

Abstract
Polyalanine (poly(A)) diseases are caused by the expansion of translated GCN triplet nucleotide sequences encoding poly(A) tracts in proteins. To date, nine human disorders have been found to be associated with poly(A) tract expansions, including congenital central hypoventilation syndrome and oculopharyngeal muscular dystrophy. Previous studies have demonstrated that unexpanded wild-type poly(A)-containing proteins localize to the cell nucleus, whereas expanded poly(A)-containing proteins primarily localize to the cytoplasm. Because most of these poly(A) disease proteins are transcription factors, this mislocalization causes cellular transcriptional dysregulation leading to cellular dysfunction. Correcting this faulty localization could potentially point to strategies to treat the aforementioned disorders, so there is a pressing need to identify the mechanisms underlying the mislocalization of expanded poly(A) protein. Here, we performed a glutathione S-transferase pulldown assay followed by mass spectrometry and identified eukaryotic translation elongation factor 1 α1 (eEF1A1) as an interacting partner with expanded poly(A)-containing proteins. Strikingly, knockdown of eEF1A1 expression partially corrected the mislocalization of the expanded poly(A) proteins in the cytoplasm and restored their functions in the nucleus. We further demonstrated that the expanded poly(A) domain itself can serve as a nuclear export signal. Taken together, this study demonstrates that eEF1A1 regulates the subcellular location of expanded poly(A) proteins and is therefore a potential therapeutic target for combating the pathogenesis of poly(A) diseases.
Keywords: Drosophila genetics, gene transcription, genetic disease, protein export, protein-protein interaction, eEF1A
Introduction
Polyalanine (poly(A)) diseases are caused by the expansion of GCN (where N refers to any of the four bases) trinucleotide repeats in the disease genes encoding the poly(A) amino acid tract in the disease proteins (1, 2). Expansion of alanine tracts has been shown to cause several human pathological conditions, including congenital malformations and/or mental retardation (1, 3). Up to the present, nine disease-associated proteins with alanine tract expansions have been identified (4). Eight of the nine poly(A) disease proteins are transcription factors with nuclear localization (1, 5). Nuclear localization is essential for transcription factors to bind to DNA and activate target gene transcription (6). It has been reported that poly(A) disease proteins, including HOXD13, HOXA13, RUNX2, SOX3, FOXL2, ARX, and PHOX2B, are mislocalized in the cytoplasm when overexpressed in mammalian cell lines (7–11). Furthermore, the degree of cytoplasmic mislocalization increases as the length of the alanine tract increases (10, 12). Polyalanine expansion in FOXL2 gives rise to its accumulation in the cytoplasm in a length-dependent manner. Expanded FOXL2 containing 19 alanine residues is found both in the nucleus and the cytoplasm. For FOXL2 with 37 alanine residues, all of the cells display cytoplasmic localization (9). Moreover, the degree of transcriptional activity of poly(A) disease protein decreases as the alanine tract increases (9). For instance, PHOX2B is a gene associated with congenital central hypoventilation syndrome (13). The majority of patients with central hypoventilation syndrome have a poly(A) repeat expansion mutation in PHOX2B protein (13). Luciferase assay results reveal an inverse correlation between trans-activation activity and the length of the poly(A) tract (7, 14). Similarly, FOXL2, which encodes a transcription factor, is the causative gene in blepharophimosis, ptosis, and epicanthus inversus syndrome (15). The wild-type FOXL2 protein has a highly conserved 14 residue C-terminal alanine tract (16). Luciferase assay and quantitative reverse transcription-polymerase chain reaction (RT-PCR) of the target genes suggest that the longer a poly(A) tract appended to the disease protein is, the lower the trans-activation activity of FOXL2 (9). These studies suggest that poly(A) tract expansion causes protein cytoplasmic localization, impairs the trans-activation activity of these poly(A) disease proteins, and results in a loss-of-function phenotype.
Eukaryotic translation elongation factor 1 α (eEF1A)3 is in charge of the delivery of aminoacylation-tRNA (aa-tRNA) to the A site of ribosome during translation elongation (17). Aside from the canonical role in translation, eEF1A is involved in the nuclear export of proteins (18). Translation elongation factor EF-1 α (Tef1) is the yeast orthologue of human eEF1A, and Tef1 was implicated in the tRNA nuclear export process in Saccharomyces cerevisiae (19). In mammalian cells, eEF1A was also reported to mediate the nuclear export of transcription-dependent nuclear export motif (TD-NEM) containing proteins, including the poly(A)-binding protein 1 (PABP1) and the von Hippel-Lindau (VHL) tumor suppressor protein (18) and SNAG-containing proteins, such as Snail1 (20). The above evidence clearly demonstrates that eEF1A plays a role in protein nuclear export. In this study, we showed that eEF1A1 regulates the localization of protein that carries a novel nuclear export signal (NES) in a poly(A) tract of disease proteins that are implicated in poly(A) disorders. Our findings provide a mechanistic explanation for how transcription factor dysregulation is triggered in poly(A) disease pathogenesis.
Results
Continuous expanded poly(A) tract alters the localization of nuclear proteins
To determine the changes in subcellular localization of proteins with an expanded poly(A) tract, the Rev nuclear export assay (21) was employed. The wild-type HIV-1 viral Rev protein carries a functional nucleolus localization signal and NES, which direct Rev protein transports between the nucleus and cytoplasm through the nuclear pore complex (22). The nuclear export reporter protein, Rev(1.4)-EGFP, is a fusion protein that carries a mutant version of Rev (Rev(1.4)) and EGFP. The Rev(1.4) mutant protein lacks nuclear export activity and thus localizes primarily to the nucleus (21). To address the effect of poly(A) tracts on the subcellular localization of Rev(1.4), we fused Rev(1.4)-EGFP with either an unexpanded (A6) or expanded (A37) poly(A) repeat (Table 1) and subsequently transfected HEK293 cells with these constructs. The subcellular localization of proteins was determined and quantified according to Ref. 21. The nuclear export reporter protein, Rev(1.4)-EGFP, without a poly(A) tract (control) was found to predominantly localize to the nuclear compartment (Fig. 1, A and B). The Rev(1.4)-NES-EGFP protein was used as the positive control to show that the insertion of a functional NES isolated from the HIV-1 Rev protein (21) caused cytoplasmic localization of the nuclear export reporter protein (Fig. 1, A and B). Similar to the Rev(1.4)-EGFP control, the Rev(1.4)-A6-EGFP protein was found mainly in the nuclear compartment (Fig. 1, A and B), which suggests that the unexpanded poly(A) tract conferred no detectable effect on the subcellular localization of the Rev(1.4)-EGFP reporter. In contrast, the Rev(1.4)-A37-EGFP protein was found to reside in both the nuclear and cytoplasmic compartments (Fig. 1, A and B). This result suggests that the expanded poly(A) tract alters the localization of nuclear proteins, directing them to the cytoplasm.
TABLE 1.
Constructs used in this study
| Control | pRev (1.4)-EGFP |
| NES | pRev (1.4)-NES-EGFP |
| A6 | pRev (1.4)-A6-EGFP |
| A22 | pRev (1.4)-A22-EGFP |
| A25 | pRev (1.4)-A25-EGFP |
| A28 | pRev (1.4)-A28-EGFP |
| A31 | pRev (1.4)-A31-EGFP |
| A34 | pRev (1.4)-A34-EGFP |
| A37 | pRev (1.4)-A37-EGFP |
| A10(PA9)3 | pRev (1.4)-A10PA9PA9PA9-EGFP |
| A7(PA6)5 | pRev (1.4)-A7PA6PA6PA6PA6PA6-EGFP |
Figure 1.

Continuous expanded poly(A) tract alters the localization of nuclear proteins. A, subcellular localization of Rev(1.4)-poly(A)-EGFP proteins in HEK293 cells. The Rev(1.4)-EGFP control protein localized to the nuclear compartment, whereas the positive control Rev(1.4)-NES-EGFP (NES), which contained a functional HIV-1 Rev NES sequence, localized to the cytoplasm. The Rev(1.4)-poly(A)-EGFP protein with a 6-alanine tract (A6) localized in the nuclear compartment, whereas that with a 37-alanine tract Rev(1.4)-A37-EGFP (A37) showed both nuclear and cytoplasmic localization. Cell nuclei were stained with Hoechst 33342. Scale bar, 10 μm. B, statistical analysis of A. C, subcellular localization of Rev(1.4)-polyAPA-EGFP proteins in HEK293 cells. The Rev(1.4)-A37-EGFP (A37) showed both nuclear and cytoplasmic localization, two Rev(1.4)-polyAPA-EGFP variants (A10(PA9)3 and A7(PA6)5) localized in the nuclear compartment. Cell nuclei were stained with Hoechst 33342. Scale bar, 10 μm. D, statistical analysis of C. E, subcellular localization of Rev(1.4)-poly(A)-EGFP protein with A22, A25, A28, A31, and A34. A22, A25, and A28 showed nuclear localization. A31 resulted in 26% of cells with both nuclear and cytoplasmic localization, whereas A34 resulted in up to 78% of cells with both nuclear and cytoplasmic localization. Scale bar, 10 μm. F, statistical analysis (E). N, nuclear; C, cytoplasmic; C + N, cytoplasmic and nuclear localization. Three independent transfection experiments were performed. At least 100 transfected cells were analyzed in each experiment. Error bars, S.E.
We next sought to investigate whether the continuity of the expanded poly(A) tract would be essential for the mislocalization. We generated two expanded polyAPA variant constructs, pRev(1.4)-A10PA9PA9PA9-EGFP and pRev(1.4)-A7PA6PA6PA6PA6PA6-EGFP, which contain three or five proline residues within the expanded 37 poly(A) tract (Table 1). As a result, the continuity of the expanded poly(A) tract is disrupted, and the long poly(A) tracts are split into shorter alanine stretches. In contrast to Rev(1.4)-A37-EGFP which showed both nuclear and cytoplasmic localization, Rev(1.4)-A10PA9PA9PA9-EGFP and Rev(1.4)-A7PA6PA6PA6PA6PA6-EGFP proteins were predominantly found in the nuclear compartment (Fig. 1, C and D). This indicates that the continuity of expanded poly(A) tract is crucial for its effect of cytoplasmic mislocalization.
Next, we seek to determine the minimal length of poly(A) tract that would lead to cytoplasmic localization of nuclear proteins. We fused Rev(1.4)-EGFP with expanded poly(A) tract of different lengths, including A22, A25, A28, A31, and A34 (Table 1), and analyzed the localization of EGFP in HEK293 cells. The A22, A25, and A28 proteins resulted in 96, 98, and 98% of the cells with pure nuclear localization, respectively, which did not appear to be different from A6 (Fig. 1, E and F). But when the expanded poly(A) tract reached 31 repeats, we started noticing a difference, because A31 resulted in 74% of the cells with pure nuclear localization, and 26% of them showed both nuclear and cytoplasmic localization (Fig. 1, E and F). When the poly(A) tract reached 34 repeats, the majority of cells possessed a nuclear + cytoplasmic localization pattern, with 78% of the cells having EGFP in both compartments and only 22% cells with pure nuclear localization (Fig. 1, E and F). From these results, we concluded that the minimal length of expanded poly(A) tract that would lead to a detectable increase of cytoplasmic localization is somewhere between 28 and 31 alanine repeats, whereas the threshold repeat length that would lead to nuclear + cytoplasmic localization in the majority of cells lies between 31 and 34 repeats.
Eukaryotic translation elongation factor 1 α1 is a novel interacting partner of protein containing expanded poly(A) tract
To identify cellular protein-interacting partners of expanded poly(A) proteins, a GST-pulldown assay followed by mass spectrometry was performed. The HEK293 cell lysate was allowed to interact with purified bacterially expressed GST-A7 or -A37 fusion proteins in vitro. Mass spectrometric analysis of the eluents unveiled eEF1A1 as an expanded poly(A) protein (GST-A37)-interacting partner (supplemental Fig. S1). To confirm this interaction, we repeated the in vitro GST-pulldown experiment in HEK293 cells and detected eEF1A1 by means of Western blotting. The endogenous eEF1A protein was only detected in eluent of the GST-A37 capture and not the GST and GST-A7 controls (Fig. 2A). We also showed that expanded poly(A) protein interacted with Myc-eEF1A1 by a GST-pulldown experiment in HEK293 cells (Fig. 2B). Taken together, our results indicate that eEF1A1 interacts specifically with protein carrying an expanded poly(A) tract but not with the unexpanded poly(A) tract.
Figure 2.

eEF1A1 interacted with protein carrying a continuous expanded poly(A) tract. A, endogenous eEF1A1 interacted with GST-A37. The GST-pulldown assay was performed to detect the interaction between endogenous eEF1A1 and GST-poly(A). The HEK293 cellular eEF1A1 protein was detected by Western blotting using anti-eEF1A antibody. The GST-poly(A) fusion proteins were detected by SDS-PAGE Coomassie Blue staining. B, the Myc-tagged eEF1A1 protein interacted with GST-A37. C, schematic diagram of eEF1A1 domains. D, domain III of eEF1A1 interacted with GST-A37 protein. The GST-pulldown assay was performed to determine the interaction between GST-poly(A) proteins and eEF1A1 domains. The eluents were detected by Western blotting using anti-Myc antibody. The GST-poly(A) fusion proteins were detected by SDS-PAGE Coomassie Blue staining. E, schematic diagram of eEF1A1 constructs with different truncated versions of domain III. F, amino acids 429–449 in eEF1A1 are important for interaction between eEF1A1 and GST-A37. The GST-pulldown assay was performed to determine the interaction region in eEF1A1 interacted with GST-A37 protein. G, continuous expanded poly(A) tract interacts with eEF1A1. Co-immunoprecipitation was performed to detected the interaction between Rev(1.4)-poly(A)-EGFP proteins and eEF1A1. The lysates of cells co-transfected with pRev(1.4)-poly(A)-EGFP and pcDNA3.1-myc-eEF1A1 constructs were subjected to a co-IP assay. IP was performed with c-Myc agarose affinity gel followed by immunoblotting with mouse anti-Myc antibody (Myc-eEF1A1) and mouse anti-GFP antibody (Rev(1.4)-poly(A)-EGFP). The interaction with eEF1A1 was abolished when continuity of the expanded poly(A) tract was disrupted by proline. Three independent experiments were performed.
The eFF1A1 protein can be structurally divided into three domains (Fig. 2C); domain I functions as a GTP-binding domain, domain II is involved in tRNA binding, and domain III is responsible for actin binding (23). To gain further insights into the expanded poly(A)/eEF1A1 interaction, we performed a GST-pulldown assay to determine which domain(s) in eEF1A1 is responsible for interacting with the expanded poly(A) tract. The result showed that eEF1A1 domain III and domain II+III, but not domain I, II, and I+II was able to interact with GST-A37 (Fig. 2D). This indicates that domain III in eEF1A1 is responsible for interacting with expanded poly(A) tract-containing protein. To further map out the expanded poly(A)-interacting region in eEF1A1, we transfected HEK293 cells with a series of Myc-tagged eEF1A1 deletion constructs (Fig. 2E) and used cell lysates to perform a GST-pulldown experiment. The results indicate that the region encompassing amino acids 429–449 of eEF1A1 is essential for the protein to interact with expanded poly(A) tracts (Fig. 2F).
To determine whether the interruption of continuity of the expanded poly(A) tract would affect the interaction between eEF1A1 and expanded poly(A) tract-containing proteins, we performed a co-immunoprecipitation experiment in HEK293 cells co-expressed with different versions of Rev(1.4)-polyAPA-EGFP variants (Table 1) and Myc-tagged eEF1A1. We found that Myc-eEF1A1 could be co-immunoprecipitated with Rev(1.4)-A37-EGFP but not the other Rev(1.4)-polyAPA-EGFP variants, including Rev(1.4)-A10PA9PA9PA9-EGFP and Rev(1.4)-A7PA6PA6PA6PA6PA6-EGFP (Fig. 2G). This result indicates that the continuity of the expanded poly(A) tract is an essential determinant for its interaction with eEF1A1.
eEF1A1 regulates the cytoplasmic localization of expanded poly(A) tract-containing protein
eEF1A is a GTP-binding protein that associates with protein synthesis (23). Besides its canonical role in protein translation, eEF1A has also been reported to be involved in nuclear export of proteins that carry the TD-NEM, such as the VHL tumor suppressor in mammalian cells (18). We thus speculated that eEF1A1 is involved in regulating the localization of expanded poly(A) tract-containing proteins.
To investigate the effect of eEF1A1 on the subcellular localization of poly(A)-containing protein, we analyzed the subcellular localization of Rev(1.4)-poly(A)-EGFP in HEK293 cells with or without knockdown of eEF1A1 using confocal microscopy. The nucleo-cytoplasmic distribution of protein was quantified by measuring the fluorescence intensity of the EGFP signals in the nuclear compartment and the cytoplasmic compartment and expressed as the nuclear/cytoplasmic (N/C) ratio. The result showed that knockdown of eEF1A1 expression caused an enrichment of the Rev(1.4)-A37-EGFP protein in the nuclear compartment but had no effect on the subcellular localization of the Rev(1.4)-A6-EGFP control protein (Fig. 3, A and B). An increase in the percentage of cells with an N/C ratio >1 indicated that more EGFP signals were detected in the nucleus. For the reporter proteins that were tagged with a functional NES from the HIV-1 Rev, the knockdown of eEF1A1 has no effect on changing the N/C ratio, suggesting that eEF1A1 is not involved in the nuclear export mediated by the classical Rev NES (Fig. 3, A and B). The efficiency of the eEF1A1 siRNA was analyzed by immunoblotting. The results showed that eEF1A1 protein levels were largely reduced after eEF1A1 siRNA treatment (Fig. 3C). Our results also showed that overexpression of eEF1A1 enhanced the cytoplasmic localization of expanded poly(A) protein, whereas overexpression of eEF1A1Δ429–449 had no such effect (Fig. 3, D–F). These data indicate that eEF1A1 regulates the subcellular localization of expanded poly(A) tract-containing protein.
Figure 3.

eEF1A1 is involved in the cytoplasmic mislocalization of expanded poly(A) tract-containing nuclear exporter reporter protein. A, subcellular localization of Rev(1.4)-poly(A)-EGFP protein in HEK293 cells after eEF1A1 knockdown. The knockdown of eEF1A1 expression caused a nuclear enrichment of Rev(1.4)-A37-EGFP protein but had no effect on the subcellular localization of Rev(1.4)-A6-EGFP protein. The cell nuclei were stained with Hoechst 33432. Scale bar, 10 μm. B, statistical analysis of A. Knockdown of eEF1A1 expression caused a statistically significant increase in the percentage of cells expressing Rev(1.4)-A37-EGFP protein with an N/C ratio of >1. C, the knockdown efficiency of eEF1A1 was measured using immunoblotting. β-Tubulin served as the loading control. D, subcellular localization of Rev(1.4)-poly(A)-EGFP protein in HEK293 cells after Myc-eEF1A1 or Myc-eEF1A1Δ429–449 overexpression. Overexpression of Myc-eEF1A1 caused Rev(1.4)-A37-EGFP protein cytoplasmic enrichment but had no effect on the localization of Rev(1.4)-A6-EGFP protein. Overexpression of Myc-eEF1A1Δ429–449 had no effect on the localization of both Rev(1.4)-A6-EGFP and Rev(1.4)-A37-EGFP. The cell nuclei were stained with Hoechst. Scale bar, 10 μm. E, statistical analysis of D. Overexpression of Myc-eEF1A1 caused a statistical significant decrease in the percentage of cells expressing Rev(1.4)-A37-EGFP protein with N/C ratio >1. F, Myc-eEF1A1 overexpression detection using immunoblotting. β-Tubulin served as the loading control. G, subcellular localization of Rev(1.4)-poly(A)-EGFP protein in HEK293 cells after eEF2 knockdown. The knockdown of eEF2 expression had no effect on the subcellular localization of both Rev(1.4)-A6-EGFP and Rev(1.4)-A37-EGFP protein. The cell nuclei were stained with Hoechst. Scale bar, 10 μm. H, statistical analysis of G. Knockdown of eEF2 expression did not change the percentage of cells expressing Rev(1.4)-poly(A)-EGFP protein with N/C ratio >1. I, the knockdown efficiency of eEF2 was measured using immunoblotting. β-Tubulin served as the loading control. Three independent transfection experiments were performed. At least 100 transfected cells were measured per experiment. Error bars, S.E. **, p < 0.01. ns, not statistically significant.
During eukaryotic protein translation elongation, eEF1A is responsible for the enzymatic delivery of aa-tRNA to the A site of the ribosome, and eukaryotic translation elongation factor 2 (eEF2) is responsible for translocating the peptidyl-tRNA out of the A site to the P site (23). To eliminate the possibility that the eEF1A1 knockdown-mediated nuclear enrichment of expanded poly(A) proteins results from a blockage of general protein translation elongation, we examined the effect of eEF2 knockdown on the nucleo-cytoplasmic distribution of expanded poly(A) tract-containing protein. We found that the knockdown of eEF2 expression had no effect on the N/C ratio of Rev(1.4)-poly(A)-EGFP protein (Fig. 3, G–I). This suggests that the role of eEF1A1 in modulating the localization of expanded poly(A) protein is not related to its protein translation function.
Knockdown of eEF1A1 expression resulted in a nuclear enrichment of expanded poly(A)-containing SoxN
To relate the mislocalization of expanded poly(A) domain to poly(A) toxicity, we extended our study to an in vivo poly(A) disease transgenic fly model. The human sex-determining region Y-box 3 (SOX3) protein is a transcription factor that is expressed in the brain and plays a role in neural development (1). A mutation in SOX3 causing the addition of an extra 7–11 alanine residues to normal 15-alanine tract induces X-linked hypopituitarism, a prevalent poly(A) disease exhibiting mental retardation, short stature, and growth hormone deficiency (11, 24, 25). The SoxNeuro (SoxN) protein is the Drosophila ortholog of SOX3, and it plays a critical role in embryogenesis, including the formation of neuroblasts (26, 27) and axonal patterning (28).
We first expressed A12-SoxN-EGFP and A34-SoxN-EGFP fusion proteins to determine the subcellular localization of overexpressed poly(A)-SoxN in SK-N-MC cells. The A12-SoxN-EGFP protein was found to be localized in the nucleus, whereas A34-SoxN-EGFP predominantly localized in the cytoplasm and formed protein aggregates (Fig. 4A). Next, we observed that the knockdown of eEF1A1 expression did not affect the distribution of the unexpanded A12-SoxN-EGFP. In contrast, the knockdown of eEF1A1 caused the nuclear enrichment of A34-SoxN-EGFP (Fig. 4A). The fluorescence intensity analysis of the two fluorescence patterns was performed along the line passed the whole cell. A significant amount of A12-SoxN-EGFP signal was found to overlap with nuclear stain in cells treated with both control siRNA and eEF1A1 siRNA. On the contrary, significantly less overlapping was observed for A34-SoxN-EGFP in cells treated with control siRNA. However, upon eEF1A1 knockdown, the overlapping was partially restored (Fig. 4A). To further investigate the effect of eEF1A1 knockdown biochemically, the nucleocytoplasmic fractionation assay was performed. The nuclear A34-SoxN protein levels rather than the A12-SoxN protein levels were found to be increased upon eEF1A1 knockdown (Fig. 4B). These results demonstrated that eEF1A1 mediates expanded poly(A) domain-containing SoxN nucleocytoplasmic distribution but does not alter wild-type SoxN localization.
Figure 4.

Knockdown of Ef1a48D expression attenuated the expanded poly(A) SoxN toxicity by mediating its mislocalization. A, the effect of knockdown of eEF1A1 on poly(A)-SoxN-EGFP localization in SK-N-MC cells. Knockdown of eEF1A1 expression caused a nuclear enrichment of A34-SoxN-EGFP protein but had no effect on the subcellular localization of A12-SoxN-EGFP protein. The cell nuclei were stained with Hoechst 33342. Confocal images were acquired and processed using Olympus Fluoview FV1000 version 3.0.2.0 software. Red and green lines were drawn successively across individual cells in the confocal image, with matching plots on the right reporting fluorescence intensity. Scale bar, 10 μm. B, representative blots of the effect of eEF1A1 knockdown on the nuclear and cytoplasmic poly(A)-SoxN protein level. Histone (H3) was used as the nuclear marker. β-Tubulin served as the loading control. C, effect of Ef1a48D knockdown on poly(A)-SoxN toxicity. A34-SoxN expression caused a rough eye phenotype, whereas A12-SoxN and GAL4 driver-alone control did not. Knockdown of Ef1a48D rescued the eye phenotype induced by A34-SoxN. The images of external eyes were taken from 2 days post-eclosion flies. D, transcript levels of poly(A)-SoxN and Ef1a48D. The transcript levels of A12-SoxN and A34-SoxN and the knockdown efficiency of Ef1a48D in flies were detected using RT-PCR at 2 days post-eclosion. The flies were raised at 28 °C. The flies were of genotypes w; gmr-GAL4/+; +/+, w; gmr-GAL4/+; UAS-A12-SoxN/+, w; gmr-GAL4/+; UAS-A34-SoxN/+, w; gmr-GAL4/+; UAS-Ef1a48D-dsRNA/+, w; gmr-GAL4/+; UAS-A12-SoxN/UAS-Ef1a48D-dsRNA, and w; gmr-GAL4/+; UAS-A34-SoxN/UAS-Ef1a48D-dsRNA. Three independent experiments were performed.
Knockdown of Ef1a48D expression in Drosophila attenuated the toxicity induced by expanded poly(A) SoxN
To gain insights into the effect of poly(A) repeats in vivo, we established transgenic fly lines, UAS-A12-SoxN and UAS-A34-SoxN, which consist of the SoxN gene with a 12-alanine tract or a 34-alanine tract. Gmr-GAL4 driver was used to overexpress A12-SoxN or A34-SoxN in the fly eye, and the external eye phenotypes were examined. The overexpression of A34-SoxN resulted in abnormal eyes, characterized by a rough surface due to the disturbance of ommatidia architecture, whereas the overexpression of A12-SoxN did not display any effect on the external eye morphology, nor did the GAL4 driver control (Fig. 4C). Depletion of Ef1a48D, which is the Drosophila homolog of human eEF1A1, by gene knockdown rescued the external eye rough surface phenotype caused by A34-SoxN, whereas it did not alter the eye formation of flies expressing A12-SoxN (Fig. 4C). Upon Ef1a48D knockdown by dsRNA, decreased levels of Ef1a48D expression were detected by RT-PCR. However, this decrease did not alter the transcript levels of A12-SoxN and A34-SoxN (Fig. 4D). Therefore, our results support the notion that Ef1a48D specifically modulates the toxicity induced by expanded poly(A)-containing SoxN without reducing its transcript levels.
In conclusion, the subcellular localization of expanded poly(A)-containing SoxN is tightly correlated to degeneration in poly(A) diseases. Expanded poly(A) tracts exert their toxicity by inducing the mislocalization of the disease proteins from the nucleus to the cytoplasm. Ef1a48D modifies expanded poly(A)-containing SoxN toxicity by mediating its mislocalization.
Knockdown of eEF1A1 partially restores the transcription function of poly(A)-containing protein
Eight of nine poly(A) disease genes encode for transcription factors, and such nuclear localization is essential for transcription factors to function. To determine the effect of expanded poly(A) tracts on the transcriptional activity of transcription factors, we performed an unconventional luciferase reporter assay to examine the regulation of gene expression mediated by the GAL4 transcription activator fused with either an unexpanded or expanded poly(A) tract. The conventional luciferase assay measures the enzymatic activity of luciferase through detecting the luminescence of its substrate. Because the translational machinery may be affected by the knockdown of eEF1A1, we assayed for the firefly luciferase transcript instead in our experiments. The pFR-Luc plasmid contains a promoter with five tandem repeats of the GAL4 binding sites that control the expression of the firefly luciferase gene (29, 30). When constructs containing GAL4-poly(A) were co-transfected with the pFR-Luc reporter plasmid, the transcription of the luciferase gene would be induced. The expression level of luciferase thus reflects the transcriptional activity of GAL4-poly(A). At the same time, phRL-TK, which is Renilla co-reporter vector, was also co-transfected to serve as an internal control for normalization of differences in transfection efficiency (31). The result showed that GAL4-A37-FLAG caused a significant decrease in firefly luciferase expression level compared with the GAL4-A6-FLAG control (Fig. 5). We detected the localization of GAL4-poly(A)-FLAG proteins. The GAL4-A6-FLAG was found diffusely localized in both nucleus and cytoplasm, whereas GAL4-A37-FLAG was found to be mainly localized in the cytoplasmic component (supplemental Fig. S2). The subcellular localization result could explain why expanded GAL4-A37-FLAG protein demonstrated a reduced transcriptional activity. We have shown above that the knockdown of eEF1A1 expression partially corrected the cytoplasmic mislocalization of expanded poly(A) tract-containing protein (Fig. 3, A and B). Here, we speculated that eEF1A1 knockdown would restore the transcriptional activity of the expanded poly(A)-containing GAL4. As expected, the reduction of firefly luciferase transcription level in GAL4-A37-FLAG-transfected cells was partially restored upon eEF1A1 knockdown (Fig. 5).
Figure 5.
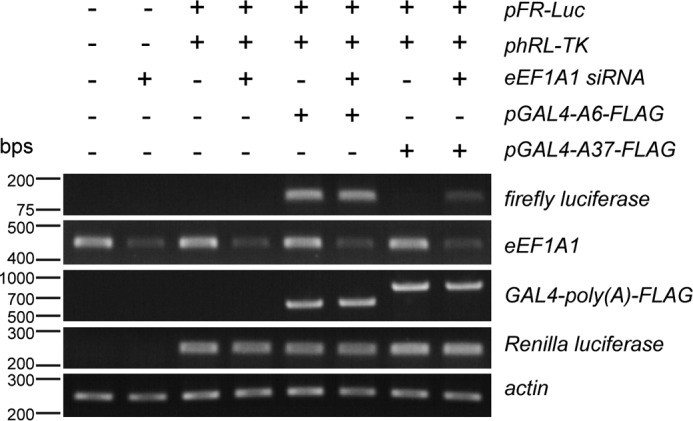
Knockdown of eEF1A1 expression partially restores transcription activity of expanded poly(A) tract-containing transcription factor. Shown is luciferase expression detection using RT-PCR. When the transcription factor, GAL4, was combined with a 37-alanine tract (GAL4-A37-FLAG), it caused a significant reduction of firefly luciferase expression level compared with that when it was combined with a 6-alanine tract control (GAL4-A6-FLAG). Knockdown of eEF1A1 expression restored the transcription activity of GAL4-A37-FLAG. The firefly luciferase expression level reflects transcription activity of GAL4-poly(A)-FLAG. Renilla luciferase served as the transfection control, and actin served as the loading control. Three independent experiments were performed.
Expanded poly(A) tract possesses nuclear export activity mediated by nuclear eEF1A1
We hypothesized that the expanded poly(A) tract may serve as a novel nuclear export signal that causes the cytoplasmic localization of poly(A) disease proteins. To determine whether the Rev(1.4)-A37-EGFP protein detected in the cytoplasm was exported out from the nuclei, we performed the fluorescence loss in photobleaching (FLIP) nuclear export assay in living cells (18). In brief, a small region of interest (ROI) of the cytoplasmic compartment of the target cell was repeatedly photobleached using a high-power 488-nm laser and followed by an immediate postbleach capture using a low-power 488-nm laser. The Rev(1.4) possesses only nuclear import activities but lacks nuclear export activities. Thus, if the expanded poly(A) tract mediates nuclear export activities, then the nuclear export of the reporter protein would result in the dilution of the remaining fluorescence in the nuclear compartment. The nuclear export activity of the target protein was determined by comparing the relative fluorescence loss of protein in the nuclear compartment over time. After photobleaching of the cytoplasmic compartment, a statistically significant decrease in the relative nuclear fluorescence intensity of the Rev(1.4)-A37-EGFP protein was observed when compared with the Rev(1.4)-A6-EGFP control (Fig. 6, A and B). This result demonstrates nuclear export activity of Rev(1.4)-A37-EGFP protein from the nuclear compartment to the cytoplasmic compartment.
Figure 6.

Expanded poly(A) tract possesses nuclear export activity mediated by nuclear eEF1A1. A, FLIP nuclear export assay. ER-Tracker Red was used as a cytoplasmic marker (gray channel). White square boxes, selected cytoplasmic regions for photobleaching. There was no obvious drop in intensity of Rev(1.4)-A6-EGFP protein (A6) in the nucleus postbleach. In contrast, Rev(1.4)-A37-EGFP protein (A37) showed a significant drop in both nuclear and cytoplasmic intensities. The intensity bars show the relative fluorescence intensity with white being the maximum and black being the minimum. Scale bar, 10 μm. B, statistical analysis of A was performed using two-way analysis of variance. A statistically significant drop in nuclear fluorescence intensity of A37 protein was observed postbleach when compared with the A6 control. The prebleach fluorescence intensity of nuclear compartment was normalized as 1. At least 14 transfected cells from three independent transfection experiments were analyzed. Error bars, S.E. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. C, PA-GFP assay. HEK293 cells transfected with pRev(1.4)-poly(A)-PA-GFP constructs were stained with ER-Tracker Red. The ROIs for photoactivation are indicated by white circles. In contrast to the control protein (A6), photoactivated nuclearly localized expanded poly(A) protein (A37) was exported to the cytoplasm starting at 1 min postactivation. At least 36 transfected cells from three independent transfection experiments were analyzed. Scale bar, 10 μm. D, Western blotting of eEF1A1 using cytoplasmic or nuclear fractions of HEK293 and SK-N-MC cells. The majority of eEF1A1 was cytoplasmic, but eEF1A1 was detected in the nucleus as well. Histone (H3) was used as the nuclear marker. β-Tubulin served as the loading control. E, subcellular localization of Rev(1.4)-poly(A)-EGFP protein in HEK293 cells after eEF1A1-NES and eEF1A1-NLS overexpression. Both constructs resulted in increased cytoplasmic localization of Rev(1.4)-A37-EGFP protein but had no effect on the subcellular localization of Rev(1.4)-A6-EGFP protein. The cell nuclei were stained with Hoechst 33432. Scale bar, 10 μm. F, statistical analysis of E. Three independent experiments were performed. At least 100 transfected cells were measured per experiment. Error bars, S.E. **, p < 0.01.
To further obtain a direct visualization of a nuclear export event, we next performed the photoactivatable GFP (PA-GFP) nuclear export assay. PA-GFP (32) is a photoactivatable variant form of EGFP (33) that has a single amino acid substitution at the 203 position from threonine to histidine (T203H) in EGFP. PA-GFP proteins only emit green fluorescence after photoactivation at 405 nm. The photoactivation property of PA-GFP allows the capturing of nucleocytoplasmic trafficking of proteins in living cells (34). In the PA-GFP nuclear export assay, the nucleolus was selected as the ROI for photoactivation using the 405-nm laser. Because the newly synthesized protein in the cytoplasm was not photoactivated, the cytoplasmic fluorescence signal detected would solely represent the photoactivated Rev(1.4)-poly(A)-PA-GFP that was being exported out from the nuclear compartment. Upon photoactivation, the photoactivated Rev(1.4)-A6-PA-GFP protein remained in the nuclear compartment (Fig. 6C). In contrast, the nuclear photoactivated Rev(1.4)-A37-PA-GFP protein was found in the cytoplasm starting from 1 min post-photoactivation (Fig. 6C). This strongly suggests that the photoactivated Rev(1.4)-A37-PA-GFP protein was exported out from the nuclear compartment to the cytoplasmic compartment. These results clearly demonstrate that the expanded poly(A) tract possesses NES activity.
If eEF1A1 is a mediator of expanded poly(A) tract-induced nuclear export, it should be present in the nucleus to interact with the poly(A) tract. However, it has been previously reported in a number of studies that eEF1A is exclusively localized to the cytoplasm (35, 36); thus, there is a need to verify the presence of eEF1A1 in the nucleus. We performed Western blotting analysis on cytoplasmic and nuclear fractions extracted from HEK293 and SK-N-MC cells to determine the presence of nuclear eEF1A1. Despite the majority of eEF1A1 being in the cytoplasm, there was a detectable amount of eEF1A1 in the nucleus (Fig. 6D). This result is consistent with the findings of a recent study, which demonstrated that eEF1A is capable of shuttling between the nucleus and the cytoplasm (20).
Because eEF1A1 has been detected in both the nucleus and the cytoplasm, we would like to determine which compartment's eEF1A1 mediates the cytoplasmic localization of poly(A)-containing protein. Hence, we co-expressed eEF1A1-NLS or eEF1A1-NES in HEK293 cells with Rev(1.4)-A6-EGFP or Rev(1.4)-A37-EGFP. eEF1A1-NLS was found to localize to the nucleus, whereas eEF1A1-NES localized to the cytoplasm (Fig. 6E). To our surprise, both eEF1A1-NLS and eEF1A1-NES resulted in increased cytoplasmic localization of Rev(1.4)-A37-EGFP (Fig. 6, E and F). Thus, our results suggest that nuclear eEF1A1 probably mediates nuclear export of poly(A)-containing proteins, whereas cytoplasmic eEF1A1 may also mediate cytoplasmic retention of poly(A)-containing proteins.
In summary, we demonstrated that the expanded poly(A) tract can serve as a novel NES, leading to nuclear export of poly(A)-containing nuclear proteins. Nuclear eEF1A1 is probably involved in regulating this nuclear export process, whereas cytoplasmic eEF1A1 may also participate in the cytoplasmic retention of poly(A)-containing proteins. These results suggest that eEF1A1 plays dual roles in both the nucleus and the cytoplasm to mediate the cytoplasmic mislocalization of proteins with an expanded poly(A) tract. Because the cytoplasmic mislocalization of poly(A)-containing proteins is a major cause of poly(A) diseases, eEF1A1 may be a potent therapeutic target in treating these diseases.
Discussion
The main finding of this study is the demonstration of the continuous expanded poly(A) tract as a novel NES and the regulation of poly(A) protein localization by eEF1A1, which provides new insights into the underlying mechanisms of poly(A) diseases. Our study identifies eEF1A1 as an interacting partner of the expanded poly(A) domain, and it serves as a modulator of the poly(A)-induced mislocalization of transcription factors. The depletion of eEF1A1 mitigates the nuclear loss of poly(A)-containing transcription factors and partially restores their transcriptional activities.
Previous studies have noted that the poly(A) disease symptoms are linked to loss of function of the poly(A) disease proteins (15, 37–39). These wild type transcription factors localize in the nucleus, whereas the expanded poly(A) mutant proteins are mislocalized in the cytoplasm (7–11). Furthermore, the mislocalization of poly(A) disease proteins leads to loss of function of these poly(A) disease proteins and reduces its transcriptional activity (7, 9, 40, 41). In addition, the expanded poly(A) tract is sufficient to confer cytoplasmic localization to a YFP reporter protein (12). Therefore, it is believed that the expanded alanine tract per se contributes to the cytoplasmic localization of poly(A) disease proteins. Altogether, current studies suggest that the pathogenesis of poly(A) diseases is closely related to the cytoplasmic mislocalization of poly(A) disease proteins. Therefore, the aim of our study is to uncover the mechanisms that underlie this mislocalization.
In this study, we performed a GST-pulldown assay and mass spectrometry analysis and identified eEF1A1 as an expanded poly(A) protein-interacting partner that binds specifically to the expanded poly(A) sequences (Fig. 2, A and B). At the structural level, human eEF1A1 is divided into three domains, referred to as domains I, II, and III (42), and binds GTP, tRNA, and actin, respectively (23). eEF1A1 binds to exportin 5 in a complex with aa-tRNA (35), which involves domain II (23). But unlike the eEF1A1/exportin 5 interaction, expanded poly(A) tract interacts only with domain III of eEF1A1 (Fig. 2D), indicating that tRNA is not required in the interaction between eEF1A1 and expanded poly(A) domain. Our data further show that amino acids 429–449 of human eEF1A1 are essential for interaction with the expanded poly(A) domain (Fig. 2F). Our results also demonstrate that the continuity of the expanded poly(A) tract is essential to the poly(A)-containing protein's interaction with eEF1A1 (Fig. 2G), and this interaction is a major determinant of the poly(A)-containing protein's mislocalization in the cytoplasm (Fig. 3, D and E).
The canonical role of eEF1A is protein synthesis in the cytoplasm (43), but many studies have reported its involvement in nuclear export functions as well. Previous mammalian studies have demonstrated eEF1A as a novel component of the nuclear protein export machinery, participating in the nuclear export of selected RNAs and proteins, such as tRNAs (17), SNAG-containing proteins (20), the VHL tumor suppressor, and PABP1 (18). However, it has been postulated that the nuclear export function of eEF1A may be executed in the cytoplasm instead of the nucleus (17, 18, 44). This is because eEF1A has been found to be exclusively localized to the cytoplasm in immunocytochemistry experiments (35, 36). Interestingly, a recent study demonstrated that eEF1A is localized in the nucleus when it is in a complex with Snail1 but localized in the cytoplasm when it is alone (20). Thus, eEF1A can be shuttled between the nucleus and the cytoplasm, depending on what proteins it is interacting with. Our Western blotting results show that despite the majority of eEF1A1 being cytoplasmic, there is still a detectable amount of nuclear eEF1A1 in HEK293 and SK-N-MC cells (Fig. 6D), which is likely to mediate nuclear export of poly(A)-containing proteins. This is supported by the fact that the overexpression of nuclear eEF1A1 resulted in increased cytoplasmic localization of expanded poly(A)-containing Rev(1.4)-EGFP protein (Fig. 6, E and F). However, overexpression of cytoplasmic eEF1A1 also had similar effects (Fig. 6, E and F); thus cannot exclude the possibility that cytoplasmic eEF1A1 may contribute to the cytoplasmic retention of expanded poly(A)-containing proteins as well.
Apart from its canonical role in translation, eEF1A is associated with protein degradation as well (23). Thus, we sought to determine whether eEF1A1 could be involved in the degradation of expanded poly(A)-containing proteins. However, we found that the knockdown of eEF1A1 expression did not affect the transcript and protein levels of poly(A)-containing proteins (supplemental Fig. S3, A and B), indicating that its effects on poly(A)-containing proteins are unlikely to be associated with protein degradation.
Our findings in this study demonstrate that the expanded poly(A) tract can function as a novel NES. We used the Rev(1.4)-EGFP reporter protein to investigate the nuclear export property of expanded poly(A) domain. Our results show that the insertion of a 37-alanine tract causes cytoplasmic localization of the Rev(1.4)-EGFP protein (Fig. 1, A and B). This cytoplasmic localization is in line with previous poly(A) disease studies (7, 9–11, 24, 40, 45). For instance, it has been reported that the expanded poly(A) disease proteins, including HOXD13, HOXA13, RUNX2, and SOX3, are cytoplasmically localized when overexpressed in COS-1 cells (10). In addition, it has also been reported that YFP-poly(A) protein of the 29-alanine tract is predominantly localized in the cytoplasm (12). Our FLIP nuclear export assay then demonstrates that the expanded poly(A) tract mediates nuclear export of Rev(1.4)-A37-EGFP protein (Fig. 6, A and B). The PA-GFP nuclear export assay allows a continuous monitoring of nucleocytoplasmic trafficking and illustrates the nuclear export event of expanded poly(A) protein in living cells (Fig. 6C). The data from these different nuclear export assays all support the notion that the expanded poly(A) tract possesses nuclear export activity. Therefore, although we cannot exclude the possibility of nuclear import impairment and/or cytoplasmic retention, we have at least demonstrated its function as a novel NES, mediating nuclear export.
The classical NES is a hydrophobic leucine-rich sequence, whose nuclear export activity is mediated by chromosome region maintenance 1 (CRM1), also known as exportin 1 (XPO1) (46, 47). The knockdown of eEF1A1 does not alter classical NES-mediated nuclear export (Fig. 3, A–C), suggesting that eEF1A1 and CRM1 operate independently and that eEF1A1 is not a general regulator for protein nuclear export. From our results, although the eEF1A1 protein level was reduced about 90% after eEF1A1 siRNA treatment (Fig. 3C), only about 40% of nuclear export of Rev(1.4)-A37-EGFP was suppressed. There are at least two possible interpretations to explain this result. First, eEF1A1 may be only a factor involved in the nuclear export but not the actual receptor. Genome-wide siRNA screening may further be performed to identify components of the expanded poly(A) tract nuclear export pathway(s). Second, multiple receptors may contribute to the nuclear export of the expanded poly(A) tract. It is not uncommon for more than one receptor to regulate the nuclear export of a particular type of protein. For instance, the nuclear export of peroxisome proliferator-activated receptor is regulated by calreticulin and XPO1 (48). One of the major isoforms of thyroid hormone receptor, TRα1, harbors multiple NESs and exports from the nucleus via an XPO1-dependent pathway in which calreticulin plays a role as a chaperone (49). Both XPO1-dependent and XPO1-independent pathways contribute to the nuclear export of constitutive androstane receptor, although it harbors only an XPO1-independent NES (50–52). Although we did not perform a genome-wide siRNA screen, we carried out a screen using a candidate gene siRNA knockdown approach to identify other molecules that may regulate the nuclear export of poly(A)-containing proteins (supplemental Fig. S4, A and B). The candidates in our screen include multiple exportins, such as XPO1, XPO4, XPO5, XPO6, XPO7, and XPOT. It has been proposed in previous studies that XPO5 may mediate the export of eEF1A protein from the nucleus (36). However, our candidate gene siRNA knockdown data show that the knockdown of XPO5 and related genes does not alter the subcellular localization of the Rev(1.4)-A37-EGFP protein (supplemental Fig. S4, A and B). This result is consistent with a previous study indicating that eEF1A mediates nuclear export of TD-NEM-containing proteins, such as VHL tumor suppressor protein, via an XPO5-independent pathway (18).
Our results show that overexpression of eEF1A1 enhances the cytoplasmic localization of expanded poly(A) protein, whereas overexpression of eEF1A1Δ429–449 has no such effect (Fig. 3, D–F). This effect on poly(A)-containing protein localization is not due to the enhancement of general protein translation, because we do not observe changes in the levels of protein translation when eEF1A1 is overexpressed (supplemental Fig. S5), which is consistent with the findings of a previous study (53). In fact, even the overexpression of eEF1A1Δ429–449 has no detectable effect on general protein translation (supplemental Fig. S5).
Whereas eEF1A1 is responsible for the enzymatic delivery of aa-tRNA to the A site of the ribosome, eEF2 is responsible for translocating the peptidyl-tRNA out of the A site to P site (23). Our results show that the knockdown of eEF2 does not affect the subcellular localization of Rev(1.4)-A37-EGFP protein (Fig. 3, G–I), which indicates that the knockdown of eEF1A1 expression suppresses the cytoplasmic mislocalization of expanded poly(A) tract specifically and that the cytoplasmic localization of expanded poly(A) protein was not due to the blockage of general protein translation.
Consistent with these findings, expanded poly(A) protein expression does not perturb the cellular function of eEF1A1 in protein translation (supplemental Fig. S6); nor does it affect the nuclear export function of eEF1A1, because VHL is still capable of exiting the nucleus when Rev(1.4)-A37-EGFP is being expressed (supplemental Fig. S6). Taken together, the canonical translation functions and nuclear export functions of eEF1A1 are unaffected by its interaction with expanded poly(A) proteins.
Protein misfolding and aggregation are also common characteristics of poly(A) expansion proteins (54). Expression of EGFP fusion with 19 alanine residues in COS-7 cells forms aggregates and induces cell death (54). The A37-EGFP protein forms cytoplasmic aggregates in the eye imaginal disc of Drosophila third instar larvae and causes smaller and rough eye phenotype (55). These studies suggest that aggregate formation in cytoplasm also contributes to poly(A) pathogenesis (10, 54–58). In line with the aforementioned findings, we find that overexpression of A34-SoxN-EGFP results in the formation of aggregates in the cytoplasm in SK-N-MC cells (Fig. 4A). Because protein misfolding is a common feature among poly(A) expansion proteins, we wondered whether it underlies the effect of how eEF1A1 regulates the subcellular localization of poly(A) expansion proteins. However, our results show that the knockdown of eEF1A1 does not induce changes in the transcript level of binding immunoglobulin protein (BiP), a chaperone protein that regulates protein folding (supplemental Fig. S3A). This indicates that the regulation of subcellular localization of poly(A) expansion proteins by eEF1A1 is not related to protein misfolding.
To correlate the nuclear export activity of the expanded poly(A) domain to poly(A) toxicity in an in vivo model, we assessed the effect of Ef1a48D on eye degeneration of poly(A)-SoxN transgenic flies. Our data show expanded poly(A) induced toxicity in the fly eye, which is consistent with the previous reports that A37 is toxic in cell culture (59), and its expression in the fly eye results in eye degeneration (55). Our results demonstrate that the knockdown of Ef1a48D expression attenuates the expanded poly(A)-SoxN-induced toxicity and eye degeneration in vivo (Fig. 4C). This suggests that Ef1a48D is a genetic modifier of expanded poly(A)-SoxN-induced toxicity and degeneration, possibly by limiting the cytoplasmic localization of poly(A)-SoxN.
Nuclear localization is essential for transcription factors to function. In our study, we combined a transcription factor, GAL4, with an unexpanded or expanded poly(A) tract to investigate the transcriptional activities of poly(A)-containing protein. Our results suggest that the transcription activity of GAL4-A37-FLAG decreases significantly compared with GAL4-A6-FLAG control (Fig. 5). Intriguingly, siRNA-silencing eEF1A1 rescues the transcriptional activity of GAL4-A37-FLAG (Fig. 5), which indicates that the knockdown of eEF1A1 partially restores the cellular function of the poly(A)-containing transcription factors. It has recently been reported that the expanded poly(A) PHOX2B disease protein adopts a cytoplasmic localization, and it impairs the wild-type PHOX2B protein function by heterodimerizing with the wild-type protein, which inhibits the nuclear import of the wild-type protein (60). These reports are consistent with our findings, and both studies provide evidence that the nuclear localization is essential for poly(A) proteins to function as transcription factors.
In our working model of poly(A) disease pathogenesis, the expanded poly(A) domain serves as a nuclear export signal and causes cytoplasmic localization of poly(A) disease-associated transcription factors. This in turn reduces their cellular transcriptional activities, resulting in gene dysregulation and loss-of-function toxicity (Fig. 7). Simultaneously, cytoplasmic expanded poly(A)-containing proteins form aggregates and may exert the gain-of-function toxic effect. eEF1A1 interacts with expanded poly(A) domain and is a mediator for the cytoplasmic translocation of expanded poly(A)-containing proteins. Depletion of eEF1A1 suppresses the cytoplasmic translocation of expanded poly(A) tract-containing proteins, which partially restores the cellular transcriptional activities of expanded poly(A) transcription factors in the nucleus. Therefore, the knockdown of eEF1A1 expression can partially rescue toxicities of poly(A) caused by cytoplasmic mislocalization of the disease proteins (Fig. 7).
Figure 7.

Working model of eEF1A1 regulation of localization of expanded poly(A) tract-containing proteins in the cytoplasm. Expanded poly(A) tract is a NES, signaling the export of expanded poly(A) proteins from the nucleus to the cytoplasm with the assistance of nuclear eEF1A1. This results in gene dysregulation inside the nucleus. In the cytoplasm, eEF1A1 may be involved in the cytoplasmic retention of expanded poly(A) proteins, which aggregate and exert toxic effects. When eEF1A1 expression is knocked down, expanded poly(A) proteins are retained in the nucleus to regulate gene transcription. In the cytoplasm, the toxicity induced by cytoplasmic expanded poly(A) proteins is reduced. In conclusion, our study connected the expanded poly(A) protein nuclear export to its neurotoxicity and identified eEF1A1 as a regulator for expanded poly(A) nuclear export and toxicity in poly(A) diseases.
In conclusion, expanded poly(A) tracts possess NES activity and contribute to the cytoplasmic localization of poly(A)-containing proteins. Our study uncovers the role of eEF1A1 in modulating expanded poly(A) protein subcellular localization and identifies it as a novel therapeutic target for poly(A) diseases.
Experimental procedures
Molecular cloning
For the generation of pRev(1.4)-A6-EGFP constructs, an A6 double-stranded oligonucleotide was generated using oligo_A6_F (5′-GAT CCA GCA GCC GCC GCT GCC GCT CCA-3′) and oligo_A6_R (5′-CCG GTG GAG CGG CAG CGG CGG CTG CTG-3′) and then ligated with pRev(1.4)-EGFP (21) using BamHI and AgeI enzymes. For the generation of pRev(1.4)-A22-EGFP, pRev(1.4)-A25-EGFP, pRev(1.4)-A28-EGFP, pRev(1.4)-A31-EGFP, and pRev(1.4)-A34-EGFP constructs, the double-stranded oligonucleotides were generated using oligo_A22_F (5′-GAT CCA GCA GCA GCC GCC GCT GCC GCT GCT GCC GCC GCT GCA GCA GCC GCC GCT GCT GCA GCA GCT GCC GCT CCA-3′) and oligo_A22_R (5′-CCG GTG GAG CGG CAG CTG CTG CAG CAG CGG CGG CTG CTG CAG CGG CGG CAG CAG CGG CAG CGG CGG CTG CTG CTG-3′), oligo_A25_F (5′-GAT CCA GCA GCA GCC GCC GCT GCC GCT GCT GCC GCC GCT GCA GCA GCC GCC GCT GCT GCA GCA GCT GCC GCT GCT GCC GCA CCA-3′) and oligo_A25_R (5′-CCG GTG GTG CGG CAG CAG CGG CAG CTG CTG CAG CAG CGG CGG CTG CTG CAG CGG CGG CAG CAG CGG CAG CGG CGG CTG CTG CTG-3′), oligo_A28_F (5′-GAT CCA GCA GCA GCC GCC GCT GCC GCT GCT GCC GCC GCT GCA GCA GCC GCC GCT GCT GCA GCA GCT GCC GCT GCT GCC GCC GCA GCA GCT CCA-3′) and oligo_A28_R (5′-CCG GTG GAG CTG CTG CGG CGG CAG CAG CGG CAG CTG CTG CAG CAG CGG CGG CTG CTG CAG CGG CGG CAG CAG CGG CAG CGG CGG CTG CTG CTG-3′), oligo_A31_F (5′-GAT CCA GCA GCA GCC GCC GCT GCC GCT GCT GCC GCC GCT GCA GCA GCC GCC GCT GCT GCA GCA GCT GCC GCT GCT GCC GCC GCA GCA GCC GCA GCA GCT CCA-3′) and oligo_A31_R (5′-CCG GTG GAG CTG CTG CGG CTG CTG CGG CGG CAG CAG CGG CAG CTG CTG CAG CAG CGG CGG CTG CTG CAG CGG CGG CAG CAG CGG CAG CGG CGG CTG CTG CTG-3′), and oligo_A34_F (5′-GAT CCA GCA GCA GCC GCC GCT GCC GCT GCT GCC GCC GCT GCA GCA GCC GCC GCT GCT GCA GCA GCT GCC GCT GCT GCC GCC GCA GCA GCC GCA GCC GCT GCA GCA GCT CCA-3′) and oligo_A34_R (5′-CCG GTG GAG CTG CTG CAG CGG CTG CGG CTG CTG CGG CGG CAG CAG CGG CAG CTG CTG CAG CAG CGG CGG CTG CTG CAG CGG CGG CAG CAG CGG CAG CGG CGG CTG CTG CTG-3′) and then ligated with pRev(1.4)-EGFP using BamHI and AgeI enzymes. The A37 DNA fragment was amplified by PCR using pEGFP-C1-A37 (61) as template and then ligated with pRev(1.4)-EGFP using BamHI and AgeI enzymes. For the pRev(1.4)-poly(A)-PAGFP, Rev(1.4)-poly(A) (A6 and A37) fragments were subcloned from pRev(1.4)-poly(A)-EGFP to pPAGFP-N1 vector (32) using EcoRI and AgeI enzymes. For the generation of pGEX-4T3-poly(A) constructs, an A7 double-stranded oligonucleotide was generated using oligo_A7_F (5′-GAT CCG CAG CAG CCG CCG CTG CCG CTC AAG CTT CGA ATT CTG CAG-3′) and oligo_A7_R (5′-TCG ACT GCA GAA TTC GAA GCT TGA GCG GCA GCG GCG GCT GCT GCG-3′) and then ligated with pGEX-4T3 vector using BamHI and SalI enzymes. The pEGFP-C1-A37 plasmid was digested with BglII and SalI enzymes, and the A37 insert was subcloned into pGEX-4T3 vector using BamHI and SalI enzymes. The GAL4 DNA fragment was amplified by PCR using single fly genomic DNA prepared from a gmr-GAL4 fly (Bloomington Drosophila Stock Center) as template. To generated the pGAL4-poly(A)-FLAG constructs, the GAL4 coding region was used to replace the Rev(1.4) coding region of pRev(1.4)-poly(A)-EGFP plasmids using EcoRI and BamHI enzymes. Further, the EGFP sequence was replaced by a synthetic FLAG tag sequence (oligo_FLAG_F, 5′-CCG GTC GCC ACC ATG GAC TAC AAG GAT GAC GAT GAC AAA TAG GC-3′; oligo_FLAG_R, 5′-GGC CGC CTA TTT GTC ATC GTC ATC CTT GTA GTC CAT GGT GGC GA-3′) using AgeI and NotI enzymes. The Myc-eEF1A1 fragment was amplified by PCR using pCMV-XL5-eEF1A1 (OriGene) as template and then inserted into pcDNA3.1(+) vector using EcoRI and XhoI enzymes. The Myc-eEF1A1 truncated fragments were amplified by PCR using pcDNA3.1-myc-eEF1A1 as template and then inserted into pcDNA3.1(+) vector using EcoRI and XhoI enzymes. The pFR-Luc plasmid was purchased from Stratagene, and phRL-TK was purchased from Promega. The pRev(1.4)-EGFP and pRev(1.4)-NES-EGFP constructs were gifts from Professor Beric Henderson (Westmead Institute for Cancer Research, Westmead Hospital, University of Sydney, Westmead, Australia). pEGFP-C1-A37 was a gift from Professor David Rubinsztein (Department of Medical Genetics, University of Cambridge, Cambridge, UK)). pPAGFP-N1 vector was a gift from Professor Jennifer Lippincott-Schwartz (Cell Biology and Metabolism Branch, National Institutes of Health, Bethesda, MD).
Cell culture and transfection
HEK293 and SK-N-MC cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Inc.) supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin-streptomycin (Life Technologies) at 37 °C in a 5% CO2 incubator. For transfection experiments, cells were seeded at a density of 0.8 × 105 cells/24-well plate. Twenty-four hours later, 1 μg of plasmid DNA was used to transfect cells using Lipofectamine 2000 reagent (Invitrogen).
siRNA
For siRNA-mediated gene silencing, Dharmacon ON-TARGETplus® SMARTpool or siGENOME siRNAs were used to transfect HEK293 cells with Lipofectamine® RNAiMAX reagent (Invitrogen). In brief, HEK293 cells were transfected with 40 pmol of siRNA for CALR (L-008197-00-0005), CSE1L (L-004413-00-0005), IPPO13 (L-020212-01-0005), XPO1 (L-003030-00-0005), XPO4 (L-027196-01-0005), XPO5 (L-014000-00-0005), XPO6 (L-022515-02-0005), and XPOT (L-010356-00-0005) and 6 pmol of siRNA for eEF1A1 (M-017199-02-0005), eEF2 (M-007245-02-0005), and control (D-001210-01-20) siRNA for 72 h before assays.
Reverse transcription-polymerase chain reaction
Total RNA from cells and flies was extracted using TRIzol® reagent (Invitrogen). RT-PCR was performed using the ImProm-IITM reverse transcription system (Promega) according to the manufacturer's instructions. Primers used were as follows: actin (forward, 5′-ATG TGC AAG GCC GGT TTC GC-3′; reverse, 5′-CGA CAC GCA GCT CAT TGT AC-3′), CALR (forward, 5′-GTT TCG AGC CTT TCA GCA AG-3′; reverse, 5′-TCC TCA GGC TTG GAG TCT GT-3′), CSE1L (forward, 5′-TCT GTA TCC CCA CGA GAT CC-3′; reverse, 5′-AAG TCA GGC CAT TTC TGT GG-3′), IPO13 (forward, 5′-ATG CCC AGA GGT ACG TGA AC-3′; reverse, 5′-TGA GCG TGT CTG AGA TGT CC-3′), XPO1 (forward, 5′-TGG GCA ATA GGC TCC ATT AG-3′; reverse, 5′-CAC CAA TCA TGT ACC CCA CA-3′), XPO4 (forward, 5′-ACC AAA GGG CAC TGA AAC AC-3′; reverse, 5′-CAG GCA AAC ATG GAC ACA AC-3′), XPO5 (forward, 5′-CCA CCT CAC TTT CCA CCA CT-3′; reverse, 5′-GCC CAA AGC AAA GTT GAG AG-3′), XPO6 (forward, 5′-GCC CAT GTT CTA CCA CGA CT-3′; reverse, 5′-GTA AGG AGG GAT GGG GTG AT-3′), XPO7 (forward, 5′-GAG GGG TTG TGT ATG CGA GT-3′; reverse, 5′-TGG TCT CTG CCT TCA CAC AG-3′), XPOT (forward, 5′-TGC GGA GGA GTT ACT TTG CT-3′; reverse, 5′-GAA GCG CTT GAC AAA ACT CC-3′), firefly luciferase (forward, 5′-CCA GGG ATT TCA GTC GAT GT-3′; reverse, 5′-AAT CTG ACG CAG GCA GTT CT-3′), Renilla (forward, 5′-CGA GCA CCA AGA CAA GAT CA-3′; reverse, 5′-GTA GGC AGC GAA CTC CTC AG-3′), GAL4-poly(A)-FLAG (forward, 5′-CCT TCA CCT GTG CCA TTG-3′; reverse, 5′-GGC CGC CTA TTT GTC ATC GTC ATC CTT GTA GTC CAT GGT GGC GA-3′), eEF1A1 (forward, 5′-CCG GAA TTC ATG GAA CAA AAA CTC ATC TCA GAA GAG GAT CTG CCA CCA ATG GAA GCA GC-3′; reverse, 5′-CCG CTC GAG TCA GCC AGC TCC AGC AGC C-3′), poly(A)-SoxN (forward, 5′-CCG GTA TGG AGC AGA AAC TCA TCT CTG AAG AGG ATC TGT AGG C-3′; reverse, 5′-CAT ATC CTG GCG CGC CTG TTG-3′), Ef1a48D (forward, 5′-CAT GTT GGA ACC CTC TAC CAA C-3′; reverse, 5′-GAA GAC CAC AAC GGT ACC GGG-3′), and BiP (forward, 5′-TAG CGT ATG GTG CTG CTG TC-3′; reverse, 5′-TTT GTC AGG GGT CTT TCA CC-3′).
Immunofluorescent staining
The immunofluorescent staining was performed as described previously (62). Primary antibodies used were anti-VHL (1:1,000; 556347; BD Biosciences) or anti-FLAG (1:100; M2; Sigma). Secondary antibodies used were goat anti-mouse with Cy3 conjugate (1:400; 81-6525; Zymed Laboratories Inc.) or goat anti-mouse with FITC conjugate (1:400; 81-6511; Zymed Laboratories Inc.). The cell images were captured using an Olympus IX-81 FV1000 confocal microscope.
FLIP nuclear export assay
HEK293 cells were seeded onto a 35-mm μ-Dish (ibidi) at a density of 1.8 × 105. Twenty-four hours later, the cells were transfected with 1 μg of pRev(1.4)-poly(A)-EGFP constructs for 12 h. The cells were subsequently stained with ER-TrackerTM Red (1:1,000; Life Technologies) at 37 °C for 15 min. Then cells were maintained in DMEM without phenol red (Life Technologies). In the FLIP experiment, the subcellular localization of EGFP fusion proteins was monitored using 10% power of a 488-nm laser at 4 μs/pixel. All of the prebleach, photobleaching, and postbleach steps were controlled automatically by the time controller module of Olympus Fluoview version 2.0c software. In the prebleach step, a single image was taken with 10% power of the 488-nm laser at 4 μs/pixel to record the initial subcellular localization of the EGFP fusion proteins. Then square ROIs of the cytoplasmic region of transfected cells were repeatedly bleached five times with 100% power of the 488-nm laser at 200 μs/pixel, and this was followed by an immediate capture of a postbleach image with 10% power of the 488-nm laser at 4 μs/pixel. Quantification of fluorescence intensity was performed using ImageJ version 1.45s software (National Institutes of Health, Bethesda, MD).
Photoactivatable GFP nuclear export assay
HEK293 cells were prepared as described under “FLIP nuclear export assay.” Cells were transfected with pRev(1.4)-poly(A)-PA-GFP constructs for 48 h before the PA-GFP assay. The preactivation, photoactivation, and postactivation steps were controlled automatically by the time controller module of Olympus Fluoview version 2.0c software. At preactivation, a single image was taken with 10% 488-nm laser at 4 μs/pixel to show that no fluorescence was observed before photoactivation. Then circular ROIs at the nucleolus of transfected cells were selected, and 20% power of the 405-nm laser at 100 μs/pixel was used to photoactivate the selected ROIs. The subcellular localization of activated Rev(1.4)-poly(A)-PA-GFP proteins were monitored for 15 min at 1-min intervals using 10% power of the 488-nm laser at 4 μs/pixel.
Western blotting
Cells were lysed with 2× SDS sample buffer containing 100 mm Tris-HCl (pH 6.8), 2% SDS, 20% glycerol (w/v), 5% β-mercaptoethanol, and 0.02% bromphenol blue and boiled for 10 min. Cell lysates were subjected to SDS-PAGE and immunoblotting. Primary antibodies used were anti-eEF1A1 (1:1,000; 2551; Cell Signaling), anti-eEF2 (1:1,000; 2332; Cell Signaling), anti-Myc (1:2,000; 2276; Cell Signaling), anti-β-tubulin (1:10,000; E7; Developmental Studies Hybridoma Bank), anti-GFP (1:5,000; 632380; Clontech), or anti-histone (1:10,000; ab47915; Abcam). Secondary antibodies used were peroxidase-conjugated AffiniPure goat anti-rabbit IgG (1:10,000; 111-035-045; Jackson ImmunoResearch) or peroxidase-conjugated AffiniPure goat anti-mouse IgG (1:10,000; 111-035-062; Jackson ImmunoResearch). Finally, the membranes were exposed to X-ray film (Fujifilm) using Luminata crescendo Western HRP substrate (Millipore).
GST-pulldown assay
Expression of GST-poly(A) fusion proteins in Escherichia coli strain BL21 (DE3) was induced by 0.1 mm isopropyl β-d-1-thiogalactopyranoside (Shanghai Biocolor Bioscience and Technology). A bacterial pellet containing recombinant protein was collected by centrifugation at 4,000 × g at 4 °C for 10 min. Then the pellet was resuspended in 10 ml of GST binding buffer (pH 7.4) containing 50 mm Tris-HCl, 1 mm EDTA, 50 mm NaCl, and 10% glycerol and sonicated using a Sonifier 450 sonicator (Branson). The protein sample was centrifuged at 16,000 × g at 4 °C for 15 min. A volume of 1 ml of supernatant was incubated with 50 μl of glutathione-Sepharose 4 Fast Flow (GE Healthcare) at 4 °C for 2 h with rotation. The glutathione-Sepharose 4 Fast Flow beads coupled with GST-poly(A) proteins were washed three times with GST binding buffer. Next, HEK293 total lysate was incubated with glutathione-Sepharose 4 Fast Flow beads coupled with GST-poly(A) proteins at 4 °C overnight with rotation. The glutathione-Sepharose 4 Fast Flow beads coupled with GST fusion proteins and interacting proteins were washed five times with 1 ml of GST washing buffer (pH 7.4) containing 50 mm Tris, 1 mm EDTA, 500 mm NaCl, 10% glycerol, and 1% Nonidet P-40. Finally, 60 μl of 2× SDS sample buffer was added, and the interacting proteins were eluted by boiling at 99 °C for 10 min. The protein samples were analyzed by SDS-PAGE and followed by Coomassie Blue staining to detect GST-poly(A) expression or by immunoblotting to detect eEF1A1 interacted with GST-A37.
Co-immunoprecipitation
Immunoprecipitation was essentially performed as described previously (63). In brief, the HEK293 cells were lysed using 1 ml of co-immunoprecipitation (co-IP) buffer containing 10 mm HEPES (pH 7.5), 5 mm MgCl2, 142.5 mm KCl, 1 mm EDTA, 10% glycerol, and 1% Triton X-100 supplemented with 10 μl of protease inhibitor mixture (Sigma) on ice for 10 min. The cell lysates were placed at 4 °C for 1 h with rotation and followed by centrifugation at 16,100 × g, 4 °C for 30 min. The supernatant was collected, and 40 μl of it was saved as “input.” For each sample, 450 μl of supernatant was added to the 20 μl of anti-c-Myc-agarose affinity gel antibody produced in rabbits (Sigma), 450 μl of which was added to 20 μl of protein A-agarose fast flow beads (Sigma) added with 0.1 μg/ml rabbit IgG (Vector Laboratories). The binding step was performed at 4 °C overnight with rotation. The agarose beads were washed five times with co-IP buffer and then resuspended with 30 μl of 2× SDS sample buffer and boiled at 99 °C for 10 min. Immunoprecipitates were analyzed by Western blotting.
Nucleocytoplasmic fractionation
The cells were lysed with fractionation buffer containing 10 mm Tris-HCl (pH 7.4), 50 mm NaCl, 3 mm MgCl2, and 0.5% Nonidet P-40. The cell lysate was centrifuged at 16,000 × g for 10 min at 4 °C. The supernatant was collected as cytoplasmic fraction. The pellet was washed three times with fractionation buffer. Each time the samples were centrifuged at 500 × g for 5 min at 4 °C. Finally, the pellet was resuspended with resuspension buffer containing 20 mm Tris-HCl (pH 8.0) and 2% SDS and saved as the nuclear fraction.
Drosophila stock
Fly stocks were cultured in cotton-plugged plastic vials with 15 ml of cornmeal-yeast-glucose-agar medium and maintained in temperature-controlled incubators. The fly strains used include gmr-GAL4 (Bloomington Drosophila Stock Center), UAS-A12-SoxN, and UAS-A34-SoxN (Rainbow Transgenic Flies, Inc.).
External eye phenotypic detection of adult flies
The adult fly eyes were observed under a stereomicroscope (SZX-12, Olympus, Tokyo, Japan). External eye images were captured by SPOT Insight CCD camera controlled by the SPOT Advanced software (version 5.1; Diagnostic Instruments Inc.).
Statistical analyses
Statistical analyses were performed using a two-tailed unpaired Student's t test, except in Fig. 1G. All experiments were performed at least three times. Results are presented as means ± S.E. A p value of less than 0.05 was considered statistically significant.
Author contributions
L. L., N. K. L. N., and H. Y. E. C. conceived and designed the experiments. L. L. and K. L. N. N. performed the experiments. L. L., N. K. L. N., A. C. K., and H. Y. E. C. analyzed the results and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Professors Beric Henderson, David Rubinsztein, and Jennifer Loppincott-Schwartz for sharing reagents.
This work was supported by General Research Fund of the Hong Kong Research Grants Council Grant 461013 and CUHK Gerald Choa Neuroscience Centre Grant 7105306. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S6.
- eEF1A1
- eukaryotic translation elongation factor 1 α1
- aa-tRNA
- aminoacylation-tRNA
- eEF2
- eukaryotic translation elongation factor 2
- FLIP
- fluorescence loss in photobleaching
- NES
- nuclear export signal
- PA-GFP
- photoactivatable GFP
- ROI
- region of interest
- TD-NEM
- transcription-dependent nuclear export motif
- VHL
- von Hippel-Lindau
- N/C
- nuclear/cytoplasmic
- EGFP
- enhanced green fluorescent protein
- IP
- immunoprecipitation.
References
- 1. Brown L. Y., and Brown S. A. (2004) Alanine tracts: the expanding story of human illness and trinucleotide repeats. Trends Genet. 20, 51–58 [DOI] [PubMed] [Google Scholar]
- 2. Amiel J., Trochet D., Clément-Ziza M., Munnich A., and Lyonnet S. (2004) Polyalanine expansions in human. Hum. Mol. Genet. 13, R235–R243 [DOI] [PubMed] [Google Scholar]
- 3. Hatters D. M., and Hannan A. J. (eds) (2013) Tandem Repeats in Genes, Proteins, and Disease, Humana Press, New York [Google Scholar]
- 4. Hughes J. N., and Thomas P. Q. (2013) Molecular pathology of polyalanine expansion disorders: new perspectives from mouse models. Methods Mol. Biol. 1017, 135–151 [DOI] [PubMed] [Google Scholar]
- 5. Albrecht A., and Mundlos S. (2005) The other trinucleotide repeat: polyalanine expansion disorders. Curr. Opin. Genet. Dev. 15, 285–293 [DOI] [PubMed] [Google Scholar]
- 6. Lee T. I., and Young R. A. (2000) Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 34, 77–137 [DOI] [PubMed] [Google Scholar]
- 7. Bachetti T., Matera I., Borghini S., Di Duca M., Ravazzolo R., and Ceccherini I. (2005) Distinct pathogenetic mechanisms for PHOX2B associated polyalanine expansions and frameshift mutations in congenital central hypoventilation syndrome. Hum. Mol. Genet. 14, 1815–1824 [DOI] [PubMed] [Google Scholar]
- 8. Strømme P., Mangelsdorf M. E., Shaw M. A., Lower K. M., Lewis S. M., Bruyere H., Lütcherath V., Gedeon A. K., Wallace R. H., Scheffer I. E., Turner G., Partington M., Frints S. G., Fryns J. P., Sutherland G. R., et al. (2002) Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 30, 441–445 [DOI] [PubMed] [Google Scholar]
- 9. Moumné L., Dipietromaria A., Batista F., Kocer A., Fellous M., Pailhoux E., and Veitia R. A. (2008) Differential aggregation and functional impairment induced by polyalanine expansions in FOXL2, a transcription factor involved in cranio-facial and ovarian development. Hum. Mol. Genet. 17, 1010–1019 [DOI] [PubMed] [Google Scholar]
- 10. Albrecht A. N., Kornak U., Böddrich A., Suring K., Robinson P. N., Stiege A. C., Lurz R., Stricker S., Wanker E. E., and Mundlos S. (2004) A molecular pathogenesis for transcription factor associated poly-alanine tract expansions. Hum. Mol. Genet. 13, 2351–2359 [DOI] [PubMed] [Google Scholar]
- 11. Woods K. S., Cundall M., Turton J., Rizotti K., Mehta A., Palmer R., Wong J., Chong W. K., Al-Zyoud M., El-Ali M., Otonkoski T., Martinez-Barbera J. P., Thomas P. Q., Robinson I. C., Lovell-Badge R., et al. (2005) Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am. J. Hum. Genet. 76, 833–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oma Y., Kino Y., Toriumi K., Sasagawa N., and Ishiura S. (2007) Interactions between homopolymeric amino acids (HPAAs). Protein Sci. 16, 2195–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Amiel J., Laudier B., Attié-Bitach T., Trang H., de Pontual L., Gener B., Trochet D., Etchevers H., Ray P., Simonneau M., Vekemans M., Munnich A., Gaultier C., and Lyonnet S. (2003) Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat. Genet. 33, 459–461 [DOI] [PubMed] [Google Scholar]
- 14. Weese-Mayer D. E., Berry-Kravis E. M., Ceccherini I., Keens T. G., Loghmanee D. A., Trang H., ATS Congenital Central Hypoventilation Syndrome Subcommittee (2010) An official ATS clinical policy statement: congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am. J. Respir. Crit. Care Med. 181, 626–644 [DOI] [PubMed] [Google Scholar]
- 15. De Baere E., Beysen D., Oley C., Lorenz B., Cocquet J., De Sutter P., Devriendt K., Dixon M., Fellous M., Fryns J. P., Garza A., Jonsrud C., Koivisto P. A., Krause A., Leroy B. P., et al. (2003) FOXL2 and BPES: mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. Am. J. Hum. Genet. 72, 478–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cocquet J., Pailhoux E., Jaubert F., Servel N., Xia X., Pannetier M., De Baere E., Messiaen L., Cotinot C., Fellous M., and Veitia R. A. (2002) Evolution and expression of FOXL2. J. Med. Genet. 39, 916–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grosshans H., Hurt E., and Simos G. (2000) An aminoacylation-dependent nuclear tRNA export pathway in yeast. Genes Dev. 14, 830–840 [PMC free article] [PubMed] [Google Scholar]
- 18. Khacho M., Mekhail K., Pilon-Larose K., Pause A., Côté J., and Lee S. (2008) eEF1A is a novel component of the mammalian nuclear protein export machinery. Mol. Biol. Cell 19, 5296–5308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murthi A., Shaheen H. H., Huang H. Y., Preston M. A., Lai T. P., Phizicky E. M., and Hopper A. K. (2010) Regulation of tRNA bidirectional nuclear-cytoplasmic trafficking in Saccharomyces cerevisiae. Mol. Biol. Cell 21, 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mingot J. M., Vega S., Cano A., Portillo F., and Nieto M. A. (2013) eEF1A mediates the nuclear export of SNAG-containing proteins via the Exportin5-aminoacyl-tRNA complex. Cell Rep. 5, 727–737 [DOI] [PubMed] [Google Scholar]
- 21. Henderson B. R., and Eleftheriou A. (2000) A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp. Cell Res. 256, 213–224 [DOI] [PubMed] [Google Scholar]
- 22. Pollard V. W., and Malim M. H. (1998) The HIV-1 Rev protein. Annu. Rev. Microbiol. 52, 491–532 [DOI] [PubMed] [Google Scholar]
- 23. Mateyak M. K., and Kinzy T. G. (2010) eEF1A: thinking outside the ribosome. J. Biol. Chem. 285, 21209–21213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laumonnier F., Ronce N., Hamel B. C., Thomas P., Lespinasse J., Raynaud M., Paringaux C., Van Bokhoven H., Kalscheuer V., Fryns J. P., Chelly J., Moraine C., and Briault S. (2002) Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am. J. Hum. Genet. 71, 1450–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Solomon N. M., Ross S. A., Morgan T., Belsky J. L., Hol F. A., Karnes P. S., Hopwood N. J., Myers S. E., Tan A. S., Warne G. L., Forrest S. M., and Thomas P. Q. (2004) Array comparative genomic hybridisation analysis of boys with X linked hypopituitarism identifies a 3.9 Mb duplicated critical region at Xq27 containing SOX3. J. Med. Genet. 41, 669–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Overton P. M., Meadows L. A., Urban J., and Russell S. (2002) Evidence for differential and redundant function of the Sox genes Dichaete and SoxN during CNS development in Drosophila. Development 129, 4219–4228 [DOI] [PubMed] [Google Scholar]
- 27. Buescher M., Hing F. S., and Chia W. (2002) Formation of neuroblasts in the embryonic central nervous system of Drosophila melanogaster is controlled by SoxNeuro. Development 129, 4193–4203 [DOI] [PubMed] [Google Scholar]
- 28. Girard F., Joly W., Savare J., Bonneaud N., Ferraz C., and Maschat F. (2006) Chromatin immunoprecipitation reveals a novel role for the Drosophila SoxNeuro transcription factor in axonal patterning. Dev. Biol. 299, 530–542 [DOI] [PubMed] [Google Scholar]
- 29. Laughon A., Driscoll R., Wills N., and Gesteland R. F. (1984) Identification of two proteins encoded by the Saccharomyces cerevisiae GAL4 gene. Mol. Cell. Biol. 4, 268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sadowski I., and Ptashne M. (1989) A vector for expressing GAL4(1–147) fusions in mammalian cells. Nucleic Acids Res. 17, 7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matthews J. C., Hori K., and Cormier M. J. (1977) Purification and properties of Renilla reniformis luciferase. Biochemistry 16, 85–91 [DOI] [PubMed] [Google Scholar]
- 32. Patterson G. H., and Lippincott-Schwartz J. (2002) A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297, 1873–1877 [DOI] [PubMed] [Google Scholar]
- 33. Chalfie M., Tu Y., Euskirchen G., Ward W. W., and Prasher D. C. (1994) Green fluorescent protein as a marker for gene expression. Science 263, 802–805 [DOI] [PubMed] [Google Scholar]
- 34. Patterson G. H., and Lippincott-Schwartz J. (2004) Selective photolabeling of proteins using photoactivatable GFP. Methods 32, 445–450 [DOI] [PubMed] [Google Scholar]
- 35. Bohnsack M. T., Regener K., Schwappach B., Saffrich R., Paraskeva E., Hartmann E., and Görlich D. (2002) Exp5 exports eEF1A via tRNA from nuclei and synergizes with other transport pathways to confine translation to the cytoplasm. EMBO J. 21, 6205–6215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Calado A., Treichel N., Müller E. C., Otto A., and Kutay U. (2002) Exportin-5-mediated nuclear export of eukaryotic elongation factor 1A and tRNA. EMBO J. 21, 6216–6224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brown L. Y., Odent S., David V., Blayau M., Dubourg C., Apacik C., Delgado M. A., Hall B. D., Reynolds J. F., Sommer A., Wieczorek D., Brown S. A., and Muenke M. (2001) Holoprosencephaly due to mutations in ZIC2: alanine tract expansion mutations may be caused by parental somatic recombination. Hum. Mol. Genet. 10, 791–796 [DOI] [PubMed] [Google Scholar]
- 38. Brown S. A., Warburton D., Brown L. Y., Yu C. Y., Roeder E. R., Stengel-Rutkowski S., Hennekam R. C., and Muenke M. (1998) Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat. Genet. 20, 180–183 [DOI] [PubMed] [Google Scholar]
- 39. Crisponi L., Deiana M., Loi A., Chiappe F., Uda M., Amati P., Bisceglia L., Zelante L., Nagaraja R., Porcu S., Ristaldi M. S., Marzella R., Rocchi M., Nicolino M., Lienhardt-Roussie A., et al. (2001) The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat. Genet. 27, 159–166 [DOI] [PubMed] [Google Scholar]
- 40. Brown L., Paraso M., Arkell R., and Brown S. (2005) In vitro analysis of partial loss-of-function ZIC2 mutations in holoprosencephaly: alanine tract expansion modulates DNA binding and transactivation. Hum. Mol. Genet. 14, 411–420 [DOI] [PubMed] [Google Scholar]
- 41. Alatzoglou K. S., Kelberman D., Cowell C. T., Palmer R., Arnhold I. J., Melo M. E., Schnabel D., Grueters A., and Dattani M. T. (2011) Increased transactivation associated with SOX3 polyalanine tract deletion in a patient with hypopituitarism. J. Clin. Endocrinol. Metab. 96, E685–E690 [DOI] [PubMed] [Google Scholar]
- 42. Soares D. C., Barlow P. N., Newbery H. J., Porteous D. J., and Abbott C. M. (2009) Structural models of human eEF1A1 and eEF1A2 reveal two distinct surface clusters of sequence variation and potential differences in phosphorylation. PLoS One 4, e6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vera M., Pani B., Griffiths L. A., Muchardt C., Abbott C. M., Singer R. H., and Nudler E. (2014) The translation elongation factor eEF1A1 couples transcription to translation during heat shock response. Elife 3, e03164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McGuire A. T., and Mangroo D. (2007) Cex1p is a novel cytoplasmic component of the Saccharomyces cerevisiae nuclear tRNA export machinery. EMBO J. 26, 288–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fullston T., Finnis M., Hackett A., Hodgson B., Brueton L., Baynam G., Norman A., Reish O., Shoubridge C., and Gecz J. (2011) Screening and cell-based assessment of mutations in the Aristaless-related homeobox (ARX) gene. Clin. Genet. 80, 510–522 [DOI] [PubMed] [Google Scholar]
- 46. Fornerod M., Ohno M., Yoshida M., and Mattaj I. W. (1997) CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90, 1051–1060 [DOI] [PubMed] [Google Scholar]
- 47. Stade K., Ford C. S., Guthrie C., and Weis K. (1997) Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 90, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 48. Umemoto T., and Fujiki Y. (2012) Ligand-dependent nucleo-cytoplasmic shuttling of peroxisome proliferator-activated receptors, PPARα and PPARγ. Genes Cells 17, 576–596 [DOI] [PubMed] [Google Scholar]
- 49. Grespin M. E., Bonamy G. M., Roggero V. R., Cameron N. G., Adam L. E., Atchison A. P., Fratto V. M., and Allison L. A. (2008) Thyroid hormone receptor α1 follows a cooperative CRM1/calreticulin-mediated nuclear export pathway. J. Biol. Chem. 283, 25576–25588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saporita A. J., Zhang Q., Navai N., Dincer Z., Hahn J., Cai X., and Wang Z. (2003) Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J. Biol. Chem. 278, 41998–42005 [DOI] [PubMed] [Google Scholar]
- 51. Kanno Y., Suzuki M., Miyazaki Y., Matsuzaki M., Nakahama T., Kurose K., Sawada J., and Inouye Y. (2007) Difference in nucleocytoplasmic shuttling sequences of rat and human constitutive active/androstane receptor. Biochim. Biophys. Acta 1773, 934–944 [DOI] [PubMed] [Google Scholar]
- 52. Kanno Y., Suzuki M., Nakahama T., and Inouye Y. (2005) Characterization of nuclear localization signals and cytoplasmic retention region in the nuclear receptor CAR. Biochim. Biophys. Acta 1745, 215–222 [DOI] [PubMed] [Google Scholar]
- 53. Munshi R., Kandl K. A., Carr-Schmid A., Whitacre J. L., Adams A. E., and Kinzy T. G. (2001) Overexpression of translation elongation factor 1A affects the organization and function of the actin cytoskeleton in yeast. Genetics 157, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ravikumar B., Duden R., and Rubinsztein D. C. (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117 [DOI] [PubMed] [Google Scholar]
- 55. Berger Z., Davies J. E., Luo S., Pasco M. Y., Majoul I., O'Kane C. J., and Rubinsztein D. C. (2006) Deleterious and protective properties of an aggregate-prone protein with a polyalanine expansion. Hum. Mol. Genet. 15, 453–465 [DOI] [PubMed] [Google Scholar]
- 56. Abu-Baker A., Messaed C., Laganiere J., Gaspar C., Brais B., and Rouleau G. A. (2003) Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 12, 2609–2623 [DOI] [PubMed] [Google Scholar]
- 57. Wong J., Farlie P., Holbert S., Lockhart P., and Thomas P. Q. (2007) Polyalanine expansion mutations in the X-linked hypopituitarism gene SOX3 result in aggresome formation and impaired transactivation. Front. Biosci. 12, 2085–2095 [DOI] [PubMed] [Google Scholar]
- 58. Nasrallah I. M., Minarcik J. C., and Golden J. A. (2004) A polyalanine tract expansion in Arx forms intranuclear inclusions and results in increased cell death. J. Cell Biol. 167, 411–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bao Y. P., Cook L. J., O'Donovan D., Uyama E., and Rubinsztein D. C. (2002) Mammalian, yeast, bacterial, and chemical chaperones reduce aggregate formation and death in a cell model of oculopharyngeal muscular dystrophy. J. Biol. Chem. 277, 12263–12269 [DOI] [PubMed] [Google Scholar]
- 60. Di Lascio S., Belperio D., Benfante R., and Fornasari D. (2016) Alanine expansions associated with congenital central hypoventilation syndrome impair PHOX2B homeodomain-mediated dimerization and nuclear import. J. Biol. Chem. 291, 13375–13393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rankin J., Wyttenbach A., and Rubinsztein D. C. (2000) Intracellular green fluorescent protein-polyalanine aggregates are associated with cell death. Biochem. J. 348, 15–19 [PMC free article] [PubMed] [Google Scholar]
- 62. Tsoi H., Li L., Chen Z. S., Lau K. F., Tsui S. K., and Chan H. Y. (2014) The SARS-coronavirus membrane protein induces apoptosis via interfering with PDK1-PKB/Akt signalling. Biochem. J. 464, 439–447 [DOI] [PubMed] [Google Scholar]
- 63. Zhang Z., Hartmann H., Do V. M., Abramowski D., Sturchler-Pierrat C., Staufenbiel M., Sommer B., van de Wetering M., Clevers H., Saftig P., De Strooper B., He X., and Yankner B. A. (1998) Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 395, 698–702 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.