAP1 are dispensable for chromatin opening and GCN4-mediated HIS4 activation in budding yeast. J. Biol. Chem. 276, 33257–33264 [DOI] [PubMed] [Google Scholar]
- 53. Rossetti L., Cacchione S., De Menna A., Chapman L., Rhodes D., and Savino M. (2001) Specific interactions of the telomeric protein rap1p with nucleosomal binding sites. J. Mol. Biol. 306, 903–913 [DOI] [PubMed] [Google Scholar]
- 54. Koerber R. T., Rhee H. S., Jiang C., and Pugh B. F. (2009) Interaction of transcriptional regulators with specific nucleosomes across the Saccharomyces genome. Mol. Cell. 35, 889–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao Y., McIntosh K. B., Rudra D., Schawalder S., Shore D., and Warner J. R. (2006) Fine-structure analysis of ribosomal protein gene transcription. Mol. Cell. Biol. 26, 4853–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Klein C., and Struhl K. (1994) Protein kinase A mediates growth-regulated expression of yeast ribosomal protein genes by modulating RAP1 transcriptional activity. Mol. Cell. Biol. 14, 1920–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mencía M., Moqtaderi Z., Geisberg J. V., Kuras L., and Struhl K. (2002) Activator-specific recruitment of TFIID and regulation of ribosomal protein genes in yeast. Mol. Cell 9, 823–833 [DOI] [PubMed] [Google Scholar]
- 58. Knight B., Kubik S., Ghosh B., Bruzzone M. J., Geertz M., Martin V., Dénervaud N., Jacquet P., Ozkan B., Rougemont J., Maerkl S. J., Naef F., and Shore D. (2014) Two distinct promoter architectures centered on dynamic nucleosomes control ribosomal protein gene transcription. Genes Dev. 28, 1695–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Scott E. W., and Baker H. V. (1993) Concerted action of the transcriptional activators REB1, RAP1, and GCR1 in the high-level expression of the glycolytic gene TPI. Mol. Cell. Biol. 13, 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chambers A., Tsang J. S., Stanway C., Kingsman A. J., and Kingsman S. M. (1989) Transcriptional control of the Saccharomyces cerevisiae PGK gene by RAP1. Mol. Cell. Biol. 9, 5516–5524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wade J. T., Hall D. B., and Struhl K. (2004) The transcription factor Ifh1 is a key regulator of yeast ribosomal protein genes. Nature 432, 1054–1058 [DOI] [PubMed] [Google Scholar]
- 62. Schawalder S. B., Kabani M., Howald I., Choudhury U., Werner M., and Shore D. (2004) Growth-regulated recruitment of the essential yeast ribosomal protein gene activator Ifh1. Nature 432, 1058–1061 [DOI] [PubMed] [Google Scholar]
- 63. Rudra D., Zhao Y., and Warner J. R. (2005) Central role of Ifh1p–Fhl1p interaction in the synthesis of yeast ribosomal proteins. EMBO J. 24, 533–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marion R. M., Regev A., Segal E., Barash Y., Koller D., Friedman N., and O'Shea E. K. (2004) Sfp1 is a stress-and nutrient-sensitive regulator of ribosomal protein gene expression. Proc. Natl. Acad. Sci. U.S.A. 101, 14315–14322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Reid J. L., Iyer V. R., Brown P. O., and Struhl K. (2000) Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol. Cell 6, 1297–1307 [DOI] [PubMed] [Google Scholar]
- 66. Hall D. B., Wade J. T., and Struhl K. (2006) An HMG protein, Hmo1, associates with promoters of many ribosomal protein genes and throughout the rRNA gene locus in Saccharomyces cerevisiae. Mol. Cell. Biol. 26, 3672–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kasahara K., Ki S., Aoyama K., Takahashi H., and Kokubo T. (2008) Saccharomyces cerevisiae HMO1 interacts with TFIID and participates in start site selection by RNA polymerase II. Nucleic Acids Res. 36, 1343–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reja R., Vinayachandran V., Ghosh S., and Pugh B. F. (2015) Molecular mechanisms of ribosomal protein gene coregulation. Genes Dev. 29, 1942–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Haruki H., Nishikawa J., and Laemmli U. K. (2008) The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell 31, 925–932 [DOI] [PubMed] [Google Scholar]
- 70. Ptashne M., and Gann A. A. (1990) Activators and targets. Nature 346, 329–331 [DOI] [PubMed] [Google Scholar]
- 71. Layer J. H., Miller S. G., and Weil P. A. (2010) Direct transactivator-transcription factor IID (TFIID) contacts drive yeast ribosomal protein gene transcription. J. Biol. Chem. 285, 15489–15499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nilsson M. T., and Widersten M. (2004) Repertoire selection of variant single-chain Cro: toward directed DNA binding specificity of helix-turn-helix proteins. Biochemistry 43, 12038–12047 [DOI] [PubMed] [Google Scholar]
- 73. Pfau J., Arvidson D. N., Youderian P., Pearson L. L., and Sigman D. S. (1994) A site-specific endonuclease derived from a mutant Trp repressor with altered DNA binding specificity. Biochemistry 33, 11391–11403 [DOI] [PubMed] [Google Scholar]
- 74. Gregory B. D., Nickels B. E., Darst S. A., and Hochschild A. (2005) An altered-specificity DNA-binding mutant of Escherichia coli σ70 facilitates the analysis of σ70 function in vivo: An altered-specificity mutant of σ70. Mol. Microbiol. 56, 1208–1219 [DOI] [PubMed] [Google Scholar]
- 75. Strubin M., and Struhl K. (1992) Yeast and human TFIID with altered DNA binding specificity for TATA elements. Cell 68, 721–730 [DOI] [PubMed] [Google Scholar]
- 76. Kim J., Tzamarias D., Ellenberger T., Harrison S. C., and Struhl K. (1993) Adaptability at the protein-DNA interface is an important aspect of sequence recognition by bZIP proteins. Proc. Natl. Acad. Sci. U.S.A. 90, 4513–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mader S., Kumar V., de Verneuil H., and Chambon P. (1989) Three amino acids of the oestrogen receptor are essential to its ability to distinguish an oestrogen from a glucocorticoid-responsive element. Nature 338, 271–274 [DOI] [PubMed] [Google Scholar]
- 78. Voss T. C., Schiltz R. L., Sung M.-H., Yen P. M., Stamatoyannopoulos J. A., Biddie S. C., Johnson T. A., Miranda T. B., John S., and Hager G. L. (2011) Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 146, 544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chu S. W., Noyes M. B., Christensen R. G., Pierce B. G., Zhu L. J., Weng Z., Stormo G. D., and Wolfe S. A. (2012) Exploring the DNA-recognition potential of homeodomains. Genome Res. 22, 1889–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shore D. (1994) RAP1: a protean regulator in yeast. Trends Genet. 10, 408–412 [DOI] [PubMed] [Google Scholar]
- 81. Rudra D., and Warner J. R. (2004) What better measure than ribosome synthesis? Genes Dev. 18, 2431–2436 [DOI] [PubMed] [Google Scholar]
- 82. Wolberger C., Vershon A. K., Liu B., Johnson A. D., and Pabo C. O. (1991) Crystal structure of a MATα2 homeodomain-operator complex suggests a general model for homeodomain-DNA interactions. Cell 67, 517–528 [DOI] [PubMed] [Google Scholar]
- 83. Fraenkel E., Rould M. A., Chambers K. A., and Pabo C. O. (1998) Engrailed homeodomain-DNA complex at 2.2 A resolution: a detailed view of the interface and comparison with other engrailed structures. J. Mol. Biol. 284, 351–361 [DOI] [PubMed] [Google Scholar]
- 84. An Y., Ji J., Wu W., Lv A., Huang R., and Wei Y. (2005) A rapid and efficient method for multiple-site mutagenesis with a modified overlap extension PCR. Appl. Microbiol. Biotechnol. 68, 774–778 [DOI] [PubMed] [Google Scholar]
- 85. Brennan M. B., and Struhl K. (1980) Mechanisms of increasing expression of a yeast gene in Escherichia coli. J. Mol. Biol. 136, 333–338 [DOI] [PubMed] [Google Scholar]
- 86. Kurtz S., and Shore D. (1991) RAP1 protein activates and silences transcription of mating-type genes in yeast. Genes Dev. 5, 616–628 [DOI] [PubMed] [Google Scholar]
- 87. Yarragudi A., Parfrey L. W., and Morse R. H. (2007) Genome-wide analysis of transcriptional dependence and probable target sites for Abf1 and Rap1 in Saccharomyces cerevisiae. Nucleic Acids Res. 35, 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sun M., Schwalb B., Schulz D., Pirkl N., Etzold S., Larivière L., Maier K. C., Seizl M., Tresch A., and Cramer P. (2012) Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 22, 1350–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sun M., Schwalb B., Pirkl N., Maier K. C., Schenk A., Failmezger H., Tresch A., and Cramer P. (2013) Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol. Cell 52, 52–62 [DOI] [PubMed] [Google Scholar]
- 90. Schulz D., Pirkl N., Lehmann E., and Cramer P. (2014) Rpb4 subunit functions mainly in mRNA synthesis by RNA polymerase II. J. Biol. Chem. 289, 17446–17452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chow J., and Dennis P. P. (1994) Coupling between mRNA synthesis and mRNA stability in Escherichia coli. Mol. Microbiol. 11, 919–931 [DOI] [PubMed] [Google Scholar]
- 92. Haimovich G., Medina D. A., Causse S. Z., Garber M., Millán-Zambrano G., Barkai O., Chávez S., Pérez-Ortín J. E., Darzacq X., and Choder M. (2013) Gene expression is circular: factors for mRNA degradation also foster mRNA synthesis. Cell 153, 1000–1011 [DOI] [PubMed] [Google Scholar]
- 93. Bonnet J., Wang C.-Y., Baptista T., Vincent S. D., Hsiao W.-C., Stierle M., Kao C.-F., Tora L., and Devys D. (2014) The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev. 28, 1999–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Duffy E. E., Rutenberg-Schoenberg M., Stark C. D., Kitchen R. R., Gerstein M. B., and Simon M. D. (2015) Tracking distinct RNA populations using efficient and reversible covalent chemistry. Mol. Cell 59, 858–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sussel L., and Shore D. (1991) Separation of transcriptional activation and silencing functions of the RAP1-encoded repressor/activator protein 1: isolation of viable mutants affecting both silencing and telomere length. Proc. Natl. Acad. Sci. U.S.A. 88, 7749–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Layer J. H., and Weil P. A. (2013) Direct TFIIA-TFIID protein contacts drive budding yeast ribosomal protein gene transcription. J. Biol. Chem. 288, 23273–23294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chambers A., Packham E. A., and Graham I. R. (1995) Control of glycolytic gene expression in the budding yeast (Saccharomyces cerevisiae). Curr. Genet. 29, 1–9 [DOI] [PubMed] [Google Scholar]
- 98. Lempiäinen H., and Shore D. (2009) Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 21, 855–863 [DOI] [PubMed] [Google Scholar]
- 99. Kuras L., Kosa P., Mencia M., and Struhl K. (2000) TAF-containing and TAF-independent forms of transcriptionally active TBP in vivo. Science 288, 1244–1248 [DOI] [PubMed] [Google Scholar]
- 100. Li X.-Y., Bhaumik S. R., and Green M. R. (2000) Distinct classes of yeast promoters revealed by differential TAF recruitment. Science 288, 1242–1244 [DOI] [PubMed] [Google Scholar]
- 101. Shen W.-C., Bhaumik S. R., Causton H. C., Simon I., Zhu X., Jennings E. G., Wang T.-H., Young R. A., and Green M. R. (2003) Systematic analysis of essential yeast TAFs in genome-wide transcription and preinitiation complex assembly. EMBO J. 22, 3395–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li X.-Y., Bhaumik S. R., Zhu X., Li L., Shen W.-C., Dixit B. L., and Green M. R. (2002) Selective recruitment of TAFs by yeast upstream activating sequences: implications for eukaryotic promoter structure. Curr. Biol. 12, 1240–1244 [DOI] [PubMed] [Google Scholar]
- 103. Shen W.-C., and Green M. R. (1997) Yeast TAF II 145 functions as a core promoter selectivity factor, not a general coactivator. Cell. 90, 615–624 [DOI] [PubMed] [Google Scholar]
- 104. Singh M. V., Bland C. E., and Weil P. A. (2004) Molecular and genetic characterization of a Taf1p domain essential for yeast TFIID assembly. Mol. Cell. Biol. 24, 4929–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Papai G., Tripathi M. K., Ruhlmann C., Layer J. H., Weil P. A., and Schultz P. (2010) TFIIA and the transactivator Rap1 cooperate to commit TFIID for transcription initiation. Nature 465, 956–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ohtsuki K., Kasahara K., Shirahige K., and Kokubo T. (2010) Genome-wide localization analysis of a complete set of Tafs reveals a specific effect of the taf1 mutation on Taf2 occupancy and provides indirect evidence for different TFIID conformations at different promoters. Nucleic Acids Res. 38, 1805–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tsukihashi Y., Kawaichi M., and Kokubo T. (2001) Requirement for yeast TAF145 function in transcriptional activation of the RPS5 promoter that depends on both core promoter structure and upstream activating sequences. J. Biol. Chem. 276, 25715–25726 [DOI] [PubMed] [Google Scholar]
- 108. Irvin J. D., and Pugh B. F. (2006) Genome-wide transcriptional dependence on TAF1 functional domains. J. Biol. Chem. 281, 6404–6412 [DOI] [PubMed] [Google Scholar]
- 109. Holstege F. C., Jennings E. G., Wyrick J. J., Lee T. I., Hengartner C. J., Green M. R., Golub T. R., Lander E. S., and Young R. A. (1998) Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95, 717–728 [DOI] [PubMed] [Google Scholar]
- 110. Papai G., Weil P. A., and Schultz P. (2011) New insights into the function of transcription factor TFIID from recent structural studies. Curr. Opin. Genet. Dev. 21, 219–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mitchell P. J., and Tjian R. (1989) Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science 245, 371–378 [DOI] [PubMed] [Google Scholar]
- 112. Titz B., Thomas S., Rajagopala S. V., Chiba T., Ito T., and Uetz P. (2006) Transcriptional activators in yeast. Nucleic Acids Res. 34, 955–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Regier J. L., Shen F., and Triezenberg S. J. (1993) Pattern of aromatic and hydrophobic amino acids critical for one of two subdomains of the VP16 transcriptional activator. Proc. Natl. Acad. Sci. U.S.A. 90, 883–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Drysdale C. M., Dueñas E., Jackson B. M., Reusser U., Braus G. H., and Hinnebusch A. G. (1995) The transcriptional activator GCN4 contains multiple activation domains that are critically dependent on hydrophobic amino acids. Mol. Cell. Biol. 15, 1220–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Deng Z., Chen C.-J., Zerby D., Delecluse H.-J., and Lieberman P. M. (2001) Identification of acidic and aromatic residues in the Zta activation domain essential for Epstein-Barr virus reactivation. J. Virol. 75, 10334–10347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lin J., Chen J., Elenbaas B., and Levine A. J. (1994) Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 8, 1235–1246 [DOI] [PubMed] [Google Scholar]
- 117. Brzovic P. S., Heikaus C. C., Kisselev L., Vernon R., Herbig E., Pacheco D., Warfield L., Littlefield P., Baker D., Klevit R. E., and Hahn S. (2011) The acidic transcription activator Gcn4 binds the mediator subunit Gal11/Med15 using a simple protein interface forming a fuzzy complex. Mol. Cell 44, 942–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Iñiguez-Lluhí J. A., Lou D. Y., and Yamamoto K. R. (1997) Three amino acid substitutions selectively disrupt the activation but not the repression function of the glucocorticoid receptor N terminus. J. Biol. Chem. 272, 4149–4156 [DOI] [PubMed] [Google Scholar]
- 119. Warfield L., Tuttle L. M., Pacheco D., Klevit R. E., and Hahn S. (2014) A sequence-specific transcription activator motif and powerful synthetic variants that bind Mediator using a fuzzy protein interface. Proc. Natl. Acad. Sci. U.S.A. 111, E3506–E3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kubik S., Bruzzone M. J., Jacquet P., Falcone J.-L., Rougemont J., and Shore D. (2015) Nucleosome stability distinguishes two different promoter types at all protein-coding genes in yeast. Mol. Cell 60, 422–434 [DOI] [PubMed] [Google Scholar]
- 121. Rohde J. R., and Cardenas M. E. (2003) The Tor pathway regulates gene expression by linking nutrient sensing to histone acetylation. Mol. Cell. Biol. 23, 629–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Uprety B., Sen R., and Bhaumik S. R. (2015) Eaf1p is required for recruitment of NuA4 in targeting TFIID to the promoters of the ribosomal protein genes for transcriptional initiation in vivo. Mol. Cell. Biol. 35, 2947–2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., and Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 124. Kalderon D., Roberts B. L., Richardson W. D., and Smith A. E. (1984) A short amino acid sequence able to specify nuclear location. Cell 39, 499–509 [DOI] [PubMed] [Google Scholar]
- 125. Heckman K. L., and Pease L. R. (2007) Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932 [DOI] [PubMed] [Google Scholar]
- 126. Sezonov G., Joseleau-Petit D., and D'Ari R. (2007) Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 189, 8746–8749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Murthy M. S., Rao B. S., Reddy N. M., Subrahmanyam P., and Madhvanath U. (1975) Non-equivalence of YEPD and synthetic complete media in yeast reversion studies. Mutat. Res. 27, 219–223 [DOI] [PubMed] [Google Scholar]
- 128. Kushnirov V. V. (2000) Rapid and reliable protein extraction from yeast. Yeast 16, 857–860 [DOI] [PubMed] [Google Scholar]
- 129. Köhrer K., and Domdey H. (1991) Preparation of high molecular weight RNA. Methods Enzymol. 194, 398–405 [DOI] [PubMed] [Google Scholar]
- 130. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC, New York [Google Scholar]