

Abstract
Skewing of the human oral microbiome causes dysbiosis and preponderance of bacteria such as Porphyromonas gingivalis, the main etiological agent of periodontitis. P. gingivalis secretes proteolytic gingipains (Kgp and RgpA/B) as zymogens inhibited by a pro-domain that is removed during extracellular activation. Unraveling the molecular mechanism of Kgp zymogenicity is essential to design inhibitors blocking its activity. Here, we found that the isolated 209-residue Kgp pro-domain is a boomerang-shaped all-β protein similar to the RgpB pro-domain. Using composite structural information of Kgp and RgpB, we derived a plausible homology model and mechanism of Kgp-regulating zymogenicity. Accordingly, the pro-domain would laterally attach to the catalytic moiety in Kgp and block the active site through an exposed inhibitory loop. This loop features a lysine (Lys129) likely occupying the S1 specificity pocket and exerting latency. Lys129 mutation to glutamate or arginine led to misfolded protein that was degraded in vivo. Mutation to alanine gave milder effects but still strongly diminished proteolytic activity, without affecting the subcellular location of the enzyme. Accordingly, the interactions of Lys129 within the S1 pocket are also essential for correct folding. Uniquely for gingipains, the isolated Kgp pro-domain dimerized through an interface, which partially overlapped with that between the catalytic moiety and the pro-domain within the zymogen, i.e. both complexes are mutually exclusive. Thus, pro-domain dimerization, together with partial rearrangement of the active site upon activation, explains the lack of inhibition of the pro-domain in trans. Our results reveal that the specific latency mechanism of Kgp differs from those of Rgps.
Keywords: cysteine protease, protein crystallization, protein degradation, protein domain, protein structure, proteinase, X-ray crystal structure, host-microbiome interaction, oral microbiota, zymogenic latency
Introduction
Collectively, the human microbiome encompasses hundreds of bacterial, fungal, and archaeal species, which roughly equal our body cells in number (1). The microbiota is mainly commensal, either symbiotic or mutualistic (2), and colonizes the inner and outer surfaces of our body at distinct niches, where it acquires nutrients to thrive and persist within the host (3). This colonization is constantly monitored by the innate immune system, which prevents bacterial infection of subjacent tissues. The most studied niches are skin, conjunctiva, gastrointestinal tract, vagina, placenta, uterus, lung, and oral cavity. In particular, the gut microbiome has been holistically dubbed “an organ” that participates in nutrient acquisition, energy balance, and immune response, among several other functions (4, 5). The oral cavity, in turn, is lubricated by saliva at rather constant temperature and close to neutral pH, which are ideal dwelling conditions for the highly complex human oral microbiome. The latter encompasses an estimated 6 billion bacteria from over 600 species or phylotypes, which arrange in a stable microbial “climax community” (6, 7). Disruption of homeostasis by intrinsic or extrinsic factors, e.g. treatment with broad spectrum antibiotics, leads to dysbiosis (2). This results from the replacement of facultative, fermentative Gram-positive species by anaerobic, proteolytic Gram-negative species, which cause tissue destruction and inflammation (8). In the oral cavity this causes inflammation of the gums (gingivitis) and the periodontium (periodontitis) (6), which erodes the alveolar bone support of the teeth. Bacterial species infecting the periodontium include Actinobacillus actinomycetemcomitans, Tannerella forsythia, Prevotella intermedia, Fusobacterium nucleatum, and Porphyromonas gingivalis. P. gingivalis is a chief component of the dysbiotic oral microbiome and the major etiologic agent of chronic periodontitis (CP),5 as revealed by comparative studies between healthy individuals and CP patients (6, 9). Routine treatment of severe CP consists of mechanical debridement of the teeth surface below the gum line, which is laborious, repetitive, painful, and incompletely effective (10). Accordingly, there is an unmet need for the development of novel therapeutic approaches against CP, which is among the most prevalent infection-driven inflammatory diseases (11), and P. gingivalis is a prime target (6).
P. gingivalis persistently colonizes the human oral cavity, as indicated by its detection in several paleomicrobiological samples, which include the wet mummy of the Tyrolean Iceman “Ötzi” dated to ∼5,300 years ago (12–14). During this multimillenial colonization of our mouth, the bacterium has evolved to deactivate our innate immune and inflammatory defense mechanisms and to keep bacterial competitors in the gingival crevice in check through a panel of virulence factors, which include peptidases (15–17). Among the latter are the gingipains K (Kgp) and R (RgpA and RgpB), which specifically cleave substrates after lysines and arginines (18), respectively. They are soluble or outer membrane-anchored cysteine peptidases responsible for up to 85% of the total extracellular proteolytic activity of the bacterium (19–21) and can be found at very high concentrations in gingival crevicular fluid from CP patients (22). Kgp accounts for most of this activity (23) and is essential for bacterial survival and progression of CP (18). Accordingly, blocking Kgp may be a promising approach to combating P. gingivalis (24, 25).
Kgp is a multidomain protein consisting of a signal peptide, an N-terminal pro-domain (NPD), a catalytic domain (CD), an immunoglobulin superfamily domain (IgSF), between three and five hemagglutinin/adhesion domains, and a C-terminal domain, thus spanning up to 1,732 residues in total. It is secreted through a C-terminal domain-dependent type-9 secretion system, which is also called a “Por secretion system” and has so far only been found in the Bacteroidetes phylum (26–33). Similar to other secretory peptidases, Kgp is produced as a zymogen to prevent undesired intracellular proteolysis and only achieves full activity once secreted. The NPD exerts zymogenic latency and is proteolytically removed during activation of Kgp (34–37). In addition to maintaining latency, NPDs often fold independently and fulfill a chaperone-like function on the downstream CDs to facilitate their correct folding during biosynthesis (38). They may also participate in intracellular sorting of zymogens (35) and inhibit the mature enzymes when added in trans, as described for funnelin metallocarboxypeptidases, for example (39, 40).
Unraveling the biochemical and structural determinants of zymogenicity is essential to understand its pathophysiological function and to facilitate the design of inhibitors to block its proteolytic activity (41). We recently reported the crystal structure of the linked CD and IgSF domains of Kgp (Kgp-CD + IgSF) (42) and of the complex between active RgpB-CD + IgSF and its cognate NPD (RgpB-NPD) (43). To determine the mechanism of Kgp zymogenicity, we crystallized the isolated NPD of Kgp and solved its structure, which revealed a fold similar to Rgp-NPD despite 20% sequence identity and allowed modeling of its interaction with CD in the zymogen. Mutagenesis studies further suggest that the NPD of Kgp is an essential chaperone for the folding of the CD and perhaps other domains.
Results and Discussion
Overall Structure of the Kgp Pro-domain
Over several years, our attempts to produce crystals of the Kgp zymogen spanning domains NPD, CD, and IgSF have failed, which contrasts with our success with RgpB (43). Accordingly, we followed the divide and conquer approach and managed to get the structures of Kgp-CD + IgSF (42) and Kgp-NPD (this work) separately. The latter is visible in the final Fourier map from residue Gln20 to Gln199 or Ala201 (Kgp numbering as superscripts) of molecules B and A, respectively, within the asymmetric unit of the crystal.
The molecule is boomerang-shaped, with approximate maximal dimensions of 55 × 40 × 30 Å (Fig. 1, A and B). Its core is a central, 12-stranded, strongly bent β-structure (strands βI′, βI–βIV, βIV′, βV, βVI, βVII + βVIII, βIX–βXI; strand numbering based on the structure of RgpB-NPD; see Fig. 1 in Ref. 43) split in β-sandwiches 1 and 2, which are approximately perpendicular to each other (reference view according to Fig. 1, C and D). Two 310-helices, ηI and ηII, are laterally attached to sandwich 2. The Kgp-NPD moiety is held together by a central hydrophobic core traversing the molecule, which glues the two sandwiches and reaches from Val92, Pro93, and Ala96 on the rightmost face of sandwich 1 to Phe64, Phe138, and Tyr145 on the leftmost face of sandwich 2. Sandwich 1 is made up by antiparallel front and back sheets, respectively, featuring four strands (βI, βII, βXI, and βVI) and three strands (βV, βIX, and βVII + βVIII). Front sheet strands βXI and βVI are, respectively, N- and C-terminally extended beyond the limits of the sandwich and bent by ∼50–60°. In this way, they contribute to the antiparallel three-stranded front sheet (strands βVI, βXI, and βX) of sandwich 2. The back sheet of the latter is five-stranded (βI′, βIII, βIV, βV, and βIV′) and mixed parallel-antiparallel.
FIGURE 1.

Structure of the Kgp pro-domain. A, ribbon-type plot of Kgp-NPD showing the regular secondary-structure elements (310-helices in magenta, labeled ηI-ηII; β-strands as blue arrows, labeled βI′ + βI–βIV + βIV′ + βV–βXI; numbering based on the structure of RgpB-NPD; see Fig. 1 in Ref. 43). The inhibitory loop includes residue Lys129, which is depicted for its side chain and labeled. B and C, two orthogonal views of A. D, topology scheme of Kgp-NPD roughly in the same orientation as in C. Each regular secondary structure element is labeled and marked with its limiting residues.
The N-terminal part of the molecule shapes the outermost strand of the back sheet of sandwich 2 (βI′) before entering a first β-ribbon, which gives rise to one edge of the front sheet of sandwich 1 (ribbon βIβII). The chain rejoins sandwich 2 through two consecutive β-ribbons (βIIIβIV and βIV′βV), which with βI′ features the back sheet. The C-terminal extension of strand βV shapes through its extra part one edge of the back sheet of sandwich 1. At this point, the chain enters edge strand βVI of the front sheet of sandwich 1. The C-terminal elongation of βVI, in turn, gives rise to an edge strand of the front sheet of sandwich 2. After this strand, a 45-residue loop, the “inhibitory loop,” connects strands βVI and βVII + βVIII. It contains helices ηI and ηII and largely protrudes from the Kgp-NPD moiety (Fig. 1, A and B). This extended loop leads to a β-ribbon that creates the two edge strands of the back sheet of sandwich 1 (termed βVII + βVIII and βIX). Thereafter, a loop leads to another β-ribbon (βXβXI) that completes the front sheet of sandwich 2. Finally, the C-terminal extension of βXI contributes to the front sheet of sandwich 1 as a central strand before reaching the C terminus of the molecule (Fig. 1D).
Structural Similarity with the B Pro-domain
The gingipain pro-domain constructs we expressed and crystallized in the present and past work comprised, respectively, fragments Gln20–Arg228 of Kgp and Gln25–Arg229 of RgpB, because the N-terminal 19 and 24 residues, respectively, correspond to signal peptides for secretion. Although the structure of Kgp is well ordered in the final Fourier map from Gln20 on, RgpB is only ordered from Gly30/Arg31 of RgpB onward (there are four molecules in the crystal asymmetric unit; see PDB code 4IEF and Ref. 43). On the opposite C terminus, Kgp is only ordered until maximally Ala201 and flexible thereafter (Asp202–Arg228), whereas RgpB is ordered until the end of the construct at Glu226/Arg229 (except for flexible segment Leu205/Val206–Ser209/Thr210). The C-terminal flexibility of Kgp in the crystal structure is supported by the likely absence of regular secondary structure within Gln199–Arg243 according to bioinformatics predictions (data not shown). Both NPDs share a grossly similar central core but differ at both termini.
Initial automatic superposition of the coordinates of Kgp-NPD and RgpB-NPD confirmed the similarity of the structures (Fig. 2, A and B). There were 147 aligned residues, which showed a core root mean square deviation of 2.4 Å. Both structures coincided at Arg32–Ala201 of Kgp and Gln34–Glu197 of RgpB, with just six extra residues in the insertion loop of the former: Asn113–Ser117 within the loop connecting strand βVI with helix ηI (LβVIηI) and Asn141 within LηIηII (Fig. 2C). Starting from this superposition, we performed an accurate sequence alignment based on visual inspection of the structures (Fig. 2, A–C), which revealed rather 164 common residues and just 21% sequence identity (in comparison, RgpA-NPD and RgpB-NPD are 75% identical; (44)). The low sequence identity and the large root mean square deviation value of the superposed structures provide an explanation a posteriori for the fact that the structure of Kgp-NPD could not be solved by molecular replacement using the coordinates of RgpB-NPD. Structural superposition further unveiled that Kgp possesses an extra β-strand, βI′, which contributes to sandwich 2 (see above) and is missing in RgpB-NPD. The latter, in turn, has an extra C-terminal helix, αΙ, missing in Kgp-NPD (see Fig. 1 in Ref. 43 and Fig. 2C). Moreover, Kgp-NPD has an extra β-strand between βIV and βV, termed βIV′, and strands βVII and βVIII, separated in RgpB, are joined to a single continuous strand in Kgp (Figs. 1, C and D, and 2C).
FIGURE 2.

Comparison of gingipain pro-domains. A, superposition in cross-eye stereo of Kgp-NPD (blue) and RgpB-NPD (yellow; PDB code 4IEF) (43) in the orientation of Fig. 1C. The respective N and C termini are labeled. B, orthogonal view of A. Here, the proven S1-intruding residue of RgpB (R126) and the putative one of Kgp (K129) are shown for their side chains and labeled. C, structure-based sequence alignment of the NPDs of RgpB and Kgp. Residues not defined in the respective structure are in light gray, structurally aligned residues are in black (when differing) or red (when identical), and residues defined but structurally non-equivalent in the two structures are in light salmon. The residue (potentially) intruding the S1 pocket of the respective CD is framed. Numbering and secondary structure elements (arrows for strands; kringles for helices) above the alignment in green correspond to Kgp; those in blue below the alignment correspond to RgpB.
Furthermore, superposition revealed that Lys129 within the inhibitory loop of Kgp likely corresponds to Arg126 of RgpB as the “intruding residue” penetrating the S1 pocket of the active site cleft, thereby contributing to inhibitory potency (see Refs. 43 and 44 and Fig. 2, B and C). To assess the importance of Lys129 for Kgp, we constructed point mutants (K129A, K129E, and K129R) and analyzed their expression at mRNA, protein, and activity levels using Rgps and wild-type Kgp as controls. Mutations had no effect on transcription as determined by qRT-PCR (data not shown). In contrast, Western blotting analysis revealed no protein for mutant K129E in stationary bacterial cultures (fraction BC), the washed cell fraction (WC), the soluble intracellular protein fraction (CP + PP), the cell envelope (CE) fraction, and the cell-free growth medium (GM) (Fig. 3A). Mutant K129R only revealed trace amounts in the first two fractions, whereas the wild type and K129A were detected in all fractions except the intracellular fraction, as expected for secreted proteins. The respective presence of protein correlated with enzymatic activity, which was absent for K129E and only residual for K129R (Fig. 3C). K129A displayed ∼45% of the wild-type activity (Fig. 3C) but only a slightly lower signal in Western blotting analysis (Fig. 3A). Mutations in the Kgp gene had no effect on expression, processing, and activity of Rgps (Fig. 3, B and D).
FIGURE 3.

Effect of Lys129 mutation on Kgp expression and activity. Western blotting analysis of parental strain P. gingivalis W83 and mutants K129A, K129E, and K129R is shown. Late exponential/early stationary bacterial cultures (BC) were separated by centrifugation into cell-free growth medium (GM) and cell pellet. The latter was washed, giving rise to the whole cell (WC) fraction, which was further fractionated into the soluble intracellular protein fraction (CP + PP) and the cell envelope fracion (CE) by sonication and ultracentrifugation. A and B, all fractions were standardized to the initial volume of the culture subjected to centrifugation and analyzed by Western blotting to detect Kgp forms (A) and Rgps (control) (B). C and D, Kgp (C) and Rgp (D) gingipain activities determined in whole cultures, cell-free growth medium, and washed cells using specific substrates. The whole-culture activity of the wild-type strain was arbitrarily taken as 100%.
Taken together, these findings suggest that occupancy of the S1 specificity pocket of the CD by Lys129 not only imposes latency to the Kgp zymogen (44) but also contributes significantly to the chaperone function of the NPD, similar to what was previously found for Rgps (45). The deep, negatively charged pocket of Kgp is optimally designed to accommodate a lysine side chain (42). Consistently, a negative charge (mutant K129E), as well as extended or reduced side chains (mutants K129R and K129A, respectively), lead to interaction impairment or inefficiency, which apparently disturbs the folding process. This results in subsequent degradation of misfolded proteins by quality control proteases in the periplasm.
Dimerization of Kgp-NPD
The two molecules in the asymmetric unit of the Kgp-NPD crystal associate through a large interface, which gives rise to a pseudo-continuous antiparallel eight-stranded β-sheet mediated by the respective edge strands βI of the upper sheets of sandwiches 1 (Fig. 4, A–C). The hydrophobic cores cohering the monomers (see above) are likewise joined at the interface. Among the internal hydrophobic residues, two free cysteines (Cys35) within strands βI are close to the dimer interface but too far apart from each other for bonding (∼7 Å), thus indicating that dimerization is non-covalent (Fig. 4C). Instead, the respective Sγ atoms are bridged through electrostatic interactions with a (tentatively assigned) azide molecule from the buffer.
FIGURE 4.
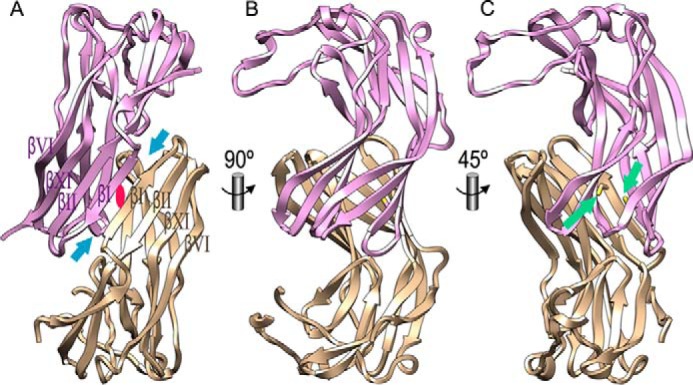
Dimer of Kgp pro-domains. A, two Kgp-NPD moieties, shown as plum and tan ribbons, respectively, associate through lateral attachment of the respective upper sheets of their sandwiches 1 (strands βI, βII, βXI, and βVI; see also Fig. 1D) via a local 2-fold axis (red ellipse) perpendicular to the sheets. The distance between the tips of the two blue arrows is ∼25 Å. B, orthogonal view of A. C, view providing insight into the central part of the hydrophobic core below the upper sheets of sandwiches 1. The side chains of Cys35 of each monomer are depicted and pinpointed by green arrows. The distance between the respective Sγ atoms is ∼7 Å.
Computational analysis of the dimer revealed an interface of 1060 Å2, which is quasisymmetrically shaped by 27 residues from either molecule A and B and includes nine hydrogen bonds and two salt bridges in total (Table 1). The calculated solvation free energy gain upon interface formation is −11.7 kcal/mol, with an associated p value of 0.230. The extensive nature of the interface suggests the functional relevance of dimerization. In addition, the complexation significance score, which estimates the relevance of an interface for assembly formation (46), is 97.7%. Altogether, these results strongly suggest that the oligomerization state in solution is a stable dimer. To verify this hypothesis, we performed size exclusion chromatography experiments at three pH values (5.5, 6.5, and 8.0), which reproduce, respectively, values of the crystallization conditions and of human gingival crevicular fluid in inflamed sites (47) plus an intermediate value. Consistent with the observations in the crystals (Fig. 4B), we found that Kgp-NPD elutes as a dimer at all three pH values (Fig. 5), thus supporting the notion that the NPD dimerizes upon Kgp activation. This dimerization is likely to provide a mechanism to prevent rejoining of the NPD and the CD, which would lead to inhibition in trans of the secreted gingipain.
TABLE 1.
Electrostatic interactions at the interface of dimeric Kgp-NPD
| Molecule A | Molecule B | Distance (Å) |
|---|---|---|
| Hydrogen bonds | ||
| Thr31 O | Asn37 N | 3.69 |
| Thr33 O | Cys35 N | 2.79 |
| Cys35 O | Thr33 N | 2.79 |
| Leu161 O | Arg165 Nϵ | 3.44 |
| Asn37 N | Thr31 O | 3.41 |
| Cys35 N | Thr33 O | 2.79 |
| Thr33 N | Cys35 O | 2.84 |
| Arg32 Nη1 | Thr36 Oγ1 | 3.72 |
| Arg165 Nϵ | Leu161 O | 3.44 |
| Salt bridges | ||
| Glu159 Oϵ2 | Arg165 Nη1 | 2.80 |
| Arg165 Nη1 | Glu159 Oϵ2 | 2.79 |
FIGURE 5.

Size exclusion chromatography of Kgp-NPD. Recombinant Kgp-NPD (1 mg/ml) was resolved on a SuperdexTM 75 increase 10/300 GL column equilibrated with 50 mm sodium acetate, 150 mm NaCl, pH 5.5 or 6.5, or with 50 mm Tris-HCl, 150 m NaCl, pH 8.0, with each buffer freshly supplemented with 2 mm dithiothreitol. The column was calibrated using conalbumin (75 kDa), ovoalbumin (43 kDa), carbonic anhydrase (29 kDa), and ribonuclease A (13.7 kDa), the respective elution volumes are indicated by vertical arrows above the chromatography profiles. For clarity, only the elution profiles at pH 5.5 and 8.0 are shown.
Homology Model of the Kgp Zymogen
To gain insight into the structure of the full Kgp zymogen, we constructed a homology model spanning domains NPD, CD, and IgSF based on the structural information available on Kgp and RgpB fragments (see “Experimental Procedures” and Fig. 6, A–D). As an independent validation of the model, the distance between the last NPD residue (Ala201) and the first CD residue (Asp229) spans ∼22 Å, which is sufficient to accommodate the missing 28 residues. Model building required rearrangement of six segments from their conformation observed in the unbound NPD and CD structures, three from each moiety, to avoid clashes (Fig. 6D). The largest changes in the NPD involved the inhibitory loop, and in particular, Lys129, which was rebuilt to match the position of a l-lysylmethyl moiety found attached to the catalytic cysteine Cys477 in mature Kgp (Fig. 6C), as well as strands βVII+βVIII and βIX plus the intervening loop, which were folded outward to avoid clashes. Rearrangement of the CD, in turn, mainly involved the N terminus, helices α11 and α12, and Lα11α12, which are close to the active site (see Ref. 42 and Fig. 6D) and produced severe clashes with the NPD (Fig. 6D), suggesting that the CD must adopt distinct conformations in the zymogenic and activated forms.
FIGURE 6.

Homology model of zymogenic Kgp. A, ribbon-type plot in cross-eye stereo of the homology model of Kgp-NPD + CD + IgSF in the reference orientation chosen for the RgpB zymogen (see Fig. 2A in Ref. 43). The NPD is in blue, the CD is in hot pink, and the IgSF is in green. The structural calcium and sodium ions from the CD are displayed as red and blue spheres, respectively. Blue and pink arrows pinpoint, respectively, the C-terminal residue of the NPD and the first residue of the CD, which are ∼22 Å apart. The catalytic histidine (His444) and the putative inhibitory lysine at the tip of the inhibitory loop (Lys129) are displayed for their side chains and labeled, as are the N and C termini of the whole model. B, close-up view of A in mono after a vertical 200° rotation to provide insight into the interaction of the inhibitory loop with the active site cleft of Kgp. The side chain of catalytic cysteine Cys477 is further labeled. C, close-up view in mono of B after a horizontal 30° rotation showing the putative inhibitory lysine Lys129 penetrating the S1 pocket of the CD in a similar manner as a l-lysylmethyl moiety (LM) covalently attached to Cys477 Sγ (carbon atoms in green) does in the structure of mature Kgp (PDB code 4RBM) (42). D, close-up view of B in cross-eye stereo to highlight the segments putatively rearranged during activation and NPD removal. The parts of the experimental structures of the NPD (cyan) and CD (magenta) deviating significantly are superimposed on the homology model of zymogenic Kgp (encircled 1, Thr75–Pro81; encircled 2, Ser126–Lys135; encircled 3, Glu159–Ile169; encircled 4, Asp229–Asp236; encircled 5, Gly522–Val526; and encircled 6, Ala563–His579). In particular, loop Ser126–Lys135 was adapted so that Lys129 matches the l-lysylmethyl moiety shown in C. Green curved arrows highlight the transition from the experimental structures to the homology model.
According to this model, the NPD would attach laterally to the catalytic moiety through a large concave surface, distal to the IgSF (Fig. 6A) and overlapping with the NPD dimerization surface. Latency of the zymogen would be conferred by the inhibitory loop, by blocking access to the active site cleft through the insertion of Lys129 into the S1 pocket. The lysine residue would be sandwiched for its aliphatic part between Trp513 and Cys477 and bound for its Nζ group by the side chains of Asp516 and Thr442, and the main chain carbonyl of Asn475 at the bottom of the pocket.
Conclusions
By following a concerted biophysical and biochemical approach, which included protein crystallography, bioinformatics with homology modeling, oligomerization studies in solution, and mutagenesis studies, we have unveiled the molecular mechanism of zymogenic latency maintenance for Kgp, an essential proteolytic virulence factor of a major disease-causing pathogen for oral dysbiosis and CP. According to our working model, Kgp-NPD, which comprises an all-β scaffold, would inhibit cognate Kgp-CD within the zymogen through steric hindrance of the active-site cleft and specificity pocket, with a key feature being the insertion of Lys129 into the S1 pocket. This part of the latency mechanism is shared with related gingipains RgpA and RgpB.
Activation is conferred by cleavage at the hinge between Kgp-NPD and Kgp-CD, resulting in conformational changes in the active site of Kgp-CD and of surface segments of Kgp-NPD, followed by release and dimerization of the Kgp-NPD. Because dimerization occurs through a surface that overlaps with the NPD/CD interface in the zymogen, dimerized Kgp-NPD is no longer inhibitory. In contrast, there is no evidence for NPD dimerization in RgpA/B, and no significant structural differences are found in RgpB-CD between the zymogenic complex and the mature enzyme (see Fig. 3 in Ref. 43). Collectively, these findings explain that although RgpA and RgpB are strongly inhibited by their respective NPDs when added in trans, with inhibition constants in the nanomolar range, Kgp is not inhibited by addition of its NPD (44). The importance of the intruding residue in Rgp inhibition is reflected by the fact that its mutation to lysine reduced the inhibitory potency by 1 order of magnitude. Replacement with alanine, glutamine, or glycine even totally abolished inhibition (44). This inhibitory capacity in trans in turn poses the requirement that Rgp-NPDs be completely degraded after NPD cleavage, so Rgp-CD activity can be fully liberated. Highly purified RgpB zymogen does not auto-activate, possibly because of NPD inhibition, and the addition of active enzyme in catalytic amounts only very slowly generates active RgpB. Along this line, the pI value of RgpB-NPD is 8.06, which is close to the pH value of human gingival crevicular fluid in inflamed sites (8.0) and thus might destabilize the NPD and render it prone to degradation. In contrast Kgp-NPD has a pI of 5.95 and should be stable at pH 8. Kgp-NPD may therefore remain intact, requiring dimerization to prevent inhibition of Kgp after activation, and the dimer might also have other functions in the colonization of P. gingivalis in the gingival crevice.
Experimental Procedures
Protein Production
Kgp-NPD of P. gingivalis strain W83 (sequence Gln20–Arg228; see UniProt database access code Q51817) was produced as a fusion construct with glutathione S-transferase by recombinant overexpression in Escherichia coli and purified as described previously (44). The cloning strategy resulted in the retention of a pentapeptide, −5GPLGS−1, from the expression vector on the N terminus of the recombinant protein after excision of the fusion protein. Kgp-NPD was then concentrated and dialyzed against 5 mm Tris-HCl, pH 7.4, 0.02% sodium azide for crystallization.
Oligomerization Studies in Solution
Size exclusion chromatography analysis of Kgp-NPD was performed in a Superdex 75 column equilibrated with 50 mm sodium acetate, 150 mm sodium chloride, pH 5.5/6.5, or 50 mm Tris-HCl, 150 mm sodium chloride, pH 8.0, at 0.5 ml/min. Both buffers were supplemented with 0.02% sodium azide and 2 mm 1,4-dithiothreitol, with the latter added to prevent formation of intermolecular disulfide bridges.
Generation of P. gingivalis Mutant Strains
Strains incorporating point mutations of Kgp-NPD residue Lys129 (K129A, K129E, and K129R) were generated from P. gingivalis strain W83. Therefore, master plasmid Kgp-CepA was first obtained through PCR/restriction digestion methods. Briefly, a 0.8-kb region upstream of the kgp gene, the CepA ampicillin resistance, the 2.8-kb fragment of the kgp gene, and the CepA ampicillin resistance cassette were amplified by PCR using Phusion polymerase (Thermo Fisher) and appropriate primers (Table 2); digested with SphI, HindIII, BglI, and BamHI restriction enzymes (Thermo Fisher); and cloned into the pUC19 plasmid. The correct placement and orientation of DNA segments in resulting plasmid Kgp-CepA were confirmed by sequencing. The wild-type plasmid construct of Kgp-NPD (Kgp-CepA) was subsequently used to produce K129A, K129E, and K129R mutations by the SLIM method (48). The mutated constructs were verified by DNA sequencing.
TABLE 2.
Primers employed in this study
| Name | Sequence |
|---|---|
| Primers for Kgp mutagenesis | |
| Kg_K129_Fs | ATCCCGAAAAGGTTCCCTT |
| Kg_K129_Rs | AGGGTTGATGTGGCATGAG |
| Kg_K129A_Rl | CATCACTGGCGCTCATAGAGGGTTGATGTGGCATGAG |
| Kg_K129A_Fl | CTATGAGCGCCAGTGATGATCCCGAAAAGGTTCCCTT |
| Kg_K129R_Rl | CATCACTACGGCTCATAGAGGGTTGATGTGGCATGAG |
| Kg_K129R_Fl | CTATGAGCCGTAGTGATGATCCCGAAAAGGTTCCCTT |
| Kg_K129E_Rl | CATCACTTTCGCTCATAGAGGGTTGATGTGGCATGAG |
| Kg_K129E_Fl | CTATGAGCGAAAGTGATGATCCCGAAAAGGTTCCCTT |
| Primers for Kgp-CepA plasmid construction | |
| pUC_SphI_R | TGTGTGCATGCAAGCTTGGCGTAATCAT |
| pUC_BamHI_F | TAGAAGGATCCCCGGGTACCGAGCT |
| Kg_Up_F | AGCTTGCATGCACACACCCCGAT |
| Kg_Up_R | ATGGAAGCTTAAGTCAGTCCAGCATGAGGAAG |
| CepA_F | GACTTAAGCTTCCATAGACGATGCCACACTG |
| CepA_R | GTTAAGATCTCAAGTCACCGATAGTGATAGTG |
| Kg_Dw_F | ACTTGAGATCTTAACCTTGGTCTGCTCTAC |
| Kg_Dw_R | CCGGGGATCCTTCTACCGTAACGTC |
| Primers for qRT-PCR analysis | |
| 16S_F | AGGCAGCTTGCCATACTGCG |
| 16S_R | ACTGTTAGYAACTACCGATGT |
| HrgpAForqPCR1 | ACGCTTCCCATTCTATCACG |
| HrgpARevqPCR2 | CTCCCGGGAAGAATTTGTTT |
| KgpForqPCR1 | AGCGCATGGATCTGAGACCGC |
| KgpRevqPCR2 | CGGCATTAGCACCCACACTCCA |
Chromosomal integration of the mutated regions into the P. gingivalis genome was achieved via double homologous recombination as described previously (49). Briefly, 1 μg of purified plasmid DNA was electroporated into P. gingivalis strain W83 competent cells (2.5 kV, 4 ms; Bio-Rad Micropulser). Bacteria were grown for 5–7 days on enriched tryptic soy broth blood agar (30 g of trypticase soy broth, 5 g of yeast extract, 5 mg of hemin, 15 g of agar, pH 7.5, per liter containing 4% defibrinated sheep blood) supplemented with 5 μg/ml ampicillin for antibiotic resistance selection. Resistant clones were further subcultured and analyzed by PCR and sequencing to confirm mutations.
Growth of P. gingivalis Strains and Cell Fractionation
Wild-type and mutant strains of P. gingivalis were grown under anaerobic conditions (85% nitrogen, 5% hydrogen, and 10% carbon dioxide) in liquid enriched tryptic soy broth (30 g of trypticase soy broth, 5 g of yeast extract, 5 mg of hemin, pH 7.5, per liter; further supplemented with 0.5 g of l-cysteine and 2 mg of menadione). For the mutants, the medium was additionally supplemented with 5 μg/ml ampicillin. The cultures were harvested at the beginning of the stationary growth phase (A600 = 1.0–1.3; on average after 24 h), adjusted to A600 = 1.0 with enriched tryptic soy broth and centrifuged (6,000 × g, 15 min, 4 °C) to separate bacterial cells from the medium. The pellets were washed with PBS, and bacterial cells were resuspended in the same buffer to be disrupted by ultrasonication (A = 70%, 50 s, 5 s pulse, 2 s off). The resulting homogenate was subjected to ultracentrifugation (150,000 × g, 60 min, 4 °C) to obtain the particle-free soluble cytoplasmic/periplasmic proteins fraction (supernatant) and the pellet. The latter consisted of the cell envelope fraction, thus encompassing inner and outer membranes, and was resuspended in PBS for further fractionation.
Analysis of Gingipain Expression
Expression levels of gingipains (Rgps and Kgp) were determined by activity assays, Western blotting analyses and qRT-PCR. The activities of Rgps and Kgp were determined in the whole cell culture, cell-free growth medium, and suspension of washed bacterial cells using the chromogenic p-nitroanillide (pNA) substrates benzoyl-Arg-pNA and acetyl-Lys-pNA, respectively. Briefly, 10 μl of cell fraction was added to 190 μl of TNCT buffer (50 mm Tris-HCl, pH 7.5, 5 mm calcium chloride, 150 mm sodium chloride, 0.05% Tween 20; supplemented with 10 mm l-cysteine-HCl neutralized with 10 mm sodium hydroxide). After 10 min of preincubation at 37 °C, the reaction was initiated by adding 10 μl of substrate (final concentration, 0.5 mm), and the increase of A405 was recorded using a Spectromax Flexstation 3 (Molecular Devices) microplate reader. Gingipain activity was expressed as mOD/min/μl, and the activity in the full-culture of the wild-type strain was taken as 100%.
For Western blotting analysis, whole bacterial cultures (BC), washed-cell lysates (WC), the soluble cytoplasmic/periplasmic fraction (CP + PP), the CE fraction, and GM were separately resolved by SDS-PAGE and proteins transferred to nitrocellulose membranes. The membranes were stained with Coomassie Brilliant Blue, photographed to document the protein load, and blocked for nonspecific binding sites with 2% albumin. Mouse monoclonal anti-Kgp and rabbit polyclonal anti-Rgp primary antibodies were used at 1:1,000 dilution. Secondary anti-mouse horseradish peroxidase-conjugated antibodies from rabbit (BD Pharmingen) and anti-rabbit antibodies from goat (Amersham Biosciences) were used at 1:15,000 dilution.
The expression of Kgp variants was further tested at the mRNA level by quantitative RT-PCR. All strains were grown from initial A600 = 0.1 until they reached 1.0. Total RNA was isolated using the TRI-reagent (Invitrogen) and incubated with DNase I (Ambion) to eliminate genomic DNA contamination. For the subsequent reverse transcriptase reaction (Applied Biosystems), 400 ng of RNA were used. Quantitative RT-PCR was performed with a Bio-Rad CFX96 real time PCR detection system using SYBR Green Jump Start (Sigma) and the following steps: 95 °C for 5 min; 40 cycles at 95 °C for 30 s; 56 °C for 30 s; and 72 °C for 45 s. The final elongation step consisted in incubation at 72 °C for 10 min. Melting curves were acquired on a SYBR channel from 60 to 95 °C (0.5 °C increment). The expression level of each variant was normalized to 16S rRNA by the ΔΔCT method and represented in relation to the wild type.
Crystallization and Diffraction Data Collection
Crystallization trials were set up at 20 °C as sitting drop vapor diffusion experiments by an Innovadyne Screenmaker 96 + 8 Xtal robot with 96-well MRC plates. Crystals were obtained in conditions 1–2 of the MIDAS screen (Molecular Dimensions), which contained 0.1 m MES, 12% polyvinylpyrrolidone K15, pH 5.5. Optimization screens were performed by the hanging drop vapor diffusion method in 24-well plates. The best crystals were obtained with protein solution (at 12 mg/ml in 5 mm Tris-HCl, pH 7.4, 0.02% sodium azide) and 14% polyvinylpyrrolidone K15, 0.1 m Bis-Tris, pH 5.5, as reservoir solution from 1:1-μl drops at 20 °C. The crystals were cryo-protected by soaking for 2 min in 50% polyvinylpyrrolidone K15, 0.1 m Bis-Tris, pH 5.5, and flash vitrified in liquid nitrogen. The crystals used for phasing experiments were immersed for 2–3 min in a solution of 0.1 m Bis-Tris, pH 5.5, 50% polyvinylpyrrolidone K15 plus either 0.5 or 1 m potassium iodide prior to flash vitrification. A native data set was collected at 0.9795 Å wavelength, and several data sets for sulfur and iodide SAD/SIRAS phasing experiments were collected at 1.7 and 1.8 Å wavelength, respectively, at Diamond Light Source Beamline I04 (Didcot).
Data Processing and Structure Solution
After analyzing several data set combinations and methods, the structure of Kgp-NPD was eventually solved by SIRAS employing a native and a iodide-derivative data set processed to 1.8 and 2.6 Å resolution, respectively, with the programs XDS and XSCALE (Ref. 50 and Table 3). The correct space group was identified as P212121 based on the systematic absences. SIRAS calculations with program SHELXD (51) and data cut to a resolution limit of 3.7 Å enabled us to identify five sites with occupancies above 20%. This solution stood out because of the clear discrimination of the correlation coefficient (CC; 32% for all data; 25% for weak reflections with E<1.5). Subsequently, phasing with program SHELXE (52) using a data set obtained by merging native and derivative data plus the iodide sites yielded calculated phase shifts. These showed small though clear discrimination in contrast, connectivity, and CCfree between the original and inverted substructures and gave an interpretable map. Phasing and main chain tracing was done with a beta version of SHELXE, which enforces goodness of tertiary structure on strand tracing. This led to a polyalanine model of 245 residues, which was subsequently refined with the REFMAC5 program (53). Two Kgp-NPD molecules (A and B) were contained in the crystal asymmetric unit and were completed in successive rounds of manual model building with program COOT (54) and crystallographic refinement with program BUSTER/TNT (55), which included TLS refinement. The final model, which was validated with program MolProbity (Ref. 56 and Table 3), contained Kgp residues Gln20–Ala201 of molecule A and G−2-Ser−1–Gln20–Gln199 of molecule B (except Ser128—Asp132) plus one azide molecule and 307 solvent molecules. Overall, molecule A was more rigid and better defined in the final Fourier map than molecule B as suggested by a lower average thermal displacement parameter (31.9 Å2 versus 41.6 Å2; Table 3), so the former molecule was taken as a reference for the “Results and Discussion” if not otherwise stated.
TABLE 3.
Crystallographic data
| Data set | Native | Iodide-derivative |
|---|---|---|
| Space group/cell constants (a, b, and c in Å) | P212121/63.65, 66.25, 96.24 | P212121/64.07, 65.98, 96.18 |
| Wavelength (Å) | 0.9795 | 1.8000 |
| No. of measurements/unique reflections | 169,919/38,292 | 81,981/12,006 |
| Resolution range (Å) (outermost shell) | 96.2–1.80 (1.91–1.80) | 48.1–2.63 (2.77–2.63) |
| Completeness (%) | 99.7 (98.7) | 94.3 (67.0) |
| Rmergea | 0.054 (0.830) | 0.116 (0.867) |
| Rr.i.m. (Rmeas)a/CC½b | 0.061 (0.943)/0.999 (0.651) | 0.124 (1.923)/0.994 (0.186) |
| Average intensityc | 19.8 (1.95) | 49.1 (3.39) |
| B-factor (Wilson) (Å2)/average multiplicity | 33.5/4.4 (4.4) | 58.9/6.4 (2.3) |
| Resolution range used for refinement (Å) | 25.8–1.80 | |
| No. of reflections in working set/in test set | 37,536/742 | |
| Crystallographic Rfactor (free Rfactor)d | 0.197 (0.240) | |
| No. of protein atoms/solvent molecules/ligands | 2,758/307/1 azide | |
| Root mean square deviation from target values | ||
| Bond lengths (Å) | 0.010 | |
| Bond angles (°) | 1.10 | |
| Average B-factor (Å2) protein atoms overall/molecule A/molecule B | 36.7/31.9/41.6 | |
| All-atom contacts and geometry analysise | ||
| Residues | ||
| In favored regions/outliers/all residues | 352/0/353 | |
| With poor rotamers/bad bonds/bad angles | 9/0/0 | |
| With Cβ deviations >0.25 Å/clashscore | 0/3.81 (98th percentile) | |
| MolProbity score | 1.53 (93rd percentile) |
Bioinformatics
A homology model of the zymogen fragment of Kgp comprising NPD + CD + IgSF was obtained as follows: the structure of RgpB-CD + IgSF in complex with its NPD (PDB code 4IEF and Ref. 43) was superimposed onto Kgp-NPD (molecule A) with the SSM program (57) using the NPDs only. Afterward, the structure of Kgp-CD + IgSF (PDB code 4RBM and Ref. 42) was superimposed on the equivalent parts of RgpB. This provided an initial model for Kgp-NPD+CD+IgSF. Detailed visual inspection of this model with COOT revealed some loop regions involved in clashes. These regions were corrected in two rounds of manual remodeling with COOT and energy minimization with the Chimera program employing default parameters (58) plus a final regularization step with the geometry_minimization routine of the PHENIX suite (59). The latter included restraining metal-binding distances to tabulated values. The final homology model was assessed with MolProbity, which revealed the following parameters of protein geometry and all-atom contacts: clash-score for all atoms, 1.76 (99th percentile); poor rotamers, 2 (0.38% of 524 residues); Ramachandran outliers, 1 (0.16% of 630 residues); Ramachandran favored, 612 residues (97.1%); Cβ deviations >0.25 Å, 0; residues with bad bond/angles, 0/0; MolProbity score, 1.09 (100th percentile). The regularized homology model of the Kgp zymogen can be obtained from the corresponding authors.
Secondary structure predictions were calculated with JPRED-4 (60). The sequence alignment in Fig. 2C was performed manually based on visual inspection with COOT of the structures previously superposed with the SSM program. Structure figures were prepared with Chimera and protein interfaces were analyzed with the PISA server (46). The coordinates of the final experimental model of Kgp-NPD from P. gingivalis strain W83 were deposited with the PDB under access code 5MUN.
Author Contributions
F. X. G.-R. and J. P. conceived the work; Z. N., F. V., M. N. S., D. M., M. N., and B. P. produced proteins and carried out the molecular biology manipulations and mutant analysis; A. P., I. U., Th. G., Ti. G., F. X. G.-R., and J. A. H. performed the structural studies; and F. X. G.-R., J. A. H., and J. P. wrote the paper with contributions from all authors.
Acknowledgments
We thank Joan Pous and Xandra Kreplin from the Protein Crystallography Platform shared between the Molecular Biology Institute of Barcelona and the Institute for Research in Biomedicine for excellent technical assistance during crystallization experiments. We also acknowledge the help provided by Diamond Light Source local contacts.
This work was supported in part by Grant FP7-HEALTH-2012-306029-2 “TRIGGER” from the Seventh Framework Programme; Grants BFU2015-64487R, MDM-2014-0435, BIO2013-49320-EXP, BIO2015-64216-P, and BIO2013-49604-EXP from the Spanish Ministerio de Economía y Competitividad; Grant 2014SGR9 from the Departament d'Innovació, Universitats i Empresa, Generalitat de Catalunya; and Diamentowy Grant 0218/DIA/2012/41 from the Polish Ministerstwo Nauki i Szkolnictwa Wyzszego. The Structural Biology Unit of Molecular Biology Institute of Barcelona (IBMB) is a “María de Maeztu” Unit of Excellence from the Spanish Ministry of Economy, Innovation and Competitiveness. Funding for travelling and synchrotron data collection was provided in part by Diamond Light Source. The Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University is a partner of the Leading National Research Center supported by the Ministry of Science and Higher Education. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5MUN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CP
- chronic periodontitis
- CD
- catalytic domain
- CTD
- C-terminal domain
- IgSF
- immunoglobulin superfamily-like domain
- Kgp
- lysine-specific gingipain
- NPD
- N-terminal pro-domain
- PDB
- Protein Data Bank
- RgpA/RgpB
- arginine-specific gingipains R1 and R2
- qRT-PCR
- quantitative RT-PCR
- BC
- bacterial cultures
- WC
- washed cell fraction
- PP
- cytoplasmic/periplasmic fraction
- CE
- cell envelope
- GM
- cell-free growth medium.
References
- 1. Sender R., Fuchs S., and Milo R. (2016) Are we really vastly outnumbered?: revisiting the ratio of bacterial to host cells in humans. Cell 164, 337–340 [DOI] [PubMed] [Google Scholar]
- 2. Littman D. R., and Pamer E. G. (2011) Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 10, 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abby S. S., Cury J., Guglielmini J., Néron B., Touchon M., and Rocha E. P. (2016) Identification of protein secretion systems in bacterial genomes. Sci. Rep. 6, 23080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O'Hara A. M., and Shanahan F. (2006) The gut flora as a forgotten organ. EMBO Rep. 7, 688–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Subramanian S., Blanton L. V., Frese S. A., Charbonneau M., Mills D. A., and Gordon J. I. (2015) Cultivating healthy growth and nutrition through the gut microbiota. Cell 161, 36–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. How K. Y., Song K. P., and Chan K. G. (2016) Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front. Microbiol. 7, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paster B. J., Boches S. K., Galvin J. L., Ericson R. E., Lau C. N., Levanos V. A., Sahasrabudhe A., and Dewhirst F. E. (2001) Bacterial diversity in human subgingival plaque. J. Bacteriol. 183, 3770–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eloe-Fadrosh E. A., and Rasko D. A. (2013) The human microbiome: from symbiosis to pathogenesis. Annu. Rev. Med. 64, 145–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ximénez-Fyvie L. A., Haffajee A. D., and Socransky S. S. (2000) Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J. Clin. Periodontol. 27, 648–657 [DOI] [PubMed] [Google Scholar]
- 10. Haffajee A. D., Socransky S. S., and Gunsolley J. C. (2003) Systemic anti-infective periodontal therapy: a systematic review. Ann. Periodontol. 8, 115–181 [DOI] [PubMed] [Google Scholar]
- 11. Seymour G. J., Ford P. J., Cullinan M. P., Leishman S., and Yamazaki K. (2007) Relationship between periodontal infections and systemic disease. Clin. Microbiol. Infect. 13, 3–10 [DOI] [PubMed] [Google Scholar]
- 12. de la Fuente C., Flores S., and Moraga M. (2013) DNA from human ancient bacteria: a novel source of genetic evidence from archaeological dental calculus. Archaeometry 55, 766–778 [Google Scholar]
- 13. Maixner F., Thomma A., Cipollini G., Widder S., Rattei T., and Zink A. (2014) Metagenomic analysis reveals presence of Treponema denticola in a tissue biopsy of the Iceman. PLoS One 9, e99994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Warinner C., Rodrigues J. F., Vyas R., Trachsel C., Shved N., Grossmann J., Radini A., Hancock Y., Tito R. Y., Fiddyment S., Speller C., Hendy J., Charlton S., Luder H. U., Salazar-García D. C., et al. (2014) Pathogens and host immunity in the ancient human oral cavity. Nat. Genet. 46, 336–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Potempa J., and Pike R. N. (2005) Bacterial peptidases. Contrib. Microbiol. 12, 132–180 [DOI] [PubMed] [Google Scholar]
- 16. Mallorquí-Fernández N., Manandhar S. P., Mallorquí-Fernández G., Usón I., Wawrzonek K., Kantyka T., Solà M., Thøgersen I. B., Enghild J. J., Potempa J., and Gomis-Rüth F. X. (2008) A new autocatalytic activation mechanism for cysteine proteases revealed by Prevotella intermedia interpain A. J. Biol. Chem. 283, 2871–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dubin G., Koziel J., Pyrc K., Wladyka B., and Potempa J. (2013) Bacterial proteases in disease: role in intracellular survival, evasion of coagulation/ fibrinolysis innate defenses, toxicoses and viral infections. Curr. Pharm. Des. 19, 1090–1113 [DOI] [PubMed] [Google Scholar]
- 18. Yongqing T., Potempa J., Pike R. N., and Wijeyewickrema L. C. (2011) The lysine-specific gingipain of Porphyromonas gingivalis: importance to pathogenicity and potential strategies for inhibition. Adv. Exp. Med. Biol. 712, 15–29 [DOI] [PubMed] [Google Scholar]
- 19. Lamont R. J., and Jenkinson H. F. (1998) Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62, 1244–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bostanci N., and Belibasakis G. N. (2012) Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol. Lett. 333, 1–9 [DOI] [PubMed] [Google Scholar]
- 21. Potempa J., Pike R., and Travis J. (1997) Titration and mapping of the active site of cysteine proteinases from Porphyromonas gingivalis (gingipains) using peptidyl chloromethanes. Biol. Chem. 378, 223–230 [DOI] [PubMed] [Google Scholar]
- 22. Guentsch A., Kramesberger M., Sroka A., Pfister W., Potempa J., and Eick S. (2011) Comparison of gingival crevicular fluid sampling methods in patients with severe chronic periodontitis. J. Periodontol. 82, 1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pathirana R. D., O'Brien-Simpson N. M., Brammar G. C., Slakeski N., and Reynolds E. C. (2007) Kgp and RgpB, but not RgpA, are important for Porphyromonas gingivalis virulence in the murine periodontitis model. Infect. Immun. 75, 1436–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pike R. N., and Potempa J. (2013) Chapter 523: gingipain K. In Handbook of Proteolytic Enzymes (Rawlings N. D., and Salvesen G., eds) pp. 2337–2344, Academic Press, Oxford [Google Scholar]
- 25. Kataoka S., Baba A., Suda Y., Takii R., Hashimoto M., Kawakubo T., Asao T., Kadowaki T., and Yamamoto K. (2014) A novel, potent dual inhibitor of Arg-gingipains and Lys-gingipain as a promising agent for periodontal disease therapy. FASEB J. 28, 3564–3578 [DOI] [PubMed] [Google Scholar]
- 26. Sato K., Naito M., Yukitake H., Hirakawa H., Shoji M., McBride M. J., Rhodes R. G., and Nakayama K. (2010) A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McBride M. J., and Zhu Y. (2013) Gliding motility and Por secretion system genes are widespread among members of the phylum Bacteroidetes. J. Bacteriol. 195, 270–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chagnot C., Zorgani M. A., Astruc T., and Desvaux M. (2013) Proteinaceous determinants of surface colonization in bacteria: bacterial adhesion and biofilm formation from a protein secretion perspective. Front. Microbiol. 4, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kharade S. S., and McBride M. J. (2015) Flavobacterium johnsoniae PorV is required for secretion of a subset of proteins targeted to the type IX secretion system. J. Bacteriol. 197, 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakayama K. (2015) Porphyromonas gingivalis and related bacteria: from colonial pigmentation to the type IX secretion system and gliding motility. J. Periodontal. Res. 50, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kadowaki T., Yukitake H., Naito M., Sato K., Kikuchi Y., Kondo Y., Shoji M., and Nakayama K. (2016) A two-component system regulates gene expression of the type IX secretion component proteins via an ECF sigma factor. Sci. Rep. 6, 23288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Diego I., Ksiazek M., Mizgalska D., Koneru L., Golik P., Szmigielski B., Nowak M., Nowakowska Z., Potempa B., Houston J. A., Enghild J. J., Thøgersen I. B., Gao J., Kwan A. H., Trewhella J., et al. (2016) The outer-membrane export signal of Porphyromonas gingivalis type IX secretion system (T9SS) is a conserved C-terminal β-sandwich domain. Sci. Rep. 6, 23123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lasica A. M., Goulas T., Mizgalska D., Zhou X., de Diego I., Ksiazek M., Madej M., Guo Y., Guevara T., Nowak M., Potempa B., Goel A., Sztukowska M., Prabhakar A. T., Bzowska M., et al. (2016) Structural and functional probing of PorZ, an essential bacterial surface component of the type-IX secretion system of human oral-microbiomic Porphyromonas gingivalis. Sci. Rep. 6, 37708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holzer H., and Heinrich P. C. (1980) Control of proteolysis. Annu. Rev. Biochem. 49, 63–91 [DOI] [PubMed] [Google Scholar]
- 35. Khan A. R., and James M. N. (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7, 815–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bryan P. N. (2002) Prodomains and protein folding catalysis. Chem. Rev. 102, 4805–4816 [DOI] [PubMed] [Google Scholar]
- 37. Wiederanders B. (2003) Structure-function relationships in class CA1 cysteine peptidase propeptides. Acta Biochim. Pol. 50, 691–713 [PubMed] [Google Scholar]
- 38. Tao K., Stearns N. A., Dong J., Wu Q. L., and Sahagian G. G. (1994) The proregion of cathepsin L is required for proper folding, stability, and ER exit. Arch. Biochem. Biophys. 311, 19–27 [DOI] [PubMed] [Google Scholar]
- 39. Vendrell J., Querol E., and Avilés F. X. (2000) Metallocarboxypeptidases and their protein inhibitors: structure, function and biomedical properties. Biochim. Biophys. Acta 1477, 284–298 [DOI] [PubMed] [Google Scholar]
- 40. Gomis-Rüth F. X. (2008) Structure and mechanism of metallocarboxypeptidases. Crit. Rev. Biochem. Mol. Biol. 43, 319–345 [DOI] [PubMed] [Google Scholar]
- 41. Mittl P. R., and Grütter M. G. (2006) Opportunities for structure-based design of protease-directed drugs. Curr. Opin. Struct. Biol. 16, 769–775 [DOI] [PubMed] [Google Scholar]
- 42. de Diego I., Veillard F., Sztukowska M. N., Guevara T., Potempa B., Pomowski A., Huntington J. A., Potempa J., and Gomis-Rüth F. X. (2014) Structure and mechanism of cysteine peptidase gingipain K (Kgp), a major virulence factor of Porphyromonas gingivalis in periodontitis. J. Biol. Chem. 289, 32291–32302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Diego I., Veillard F. T., Guevara T., Potempa B., Sztukowska M., Potempa J., and Gomis-Rüth F. X. (2013) Porphyromonas gingivalis virulence factor gingipain RgpB shows a unique zymogenic mechanism for cysteine peptidases. J. Biol. Chem. 288, 14287–14296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Veillard F., Sztukowska M., Mizgalska D., Ksiazek M., Houston J., Potempa B., Enghild J. J., Thogersen I. B., Gomis-Rüth F. X., Nguyen K. A., and Potempa J. (2013) Inhibition of gingipains by their profragments as the mechanism protecting Porphyromonas gingivalis against premature activation of secreted proteases. Biochim. Biophys. Acta 1830, 4218–4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mikolajczyk J., Boatright K. M., Stennicke H. R., Nazif T., Potempa J., Bogyo M., and Salvesen G. S. (2003) Sequential autolytic processing activates the zymogen of Arg-gingipain. J. Biol. Chem. 278, 10458–10464 [DOI] [PubMed] [Google Scholar]
- 46. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 47. Bickel M., Munoz J. L., and Giovannini P. (1985) Acid-base properties of human gingival crevicular fluid. J. Dent. Res. 64, 1218–1220 [DOI] [PubMed] [Google Scholar]
- 48. Chiu J., March P. E., Lee R., and Tillett D. (2004) Site-directed, ligase-independent mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 32, e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nguyen K. A., Travis J., and Potempa J. (2007) Does the importance of the C-terminal residues in the maturation of RgpB from Porphyromonas gingivalis reveal a novel mechanism for protein export in a subgroup of Gram-negative bacteria? J. Bacteriol. 189, 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kabsch W. (2010) XDS. Acta Crystallogr. D 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sheldrick G. M., Gilmore C. J., Hauptman H. A., Weeks C. M., Miller R., and Usón I. (2011) Part 16: direct methods. 16.1 ab initio phasing. In International Tables for Crystallography. (Arnold E., Himmel D. M., and Rossmann M. G., eds) pp. 413–429, Kluwer Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
- 52. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., and Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 56. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 58. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 59. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Drozdetskiy A., Cole C., Procter J., and Barton G. J. (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. García-Castellanos R., Marrero A., Mallorquí-Fernández G., Potempa J., Coll M., and Gomis-Ruth F. X. (2003) Three-dimensional structure of MecI: molecular basis for transcriptional regulation of staphylococcal methicillin resistance. J. Biol. Chem. 278, 39897–39905 [DOI] [PubMed] [Google Scholar]
- 62. Weiss M. S. (2001) Global indicators of X-ray quality. J. Appl. Crystallogr. 34, 130–135 [Google Scholar]
- 63. Karplus P. A., and Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]