

Abstract
All-trans-retinoic acid (RA), a bioactive derivative of vitamin A, exhibits diverse effects on gene transcription and non-genomic regulatory pathways. The steady-state levels of RA are therefore tightly controlled, but the mechanisms responsible for RA homeostasis are not fully understood. We report a molecular mechanism that allows cells to maintain a stable rate of RA biosynthesis by utilizing a biological circuit generated by a bifunctional retinoid oxidoreductive complex (ROC). We show that ROC is composed of at least two subunits of NAD+-dependent retinol dehydrogenase 10 (RDH10), which catalyzes the oxidation of retinol to retinaldehyde, and two subunits of NADPH-dependent dehydrogenase reductase 3 (DHRS3), which catalyzes the reduction of retinaldehyde back to retinol. RDH10 and DHRS3 also exist as homo-oligomers. When complexed, RDH10 and DHRS3 mutually activate and stabilize each other. These features of ROC ensure that the rate of RA biosynthesis in whole cells is largely independent of the concentration of the individual ROC components. ROC operates in various subcellular fractions including microsomes, mitochondria, and lipid droplets; however, lipid droplets display weaker mutual activation between RDH10 and DHRS3, suggesting reduced formation of ROC. Importantly, disruption of the ROC-generated circuit by a knockdown of DHRS3 results in an increased flux through the RA biosynthesis pathway and elevated RA levels despite the decrease in RDH10 protein destabilized by the absence of DHRS3, hence demonstrating a loss of control. Thus, the bifunctional nature of ROC provides the RA-based signaling system with robustness by safeguarding appropriate RA concentration despite naturally occurring fluctuations in RDH10 and DHRS3.
Keywords: dehydrogenase, homeostasis, reductase, retinoic acid, retinol, vitamin A
Introduction
All-trans-retinoic acid (RA)2 is a bioactive derivative of vitamin A that regulates gene transcription through binding to nuclear retinoic acid receptors (α, β, and γ) and also exhibits numerous non-genomic effects (reviewed in Ref. 1). It is well established that during embryonic development, the concentration of RA is tightly controlled in a spatial and temporal manner (reviewed in Ref. 2), whereas in adult tissues, RA levels are maintained within a very narrow range that is specific for each given tissue. However, the molecular mechanisms responsible for the maintenance of RA homeostasis are not yet fully understood.
RA is synthesized from all-trans-retinol in two sequential steps. First, retinol is oxidized reversibly to retinaldehyde, and then retinaldehyde is oxidized irreversibly to RA (reviewed in Ref. 3). RA can maintain its own homeostasis via feedback regulation of cytochrome P450 enzymes CYP26A1, CYP26B1, and CYP26C1 (reviewed in Ref. 4), which catalyze its degradation, and through induction of lecithin retinol acyl transferase, the enzyme that converts retinol to its storage forms, retinyl esters (5). It was also reported that the expression of retinaldehyde dehydrogenases (RALDH 1–3), which oxidize retinaldehyde to RA, can be inhibited by excessive levels of RA in mice (6). However, very little is known about the mechanisms that control the rate of RA biosynthesis at the level of retinol dehydrogenases, the enzymes that catalyze the first step in RA pathway: the generation of retinaldehyde from retinol.
As demonstrated previously, the oxidation of retinol to retinaldehyde is the rate-limiting step that determines the overall rate of RA biosynthesis from retinol (7). Studies from independent laboratories established that a member of the short chain dehydrogenase/reductase (SDR) superfamily of proteins, retinol dehydrogenase 10 (RDH10, SDR16C4 in humans, and SDR16C10 in mice (3)), serves as the major retinol dehydrogenase during embryonic development (8–10). Genetic disruption of murine Rdh10 results in a marked reduction in RA synthesis that leads to a number of developmental abnormalities, including hypoplastic forelimb buds and altered hindbrain and craniofacial patterning. These embryos ultimately expire during mid-embryogenesis (8–10).
Remarkably, RDH10 is not the only member of the SDR superfamily that is essential for RA biosynthesis during development. Recent studies demonstrated that a targeted gene knock-out of dehydrogenase reductase 3 (DHRS3, SDR16C1 in humans, SDR16C9 in mice), which belongs to the same family of SDRs as RDH10 (3), results in overproduction of RA in mouse embryos, leading to late gestational/early postnatal lethality (11, 12). DHRS3-null embryos have eyes of reduced size, cleft palates, skeletal defects, and alterations to heart morphology (11, 12).
Recently, we discovered that DHRS3 displays a robust retinaldehyde reductive activity when co-expressed in the same cells with RDH10 (12). In turn, the activity of RDH10 is significantly enhanced by the presence of DHRS3. Interestingly, catalytically inactive Tyr mutants of RDH10 or DHRS3 are as efficient as wild-type proteins in activating their partner. Moreover, when RDH10 is co-expressed in living cells with a catalytically inactive Y188A mutant of DHRS3, the cells produce severalfold higher amounts of RA, because the mutant still activates RDH10 but is unable to convert retinaldehyde produced by RDH10 back to retinol. The unopposed and enhanced catalytic activity of RDH10 thus results in higher rates of retinaldehyde and RA biosynthesis. Collectively, these observations suggest that RDH10 and DHRS3 proteins physically interact. The work presented here provides evidence for the existence of a higher order hetero-oligomeric complex of RDH10 and DHRS3, explores the composition and properties of RDH10-DHRS3 complex, and uncovers the critical role of the biological circuit generated by this antagonistically bifunctional complex in the maintenance of RA homeostasis.
Results
RDH10 and DHRS3 form homo- and hetero-oligomers
The results of our previous work indicated that RDH10 and DHRS3 proteins activate each other when co-expressed in the same cells (12). Here, we used a co-immunoprecipitation approach to determine whether the activation is mediated through direct protein-protein interaction between RDH10 and DHRS3. For reproducibility, these experiments were conducted using RDH10 and DHRS3 proteins expressed in two different systems: insect Sf9 cells and human HEK 293 cells. RDH10 was expressed as a C-terminal fusion with HA tag and was used as a bait in pulldown experiments. DHRS3 was expressed as a C-terminal fusion to FLAG tag. In control experiments, we included other members of the SDR superfamily of proteins, retinol dehydrogenase epidermal 2 (RDHE2) and retinol dehydrogenase 11 (RDH11). RDHE2 and RDH11 were both tagged with FLAG on their corresponding C termini. Solubilized microsomes from Sf9 cells expressing DHRS3-FLAG, RDHE2-FLAG, and RDH11-FLAG individually or in combination with RDH10-HA were incubated with anti-HA-agarose, and the proteins pulled down with the beads were analyzed by Western blotting using RDH10 antibodies or FLAG antibodies (Fig. 1A). Neither RDHE2-FLAG nor RDH11-FLAG were detected in the pulldown fractions, whereas DHRS3-FLAG was consistently co-immunoprecipitated in the presence of RDH10-HA, but not in its absence. This result indicated that RDH10-HA physically binds to DHRS3-FLAG, and this binding is specific for DHRS3 protein.
FIGURE 1.

RDH10 and DHRS3 form hetero-oligomers and homo-oligomers. A, Sf9 microsomes (30 μg) containing the indicated proteins were solubilized and processed for immunoprecipitation of HA-tagged RDH10. 2 μg of microsomes were used for input. RDHE2 and RDH11 served as negative controls. Binding of RDH10-HA to agarose was confirmed by staining with RDH10 antibodies (1:3000). FLAG-tagged binding partners were detected using FLAG antibody (1:3000) after separation in 12% SDS-PAGE. B, homo-oligomers of RDH10 and DHRS3 in Sf9 cells microsomes were detected as described in A. Note that RDH10-FLAG and DHRS3-FLAG do not associate with anti-HA-agarose in the absence of HA-tagged partners. Microsomes containing RDH10-HA and DHRS3-FLAG were utilized as positive controls for protein-protein interactions. C and D, HEK 293 cells were transfected with combinations of HA- and FLAG-tagged constructs as indicated. Pulldowns were performed as described in A. RDH10 and DHRS3 oligomers were detected using RDH10 or DHRS3 antibodies (C), which allowed simultaneous visualization of both HA-tagged and FLAG-tagged variants. Note that RDH10-HA was expressed at lower levels than RDH10-FLAG (C) and was below the detection limit in the input (∼11 μg) from total cell lysate (D). Negative control DHRS3-FLAG expressed alone did not bind to agarose and served as a control for nonspecific binding (D). I, input; IB, immunoblot; IP, immunoprecipitation; P, pulldown. Pulled down proteins are indicated by asterisks.
The majority of SDR enzymes that have been characterized function as homodimers or homotetramers (13, 14). To investigate whether RDH10 forms homo-oligomers, we co-expressed RDH10-HA with RDH10-FLAG. Anti-HA-agarose pulldown assays showed that RDH10-FLAG was detected in the eluate from the agarose only when co-expressed with RDH10-HA (Fig. 1B). Similarly, DHRS3-FLAG was pulled down in the presence of DHRS3-HA but not in its absence. These results demonstrated that both human RDH10 and human DHRS3 expressed in microsomes of Sf9 cells were capable of forming homo-oligomers.
To determine whether DHRS3 and RDH10 form homo- and hetero-oligomers in human cells, we performed pulldown assays on DHRS3 and RDH10 proteins expressed in HEK 293 cells. First, RDH10-HA was co-expressed with RDH10-FLAG, and DHRS3-HA was co-expressed with DHRS3-FLAG. Immunoprecipitation of RDH10-HA or DHRS3-HA from total cellular lysates followed by staining with RDH10 or DHRS3 antibodies allowed us to simultaneously detect both HA- and FLAG-tagged species of the corresponding proteins. The HA-tagged proteins appeared as protein bands with the higher molecular mass and FLAG-tagged with the lower molecular mass (Fig. 1C). This indicated that, as was the case with the proteins expressed in insect cells, in human cells, RDH10 and DHRS3 also formed homo-oligomers.
Next, the cells were co-transfected with RDH10-HA and DHRS3-FLAG constructs. In the absence of RDH10-HA bait, virtually no FLAG-tagged protein was detected in the pelleted anti-HA-agarose, whereas in the presence of RDH10-HA, a fraction of DHRS3-FLAG was reproducibly associated with anti-HA-agarose beads (Fig. 1D). The active site mutant of DHRS3, Y188A DHRS3-FLAG, was also detected in the pelleted anti-HA-agarose when co-expressed with wild-type RDH10, consistent with our previous observation that this mutant can activate RDH10 despite being itself catalytically inactive. Collectively, these experiments demonstrated that RDH10 and DHRS3 form both homo- and hetero-oligomers when expressed in either insect Sf9 cells or in human HEK 293 cells.
DHRS3 and RDH10 oligomers are larger than dimers
To better understand the structural assembly of RDH10-DHRS3 hetero-oligomer, we investigated the subunit composition of the complex. To determine whether RDH10-DHRS3 complex contains more than one subunit of each protein, DHRS3-FLAG was co-expressed with DHRS3-HA and RDH10-His6 in Sf9 cells. Microsomes containing the three proteins were isolated, solubilized, and subjected to the first round of immunoprecipitation using anti-FLAG M2 affinity gel to select for protein complexes containing DHRS3-FLAG (Fig. 2A). The protein species that bound to anti-FLAG M2 affinity gel were eluted with FLAG peptide and incubated with anti-HA-agarose to select for species containing DHRS3-HA. Proteins bound to anti-HA-agarose were solubilized and analyzed by Western blotting for the presence of RDH10-His6. Probing with RDH10 antibodies showed that RDH10 was present in the eluate after two rounds of sequential immunoprecipitations that selected first for DHRS3-FLAG, and then for DHRS3-HA (Fig. 3A). The result of this experiment indicated that RDH10-DHRS3 complex consists of at least three subunits, two of which are DHRS3. The same sequence of pulldown steps was performed on microsomes from Sf9 cells co-expressing RDH10-FLAG, RDH10-HA, and DHRS3-His6 to determine whether the complex contains more than one subunit of RDH10 (Fig. 2B). Western blotting analysis using DHRS3 antibody revealed the presence of DHRS3 in the final eluate from anti-HA-agarose, after selection for RDH10-FLAG and then for RDH10-HA (Fig. 3B). Together, these results indicated that RDH10-DHRS3 complex contains at least two subunits of each RDH10 and DHRS3.
FIGURE 2.

A diagram of sequential co-immunoprecipitation scheme. Proteins carrying three different tags (DHRS3-FLAG, DHRS3-HA, and RDH10-His6 in A and RDH10-FLAG, RDH10-HA, and DHRS3-His6 in B) were co-expressed in Sf9 cells. Lysates of Sf9 microsomes were immunoprecipitated with anti-FLAG agarose beads. The bound fraction was eluted from beads and subjected to the second round of immunoprecipitation with anti-HA-agarose. The eluate from anti-HA-agarose was analyzed by Western blotting for the presence of the protein carrying the third tag using RDH10 or DHRS3 antibodies. The presence of the His-tagged proteins in the eluates from anti-HA-agarose after two rounds of immunoprecipitation indicates that the complex contains at least three subunits.
FIGURE 3.
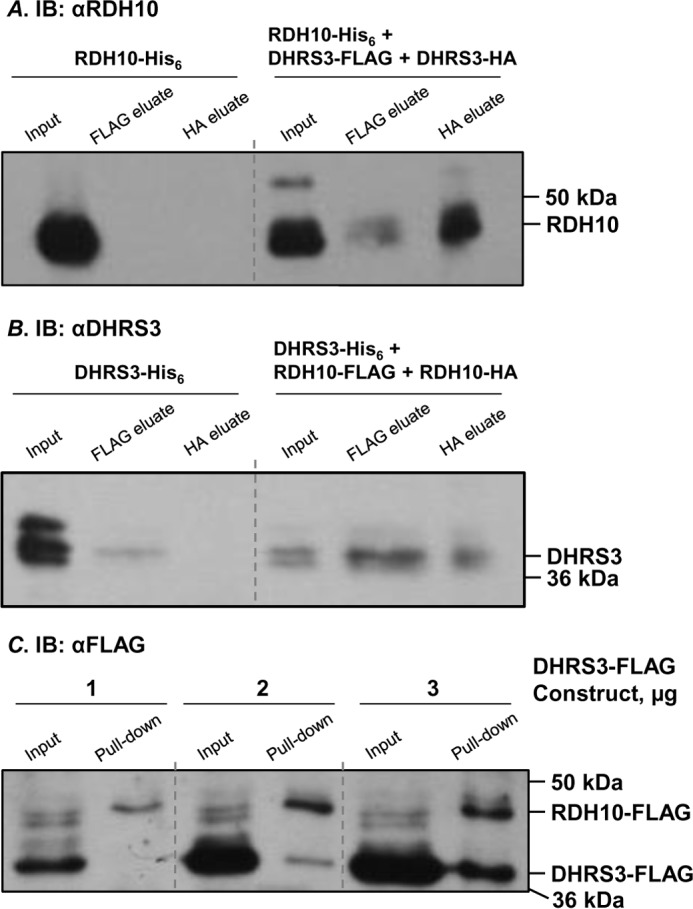
RDH10-DHRS3 hetero-oligomer is larger than a dimer. A, DHRS3-His6 was expressed in Sf9 cells alone or in combination with RDH10-FLAG and RDH10-HA. Solubilized microsomes (200 μg) were incubated with ANTI-FLAG M2 affinity gel. Bound proteins were eluted with FLAG peptide and applied to anti-HA-agarose. Final eluate was probed with DHRS3 antibodies after separation in 10% SDS-PAGE. B, RDH10-His6 was expressed in Sf9 cells alone or in combination with DHRS3-FLAG and DHRS3-HA. Two sequential rounds of immunoprecipitation were performed as described in A. Final eluate was probed with RDH10 antibodies. For immunoblotting in A and B, 2 μg of microsomes were loaded for input; ∼20% of eluate from anti-FLAG M2 affinity gel for FLAG eluate; and 100% of the eluate from anti-HA-agarose for HA eluate. C, HEK 293 cells were co-transfected with 1 μg of RDH10-HA, 1 μg of RDH10-FLAG, and increasing amounts of DHRS3-FLAG expressing construct (1, 2, and 3 μg as indicated). Both RDH10-FLAG and DHRS3-FLAG fusion proteins were detected using FLAG antibody. Pulldown assays of RDH10-HA show that a large excess of DHRS3-FLAG protein is required for efficient formation of DHRS3-RDH10 hetero-oligomers. Note the increase in the ratio between DHRS3-FLAG/RDH10-FLAG in the pulldown fraction from 1 to 3 μg of DHRS3 construct. The increase in RDH10-FLAG is likely due to the stabilizing effect of DHRS3 protein as shown in Fig. 6. IB, immunoblot.
Because we noticed that the homo- and hetero-oligomers of RDH10 and DHRS3 are both present at the same time, we wanted to determine whether there is any preferential formation of one species over the other. HEK 293 cells were co-transfected with vectors expressing RDH10-HA, RDH10-FLAG, and increasing amounts of DHRS3-FLAG construct. The HA-tagged RDH10 was precipitated from total cell lysates using anti-HA-agarose. Western blotting analysis with FLAG antibody revealed that a large excess of DHRS3-FLAG over RDH10-FLAG protein was required for efficient formation of hetero-oligomers, at least under these specific experimental conditions in HEK293 cells (Fig. 3C). This result suggested that homo-oligomers of RDH10 are formed preferentially over RDH10-DHRS3 hetero-oligomers, and the two species may co-exist to some degree when overexpressed in HEK 293 cells.
The interaction between RDH10 and DHRS3 occurs in various subcellular fractions
Previously, the membrane-associated RDH10 and DHRS3 proteins were detected in the microsomal and mitochondrial fractions of cell membranes and also in lipid droplets (12, 15–17). Hence, we asked whether the protein-protein interaction between RDH10 and DHRS3 occurs equally well in different subcellular fractions or is limited to certain types of cell membranes. HEK 293 cells were co-transfected with RDH10-HA and DHRS3-FLAG, and the binding between the two proteins in isolated microsomes and mitochondria was analyzed by pulldown assays. Immunoprecipitation using anti-HA-agarose followed by Western blotting with FLAG antibodies showed that DHRS3-FLAG co-precipitated with RDH10-HA from both mitochondria and microsomes (Fig. 4A).
FIGURE 4.
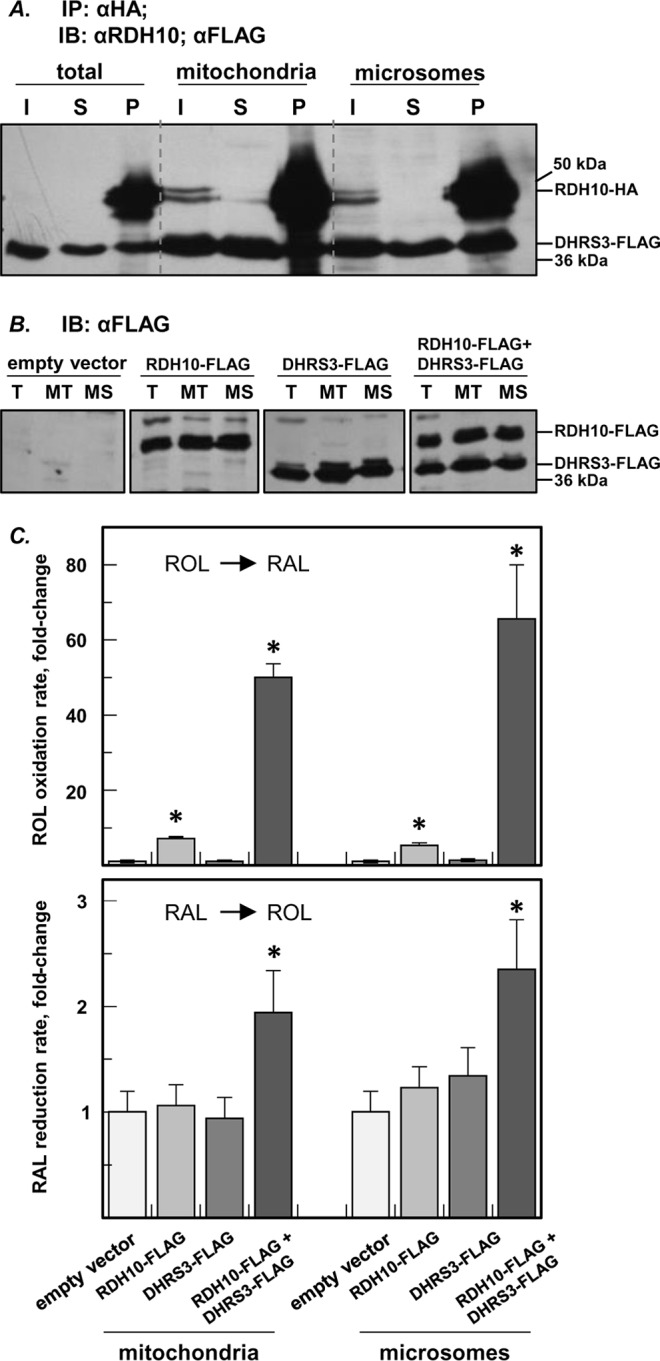
RDH10-DHRS3 hetero-oligomerization occurs in various subcellular fractions. A, the interaction between RDH10-HA and DHRS3-FLAG proteins in mitochondria (∼10 μg) and microsomes (∼10 μg) from HEK 293 cells was analyzed by pulldown assays. I, input; S, supernatant (unbound); P, pulldown. Note that in lanes I of A, RDH10-HA (upper band) is below detection limit in the input (∼11 μg) from total cell lysate but is much more intense in lane P, because all of HA eluate (including all of the bound RDH10-HA bait) was loaded on the gel to ensure reliable detection of the co-immunoprecipitated DHRS3-FLAG (lower band). B, Western blotting analysis of RDH10-FLAG and DHRS3-FLAG expressed separately or together in HEK 293 cells for activity assays of subcellular fractions. T, total lysate; MT, mitochondria; MS, microsomes. C, oxidation of retinol (ROL) to retinaldehyde (RAL) and reduction of retinaldehyde to retinol by HEK 293 mitochondria and microsomes containing RDH10-FLAG or DHRS3-FLAG alone or in combination. The rates are normalized to cells transfected with empty vector (means ± S.D.). *, p < 0.05, n = 3. Note that in both subcellular fractions co-expression of RDH10 and DHRS3 results in an increase of retinol oxidizing activity in comparison with cells expressing RDH10 alone, and overexpression of DHRS3 results in the increase of retinaldehyde reductive activity only in the presence of RDH10. This mutual activation confirms the interaction between RDH10 and DHRS3 in both fractions. IB, immunoblot; IP, immunoprecipitation.
The interaction between the two proteins in the microsomes and mitochondria was further confirmed by co-activation assays (Fig. 4, B and C). HEK 293 cells expressing RDH10-HA alone, DHRS3-FLAG alone, or both proteins together were fractionated by differential centrifugation, and the membrane fractions were tested for their ability to convert retinoids in vitro. The retinol oxidizing activity in the presence of NAD+, the preferred co-factor of RDH10, was higher in both mitochondrial and microsomal fractions of the cells co-expressing RDH10 and DHRS3 in comparison with cells expressing RDH10 alone. In the reductive direction, in both mitochondrial and microsomal fractions DHRS3 exhibited a retinaldehyde reductive activity in the presence of NADPH as co-factor only when co-expressed with RDH10.
HEK 293 cells produce very few lipid droplets. To increase the number of lipid droplets, 1 day prior to transfection with RDH10-HA and DHRS3-FLAG constructs, HEK 293 cells were transfected with expression vector encoding diacylglycerol acyltransferase 2 (DGAT2) and treated with 0.4 mm sodium oleate overnight (17). After transfection with RDH10 and DHRS3, the treatment with sodium oleate was repeated. Western blotting analysis confirmed the localization of RDH10 and DHRS3 in lipid droplets (Fig. 5A). As determined by activity assays, the retinol oxidizing activity in the lipid droplet fraction of cells co-transfected with DHRS3 and RDH10 was ∼1.8-fold higher than in the lipid droplets of cells transfected with RDH10 alone (Fig. 5B). Similarly, the retinaldehyde reductive activity increased 2.5-fold in lipid droplets containing both proteins compared with the background or DHRS3 alone. These results indicated that the mutual activation of DHRS3 and RDH10 proteins did occur in lipid droplets. However, the amplitude of the DHRS3-mediated increase in the retinol oxidative activity of lipid droplets (1.8-fold) was noticeably smaller compared with mitochondria (7-fold) or microsomes (13-fold).
FIGURE 5.

The mutual activation of RDH10 and DHRS3 is reduced in lipid droplets. A, Western blotting analysis of lipid droplets from HEK 293 cells transfected with RDH10-HA or DHRS3-FLAG constructs separately and in combination. Immunoblotting was performed using RDH10 and DHRS3 antibodies, which detect endogenous (Endog.) RDH10 and DHRS3 in addition to ectopically expressed proteins. B, oxidation of retinol (ROL) and reduction of retinaldehyde (RAL) by lipid droplets (means ± S.D., n = 3). *, p < 0.05. C, proximity ligation assay (red signal) shows direct protein-protein interaction between RDH10 and DHRS3 expressed in HepG2 cells.
To detect the interaction between RDH10 and DHRS3 proteins in intact cells, we utilized a proximity ligation assay. HepG2 cells were transfected with a bicistronic vector expressing untagged RDH10 and untagged DHRS3. Upon fixation and permeabilization, the cells were incubated with primary rabbit anti-RDH10 antibody and primary mouse anti-DHRS3 antibody. Following incubation with secondary anti-mouse and anti-rabbit antibodies carrying nucleotide adapters and amplification of the ligated DNA circle formed by complementary oligonucleotides, the fluorescently labeled DNA product was visualized by fluorescence microscopy. Bright red signals observed in the transfected cells signified the protein-protein interaction between RDH10 and DHRS3 and often displayed a ring pattern characteristic of lipid droplets (Fig. 5C). The proximity ligation assay confirmed the results of pulldown assays and provided additional evidence that RDH10 and DHRS3 physically interact when expressed in living cells.
RDH10 and DHRS3 proteins stabilize each other
While investigating the behavior of co-expressed RDH10 and DHRS3, we noticed that each protein appeared to be more abundant in the cells co-expressing the two proteins compared with cells expressing RDH10 and DHRS3 separately. This observation was confirmed in three different cell lines: CHO, HepG2, and HEK 293 (Fig. 6, A–C). Quantitative PCR analysis showed that the higher amounts of co-expressed DHRS3 and RDH10 proteins were not due to higher levels of transcripts, either ectopically expressed or present endogenously (supplemental Fig. S1), but were likely due to increased protein stability of the partner proteins. Furthermore, co-expression of wild-type DHRS3 with unstable RDH10 deletion mutant lacking residues 85–109 (ΔRDH10) resulted in a noticeable decrease in DHRS3 protein (Fig. 6C and supplemental Fig. S2). Treatment of cells with proteosome inhibitor MG132 noticeably increased protein levels of both ΔRDH10 and DHRS3 (supplemental Fig. S2). To determine the half-lives of co-expressed RDH10 and DHRS3 in comparison with individually expressed proteins, HEK 293 cells were transfected with expression constructs for HA-tagged human DHRS3 and RDH10 separately or co-transfected with both plasmids. After starving in methionine/cysteine-free medium, the cells were incubated in a medium containing radiolabeled methionine and cysteine and harvested at several time points over a 24-h chase period. The HA-tagged proteins were precipitated with anti-HA-agarose, separated by denaturing polyacrylamide gel electrophoresis, and analyzed by autoradiography and image densitometry (Fig. 6D). The estimated half-life of RDH10 expressed alone was 11 h, but it increased to 16 h in the presence of co-expressed DHRS3. The half-life of DHRS3 in the presence of RDH10 increased from 8 to 22 h (Fig. 6E). Thus, in addition to mutually activating each other, the two proteins extended each other's half-lives.
FIGURE 6.

The half-lives of RDH10 and DHRS3 proteins are extended when they are co-expressed. A–C, CHO cells (A), HepG2 cells (B), and HEK 293 cells (C) were transfected in replicates with vectors expressing RDH10 or DHRS3 alone or in combination. Western blotting analysis was performed using RDH10 and DHRS3 antibodies (both at 1:2,000). GAPDH served as a loading control. ΔRDH10 is a construct that lacks amino acids 85–109 and has the same mobility as DHRS3. D, autoradiography of radiolabeled DHRS3-HA and RDH10-HA expressed in HEK 293 cells separately or together. E, the band intensities at different time points were determined by densitometry and used to calculate the half-lives. A.U., absolute units calculated as percentage of initial band intensity.
The circuit generated by RDH10-DHRS3 complex protects RA homeostasis against variations in enzyme levels
The expression of DHRS3 is known to be induced by RA (18, 19). Having established that DHRS3 forms a complex with RDH10, we wanted to determine how the increase in DHRS3 protein would affect the rate of RA biosynthesis at constant levels of RDH10. To model a gradual increase in DHRS3 protein, HEK 293 cells were co-transfected with a fixed amount of RDH10-FLAG construct and increasing amounts of DHRS3-FLAG construct. The ratio of DHRS3 to RDH10 expression plasmids varied from 0 to 2, as indicated (Fig. 7A). Western blotting analysis using FLAG antibody showed that DHRS3 protein increased gradually with increasing amount of DHRS3 expression plasmid whereas the amount of RDH10 protein remained relatively constant. To determine the rate of RA biosynthesis, the cells were treated with 2 μm retinol for 9 h, and RA was extracted and measured by normal phase HPLC. As expected, transfection of the cells with RDH10 alone resulted in a significant increase in RA production: ∼4-fold compared with cells transfected with empty vector. Surprisingly, this increase in RDH10-dependent RA production was almost completely abolished by the lowest amount of the co-expressed DHRS3 protein (Fig. 7A). Further increase in DHRS3 protein had little or no effect in terms of decreasing RA production.
FIGURE 7.

Antagonistically bifunctional activity of RDH10-DHRS3 complex provides independence from fluctuations in the levels of individual components. A, Western blotting and activity analysis of HEK 293 cells co-transfected with a fixed amount of RDH10-FLAG construct and increasing amounts of DHRS3-FLAG construct. The ratio of DHRS3/RDH10 plasmid varied from 0 to 2 (μg/μg), as indicated. RA production was determined by HPLC after incubating the cells with 2 μm retinol for 9 h (means ± S.D., n = 3). B, HEK 293 cells were transfected with increasing amounts of RDH10-expressing construct (1–3 μg as indicated) separately or together with a fixed amount of bicistronic vector (RDH10-IRES-DHRS3). RA production was measured as in A. Note that RA production levels in the cells co-transfected with RDH10 and bicistronic vector remain stable despite the gradual increase in RDH10 protein. C, Western blotting and activity analysis of HEK 293 cells co-transfected with a fixed amount of RDH10-FLAG and increasing amounts of Y188A DHRS3-FLAG. Note the gradual increase in RA production, reflecting the loss of homeostasis. IB, immunoblot.
To further test the robustness of DHRS3 effect on RA homeostasis, we kept the amount of DHRS3 protein constant but gradually increased the amount of RDH10. To ensure that all cells consistently and reproducibly received the same starting ratio of RDH10/DHRS3 proteins, we used a bicistronic vector containing both RDH10 and DHRS3 cDNAs separated by the internal ribosome entry site. To model the increase in RDH10/DHRS3 ratio, the cells were co-transfected with bicistronic RDH10/DHRS3 vector and gradually increasing amounts of RDH10 expression vector. The gradual increase in RDH10 protein was verified by Western blotting using RDH10 antibodies (Fig. 7B). The cells were treated with 2 μm all-trans-retinol, and the RA was extracted from the culture medium and quantified by HPLC. Remarkably, the steady increase in RDH10 protein had little or no effect on RA production in the cells transfected with bicistronic plasmid that produced both RDH10 and DHRS3, whereas a similar increase of RDH10 in the absence of DHRS3 resulted in a gradual increase in RA production, proportional to RDH10 protein levels (Fig. 7B). It is worth noting that the increase in RDH10 protein was accompanied by an increase in DHRS3 protein, even though the cells were transfected with the same amount of the bicistronic plasmid. This observation was consistent with the stabilizing effect of RDH10 on DHRS3 described above.
The surprising stability of RA levels over a wide range of RDH10/DHRS3 ratios prompted us to investigate whether this homeostatic effect was due to the circuit of retinol and retinaldehyde interconversion created by the opposing RDH10 and DHRS3 catalytic activities. To disrupt this circuit, we titrated the cells expressing RDH10 with increasing amounts of catalytically inactive Y188A DHRS3 (Fig. 7C). Previously, we showed that this mutant still activated RDH10 but was unable to convert retinaldehyde produced by RDH10 back to retinol (12). We also showed that this unopposed and enhanced catalytic activity of RDH10 resulted in higher rates of retinaldehyde and RA biosynthesis in living cells. In agreement with this previous observation, the gradual increase in Y188A DHRS3/RDH10 ratio led to a correspondingly gradual increase in RA biosynthesis, proportional to Y188A DHRS3 expression level. Conversely, when wild-type DHRS3 was titrated with increasing amounts of catalytically inactive Y210A RDH10, there was a gradual increase in the rate of retinaldehyde conversion to retinol, eventually reaching a plateau (supplemental Fig. S3). These experiments demonstrated that disruption of the retinoid oxidoreductive circuit resulted in a loss of the tight control over RA levels and perturbation of RA homeostasis.
The behavior of native RDH10 and DHRS3 supports the role of the oxidoreductive circuit in RA homeostasis
To determine whether our findings obtained with recombinant RDH10 and DHRS3 proteins are valid for the native, endogenously expressed RDH10 and DHRS3, we examined the impact of endogenous DHRS3 knockdown on RDH10 protein levels, RDH10 activity in subcellular fractions, and generation of RA from retinol in living cells. Immunocytochemical analysis using RDH10- and DHRS3-specific antibodies showed that the native proteins appeared in a ring pattern similar to that observed for recombinantly expressed RDH10 and DHRS3 in proximity ligation assays (Fig. 8). Importantly, the red fluorescent signal corresponding to endogenous RDH10 or DHRS3 showed a significant overlap with the green immunofluorescence produced by the lipid droplet marker protein PLIN2, thus confirming the co-localization of native RDH10 and DHRS3 in lipid droplets. The DHRS3 signal was significantly reduced in HepG2 cells stably transfected with DHRS3 shRNA (Fig. 8).
FIGURE 8.

Endogenous RDH10 and DHRS3 proteins co-localize with PLIN2 in HepG2 cells. Immunocytochemistry of wild-type HepG2 cells was performed using a mixture of rabbit DHRS3 antibodies and chicken PLIN2 antibodies (A) or rabbit RDH10 antibodies and chicken PLIN2 antibodies (B). The signal was visualized after subsequent incubation with Alexa Fluor 594-conjugated anti-rabbit antibody (red dye) and Alexa Fluor 488-conjugated anti-chicken antibody (green dye). C, controls without primary antibodies. D, immunolocalization of DHRS3 in HepG2 cells treated with RA (10 nm), untreated cells, or cells stably transfected with DHRS3 shRNA. Note the gradual decrease in signal from RA-treated cells to DHRS3-silenced cells.
Silencing of endogenous DHRS3 expression in HepG2 cells resulted in elevated steady-state levels of RA (Fig. 9A). To determine how the depletion of DHRS3 affected the endogenous retinoid activities of subcellular fractions, DHRS3-silenced HepG2 cells and control HepG2 cells stably transfected with scrambled shRNA were fractionated, and the activities of microsomes, mitochondria, and lipid droplets were measured in the oxidative (retinol plus NAD+) and reductive (retinaldehyde plus NADPH) directions. These assays revealed that the retinol dehydrogenase activity of DHRS3-depleted mitochondrial and microsomal fraction was significantly reduced (∼3-fold) (Fig. 9B). The retinol dehydrogenase activity associated with lipid droplets was also reduced but to a lesser extent. Western blotting analysis showed that the reduction in the retinol dehydrogenase activity of the fractions correlated with the decrease in the amount of RDH10 protein (Fig. 9C). This finding is consistent with our observation that the half-life of RDH10 is shortened in the absence of DHRS3 and suggests that native RDH10 behaves similarly to recombinant RDH10. Furthermore, the fact that despite its reduced levels RDH10 is able to maintain higher steady-state levels of RA illustrates the significance of a circuit created by the antagonistic activities of RDH10-DHRS3 complex. When the circuit is malfunctioning because of depletion of DHRS3, the tight control over RDH10 activity is lost, resulting in overproduction of RA and loss of homeostasis.
FIGURE 9.
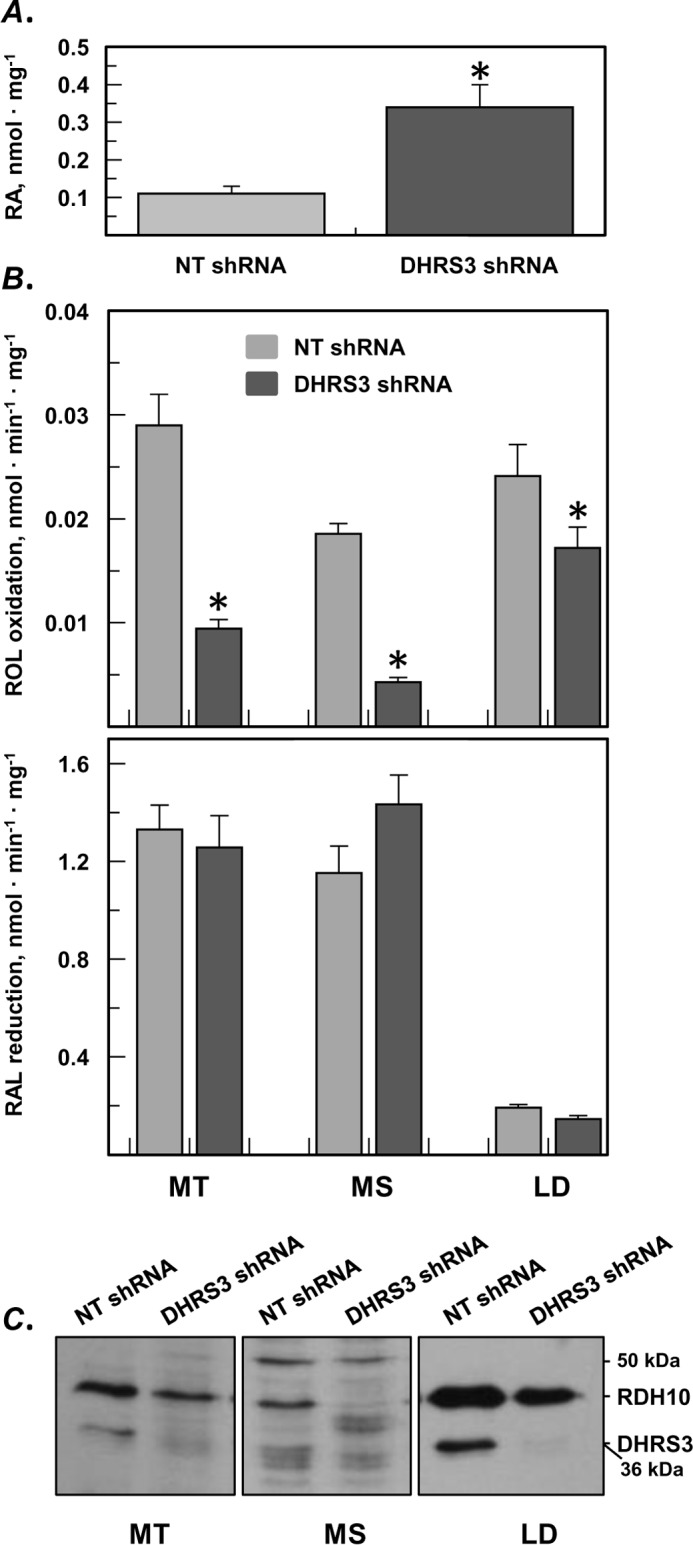
Endogenous RDH10 protein is reduced in the absence of DHRS3 but produces more RA because of disruption of the circuit. A, HepG2 cells stably transfected with shRNA targeting DHRS3 produce more RA than cells transfected with non-targeting (NT) shRNA. B, in vitro activities of subcellular fractions isolated from DHRS3-silenced HepG2 cells versus control cells (means ± S.D., n = 3). *, p < 0.05. Note the decrease in the retinol dehydrogenase activity of mitochondria (MT), microsomes (MS), and lipid droplets (LD) from DHRS3-silenced cells. C, Western blotting analysis of endogenous RDH10 and DHRS3 shows decreased levels of both proteins in subcellular fractions (50 μg each) of DHRS3-silenced HepG2 cells. ROL, retinol; RAL, retinaldehyde.
Surprisingly, the in vitro retinaldehyde reductive activities of the subcellular fractions did not change despite the reduction in DHRS3 protein, as confirmed by Western blotting analysis (Fig. 9B) and immunofluorescence (Fig. 8D). This observation suggests that the primary role of DHRS3 is to control the retinol dehydrogenase activity of RDH10 rather than to serve as a general purpose retinaldehyde reductase. The fact that DHRS3 is inactive as a retinaldehyde reductase in the absence of RDH10 also supports this conclusion.
Discussion
The work presented here provides the first evidence of the physical interaction between two proteins with antagonistic activities, RDH10 and DHRS3, and uncovers the fundamental biological significance of the hetero-oligomeric complex comprised of RDH10 and DHRS3 in the maintenance of RA homeostasis. We show that the protein-protein interaction between RDH10 and DHRS3 is highly specific for this pair of proteins, because neither human RDHE2 (SDR16C5), a retinol dehydrogenase closely related to RDH10 (SDR16C4) (20), nor human RDH11, a retinaldehyde reductase from a different family of SDRs (SDR7C1) (3), co-immunoprecipitate with RDH10 in pulldown assays. We also show that RDH10 and DHRS3 form homo-oligomers composed of at least two subunits and hetero-oligomers that include at least two subunits of each protein. Proximity ligation assay demonstrates that hetero-oligomerization occurs not only in cell-free assays but also in living cells.
The hetero-oligomeric assembly of RDH10 and DHRS3 resembles that of the recently described human 3-ketoacyl-acyl carrier protein reductase (KAR) (21). KAR is a heterotetramer composed of two subunits of 17β-hydroxysteroid dehydrogenase type 8 (HSD17B8) and two subunits of carbonyl reductase type 4. Like RDH10 and DHRS3, both components of KAR belong to the SDR family of proteins and share similarly low pair-wise protein sequence identity (35%) as RDH10 and DHRS3 (∼35%). HSD17B8 was shown to catalyze the reversible NAD(H)-dependent oxidation/reduction of steroid molecules in vitro, whereas carbonyl reductase type 4 was reported to exhibit an NADPH-dependent quinone reductase activity. In comparison, the NAD+-dependent RDH10 and the NADPH-dependent DHRS3 are known to catalyze the oxidoreductive interconversion of the same pair of substrates, retinol and retinaldehyde, and their hetero-oligomeric assembly essentially represents a bifunctional retinoid oxidoreductive complex (ROC).
Although there are many similarities between KAR and ROC, there are also notable differences. The subunits of KAR can form homotetramers, but when co-expressed in Escherichia coli, they form exclusively heterotetramers. In our hands, homo-oligomers of RDH10 and DHRS3 expressed in either insect Sf9 cells or HEK 293 cells exist simultaneously with ROC hetero-oligomers. Furthermore, it appears that RDH10 homo-oligomers form preferentially over hetero-oligomers in HEK 293 cells and that the abundance of homo- versus heterocomplex formation is controlled via the expression of DHRS3. Considering that the expression of DHRS3 is induced by RA, this could provide a mechanism for attenuation of RDH10 RA producing activity through the formation of ROC. In addition, unlike the components of KAR, each of which is catalytically active by itself, DHRS3 is completely dependent on the presence of RDH10 for its retinaldehyde reductive activity, and thus far, there is no evidence of other substrates being utilized by DHRS3.
We have also discovered that the components of ROC mutually stabilize each other. The stabilizing effect is especially noticeable with respect to DHRS3, whose half-life nearly triples in the presence of RDH10, but RDH10 also benefits from the presence of DHRS3, with its half-life extended by 5 h. The mutually stabilizing effect is very reproducible and is observed in three different cell lines: CHO, HepG2, and HEK 293 cells. As further evidence for interdependence between RDH10 and DHRS3 in ROC hetero-oligomer, ΔRDH10 mutant that is missing amino acid residues 85–109 and has reduced stability exhibits a destabilizing effect on wild-type DHRS3.
As shown by subcellular fractionation studies in combination with the analysis of mutual RDH10/DHRS3 activation and also by proximity ligation assays, ROC formation occurs throughout the cell, in membranes of endoplasmic reticulum, mitochondria, and lipid droplets. However, the mutual activation of RDH10 and DHRS3 in lipid droplets appears to be reduced in comparison with microsomes and mitochondria. Although the RDH10 retinol dehydrogenase activity of microsomes and mitochondria is increased 7–13-fold in the presence of DHRS3, in lipid droplets, the DHRS3-dependent increase in RDH10 activity is less than 2-fold. The reduced co-activation could potentially signal a weaker or less frequent protein-protein interaction between RDH10 and DHRS3 in lipid droplets and, hence, reduced formation of ROC hetero-oligomer.
In general, the observed increase in the retinol oxidizing activities of subcellular fractions containing ROC relative to fractions containing RDH10 alone is much greater than the apparent increase in their retinaldehyde reductive activities (up to 13-fold versus ∼2-fold). In part, the activation of DHRS3 could be masked by the higher baseline activity of HEK 293 cells toward the reduction of retinaldehyde as compared with the oxidation of retinol. At the same time, the high retinaldehyde reductive activity present in HEK 293 cells suggests the existence of other retinaldehyde reductases in addition to DHRS3 (3). This seems highly likely considering that DHRS3 appears to function as a designated RDH10 partner rather than as an independent retinaldehyde reductase.
A surprising finding of this study is that the rate of RA biosynthesis in whole cells containing ROC hetero-oligomer is largely independent of the concentration of ROC components. This is in contrast to the cells containing RDH10 alone, which increase their production of RA proportionally to increasing RDH10 protein. Our finding illustrates the principle of action of antagonistically bifunctional or so-called “paradoxical” components in biological circuit. The paradoxical components are known to operate in bacterial systems, but very few examples have been described so far in mammalian cells (22).
Antagonistically bifunctional components, such as RDH10 and DHRS3 in ROC, simultaneously have two opposing effects on the same biological process, i.e. interconversion of retinol and retinaldehyde in the case of ROC. As described here and in our previous study, an increase in the concentration of either component increases both the oxidation of retinol and the reduction of retinaldehyde, thus canceling out the effect on the steady-state output of RA. When the circuit is disrupted by substituting wild-type DHRS3 for its catalytically dead Y188A mutant or by silencing the expression of DHRS3 in HepG2 cells, the RA output changes dramatically, causing numerous physiological changes in the cells (12).
As described by Hart and Alon (22), the “paradoxical” components provide cell circuits with robustness: the ability to ensure a desired input-output relationship despite naturally occurring variations in the concentrations of the circuit components. The critical feature of bifunctional components necessary for providing robustness is that distinct monofunctional enzymes have to work together as a complex to carry out opposing reactions. A retinol dehydrogenase and a retinaldehyde reductase operating independently from each other would make the output of retinaldehyde and RA dependent on the concentrations of both proteins in the circuit. By analogy with ROC, we speculate that because KAR operates as antagonistically bifunctional complex, it is likely to control the homeostasis of an important metabolite, the identity of which remains to be determined.
Protein circuits are also critical for the correct patterning of tissues in development (22). The pattern formation in embryos is generally carried out by gradients of morphogens created by the diffusion of morphogen molecules away from their source. As is very well known for RA signaling during development, making too little or too much RA at its source can create a narrower or wider gradient and thus distort patterns. The bifunctional nature of the ROC appears to provide the signaling system with robustness to ensure the accurate gradient of RA despite naturally occurring fluctuations in RDH10 and DHRS3. Indeed, as shown by previous studies, genetic disruption of either RDH10 or DHRS3 results in abnormal levels of RA in embryonic tissues (11, 12).
The maintenance of RA homeostasis is critical for the health of cells and tissues, considering its numerous genomic and non-genomic effects. In this sense, proper functioning of ROC is essential for health. The RA homeostasis is known to be disrupted in various pathologies leading to cancer, metabolic dysfunction, immune deficiencies, etc. Future studies will show whether the underlying cause of perturbed RA homeostasis in some of these diseases is the disruption of ROC functionality.
Experimental procedures
Antibodies
Rabbit polyclonal anti-RDH10 and anti-DHRS3 antibody were purchased from Proteintech Group (Rosemont, IL), and anti-PLIN2 antibody was from Abcam (Cambridge, MA). Secondary goat anti-rabbit HRP-conjugated, donkey anti-rabbit Alexa Fluor 594-conjugated and donkey anti-chicken Alexa Fluor 488-conjugated antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Custom mouse polyclonal anti-DHRS3 antibodies were generated by Proteintech Group (Chicago, IL). Anti-FLAG antibodies were from Sigma-Aldrich. Secondary goat anti-mouse antibodies were from Jackson ImmunoResearch Laboratories.
Expression constructs
Primer sequences are provided in supplemental Table S1. Human wild-type DHRS3 and catalytically inactive Y188A DHRS3 FLAG-tagged constructs and RDH10 3× HA-tagged construct were described previously (12). These constructs served as templates for PCR amplication of the coding sequences using InvitrogenTM Platinum Pfx DNA polymerase (Thermo Fisher Scientific) to generate additional constructs described below. The catalytically inactive RDH10 mutant, Y210A RDH10-HA, was generated via site-directed mutagenesis using wild-type RDH10-HA construct in pIRES-hrGFP-2a vector (Stratagene, La Jolla, CA) as a template. Construct encoding DHRS3-HA fusion was generated by cloning the DHRS3 coding sequence into EcoRI-SalI sites of the pIRES-hrGFP-2a vector in frame with the C-terminal 3× HA tag. For RDH10-FLAG construct, coding sequence of RDH10 was cloned into EcoRI-XhoI sites of pCMVtag4a (Stratagene) in frame with the C-terminal FLAG tag. RDH10 mutant with deletion of amino acids 85–109 (ΔRDH10) was generated by mutagenesis of the full-length untagged RDH10 in pCMV-Tag4a using primers listed in supplemental Table S1. For RDH11-FLAG construct, coding sequence of human RDH11 was amplified and cloned into EcoRI-SalI sites of pCMV-tag4a in frame with the C-terminal FLAG tag.
For expression in insect Sf9 cells, sequences encoding HA and FLAG tag fusions in pCMV-Tag4a and pIRES-hrGFP-2a vectors were PCR-amplified and cloned into pVL1393 baculovirus transfer vector using BamHI-XbaI sites for DHRS3-HA fusion, EcoRI-PstI for RDH10-HA fusion, BamHI-XbaI for RDH10 and DHRS3-FLAG fusions, and EcoRI-BglII for RDH11-FLAG fusion. RDHE2 carrying a C-terminal FLAG-tag in pCMVtag4a was described previously (23). FLAG-tagged RDHE2 was amplified and inserted into BamHI and XbaI sites of pVL1393. The coding sequence of human DGAT2 was amplified from HepG2 cDNA, and cloned into EcoRI-XhoI sites of pCMV-tag4a in frame with the C-terminal FLAG tag. RDH10-His6 was described previously in (15). For construct encoding DHRS3-His6 in pVL1393, the coding sequence of human DHRS3 was PCR-amplified from DHRS3-FLAG construct and cloned into BamHI and NotI sites of the modified pVL1393 plasmid (originally described in Ref. 24 for RDH11-His6 construct) in-frame with the C-terminal His6 tag.
To generate a bicistronic construct expressing both RDH10 and DHRS3, the DHRS3 coding sequence was cloned into SmaI-XbaI sites in pIRESneo vector, downstream of the internal ribosome entry site (IRES) sequence. The IRES-DHRS3 cassette was amplified and cloned into EcoRI-SalI sites of the pCMV-Tag4a vector, downstream from the coding sequence of RDH10 cloned into BamHI-EcoRI sites.
Reverse transcription and quantitative PCR were performed as described previously (25). Relative gene expression levels were calculated using the comparative Ct method by normalization to reference genes.
Cell transfections
HEK 293, CHO, and HepG2 cells were routinely cultivated at 37 °C in a humid atmosphere containing 5% CO2. Transfections of mammalian cells were carried out using Lipofectamine 2000 (Thermo Fisher Scientific). For transfections with multiple plasmids, the total amount of DNA was equalized by the addition of empty vector. Transfection of Sf9 cells was performed as described previously (26).
For retinoid treatment of the intact cells, HEK 293 cells were grown and transfected at near confluency in 6-well culture dishes. The amount of plasmid constructs used for transfections varied as indicated under “Results” and in the figure legends. The ratio of Lipofectamine 2000 (μl) to the amount of plasmid DNA (μg) was 2:1. Treatment of cells with retinoids, retinoid extraction, and HPLC analysis were performed as described previously (15). Analysis of retinoid conversion in vitro was also performed as described previously (27).
For subcellular fractionation, the cells were grown in 10-cm cell culture dishes and transfected using 4 μg of RDH10-HA, 10 μg of DHRS3-FLAG constructs, and 28 μl of Lipofectamine 2000 per dish. For the induction of lipid droplets, cells in 10-cm dishes were first transfected with DGAT-expressing construct (14 μg of plasmid, 28 μl of Lipofectamine 2000 per dish), and the following day, with RDH10-HA and DHRS3-FLAG constructs as described above. To promote the growth of lipid droplets, DGAT-transfected HEK 293 cells were treated overnight with 0.4 mm sodium oleate added from 4 mm oleate, 20% bovine serum albumin stock in serum-free medium.
Subcellular fractionation
Stably transfected HepG2 cell lines or transiently transfected HEK 293 cells were rinsed with PBS and homogenized with a Dounce homogenizer in 25 mm Tricine, pH 7.8, 250 mm sucrose buffer. Cell homogenates were centrifuged at 3,000 × g for 10 min to obtain postnuclear fraction. Postnuclear fractions were centrifuged at 10,000 × g to pellet crude mitochondrial fraction. The 10,000 × g supernatants were supplemented with 2 m sucrose solution to adjust the sucrose concentration to 0.6 m. Samples (∼2.5 ml) were loaded into 5-ml ultracentrifuge tubes and overlaid with 1 ml of 0.15 m sucrose in reaction buffer (40 mm potassium phosphate, pH 7.4, 90 mm potassium chloride) and with another 1 ml of reaction buffer without sucrose. Gradients were centrifuged at 40,000 rpm in SW55Ti Beckman rotor for 1 h. Lipid droplet fraction was collected as a floating opaque layer on top of the gradients, and microsomal fractions were collected as a pellet in the bottom of the tubes. Mitochondrial and microsomal fractions were resuspended in reaction buffer with 20% glycerol.
Pulse-chase
HEK 293 cells were transfected with 1.5 μg of RDH10-HA and DHRS3-HA expression constructs, separately or in combination, in 6-well cell culture dishes. Each transfection variant was done in five separate wells. On the next day after transfection, the cells were starved for 30 min in serum-free DMEM without methionine and cysteine and pulsed with 100 μCi of Tran35S-LabelTM labeling reagent (MP Biomedicals, Santa Ana, CA) in DMEM without methionine and cysteine for 1 h. After removal of the labeling medium, the cells were rinsed with PBS and incubated in the regular growth medium for 3, 6, 12, and 24 h. The 0-h points were collected immediately after the removal of labeling medium. The cells were lysed in RIPA buffer, and lysates were frozen until all time points were collected. Lysates were incubated with 15 μl of anti-HA-agarose slurry overnight, and beads were pelleted and washed three times with 1 ml of RIPA buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40). Bound proteins were eluted by heating the beads in SDS-PAGE loading buffer, separated in 12% polyacrylamide gel, and analyzed by autoradiography. Densitometry analysis was performed using UN-SCANIT software. Protein half-lives were calculated using GraFit (Erithacus Software Ltd.).
Co-immunoprecipitation
Transfected HEK 293 cells, mitochondrial or microsomal fractions of HEK 293, or 30–40 μg of Sf9 microsomes expressing combinations of proteins of interest were solubilized with 400–500 μl of RIPA buffer for 10 min on ice. Samples were preincubated with 15 μl of Pierce recombinant protein A-agarose (Thermo Scientific) for 1 h at 4 °C. Protein A-agarose beads were pelleted in low speed centrifuge, and supernatants were supplemented with 15 μl of Pierce HA epitope tag antibody agarose conjugate (Thermo Scientific) that was preblocked with 2% BSA solution. Samples were incubated overnight at 4 °C with rotation. Beads were washed with 120 volumes of RIPA buffer supplemented with 1 m NaCl three times for 30 min at 27 °C with rotation. Bound proteins were eluted by heating beads in SDS-PAGE loading buffer supplemented with 100 mm DTT.
To differentiate between dimers and higher order oligomeric structure, 200 μg of Sf9 microsomes expressing combinations of proteins of interest were solubilized with RIPA buffer and incubated for 1 h with recombinant protein A-agarose at 4 °C and then applied to 50 μl ANTI-FLAG M2 affinity gel (Sigma-Aldrich) preblocked with 2% BSA. Agarose and solubilized microsomes were incubated at 4 °C for 3 h and then washed with 120 volumes of RIPA buffer for 90 min at 27 °C with rotation. Bound proteins were eluted with 50 μl of RIPA buffer supplemented with 1 μg/μl 3× FLAG peptide (ApexBio Technology, Houston, TX). Eluates were then applied to 40 μl of HA epitope tag antibody-agarose conjugate, and HA immunoprecipitations were performed as described above.
Protein samples were separated in 10 or 12% SDS-PAGE and transferred to PVDF Immobilon membranes (Millipore). The membranes were blocked with a 4% bovine serum albumin in Tris-buffered saline containing 0.05% Tween 20 (TBST) and incubated with different antibodies as indicated, in conjunction with the appropriate HRP-conjugated secondary antibody and Pierce enhanced chemiluminescent Western blotting substrate for detection of HRP (Thermo Scientific). Gel loading was determined by reprobing with HRP-conjugated monoclonal GAPDH antibody (1:5000; Sigma-Aldrich).
Immunocytochemistry
Wild-type HepG2 cells were grown on poly-l-lysine-coated coverslips placed in 6-well culture dishes. The cells were rinsed with PBS, fixed with 3% paraformaldehyde for 10 min, rinsed twice with PBS, permeabilized with 100 μm digitonin in PBS for 15 min, and rinsed with PBS three times. The samples were blocked with 10% heat-inactivated goat serum in PBS with 0.1% Tween 20 for 1 h at room temperature and incubated with rabbit anti-RDH10 or anti-DHRS3 antibody (1:200) and chicken anti-PLIN2 antibody (1:200) in blocking solution overnight at 4 °C. Next, samples were washed three times with PBS with 0.1% Tween 20 and incubated with fluorescently conjugated secondary antibody (1:500) in blocking solution for 1 h at room temperature. After three washes with PBS, coverslips were counterstained with 1 μg/ml DAPI and mounted in fluorescent microscopy medium. Visualization was performed using Zeiss Axioplan (Model) microscope.
Proximity ligation assay
HepG2 cells were plated on top of poly-l-lysine-coated coverslips and placed in 6-well culture dishes. The cells were transfected using 4 μg of bicistronic RDH10-IRES-DHRS3 construct and 8 μl of Lipofectamine 2000 per well. The cells on the coverslips were fixed and permeabilized as described for immunocytochemistry. The samples were blocked in 2% BSA in PBS with 0.1% Tween 20 and incubated with rabbit polyclonal anti-RDH10 (1:400) and mouse polyclonal anti-DHRS3 (1:400) antibody in blocking solution overnight at 4 °C. Detection was performed using Duolink proximity ligation kit (Sigma-Aldrich) according to the manufacturer's protocol.
Statistical analysis
Unpaired t test was used to test for statistical significance. Quantitative PCR data are presented as the means ± S.E.
Author contributions
O. V. B., M. K. A., and N. Y. K. designed the research; O. V. B., M. K. A., and L. W. performed the research; and O. V. B., M. K. A., L. W., and N. Y. K. analyzed the data and wrote the paper.
Supplementary Material
This work was supported by National Institutes of Health Grant AA12153 from the National Institute on Alcohol Abuse and Alcoholism. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1 and Figs. S1–S3.
- RA
- all-trans-retinoic acid
- ROC
- retinoid oxidoreductive complex
- RDH10
- retinol dehydrogenase 10
- DHRS3
- dehydrogenase reductase 3
- SDR
- short chain dehydrogenase/reductase
- RDHE2
- retinol dehydrogenase epidermal 2
- RDH11
- retinol dehydrogenase 11
- DGAT
- diacylglycerol acyltransferase
- KAR
- 3-ketoacyl-acyl carrier protein reductase
- IRES
- internal ribosome entry site
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- RIPA
- radioimmune precipitation assay.
References
- 1. Al Tanoury Z., Piskunov A., and Rochette-Egly C. (2013) Vitamin A and retinoid signaling: genomic and nongenomic effects. J. Lipid Res. 54, 1761–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clagett-Dame M., and Knutson D. (2011) Vitamin A in reproduction and development. Nutrients 3, 385–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kedishvili N. Y. (2013) Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 54, 1744–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ross A. C., and Zolfaghari R. (2011) Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 31, 65–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Byrne S. M., and Blaner W. S. (2013) Retinol and retinyl esters: biochemistry and physiology. J. Lipid Res. 54, 1731–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee L. M., Leung C. Y., Tang W. W., Choi H. L., Leung Y. C., McCaffery P. J., Wang C. C., Woolf A. S., and Shum A. S. (2012) A paradoxical teratogenic mechanism for retinoic acid. Proc. Natl. Acad. Sci. U.S.A. 109, 13668–13673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Napoli J. L. (1986) Retinol metabolism in UC-PKI cells: characterization of retinoic acid synthesis by an established mammalian cell line. J. Biol. Chem. 261, 13592–13597 [PubMed] [Google Scholar]
- 8. Sandell L. L., Sanderson B. W., Moiseyev G., Johnson T., Mushegian A., Young K., Rey J. P., Ma J. X., Staehling-Hampton K., and Trainor P. A. (2007) RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 21, 1113–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rhinn M., Schuhbaur B., Niederreither K., and Dollé P. (2011) Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc. Natl. Acad. Sci. U.S.A. 108, 16687–16692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sandell L. L., Lynn M. L., Inman K. E., McDowell W., and Trainor P. A. (2012) RDH10 oxidation of vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PLoS One 7, e30698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Billings S. E., Pierzchalski K., Butler Tjaden N. E., Pang X. Y., Trainor P. A., Kane M. A., and Moise A. R. (2013) The retinaldehyde reductase DHRS3 is essential for preventing the formation of excess retinoic acid during embryonic development. FASEB J. 27, 4877–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adams M. K., Belyaeva O. V., Wu L., and Kedishvili N. Y. (2014) The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J. Biol. Chem. 289, 14868–14880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kavanagh K. L., Jörnvall H., Persson B., and Oppermann U. (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol. Life Sci. 65, 3895–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhatia C., Oerum S., Bray J., Kavanagh K. L., Shafqat N., Yue W., and Oppermann U. (2015) Towards a systematic analysis of human short-chain dehydrogenases/reductases (SDR): Ligand identification and structure-activity relationships. Chem. Biol. Interact. 234, 114–125 [DOI] [PubMed] [Google Scholar]
- 15. Belyaeva O. V., Johnson M. P., and Kedishvili N. Y. (2008) Kinetic analysis of human enzyme RDH10 defines the characteristics of a physiologically relevant retinol dehydrogenase. J. Biol. Chem. 283, 20299–20308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deisenroth C., Itahana Y., Tollini L., Jin A., and Zhang Y. (2011) p53-inducible DHRS3 is an endoplasmic reticulum protein associated with lipid droplet accumulation. J. Biol. Chem. 286, 28343–28356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang W., and Napoli J. L. (2013) The retinol dehydrogenase Rdh10 localizes to lipid droplets during acyl ester biosynthesis. J. Biol. Chem. 288, 589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cerignoli F., Guo X., Cardinali B., Rinaldi C., Casaletto J., Frati L., Screpanti I., Gudas L. J., Gulino A., Thiele C. J., and Giannini G. (2002) retSDR1, a short-chain retinol dehydrogenase/reductase, is retinoic acid-inducible and frequently deleted in human neuroblastoma cell lines. Cancer Res. 62, 1196–1204 [PubMed] [Google Scholar]
- 19. Zolfaghari R., Chen Q., and Ross A. C. (2012) DHRS3, a retinal reductase, is differentially regulated by retinoic acid and lipopolysaccharide-induced inflammation in THP-1 cells and rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G578–G588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adams M. K., Lee S. A., Belyaeva O. V., Wu L., and Kedishvili N. Y. (2016) Characterization of human short chain dehydrogenase/reductase SDR16C family members related to retinol dehydrogenase 10. Chem. Biol. Interact. pii, S0009–2797(16)30494-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Venkatesan R., Sah-Teli S. K., Awoniyi L. O., Jiang G., Prus P., Kastaniotis A. J., Hiltunen J. K., Wierenga R. K., and Chen Z. (2014) Insights into mitochondrial fatty acid synthesis from the structure of heterotetrameric 3-ketoacyl-ACPreductase/3R-hydroxyacyl-CoA dehydrogenase. Nat. Commun. 5, 4805. [DOI] [PubMed] [Google Scholar]
- 22. Hart Y., and Alon U. (2013) The utility of paradoxical components in biological circuits. Mol. Cell 49, 213–221 [DOI] [PubMed] [Google Scholar]
- 23. Lee S. A., Belyaeva O. V., and Kedishvili N. Y. (2008) Effect of lipid peroxidation products on the activity of human retinol dehydrogenase 12 (RDH12) and retinoid metabolism. Biochim. Biophys. Acta 1782, 421–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Belyaeva O. V., Stetsenko A. V., Nelson P., and Kedishvili N. Y. (2003) Properties of short-chain dehydrogenase/reductase RalR1: characterization of purified enzyme, its orientation in the microsomal membrane, and distribution in human tissues and cell lines. Biochemistry 42, 14838–14845 [DOI] [PubMed] [Google Scholar]
- 25. Gough W. H., VanOoteghem S., Sint T., and Kedishvili N. Y. (1998) cDNA cloning and characterization of a new human microsomal NAD+-dependent dehydrogenase that oxidizes all-trans-retinol and 3α-hydroxysteroids. J. Biol. Chem. 273, 19778–19785 [DOI] [PubMed] [Google Scholar]
- 26. Belyaeva O. V., Lee S. A., Adams M. K., Chang C., and Kedishvili N. Y. (2012) Short chain dehydrogenase/reductase rdhe2 is a novel retinol dehydrogenase essential for frog embryonic development. J. Biol. Chem. 287, 9061–9071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee S. A., Belyaeva O. V., Wu L., and Kedishvili N. Y. (2011) Retinol dehydrogenase 10 but not retinol/sterol dehydrogenase(s) regulates the expression of retinoic acid-responsive genes in human transgenic skin raft culture. J. Biol. Chem. 286, 13550–13560 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.