

Abstract
Purpose:
Our goal was to design a customized microarray, arrEYE, for high-resolution copy number variant (CNV) analysis of known and candidate genes for inherited retinal dystrophy (iRD) and retina-expressed noncoding RNAs (ncRNAs).
Methods:
arrEYE contains probes for the full genomic region of 106 known iRD genes, including those implicated in retinitis pigmentosa (RP) (the most frequent iRD), cone–rod dystrophies, macular dystrophies, and an additional 60 candidate iRD genes and 196 ncRNAs. Eight CNVs in iRD genes identified by other techniques were used as positive controls. The test cohort consisted of 57 patients with autosomal dominant, X-linked, or simplex RP.
Results:
In an RP patient, a novel heterozygous deletion of exons 7 and 8 of the HGSNAT gene was identified: c.634-408_820+338delinsAGAATATG, p.(Glu212Glyfs*2). A known variant was found on the second allele: c.1843G>A, p.(Ala615Thr). Furthermore, we expanded the allelic spectrum of USH2A and RCBTB1 with novel CNVs.
Conclusion:
The arrEYE platform revealed subtle single-exon to larger CNVs in iRD genes that could be characterized at the nucleotide level, facilitated by the high resolution of the platform. We report the first CNV in HGSNAT that, combined with another mutation, leads to RP, further supporting its recently identified role in nonsyndromic iRD.
Genet Med 19 4, 457–466.
Keywords: copy-number variation, customized array CGH, HGSNAT, inherited retinal dystrophy, ncRNA
Introduction
Retinitis pigmentosa (RP), the most frequently inherited retinal dystrophy (iRD), is characterized by tremendous genetic heterogeneity, with mutations in more than 70 genes explaining 30 to 80% of cases, depending on the mode of inheritance.1 Although RP is clinically distinct from other iRDs, end-stage RP may be difficult to differentiate from late stages of some cone–rod dystrophies (CRD) and macular dystrophies (MDs).
To date, targeted next-generation sequencing (NGS) of gene panels and whole-exome sequencing (WES) are commonly used approaches to establish a molecular diagnosis of RP. These sequencing techniques are aimed mainly at identifying single-nucleotide variants and small insertions and deletions in coding sequences of known and candidate genes, but they are less suitable for finding coding or noncoding copy number variations (CNVs). Although a higher prevalence of CNVs has been described in a limited number of iRD genes such as PRPF31,2 EYS,3,4 KCNV2,5 and USH2A,3,4,6 CNV analysis is not systematically included in the genetic workup of iRD, probably leading to an underestimation of CNVs as a cause of iRD. Indeed, Khateb et al. recently identified subtle single-exon to multigene deletions involving EYS, MYO7A, NPHP4, RPGR, and CHM in 10% of a cohort of 60 iRD cases using WES data, emphasizing the importance of CNV screening in iRD.7
CNV analysis in iRD using WES, targeted NGS, and whole-genome sequencing (WGS) data has been proven to be increasingly successful.3,4,7,8,9 Because exons are unevenly distributed within the genome, algorithms using read depth are recommended for CNV analysis using WES and targeted NGS data. Despite the development of different CNV tools over the past years, none has the same performance of microarray-based comparative genomic hybridization (array CGH). The WES CNV analyses have variable performance, with a high false-positive rate, low sensitivity, and a duplication bias. To avoid a high false-positive rate, high-coverage data are required.7,10 By improving the design of targeted NGS approaches, high-coverage NGS data can be easily obtained.4,8,11 Despite higher sensitivity and specificity of recent tools, additional validation of identified CNVs and low-quality targets are still recommended to avoid false-positive and false-negative results, respectively.12 WGS is more reliable than WES for CNV assessment because it covers the breakpoint junctions that mostly reside in intronic regions, thus facilitating the detection of more complex CNVs, and because it performs with a greater uniform distribution of sequencing-quality parameters.3 Obtaining high-coverage data is a prerequisite for accurate CNV analysis using WGS, thus increasing its costs. A combined approach of NGS and array CGH allows identification of a wide range of mutations for heterogeneous diseases such as iRD and allows for overcoming the aforementioned issues.
Here, we developed a customized array CGH platform, called arrEYE, for copy-number analysis of coding and noncoding regions of known and candidate RP, CRD, and MD genes and of retina-expressed noncoding RNAs (ncRNAs) such as micro RNAs (miRNAs) and long intergenic noncoding RNAs (lincRNAs). The arrEYE chip was validated using seven positive control samples with eight CNVs in known RP genes, which had been identified using other techniques. Next, 57 prescreened index cases with RP underwent CNV analysis using arrEYE. We discuss the identification of three novel CNVs in iRD genes, including HGSNAT, which was recently associated with nonsyndromic iRD, USH2A, and RCBTB1, a recently discovered novel iRD gene. We demonstrate that arrEYE allows detection of subtle single-exon to larger CNVs in known and candidate iRD genes and that novel identified CNVs could be easily characterized at the nucleotide level, thus allowing unraveling of their underlying mechanisms.
Materials and Methods
Ethics statement
This study was conducted following the tenets of the Declaration of Helsinki.
Patient samples
We studied 57 patients (P1 to P57) initially diagnosed with autosomal dominant (n=39), X-linked (n = 3), or simplex (n = 15) RP and referred for molecular genetic testing by either ophthalmologists or clinical geneticists. Genomic DNA (gDNA) was extracted from leukocytes using the QIAamp DNA mini kit (Qiagen, Antwerp, Belgium), the Gentra Puregene Cell kit (Qiagen, Antwerp, Belgium), or the ReliaPrep Large-Volume HT gDNA Isolation System (Promega, Leiden, Netherlands) according to the manufacturer's descriptions. Most patients previously underwent adRP APEX chip analysis (Asper Ophthalmics, Tartu, Estonia). All patients were tested for mutations in coding exons and intron–exon boundaries of the four most prevalent adRP genes (RHO, RP1, PRPH2, PRPF31).
Seven positive controls were included (PC1 to PC7), with eight CNVs in one of the genes present on the customized array, hereafter called arrEYE.
Microarray probe design
We used the Agilent oligonucleotide-based microarray technology and the SurePrint 8 × 60K format. The arrEYE microarray design was developed using the Web-based application SureDesign (https://earray.chem.agilent.com/suredesign/; Agilent Technologies, Santa Clare, CA). We created five different probe groups, each containing a specific set of genes and miRNAs or lincRNAs. The Agilent High-Definition Database (July 2015, human reference sequence, hg19, Build GRCh37, February 2009) was used to select probes. More detailed information is available in the Supplementary data (Supplementary Table S1 online).
Microarray-based comparative genomic hybridization (array CGH) and data analysis
In brief, 200 ng of genomic DNA was labeled using the BioPrime Array CGH Genomic Labeling Module (Invitrogen, Merelbeke, Belgium) using cyanine 5 (Cy5)-labeled (control gDNA; Promega) and cyanine 3 (Cy3)-labeled (patient gDNA) deoxycytidine triphosphate (dCTPs; GE Healthcare, Diegem, Belgium). Following precipitation, hybridization with Cot-1 DNA, and washing, the array slides were scanned using an Agilent DNA Microarray Scanner and quantified with Agilent Feature Extraction Software (version 10.7.1.1). The data were further processed, analyzed, and visualized using the ViVar platform13 (October 2015; https://www.cmgg.be/vivar/) using a circular binary segmentation algorithm. Prior to the validation, we considered a region deleted or duplicated when the log2 ratio encompassing at least three probes was below −0.35 or above +0.35, respectively. The derivative log ratio (DLR), which calculated the probe-to-probe log ratio noise, was used as a quality parameter. Typically, a DLR spread of 0.20 is considered a threshold for detecting small aberrations. A DLR ≤0.19 is accepted as good array quality, a DLR between 0.20 and 0.29 is considered borderline, and a DLR ≥0.30 should be rejected.14
qPCR validation of CNVs
CNVs found in the patient samples were confirmed using qPCR (LightCycler 480; Roche, Vilvoorde, Belgium; data analysis: qbase+ 3.0, Biogazelle, Zwijnaarde, Belgium; Supplementary Table S2 online). The genomic positions of the CNVs found by array CGH were used to design primers for junction PCR and breakpoint sequencing. qPCR primers were designed using PrimerQuest (http://eu.idtdna.com/primerquest/home/index) and PrimerXL (http://www.primerxl.org). Junction primers were designed using Primer3Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi).
CNV and variant interpretation
Several databases were used to interpret the pathogenic significance of identified CNVs. The Database of Genomic Variants (http://www.ncbi.tcag.ca/varation) continuously updates CNVs found in healthy controls. The Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER; https://decipher.sanger.ac.uk/) catalogs clinically relevant CNVs. In addition, genetic disorders caused by CNVs are cataloged in the Online Mendelian Inheritance in Man (OMIM; http://www.cnbi.nlm.nih.gov/omim) database.
The functional impact of sequence variants was assessed based on the outcome of in silico predictions performed in Alamut 2.7 (Interactive Biosoftware, Rouen, France).
PCR and Sanger sequencing
All exons and exon–intron boundaries of the RCBTB1, HGSNAT, and USH2A genes were PCR-enriched and Sanger-sequenced in P4, P33, and P53, respectively. Primers were designed (Supplementary Table S3 online) using the online primer design software tools Primer3Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) and pxlence (https://pxlence.com).
Results
Customized microarray probe design
Details about the genes, number of probes, and the median probe spacing are provided in Supplementary Table S1 online.
The first probe group included 109 genes known to cause autosomal dominant, recessive, or X-linked RP, CRD, or MD: genes listed in RetNet (https://sph.uth.edu/retnet) (n = 106) and three candidate genes.
The second probe group was based on a data set generated by cone–rod homeobox (CRX) chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq) in adult mouse retina. CRX is a key transcription factor for photoreceptor differentiation and survival that regulates downstream photoreceptor transcription factors and their target genes. The CRX ChIP-seq data set provided a map of cis-regulatory CRX-binding regions (CBRs) clustering around photoreceptor genes.15 By combining the CBR data with the genomic positions of published RP candidate (RP6, RP22, RP24, RP29, RP32, RP34, RP63), a total of 36 candidate genes were included.
The third probe group contained genes that are directly connected to seven splicing factor genes that have previously been implicated in adRP (RetNet). Six of these encoded proteins of the tri-small nuclear ribonucleoprotein (tri-snRNP) complex. Gene selection was first based on the work by Benaglio et al.,16 in which four tri-snRNP splicing factor genes (EFTUD2, AAR2, NHP2L1, and PRPF4) were screened in adRP cases. Furthermore, spliceosomal proteins shown to interact with proteins previously implicated in adRP were included.17 Finally, CNOT3, known to modulate the effect of PRPF31 mutations, was included.18 Overall, probe group 3 contained 21 candidate genes.
The fourth probe group was composed of miRNAs potentially regulating gene expression in the eye. First, 220 miRNAs from miRNeye, a high-resolution expression atlas of miRNAs expressed in the developing and adult wild-type mouse eye, were included.19 Second, literature-based selection of miRNAs associated with iRD (e.g., miR-204),20 miRNAs with a differential retinal expression in RP mouse models (e.g., miR-96),21 and miRNAs expressed in human retina (e.g., miR-124)22 resulted in 27 additional miRNAs. The miRBase Tracker (http://www.mirbasetracker.org) was used to map the miRNA names to the most recent miRBase accession number. Human homologs were selected by performing a very stringent BLAST alignment (e-value <0.02) of the miRNA sequences to all human miRNAs in miRBase (See R code in Supplementary File S1 online).23
The fifth probe group contained evolutionarily conserved lincRNAs expressed in retinal tissues.24
Validation of the customized microarray
Validation of previously identified CNVs. Samples were included from seven patients (PC1-7) with eight CNVs identified using either MLPA (heterozygous CNVs) or nonamplification with conventional PCR (homozygous CNVs).25 Of these eight CNVs, seven were detected using arrEYE. The first CNV (PC1; Figure 1a, Table 1), a heterozygous duplication encompassing exon 4 to exon 13 of the USH2A gene, showed a change of 443 probes (ratio: 0.5325), thus confirming this duplication. The second CNV (PC2; Figure 1b, Table 1), a heterozygous deletion of USH2A exon 44, showed decreased signals for six probes covering exon 44 and its intron–exon boundaries (ratio: −0.5614). The deletion had previously been delineated by qPCR and Sanger sequencing of the junction product (chr1:g.(216040210_216042178)del). Using the arrEYE chip, we were able to approximately define the breakpoint boundaries (chr1:g.(216039973_216040269)_(216041753_216042428)del). A third CNV, a heterozygous deletion of exons 22–24 of USH2A (PC3; Figure 1c, Table 1), displayed lower signals for 201 probes (ratio: −0.52), thus confirming this heterozygous deletion. The fourth CNV (PC4, Figure 1d, Table 1), a heterozygous BEST1 deletion of exons 1 and 2, showed decreased signals for 15 probes encompassing these exons (ratio: −0.5393). The fifth and sixth CNVs, which were two distinct, compound, heterozygous deletions both encompassing the last exon (exon 2) of KCNV2 (PC5; Figure 1e, Table 1), were identified as a homozygous deletion by a decreased signal of 20 probes (ratio: −1.31). The sixth and seventh positive control samples performed with a poor DLR spread (DLR > 0.20), thereby hampering an accurate CNV analysis. The seventh CNV, a homozygous SPATA7 deletion of exons 1–4 (PC6; DLR = 0.2166), showed absent signals for 86 probes (probes dropped to a ratio of −1.26), thereby confirming the homozygous deletion (Supplementary Figure S1 online, Table 1). An eighth CNV, a heterozygous deletion of ABCA4 exons 20–22 (PC7; DLR = 0.4233), could not be detected due to the spread of the probes as a result of poor DNA quality.
Figure 1.

Array CGH profiles of copy number variations in positive controls. E: exon. Crossed dots indicate probes with a ratio outside the shown y-axis, with no further impact on the calculations. (a) Positive control 1. A heterozygous duplication of exon 4 to exon 13 in USH2A. (b) Positive control 2. A heterozygous deletion of exon 44 in USH2A. (c) Positive control 3. A heterozygous deletion of exons 22 to 24 in USH2A. (d) Positive control 4. A heterozygous deletion of exon 1 and exon 2 in BEST1. (e) Positive control 5. A compound heterozygous deletion of exon 2 in KCNV2.
Table 1. Copy-number variations detected using arrEYE v1.0 (GRCh37/hg19).

Defining the diagnostic thresholds. To assess the DLR cut-off of our arrEYE platform, a total of 36 exonic putative CNVs found in our RP test cohort (regardless of the DLR value and the number of altered probes) were validated using qPCR (Supplementary Tables S2 and S4 online). Of these, three CNVs could be validated as true CNVs. For each CNV, including the seven validated CNVs of the positive control samples, the number of altered probes was plotted against the DLR ratio of the sample (42 CNVs in total; Supplementary Figure S2 online; Supplementary Table S4 online). A clear-cut threshold of 0.1467 for the DLR ratio and a threshold of more than five altered probes are needed to obtain an accurate result. One CNV, however, met the aforementioned threshold values (P51; DLR: 0.1199; 24 probes) but could not be validated using qPCR. To explain this outlier, we must take into account the dynamic range of the log2 ratio, which is another important parameter to evaluate the microarray experiment. A deletion gives a theoretical log2 ratio of −1 (log2 1/2), whereas a duplication gives a theoretical log2 ratio of 0.58 (log2 3/2). In practice, these values have a dynamic range due to nonspecific hybridization, autofluorescence, washing conditions, and background noise. The upper threshold for duplication was set at 0.35 and that for deletions was set at −0.35. The ratio value of this specific deletion is −0.36, which is close to the lower threshold set. All heterozygous deletions detected using the arrEYE platform reach a ratio value between −0.51 and −0.81 (Table 1). To exclude false-positive results, the lower threshold was set to at least −0.37 (but preferably −0.50).
Identification of CNVs in three probands with iRD
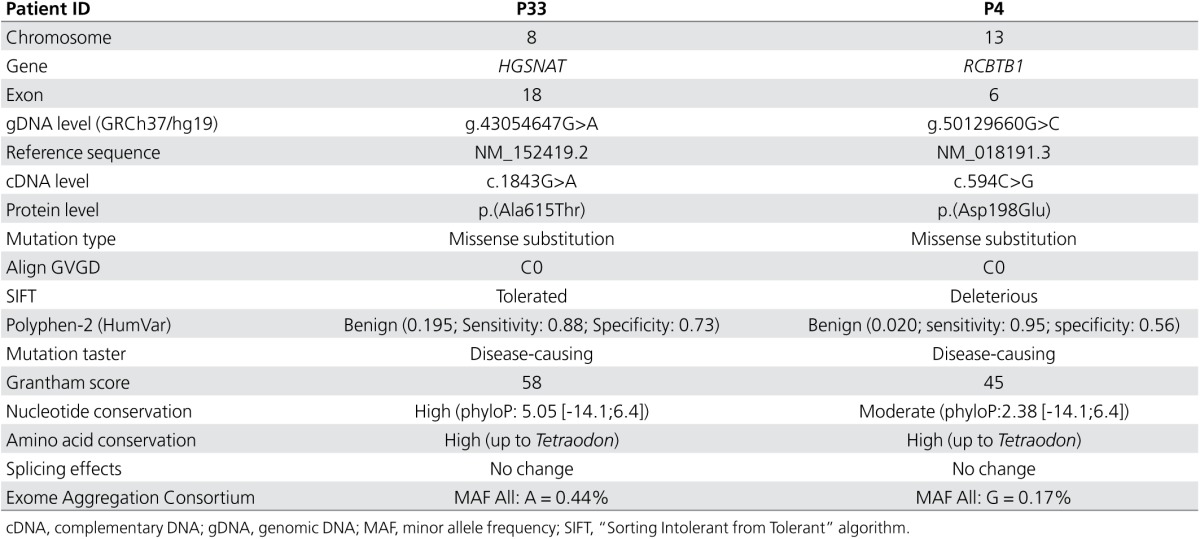
Heterozygous exons 7 and 8 deletion in novel iRD gene HGSNAT. Array CGH revealed a heterozygous 2.5-kb deletion of exons 7 and 8 of the HGSNAT gene in a male patient (P33) with sporadic RP (Figure 2). The deletion was confirmed by qPCR in both the male patient and his clinically unaffected daughter. Using primers flanking the altered probes, the breakpoints of the deletion could be determined at the nucleotide level: c.634-408_820+338delinsAGAATATG (chr8:43,025,320_43,027,867delinsAGAATATG) (Table 1, Figure 2e). This deletion creates a frameshift p.(Glu212Glyfs*2) that might be targeted by nonsense-mediated decay. Except for two deletions (spanning more than 1 Mb), no other overlapping structural variations were reported in any of the investigated databases. Sanger sequencing of the HGSNAT gene revealed missense variant c.1843G>A, p.(Ala615Thr) in exon 18. The alanine residue is highly conserved, but the change is predicted as tolerated by several in silico analyses. The variant represents a known SNP (rs112029032) with an overall minor allele frequency of 0.44% (ExAC) (Table 2). However, this missense variant has recently been described in a homozygous state in three Dutch siblings with nonsyndromic RP with just one of the two alleles in cis with the missense variant c.398G>C p.(Gly133Ala) (MIM 616544).26 In addition, the p.(Ala615Thr) variant has also been reported in patients with mucopolysaccharidosis type IIIC (MPS IIIC, MIM 252930); in this case, it was in cis with either a splice site mutation or a missense mutation.27 Segregation analysis showed that the deletion found here and p.(Ala615Thr) are located in trans (Figure 2b). The patient (P33) had late-onset RP with initial night blindness since age 47 years and slow disease progression. A characteristic hyperautofluorescent ring surrounding the central macula, midperipheral outer retinal atrophy with intraretinal pigmentation of mixed specular and nummular types, and attenuated retinal vasculature were seen (Figure 2a). No systemic disease manifestations were recorded.
Figure 2.

Heterozygous deletion encompassing exons 7 and 8 of HGSNAT. (a) From the top down: composite fundus pictures; infrared reflectance composites and optical coherence tomography (OCT) of right eye; and blue light fundus autofluorescence (BAF) image of left eye of patient P33. LE: left eye, RE: right eye. The fundoscopy shows typical signs of RP, with outer retinal atrophy and mostly spicular but some nummular intraretinal pigmentation in the midperiphery, with better preservation of retina in the far periphery and pericentral macula. Infrared reflectance composite fundus images of patient P33 (HRA2; Heidelberg, Germany) illustrates these signs further. OCT of the central macula shows severe atrophy of outer retinal layers representing retinal pigment epithelium and photoreceptors, with some remnants remaining in immediate perifoveal area and extreme thinning of fovea. On BAF image of the LE, severe outer retinal atrophy in the posterior pole with hyperautofluorescent ring, which is surrounded by a ring of outer retinal atrophy with highly visible, fibrotic, larger choroidal vessels; around that, another concentric area of hypoautofluorescence is coincident with an area of intraretinal pigmentation. (b) Segregation analysis of the identified deletion and unclassified variant c.1843G>A p.(Ala615Thr). III:1: proband patient P33. (c) Array CGH profile. E: exon. (d) qPCR plot. Exons 6 and 9 have a normal copy number (gray bars), whereas exons 7 and 8 have only 1 copy (reddish bars). E: exon. (e) Schematic representation of the HGSNAT deletion. From the top down: localization of the deletion relative to the entire HGSNAT gene; zoomed to the level of exons 7 and 8; breakpoint regions; and sequence level of the junction. The nucleotides of the breakpoint regions are indicated with a gray and pink bar. Eight nucleotides (AGAATATG) are inserted in between. Red bar: deletion; blue bar: coding region; blue line: intronic region.
Table 2. Identified sequence variations and in silico predictions.
Novel heterozygous deletion in USH2A. In a patient (P53) with late-onset RP, a heterozygous 24-kb deletion of exons 42 to 44 of the USH2A gene was found (Figure 3). Sequencing of the breakpoint junction fragment revealed the exact position of the breakpoints: c.8223+4491_8845+6927del (chr1:216033422_216057277del) p.Val2742Leufs*25 (Table 1, Figure 3d). No overlapping deletions were reported in any of the investigated databases. The USH2A gene is known to cause Usher syndrome, an autosomal recessive disorder characterized by mild to severe hearing impairment and RP (MIM 276901). The USH2A gene has been associated with nonsyndromic RP (MIM 613809).28 Targeted resequencing of USH2A and of a deep intronic region covering the location of a known pathogenic deep intronic mutation (c.7595-2144A>G)29 did not reveal a second mutation. Although other deep intronic and regulatory mutations cannot be excluded, a mutation in another gene may underlie the phenotype because both the father and the brother of the patient presented with iRD, which is rather compatible with autosomal dominant inheritance (Figure 3c).
Figure 3.

Heterozygous deletion encompassing exons 42 to 44 of USH2A. (a) Array CGH profile. E: exon. (b) qPCR plot proband (P53). Exons 41 and 45 have a normal copy number (gray bars), whereas exon 42 to 44 have only one copy (reddish bars). (c) Segregation analysis of the identified deletion. II:4: proband patient P53. (d) Schematic representation of the USH2A deletion. From the top down: localization of the deletion relative to the entire USH2A gene; zoomed to the level of exon 42 to 44; breakpoint regions; and sequence level of the junction. The nucleotides of the breakpoint regions are indicated with a gray and pink bar. The two nucleotides (GT) with microhomology at the breakpoints are indicated with a turquoise bar. Red bar: deletion; blue bar: coding region; blue line: intronic region.
Heterozygous duplication in novel iRD gene RCBTB1. A heterozygous duplication containing the first nine exons of RCBTB1 was identified in a patient (P4) with RP but with chorioretinal atrophy more pronounced in the macula; this patient had the first symptoms of night blindness at age 47 years (Supplementary Figure S3 online). He has a maternal uncle affected by typical RP and a clear macular chorioretinal dystrophy who has exhibited night blindness since the age of 26 years. The RCBTB1 duplication was not found in the maternal uncle. A duplication encompassing the same exons is present in the DECIPHER database (patient identifier: 255256). This patient inherited this allele from a healthy parent. A 68.78-kb de novo duplication on chromosome 15 was also described for this patient. The provided phenotypical features of the patient do not point to any retinal dystrophy or other eye disorder.
Sequencing of RCBTB1 in P4 revealed a heterozygous missense variant of unknown significance: c.594C>G p.(Asp198Glu) (Table 2). The affected uncle was heterozygous for the same missense variant.
Discussion
Here, we report a customized oligonucleotide array for copy-number screening in iRD. More specifically, the arrEYE platform enables identification of CNVs in known iRD genes responsible for RP, CRD, and MD, and also in several interesting candidate genes for RP and retina-expressed ncRNAs.
The arrEYE platform uncovered a novel CNV in the HGSNAT gene in a patient with RP who was found to be compound heterozygous for this CNV and a known missense variant c.1843G>A p.(Arg615Thr). HGSNAT mutations have previously been associated with MPS IIIC or Sanfilippo syndrome type C, an autosomal recessive lysosomal storage disorder caused by deficiency of heparan-α-glucosaminide N-acetyltransferase. Interestingly, mutations in HGSNAT, one of which was the missense variant found here, were recently reported in patients with nonsyndromic RP.26 The late-onset phenotype described here (Figure 2) is entirely in keeping with those reported in that study. Transient overexpression of p.(Arg615Thr) in cell lines showed a slightly reduced expression of activity (50–80% enzymatic activity) compared to the wild type.27,30 The p.(Trp403Cys) mutation is often found in cis with p.(Arg615Thr), which significantly reduces both HGSNAT protein expression and activity.30 The hypomorphic p.(Arg615Thr) variant in combination with a more severe allele might explain the nonsyndromic RP phenotype seen here and in the previously reported Dutch family. A higher expression of Hgsnat in mouse retina compared with other tissues affected in MPS IIIC suggests the need for higher HGSNAT activity to maintain proper function of the retina and might explain the retinal involvement in the case of more subtle changes in HGSNAT dosage or activity.26 This has also been described in other genes associated with multisystem lysosomal storage diseases (CLN3 and MFSD8).31,32
The smallest clinically relevant CNV identified in our patient cohort was a 1.9-kb single-exon deletion of USH2A that could be detected because of the design of consecutive probes covering the entire gene, including introns. This high-resolution probe design, covering intronic regions, also facilitated the delineation of CNVs and the establishment of their underlying mechanisms, such as nonallelic homologous recombination, transposable element insertions, or nonhomologous (NH) mechanisms. Little or no homology at the breakpoints might indicate a NH mechanism. Nonrecurrent CNVs can be caused by various events such as nonhomologous end-joining and microhomology-mediated end-joining, or by a template-switching mechanism during replication.33 No microhomology (MH) was found at the breakpoints of the HGSNAT deletion. A microinserted sequence (MI) of eight nucleotides at the deletion breakpoints suggests a template-switching mechanism (Figure 2e). A copy of the MI was found adjacent (59 bp) to the distal breakpoint sequence, corresponding to one of the two distances frequently observed between the MI template and the breakpoint. It has been suggested that the distance to template sites could be related to DNA packing in cells during replication.33 The presence of MH of two nucleotides shared between the proximal and distal sequences surrounding the breakpoints of the novel USH2A deletion suggests nonhomologous end-joining as a MH-mediated repair mechanism (Figure 3d).34
Moreover, a heterozygous duplication involving nine exons of RCBTB1 was found in a patient with RP, but not in his affected maternal uncle. Haploinsufficiency of RCBTB1 has recently been found to be associated with Coats disease (MIM 300216) and familial exudative vitreoretinopathy (FEVR, MIM 133780), both of which are characterized by abnormal retinal vascular development. The main clinical features of these congenital vitreoretinopathies are avascularization of the peripheral retina and subretinal exudation. Furthermore, there is a characteristic range for severity of retinal phenotype, from asymptomatic with preserved rod and cone responses to a flat ERG due to severe retinal detachment.35 Interestingly, our group simultaneously identified RCBTB1 as a disease gene for autosomal recessive nonsyndromic and syndromic iRD, varying from severe iRD compatible with RP to a progressive iRD with central chorioretinal atrophy and peripheral retinal dystrophy. Extraocular features include adult-onset sensorineural hearing loss, lung fibrosis (MIM 178500), thyroid involvement (MIM 138800), primary ovarian insufficiency, and mild intellectual disability.36 In the family described here, an autosomal dominant (with reduced penetrance; see individual III:5) or X-linked RP (with an affected female; see II:2) is most likely (Supplementary Figure S3b online). The phenotype of both the index patient P4 (with the RCBTB1 duplication and a missense variant) and his maternal uncle (carrier of only the RCBTB1 missense variant) does not match with the recently described autosomal dominant FEVR phenotype, however. Considering the lack of segregation of the duplication with the RP phenotype in this family, the phenotypic presentation, and the occurrence of an overlapping inherited, nonpathogenic duplication in DECIPHER, the RCBTB1 duplication found here is considered not causal for the RP phenotype observed in P4.
A number of methods have been used to identify subtle and larger CNVs in iRD, most of which only test coding regions. CNV screening based on qPCR or MLPA is restricted to a limited number of targets and can result in false-positive signals (mimicking a deletion) due to single-nucleotide variants affecting the primer or probe sequences. A reliable detection of small CNVs using NGS data can be challenging in poorly or highly variable covered exons. Array CGH is more sensitive to detect such small CNVs, particularly with a highly dense and evenly distributed set of probes,37 as in the arrEYE platform. It is recommended that NGS findings should be confirmed to complement CNV analysis in poorly covered regions using another technique.12 Genome-wide array CGH with exonic probes has been used to confirm WES findings,38 although the same shortcomings are envisaged: noncontiguous distribution of exons, low-quality probes for GC-rich exons, and no unique probes for highly repetitive exons or pseudogenes. An evenly spaced probe distribution encompassing both exonic and intronic regions can overcome these problems. Although this array CGH design facilitates the fine mapping of the breakpoint regions, it does not allow for pinpointing exact breakpoint regions or differentiating tandem from nontandem duplications, in contrast to WGS algorithms.39,40
The clinical detection rate of CNVs in iRD genes in our small RP cohort is lower than could be expected (2/57; 3.5%). Indeed, Khateb et al. showed a higher prevalence (10%) of CNVs in a cohort of 60 index cases with arRP and simplex RP.7 With the first version of arrEYE, we focused on genes for RP and overlapping phenotypes CRD and MD. A next version, however, can easily be extended with probes for other iRDs such as Leber congenital amaurosis, which could lead to a higher yield.
In conclusion, we designed the first customized array CGH platform for high-resolution CNV analysis of the full genomic region of known and candidate iRD genes and of retina-expressed ncRNAs. We identified subtle single-exon to larger CNVs in iRD genes that could be characterized at the nucleotide level. We report, for the first time, a CNV in HGSNAT that, in combination with a missense mutation, leads to arRP, thereby further supporting its recently identified role in nonsyndromic iRD. Finally, we expanded the allelic spectrum of USH2A and RCBTB1 with novel CNVs. In combination with NGS-based approaches, the arrEYE platform can provide an integrated molecular diagnosis in iRD.
Disclosure
The authors declare no conflict of interest. The sponsor or funding organizations had no role in the design or conduct of this research.
Acknowledgments
The authors gratefully acknowledge the families who participated in this study. This study was supported by Belspo IAP project P7/43, Belgian Medical Genomics Initiative to E.D.B., the Ghent University Special Research Fund (BOF15/GOA/011 to E.D.B.), and Funds for Research in Ophthalmology (FRO to C.V.C.). F.C. is the recipient of a postdoctoral fellowship from the FWO. E.D.B. and B.P.L. are Senior Clinical Investigators for the FWO. K.V.S. is doctoral fellow from the Institute for Innovation by Science and Technology (FWO/IWT).
Supplementary Material
References
- Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet 2013;84:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LS, Bowne SJ, Seaman CR, et al. Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2006;47:4579–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi KM, Tearle RG, Liu YP, et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci USA 2013;110:16139–16144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Carro R, Corton M, Sánchez-Navarro I, et al. Panel-based NGS reveals novel pathogenic mutations in autosomal recessive retinitis pigmentosa. Sci Rep 2016;6:19531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissinger B, Schaich S, Baumann B, et al. Large deletions of the KCNV2 gene are common in patients with cone dystrophy with supernormal rod response. Hum Mutat 2011;32:1398–1406. [DOI] [PubMed] [Google Scholar]
- García-García G, Aller E, Jaijo T, et al. Novel deletions involving the USH2A gene in patients with Usher syndrome and retinitis pigmentosa. Mol Vis 2014;20:1398–1410. [PMC free article] [PubMed] [Google Scholar]
- Khateb S, Hanany M, Khalaileh A, et al. Identification of genomic deletions causing inherited retinal degenerations by coverage analysis of whole exome sequencing data. J Med Genet; e-pub ahead of print 22 April 2016 [DOI] [PubMed]
- Eisenberger T, Neuhaus C, Khan AO, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One 2013;8:e78496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisschuh N, Mayer AK, Strom TM, et al. Mutation detection in patients with retinal dystrophies using targeted next generation sequencing. PLoS One 2016;11:e0145951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Sheng Q, Samuels DC, et al. Comparative study of exome copy number variation estimation tools using array comparative genomic hybridization as control. Biomed Res Int 2013;2013:915636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consugar MB, Navarro-Gomez D, Place EM, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med 2015;17:253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson LF, van Dijk F, de Boer EN, et al. CoNVaDING: Single exon variation detection in targeted NGS data. Hum Mutat 2016;37:457–464. [DOI] [PubMed] [Google Scholar]
- Sante T, Vergult S, Volders PJ, et al. ViVar: a comprehensive platform for the analysis and visualization of structural genomic variation. PLoS One 2014;9:e113800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askree SH, Chin EL, Bean LH, Coffee B, Tanner A, Hegde M. Detection limit of intragenic deletions with targeted array comparative genomic hybridization. BMC Genet 2013;14:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo JC, Lawrence KA, Karlstetter M, et al. CRX ChIP-seq reveals the cis-regulatory architecture of mouse photoreceptors. Genome Res 2010;20:1512–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benaglio P, San Jose PF, Avila-Fernandez A, et al. Mutational screening of splicing factor genes in cases with autosomal dominant retinitis pigmentosa. Mol Vis 2014;20:843–851. [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, et al. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell 2012;45:567–580. [DOI] [PubMed] [Google Scholar]
- Venturini G, Rose AM, Shah AZ, Bhattacharya SS, Rivolta C. CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet 2012;8:e1003040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karali M, Peluso I, Gennarino VA, et al. miRNeye: a microRNA expression atlas of the mouse eye. BMC Genomics 2010;11:715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte I, Hadfield KD, Barbato S, et al. MiR-204 is responsible for inherited retinal dystrophy associated with ocular coloboma. Proc Natl Acad Sci USA 2015;112:E3236–E3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundermeier TR, Palczewski K. The physiological impact of microRNA gene regulation in the retina. Cell Mol Life Sci 2012;69:2739–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KM, Dentchev T, Stambolian D. MiRNA expression in the eye. Mamm Genome 2008;19:510–516. [DOI] [PubMed] [Google Scholar]
- Van Peer G, Lefever S, Anckaert J, et al. miRBase Tracker: keeping track of microRNA annotation changes. Database (Oxford) 25 August 2014;p ii. [DOI] [PMC free article] [PubMed]
- Mustafi D, Kevany BM, Bai X, et al. Evolutionarily conserved long intergenic non-coding RNAs in the eye. Hum Mol Genet 2013;22:2992–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schil K, Naessens S, Van de Sompele S, et al. Coding and non-coding copy number variations explaining unsolved retinal dystrophies: role of genomic architectural features and underlying mechanisms (program number 1420). Annual Meeting of the American Society of Human Genetics, Seattle, WA, 1–5 May 2016. [Google Scholar]
- Haer-Wigman L, Newman H, Leibu R, et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum Mol Genet 2015;24:3742–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldhammer M, Durand S, Pshezhetsky AV. Protein misfolding as an underlying molecular defect in mucopolysaccharidosis III type C. PLoS One 2009;4:e7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet 2000;66:1975–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori A, Vaché C, Baux D, et al. Whole USH2A gene sequencing identifies several new deep intronic mutations. Hum Mutat 2016;37:184–193. [DOI] [PubMed] [Google Scholar]
- Fedele AO, Hopwood JJ. Functional analysis of the HGSNAT gene in patients with mucopolysaccharidosis IIIC (Sanfilippo C Syndrome). Hum Mutat 2010;31:E1574–E1586. [DOI] [PubMed] [Google Scholar]
- Wang F, Wang H, Tuan HF, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet 2014;133:331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosing S, van den Born LI, Sangermano R, et al. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology 2015;122:170–179. [DOI] [PubMed] [Google Scholar]
- Abyzov A, Li S, Kim DR, et al. Analysis of deletion breakpoints from 1,092 humans reveals details of mutation mechanisms. Nat Commun 2015;6:7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin H, D'haene B, Beysen D, et al. Microhomology-mediated mechanisms underlie non-recurrent disease-causing microdeletions of the FOXL2 gene or its regulatory domain. PLoS Genet 2013;9:e1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JH, Liu JH, Ko YC, et al. Haploinsufficiency of RCBTB1 is associated with Coats disease and familial exudative vitreoretinopathy. Hum Mol Genet 2016;25:1637–1647. [DOI] [PubMed] [Google Scholar]
- Coppieters F, Ascari G, Dannhausen K, et al. Isolated and syndromic retinal dystrophy caused by biallelic mutations in the RCBTB1 gene, a gene implicated in ubiquitination. Am J Hum Genet 2016;99:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasson A, Leroux C, Orhant L, et al. Custom oligonucleotide array-based CGH: a reliable diagnostic tool for detection of exonic copy-number changes in multiple targeted genes. Eur J Hum Genet 2013;21:977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarakoon PS, Sorte HS, Kristiansen BE, et al. Identification of copy number variants from exome sequence data. BMC Genomics 2014;15:661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkadi A, Bolze A, Itan Y, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci USA 2015;112:5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia M, Goes FS, Zandi PP. Whole-genome CNV analysis: advances in computational approaches. Front Genet 2015;6:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.