

Abstract
The role of Golgi apparatus during phagocytic uptake by macrophages has been ruled out in the past. Notably, all such reports were limited to Fcγ receptor-mediated phagocytosis. Here, we unravel a highly devolved mechanism for recruitment of Golgi-derived secretory vesicles during phagosome biogenesis, which was important for uptake of most cargos, except the IgG-coated ones. We report recruitment of mannosidase-II-positive Golgi-derived vesicles during uptake of diverse targets, including latex beads, Escherichia coli, Salmonella typhimurium, and Mycobacterium tuberculosis in human and mouse macrophages. The recruitment of mannosidase-II vesicles was an early event mediated by focal exocytosis and coincided with the recruitment of transferrin receptor, VAMP3, and dynamin-2. Brefeldin A treatment inhibited mannosidase-II recruitment and phagocytic uptake of serum-coated or -uncoated latex beads and E. coli. However, consistent with previous studies, brefeldin A treatment did not affect uptake of IgG-coated latex beads. Mechanistically, recruitment of mannosidase-II vesicles during phagocytic uptake required Ca2+ from both extra- and intracellular sources apart from PI3K, microtubules, and dynamin-2. Extracellular Ca2+ via voltage-gated Ca2+ channels established a Ca2+-dependent local phosphatidylinositol 1,4,5-trisphosphate gradient, which guides the focal movement of Golgi-derived vesicles to the site of uptake. We confirmed Golgi-derived vesicles recruited during phagocytosis were secretory vesicles as their recruitment was sensitive to depletion of VAMP2 or NCS1, whereas recruitment of the recycling endosome marker VAMP3 was unaffected. Depletion of both VAMP2 and NCS1 individually resulted in the reduced uptake by macrophages. Together, the study provides a previously unprecedented role of Golgi-derived secretory vesicles in phagocytic uptake, the key innate defense function.
Keywords: calcium, Golgi, macrophage, Mycobacterium tuberculosis, phagocytosis, focal exocytosis, mannosidase-II, phagosome biogenesis, VAMP3, voltage-gated calcium channel
Introduction
Phagocytosis lies at the core of innate defense mechanisms in higher eukaryotes. The process of phagocytosis involves internalization of external particles, including pathogens, cellular debris, etc., into a specialized membrane-bound organelle called phagosomes. The phagosome thus formed undergoes a series of fusion and fission events leading to acquisition of hydrolytic enzymes and microbicidal properties to mature into phagolysosomes (1–3).
It was perceived initially that the membrane for phagosomes could be solely derived from the plasma membrane (4). However, in macrophages, the professional phagocytes, which can engulf objects bigger than their own size without significantly altering their function, there is no apparent decline in the membrane surface area following phagocytosis (5). Rather, capacitance measurement experiments showed that a concomitant increase in the membrane surface area precedes resealing of the phagosome, suggesting supply of phagosome membrane from intracellular sources (5). Professional phagocytes are therefore expected to be under tremendous pressure to sustain the supply of the membrane so as to effectively phagocytize their targets. Consequently, a variety of membrane-bound organelles, including the vesicles originating from recycling endosomes and lysosomes as well as ER,4 were later shown to provide membrane for the nascent phagosome (6–8).
Vesicle recruitment during phagocytosis involves exocytosis of vesicles in the vicinity of nascent phagosomes (9). Studies in the past have shown secretion of vesicular content like lysosomal hydrolases and azurophil granules during engulfment by macrophages and neutrophils, respectively, thereby coupling the process of phagocytosis with targeted exocytosis, a process also termed as focal exocytosis (7, 9). Recycling endosome marker VAMP3, a soluble N-ethylmaleimide-sensitive factor attachment receptor protein on vesicles (v-SNARE), was shown to accumulate at the early phagosomes through focal exocytosis (7). Moreover, synaptotagmin V (SytV), a Ca2+ sensor on the recycling endosomes, was shown to regulate phagocytosis but not phagosome maturation, suggesting a key role of Ca2+ in the regulation of vesicle exocytosis (10). Similarly, SytVII, a lysosome resident Ca2+ sensor, was reported to regulate delivery of lysosome membrane to the nascent phagosomes (11). Interestingly, SytVII was also shown to regulate Ca2+-dependent exocytosis of lysosomes during plasma membrane repair (12). Recently, TRPML1, a key Ca2+ channel in the lysosomes, was shown to regulate focal exocytosis during phagocytosis of large particles (13). Phosphoinositide 3-kinase (PI3K) is yet another key regulator of phagocytosis and is believed to help pseudopod extension around the particles during phagocytosis (14). However, inhibition of PI3K by wortmannin may not limit membrane availability for phagocytosis (14). There are some reports, however, suggesting the role of PI3K in recruiting the membranes from intracellular sources (15). Yet another study revealed recruitment of ER at the site of phagocytosis to provide membrane for the newly formed phagosomes, a process that was regulated by the ER resident SNARE protein ERS24 (2, 6).
Intriguingly, despite being intricately involved with the recycling endocytic network and as one of the major sources of vesicles destined to the plasma membrane-like secretory vesicles, involvement of Golgi apparatus (GA) or vesicles derived from GA in the process of phagocytosis has been ruled out (16). However, in an interesting observation, fragmentation and reorganization of GA were reported during frustrated phagocytosis (17). Interestingly, the majority of the reports that conclude no direct involvement of GA during phagocytic uptake relied entirely on the experiments that were limited to Fcγ receptor-mediated phagocytosis (18–20). It is important to note that macrophages employ several independent receptor systems for phagocytosis, including complement-mediated, pattern receptor-mediated, and scavenger receptor-mediated phagocytosis (3, 21). Whether Golgi apparatus is dispensable even for these phagocytic pathways remains unknown.
Here, we studied the role of vesicles derived from the GA during phagocytic uptake using mannosidase-II as the marker for these vesicles. Mannosidase-II, a medial and trans-Golgi marker, was also detected within the secretory granules and at the surface of certain cell types like enterocytes, pancreatic acinar cells, and goblet cells (22). Later, using mannosidase-II as an exclusive marker of Golgi-derived vesicles, its recruitment to the cell surface was used to track the role of these vesicles during plasma membrane repair in mouse bone marrow-derived primary macrophages (22, 23). Here, we show that mannosidase-II-containing Golgi-derived vesicles are recruited during phagocytosis of a variety of targets, including inert particles (latex beads, uncoated, or serum coated), non-pathogenic bacteria (Escherichia coli), and pathogenic bacteria (Salmonella typhimurium and Mycobacterium tuberculosis) in mouse (RAW264.7 and BMDMs) and human (THP-1 and U937) macrophages. The recruitment of mannosidase-II vesicles at the site of phagocytosis occurred through the process of focal exocytosis. This process was dependent on voltage-gated Ca2+ channels that helped establish a Ca2+-dependent PIP3 gradient for uptake. Mechanistically, TGN and secretory vesicle resident protein NCS1 (neuronal calcium sensor 1) sensed and triggered the movement of vesicles to help phagosome biogenesis, which was also dependent on secretory vesicle-specific v-SNARE VAMP2. We trace here a previously unknown function of Golgi-derived vesicles during phagocytic uptake in macrophages and also provide a mechanistic basis for the same.
Results
α-Mannosidase-II (MAN2A) Localizes to GA and Golgi-derived Vesicles
To confirm the localization of mannosidase-II to GA in our experimental setup, we nucleofected THP-1 human macrophages and RAW264.7 mouse macrophages with an mCherry-tagged mannosidase-II construct (mCherry-MannII-N-10 (mCherry-MAN2A), Addgene plasmid no. 55074), and we visualized the cells under a Nikon A1R confocal microscope (see under “Materials and Methods”). Expression of mannosidase-II in these cells distinctly labeled the GA as shown in Fig. 1A. We also immunostained mannosidase-II using specific antibody, which labeled the GA (Fig. 1B). Treatment of mCherry-MAN2A-expressing cells with brefeldin A, the classical inhibitor of Golgi function, resulted in the disruption and fragmentation of the GA structure (Fig. 1C). In both transfected as well as antibody-stained cells, in addition to the main GA, we could also observe several smaller punctate structures, representing the Golgi-derived vesicles (Fig. 1, A and B). In U937 cells stably expressing mRuby-tagged β-1,4-galactosyltransferase (B4GALT1-mRuby), a mid-Golgi resident enzyme, such vesicle-like structures could rarely be seen (Fig. 1D); mannosidase-II, however, co-localized with B4GALT1-mRuby (Fig. 1D). These results confirmed that the mannosidase-II construct used in this study labeled both the GA- and Golgi-derived vesicles.
FIGURE 1.
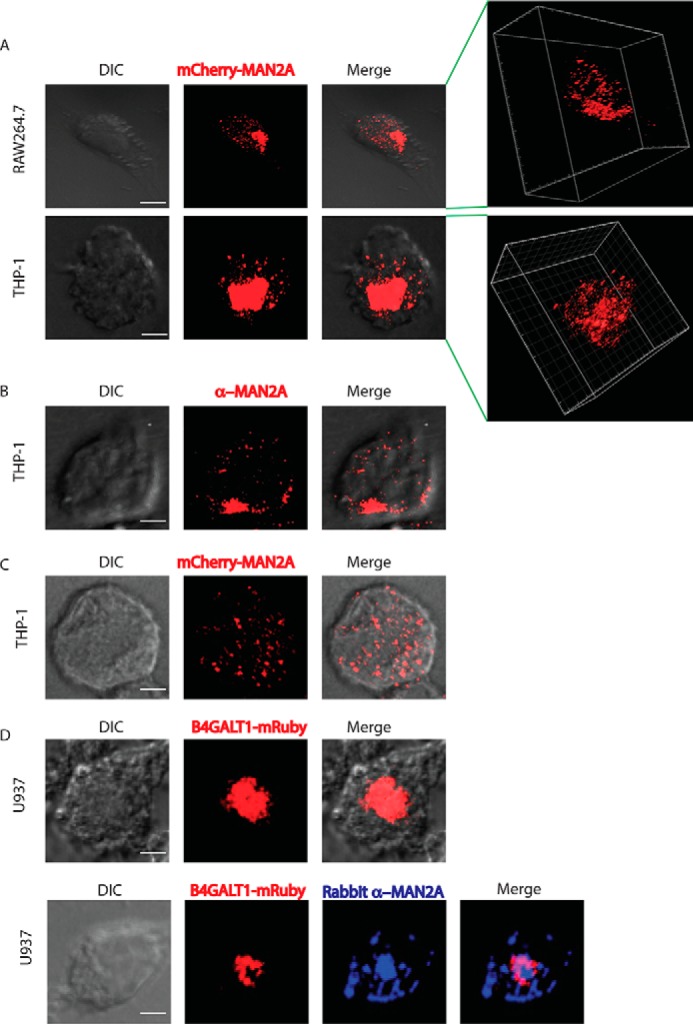
Mannosidase-II is a marker of Golgi apparatus and vesicles derived from the Golgi. A, RAW264.7 cells and THP-1-derived macrophages were nucleofected with mCherry-MAN2A. At 24 h post-nucleofection, cells were visualized under the confocal microscope (see “Materials and Methods” for detail). The 3D plots at the right were created using Imaris 7.2 software tool (scale bar, 5 μm). B, THP-1-derived macrophages were permeabilized using 0.5% Triton X-100 and stained with anti-mannosidase-II antibody followed by the secondary antibody tagged with Alexa Fluor 560, fixed, and visualized under the confocal microscope (scale bar, 5 μm). C, THP-1-derived macrophages were nucleofected with mCherry-MAN2A. At 24 h post-nucleofection, the cells were treated with brefeldin A (20 μm) for 4 h and visualized under the microscope (scale bar, 5 μm). D, U937 human macrophages, stably expressing the Golgi marker mRuby-B4GALT1 (red, β-galactosyltransferase). Lower panel, B4GALT1 (red) expressing U937 macrophages were stained with anti-mannosidase-II antibody followed by secondary antibody (Alexa Fluor 405, blue, scale bar, 5 μm). DIC, differential interference contrast.
Mannosidase-II Is Recruited at the Site of Phagocytic Uptake
RAW264.7 cells transfected with mCherry-tagged mannosidase-II were incubated with mouse serum-coated latex beads (see “Materials and Methods”). At 30 min and 1 h post-addition of beads to the macrophages, cells were fixed and visualized under the microscope. We observed the recruitment of mannosidase-II to the sites where latex beads were phagocytosed, forming a ring-like structure around the beads at many instances (Fig. 2A). Overall intensity of mannosidase-II at the bead surface had a median of ∼900 at 30 min and ∼1500 A.U. at 1 h as determined using the 3D module tool in Imaris 7.2 (see “Materials and Methods” and Fig. 2A). To check whether mannosidase-II recruitment during phagocytosis was a general phenomenon, we also monitored uptake of the non-pathogenic bacterium E. coli and the pathogenic bacterium S. typhimurium. In both cases, we could see mannosidase-II recruited at very early stages of phagocytosis. In the case of E. coli, nearly 60% of the bacteria at the site of entry showed mannosidase-II recruitment at 15 and 30 min post-infection, whereas nearly 30% of Salmonella-containing phagosomes showed mannosidase-II recruitment at 5 and 10 min (Fig. 2B). In the case of yet another pathogenic bacterium M. tuberculosis strain H37Rv, mannosidase-II was recruited at 30–40% phagosomes at the time of entry (Fig. 2C). Recruitment of mannosidase-II at the early phagosomes in the case of H37Rv was also verified by immunostaining against mannosidase-II (Fig. 2D). We also verified the recruitment of mannosidase-II in BMDMs during phagocytic uptake of E. coli, thus ensuring that it was not a phenomenon restricted only to the cell lines (Fig. 2E). To confirm that the mannosidase-II-positive phagosomes indeed represented early stages of phagosomes, we immunostained the mCherry-MAN2A-expressing cells that were either incubated with mouse serum-coated latex beads or infected with M. tuberculosis with TfR antibody. We observed significant overlap between recruited mannosidase-II and TfR around the internalized beads and M. tuberculosis (Fig. 3, A and B). TfR recruitment around phagocytized beads showed saturating levels with a median of about 4095 A.U., much higher than the intensity distribution of mannosidase-II (∼1000 A.U., Fig. 2D). In the case of M. tuberculosis, TfR showed relatively higher co-localization with the phagosomes (∼40–60%) compared with mannosidase-II (30–50%, Fig. 3B). As expected, TfR on M. tuberculosis phagosomes declined rapidly from 1 to 2 h post-infection (Fig. 3B).
FIGURE 2.

Mannosidase-II is recruited at the site of phagocytosis. A, RAW264.7 macrophages expressing mCherry-MAN2A (red) were incubated with mouse serum-coated latex beads for 30 min and 1 h. The images shown are representative of the 30-min samples. Right, total intensity of mCherry-MAN2A puncta on the bead surface was estimated using 3D spot creation module in Imaris 7.2, and the intensity distribution of the population has been plotted (see “Materials and Methods” for detail). The box plot represents data from more than 200 beads analyzed from two different experiments (values ± S.D.; scale bar, 4 μm). B, THP-1-derived macrophages were infected with GFP expressing E. coli (green, top panel) or S. typhimurium (green, lower panel) for 15 and 30 min. Cells were then stained with anti-mannosidase-II antibody followed by Alexa Fluor 568-tagged secondary antibody (red). Images shown are representative of the 15-min samples from both experiments. The bar plots at right show % co-localization of E. coli or Salmonella with mannosidase-II at the surface at 5 and 10 or 15 and 30 min, respectively, post-infection. Data represent the average of more than 200 bacteria from two different experiments (values ± S.D.; scale bar, 3 μm). C, mCherry-MAN2A (red)-expressing THP-1-derived macrophages were infected with PKH67-labeled H37Rv (green) for 1 and 2 h. Samples were fixed and visualized under the microscope. The bar plot at right shows % co-localization of H37Rv with mannosidase-II at both these time points. Data represent average of more than 200 bacteria from two different experiments (values ± S.D.; scale bar, 4 μm). D, THP-1-derived macrophages were infected with PKH67-labeled H37RV and fixed at the respective time points. These cells were then permeabilized using 0.05% Triton X-100 and stained with anti-mannosidase-II antibody followed by the secondary antibody tagged with Alexa Fluor 568, fixed, and visualized under the confocal microscope (scale bar, 3 μm). E, mouse BMDMs were infected with GFP expressing E. coli for 10 min, fixed, and stained with anti-MAN2A antibody (scale bar, 3 μm). DIC, differential interference contrast.
FIGURE 3.

Early recruitment of mannosidase-II at phagosomes and proteomic characterization. A, mCherry-MAN2A (red)-expressing RAW264.7 macrophages were incubated with mouse serum-coated latex beads for 30 min or 1 h. At the respective time points, cells were immunostained with anti-TfR antibody followed by a secondary antibody tagged with Alexa Fluor 405 (blue). Presence of TfR or mannosidase-II at the bead surface was calculated in terms of fluorescence intensity using the 3D spot creation module in Imaris 7.2 software. The box plot at right shows data from more than 100 beads from two independent experiments (scale bar, 5 μm). B, mCherry-MAN2A (red)-expressing RAW264.7 macrophages were infected with PKH67-labeled H37Rv (green) for 1 and 2 h. At the respective time points, samples were fixed and stained with anti-transferrin receptor antibody followed by Alexa Fluor 405-tagged secondary antibody (blue). The images are representative of the 1-h time point. For the plots at right, % co-localization of H37Rv with mannosidase-II, TfR, or both mannosidase-II and TfR was calculated using Imaris 7.2. The data represent the average of more than 150 bacteria from three different experiments (values ± S.D.; scale bar, 5 μm). C, preparation of latex bead phagosomes from THP-1-derived macrophages on a sucrose density gradient (see “Materials and Methods”). D, THP-1-derived macrophages were incubated with latex beads (1 μm size) for 1 h. Phagosomes were isolated using differential density ultracentrifugation, and samples were probed for indicated markers using Western blottings. Molecular mass markers are the closest markers to the band of interest. WCL, whole cell lysate. E, latex beads phagosomes isolated from RAW264.7 macrophages were lysed and resolved on a 10% SDS-PAGE. The molecules below the 25-kDa molecular mass were cut from the gel and analyzed using mass spectrometry to identify enrichment of low molecular weight proteins (see “Materials and Methods”). The list of genes identified was then searched in the AMIGO2.0 database to establish functional association. Finally the representative network was constructed using Cytoscape 2.6.1. The pink nodes and edges in the network denote association with the Golgi apparatus.
Characterization of Early Phagosomes Reveals Direct Involvement of Secretory Vesicles Derived from the GA in Phagosome Biogenesis
As shown above and by others (22, 23), mannosidase-II marks both GA- and Golgi-derived vesicles. In most of the fields observed in the previous sections, we could see mannosidase-II organized into a separate Golgi structure, underscoring the fact that the phagosome-associated mannosidase-II was derived from the recruitment of Golgi-derived vesicles at the site of infection. To further confirm the selective enrichment of mannosidase-II at the nascent phagosomes, we purified latex bead phagosomes using sucrose density gradient ultracentrifugation from macrophages within 1 h of uptake (Fig. 3C). The phagosome preparation showed the presence of expected early phagosome markers like TfR and RAB5 (Fig. 3D). They also showed the presence of previously reported markers like VAMP3 from recycling endosomes and calnexin from the ER (Fig. 3D). At the same time, the phagosome preparation was devoid of any mitochondrial, nuclear, or cytosolic contamination (Fig. 3D). In agreement with the microscopy data, phagosomes also showed the presence of mannosidase-II, the marker for Golgi-derived vesicles (Fig. 3D). In addition, we could also score the presence of another Golgi-derived secretory vesicle marker, NCS1, in the phagosomes (Fig. 3D). We verified that there was no GA contamination in the preparation as the phagosomes were devoid of B4GALT1 (Fig. 3D).
Trafficking of vesicles in the cells is regulated by a large number of proteins, including small GTPases like RABs, ADP-ribosylation factors (ARFs), and SNAREs (24–26). To understand which RABs, SNAREs or ARFs could be involved in the recruitment of mannosidase-II vesicles at the phagosomes, we performed mass spectrometry of the purified latex beads phagosome preparations, specifically to identify molecules below 25 kDa molecular mass (see under “Materials and Methods”). We were able to identify about 290 proteins from this preparation, all of them below 25 kDa (supplemental Table S1). We performed a gene ontology analysis on these proteins using Amigo2.0 database to specifically see enrichment of 11 classes, including ER, GA, secretory vesicles, recycling endosomes, endocytic vesicles, neurotransmitter release, TGN, vesicle-mediated transport, phagocytosis/engulfment, exocytosis, and endosomal transport (Fig. 3E and supplemental Table S2). A large number of proteins belonging to ER were found in the phagosomes (Fig. 3E). The phagosome mass spectrometry data revealed the presence of 17 different RABs, five out of six known ARFs, and four VAMPs. Many of these proteins are involved with Golgi apparatus, exocytosis, secretory vesicles, and neurotransmitter release (Fig. 3E). VAMP2 gets enriched in the secretory vesicles that are released from the GA and targeted to the membranes (27). It also helps in the release of neurotransmitters by regulating the exocytosis of secretory vesicles (28). Collectively, the presence of markers associated with vesicles derived from the GA reconfirmed the utility of this compartment as an additional source of membranes for phagosome biogenesis.
Requirement of Golgi Apparatus during Phagocytosis
Having observed overwhelming representation of proteins from GA in the phagosome proteome and clearly established the recruitment of MAN2A-positive Golgi-derived vesicles at the phagosomes, it was important to test whether GA could be directly involved in regulating the uptake. It must be noted that classically the role of GA during Fcγ receptor-mediated phagocytosis has been ruled out (16). Corroborating previous studies, we observed that uptake of human IgG-coated latex beads was unaffected by brefeldin A treatment, an established Golgi inhibitor at doses ranging from 10, 20, and 30–100 μg/ml (Fig. 4A). However, uptake of serum-opsonized latex beads, latex beads opsonized with complement-deactivated serum, or uncoated latex beads was significantly inhibited by brefeldin A treatment at doses as low as 10 and 20 μg/ml, respectively (Fig. 4, B–D). Similarly, uptake of E. coli was also significantly inhibited by brefeldin A treatment at 30 and 100 μg/ml (Fig. 4E). Interestingly, brefeldin A treatment also led to diminished recruitment of mannosidase-II at the latex bead phagosomes or E. coli phagosomes (Fig. 4, F and G). Mannosidase-II recruitment was not entirely absent in the case of Fcγ receptor-mediated phagocytosis and was sensitive to brefeldin A treatment but, unlike other cargos, that did not lead to any affect on uptake (Fig. 4H). Because the requirement of Golgi function varied between different modes of phagocytosis, we questioned whether that could reflect the effect of brefeldin A on the corresponding receptor expression level at the cell surface. All cell surface receptors travel through the Golgi, and their expression levels are likely to get perturbed in brefeldin A-treated cells. However, none of the receptors tested, including Fcγ receptor, complement receptor (CR3), mannose receptor, and Dectin-2, showed any noticeable decline at the doses and duration of treatment used for the uptake studies (Fig. 4I).
FIGURE 4.

Role of Golgi apparatus during phagocytosis. THP-1 macrophages were incubated with human IgG-coated FITC-labeled latex beads (A), serum-coated FITC-labeled latex beads (B), complement-deactivated serum-coated latex beads (C), uncoated latex beads (D), or E. coli (E) for 30 min in the absence or presence of brefeldin A at concentrations mentioned. BfA treatment was given 30 min prior to the addition of the beads or E. coli. Samples were acquired in a flow cytometer to measure uptake. Plots represent data from an average of three independent experiments. Error bars represent S.D. (**, p value <0.01; ***, p value <0.005). F, THP-1 macrophages were allowed to uptake serum-coated FITC-labeled latex beads for 30 min in the presence or absence of BfA. Samples were fixed and stained for mannosidase-II using anti-MAN2A antibody followed by Alexa Fluor 405 secondary antibody. Confocal images acquired were analyzed using 3D module in Imaris 7.2 software to calculate mannosidase-II intensity distribution over the latex beads (***, p value <0.001). G, GFP expressing E. coli was incubated with THP-1 macrophages for 30 min in the presence or absence of BfA. Samples were fixed and stained for mannosidase-II using anti-MAN2A antibody followed by Alexa Fluor 405 secondary antibody. Confocal images were analyzed using Imaris software to calculate percent co-localization. H, THP-1 macrophages were allowed to uptake IgG-coated FITC-labeled latex beads for 30 min in the presence or absence of BfA. Samples were fixed and stained for mannosidase-II using anti-MAN2A antibody followed by Alexa Fluor 405 secondary antibody. Confocal images acquired were analyzed using 3D module in Imaris software to calculate mannosidase-II intensity distribution over the latex beads. Co-localization (*, p value <0.05). I, U937-derived macrophages were incubated with 20 μg/ml brefeldin A for 60 min, fixed, and stained for receptors mentioned. Cells were acquired by flow cytometry and overlays analyzed using FlowJo.
Golgi-derived Vesicles Cooperate with the Vesicles Derived from the Recycling Endosomes during Phagocytosis
Recruitment of vesicles from endocytic origin (VAMP3-positive) and lysosomal origin (LAMP1-positive) during phagocytosis has been reported earlier (7, 11). We also observed the presence of VAMP3 on the proteome of early latex beads phagosome as shown above (Fig. 3D). We verified that VAMP3 compartment represented distinct population of vesicles than those represented by mannosidase-II (Fig. 5A). However, because VAMP3 is known to traffic through the Golgi apparatus, there were some overlaps between mannosidase-II and VAMP3 within the Golgi stacks (Fig. 5A). However, outside the GA, there was very limited overlap between mannosidase-II and VAMP3. In mCherry-MAN2A-expressing RAW264.7 cells, both latex beads and M. tuberculosis very early during phagocytosis (30 min and 1 h for beads; 1 and 2 h for M. tuberculosis) showed very high recruitment of and co-localization with VAMP-3 (Fig. 5, B and C). In the case of M. tuberculosis, more than 90% of the mannosidase-II-positive phagosomes were also positive for VAMP-3 (Fig. 5C). Thus, spatially and temporally, mannosidase-II recruitment at the phagosomes was strikingly similar to VAMP3 recruitment. Interestingly, unlike mannosidase-II recruitment, VAMP3 recruitment was not sensitive to brefeldin A treatment during uptake of latex beads or E. coli (Fig. 5, D and E, respectively). The selective influence of brefeldin A on mannosidase-II recruitment suggests independent pathways for the recruitment of mannosidase-II vesicles and VAMP3 vesicles. Although mannosidase-II vesicles are known to originate from the Golgi apparatus, VAMP3 vesicles are recruited from recycling endosomes (7). Thus, it seems that vesicles from different origins cooperate in aiding the biogenesis of phagosomes.
FIGURE 5.

Vesicles derived from Golgi apparatus are distinct from recycling endosome vesicles and are recruited independently. A, U937 cells expressing MAN2A-mCherry were fixed and stained with anti-VAMP3 antibody followed by Alexa Fluor 405-labeled secondary antibody (scale bar, 15 μm). White box in the merge panel identifies the area that was magnified for the zoom panel. DIC, differential interference contrast. B, mCherry-MAN2A (red)-expressing RAW264.7 macrophages were incubated with mouse serum-coated latex beads for 30 min or 1 h. At the respective time points, cells were immunostained with anti-VAMP-3 antibody followed by a secondary antibody tagged with Alexa Fluor 405 (blue). Presence of VAMP-3 or mannosidase-II at the bead surface was calculated using the 3D spot creation module in Imaris 7.2 software. The box plot at right shows data from more than 100 beads from two independent experiments (scale bar, 5 μm). C, mCherry-MAN2A (red)-expressing THP-1-derived macrophages were infected with PKH67-labeled H37Rv (green) for 1 and 2 h. At the respective time points, samples were fixed and stained with anti-VAMP-3 antibody followed by Alexa Fluor 405-tagged secondary antibody (blue). The images are representative of the 1-h time point. For the plots at right, % co-localization of H37Rv with mannosidase-II, VAMP-3, or both mannosidase-II and VAMP-3 was calculated using Imaris 7.2. The data represent an average of more than 150 bacteria from three different experiments (values ± S.D.; scale bar, 4 μm). D, U937 cells were incubated with serum-coated FITC-labeled latex beads in the presence or absence of BfA for 30 min. Samples were fixed and stained with anti-VAMP3 antibody followed by Alexa Fluor 405-labeled secondary antibody. Images were analyzed using 3D module in Imaris 7.2, and VAMP3 intensity distribution in the latex beads was calculated. E, U937 cells were incubated with GFP expressing E. coli in the presence or absence of BfA for 30 min. Samples were fixed and stained with anti-VAMP3 antibody followed by Alexa Fluor 405-labeled secondary antibody. Images were analyzed using 3D module in Imaris 7.2, and VAMP3 intensity distribution in the latex beads was calculated.
Mannosidase-II-positive Golgi-derived Vesicles Are Recruited at the Phagosomes through Focal Exocytosis
We next wanted to understand how mannosidase-II vesicle recruitment was regulated during phagocytosis. Previous studies show the recruitment of VAMP-3-positive vesicles from the recycling endosomes is dependent on focal exocytosis (7). It was previously reported that mannosidase-II-positive Golgi-derived vesicles get recruited at the site of membrane damage during repair (23). It is also well established that vesicular movement during membrane repair occurs through focal exocytosis (12). One of the key regulators of focal exocytosis, specifically for post-Golgi transport vesicles toward the plasma membrane, is dynamin (29). Dynamins are the members of a large GTPase family, and dynamin-2 is the ubiquitously expressed isoform, whereas dynamin-1 is mostly neuronal (30, 31). Their involvement in budding of transport vesicles from the Golgi is also well documented along with their role in regulating focal exocytosis (13, 32). Additionally, dynamins are also involved in the endocytosis where they regulate scission of endocytic vesicles from the membrane (77). In mCherry-MAN2A-expressing RAW264.7 cells, we observed significant overlap of mannosidase-II with dynamin-2 on the phagosomes containing either latex beads or M. tuberculosis (Fig. 6, A and B). Again as in the case of TfR, dynamin-2 recruitment on the nascent phagosomes was relatively higher than mannosidase-II recruitment for both latex beads and M. tuberculosis (Fig. 6, A and B). Presence of dynamin-2 at the phagosomes was consistent with previous reports, which showed their recruitment at early phagosomes during phagocytosis (33).
FIGURE 6.

Golgi-derived vesicles are recruited through focal exocytosis. A, mCherry-MAN2A (red)-expressing RAW264.7 macrophages were incubated with mouse serum-coated latex beads for 30 min or 1 h. At the respective time points, cells were immunostained with anti-dynamin2 antibody followed by a secondary antibody tagged with Alexa Fluor 405 (blue). Presence of dynamin2 (Dyn-2) or mannosidase-II at the bead surface was calculated using the Imaris 7.2 software. The box plot at right shows data from more than 100 beads from two independent experiments (values ± S.D.; scale bar, 4 μm). B, mCherry-MAN2A (red)-expressing THP-1 macrophages were infected with PKH67-labeled H37Rv (green) for 1 and 2 h. At the respective time points, samples were fixed and stained with anti-dynamin2 antibody followed by Alexa Fluor 405-tagged secondary antibody (blue). The images are representative of the 1-h time point. For the plots at right, % co-localization of H37Rv with mannosidase-II, dynamin2 or both mannosidase-II and dynamin2 was calculated using Imaris 7.2. The data represent the average of more than 150 bacteria from three different experiments (values ± S.D.; scale bar, 4 μm). C, mCherry-MAN2A (red)-expressing RAW264.7 macrophages were pretreated with 80 μm Dynasore and incubated with mouse serum-coated latex beads for 30 min or 1 h. At 30 min, samples were fixed and stained with anti-dynamin2 antibody followed by Alexa Fluor 405-tagged secondary antibody (blue). Intensity of mannosidase-II and dynamin at the bead surface was calculated using the 3D spot creation module in Imaris 7.2 software. The box plot shows data from more than 100 beads from two independent experiments (*, p value <0.05). D, THP-1-derived macrophages were treated with the respective dynasore concentrations for 4 h and incubated with FITC-labeled latex beads (1 μm). At the respective time points, cells were fixed and analyzed by flow cytometry. To calculate % uptake, data for the uptake of each cargo for a given time point in the dynasore-treated set was normalized against the respective untreated control set (values ± S.D.). E, THP-1-derived macrophages were pretreated with 40 or 80 μm dynasore for 4 h and infected with PKH67-labeled H37RV. The cells were fixed at respective points and analyzed by flow cytometry (values ± S.D.). F, THP-1-derived macrophages expressing mCherry-MAN2A were pretreated with 40 μm Dynasore for 4 h and infected with PKH67-labeled H37RV. The cells were fixed at respective time points and analyzed by a confocal microscope. The percent co-localization was determined by Imaris 7.2. Data represent an average of more than 200 bacteria from two different experiments (values ± S.D.,**, p value <0.01). G, THP-1-derived macrophages were pretreated with 25 μm nocodazole for 2.5 h and infected with PKH67-labeled H37RV. The cells were fixed at respective points and analyzed by flow cytometry (values ± S.D.). H, THP-1-derived macrophages expressing mCherry-MAN2A were pretreated with 25 μm nocodazole for 2.5 h and infected with 1-μm of FITC-labeled latex beads. The cells were fixed at respective points and analyzed by flow cytometry (values ± S.D.). I, THP-1-derived macrophages expressing mCherry-MAN2A were pretreated with 25 μm nocodazole for 2.5 h and infected with PKH67-labeled H37RV. The cells were fixed at respective points and analyzed by confocal microscopy. The percent co-localization was determined using Imaris 7.2. Data represent an average of more than 200 bacteria from two different experiments (values ± S.D.; **, p value <0.01). J, RAW264.7 macrophages expressing mCherry-MAN2A were pretreated with 25 μm nocodazole for 2.5 h and infected with 4-μm latex beads. The cells were fixed at respective points and analyzed by a confocal microscope. 3D creation of bead was done to determine the maximum fluorescence intensity of mannosidase II on the bead, via spot module in Imaris 7.2. The box plot at right shows data from more than 100 beads from two independent experiments (**, p value <0.01).
Dynamin-mediated focal exocytosis can be inhibited by dynasore, a specific dynamin inhibitor (13). We next inhibited dynamin-2 by dynasore treatment for varying periods of time and observed its effect on mannosidase-II recruitment and phagocytosis. In the case of latex beads, 4 h of pretreatment with dynasore (at 40 and 80 μm) resulted in a significant inhibition in the recruitment of mannosidase-II at the phagosomes (Fig. 6C). It also markedly reduced the phagocytosis of latex beads (Fig. 6D). Dynasore treatment also inhibited uptake of M. tuberculosis in THP-1 macrophages (Fig. 6E). There was noticeable decline in the recruitment of mannosidase-II at the M. tuberculosis phagosomes (Fig. 6F). The reduction in uptake upon dynasore treatment was not due to the inhibition of endocytic functions of dynamins because dynasore, unlike mannosidase-II, did not inhibit recruitment of recycling endosome marker VAMP3 to the phagosomes. Dynamin-assisted movement of vesicles also requires microtubules (29). Inhibition of microtubules by nocodazole treatment resulted in a transient decline in the uptake of M. tuberculosis and latex beads (Fig. 6, G and H). Furthermore, nocodazole treatment resulted in a marked decline in the recruitment of mannosidase-II to the phagosomes containing M. tuberculosis and latex beads (Fig. 6, I and J). These results together established that the mannosidase-II-positive Golgi-derived vesicles were recruited at the phagosomes through focal exocytosis and were assisted by dynamins.
PI3K Is Required for Mannosidase-II Recruitment at the Nascent Phagosomes and Phagocytosis
The role of PI3K in regulating phagocytosis is well known where its function is believed to be mostly required for pseudopod extension around the cargo during phagocytosis (14). We wanted to test whether some of the effects of PI3K on phagocytosis were due to its involvement in the recruitment of mannosidase-II vesicles for phagocytosis. Inhibition of PI3K by wortmannin led to nearly 80% decline in the uptake of latex beads at 30 and 60 min (Fig. 7A). In the case of M. tuberculosis, the decline in uptake was ∼85% at 1 and 2 h post-infection and 70% at 4 h post-infection (Fig. 7B). Curiously, PI3K inhibition also resulted in a marked decrease in the recruitment of mannosidase-II at the nascent phagosomes (Fig. 7, C and D). Thus, at least to some extent, it seems the effect of inhibition of PI3K on phagocytosis may be due to a reduced recruitment of mannosidase-II-positive Golgi-derived vesicles at the phagosomes. A similar effect of PI3K inhibition on the membrane recruitment for the newly forming phagosomes has been discussed previously (15).
FIGURE 7.

Role of PIP3 and calcium from intra- and extracellular sources in recruitment of mannosidase II vesicles. A, THP-1-derived macrophages were pretreated with 5 μm wortmannin for 4 h. FITC latex beads (1 μm) were added to these. The cells were fixed at respective time points and analyzed by flow cytometry. Data shown are average from three different experiments and represented as % uptake in treated cells with respect to the untreated cells (values ± S.D.). B, THP-1-derived macrophages were pretreated with 5 μm wortmannin for 4 h. They were infected with PKH-labeled H37Rv. At the respective time points, the cells were fixed and analyzed by flow cytometry. Data shown are average of three different experiments and represented as % uptake in treated cells with respect to the untreated cells (values ± S.D.). C, mCherry-MAN2A (red)-expressing RAW264.7 macrophages were pretreated with 5 μm wortmannin or EGTA (3 mm) + TMB8 (100 μm) for 30 min, respectively. The cells were incubated with mouse serum-coated latex beads for 30 min or 1 h. At the respective time points, cells were fixed and analyzed by confocal microscopy. The images shown are representative of the 30-min samples. The 3D construction of latex bead was done using spot creation module of Imaris 7.2. D, RAW264.7 macrophages expressing mCherry-MAN2A (red) were incubated with mouse serum-coated latex beads for 30 min and 1 h. The total intensity of mCherry-MAN2A puncta on the bead surface was determined using 3D spot creation module in Imaris 7.2, and the intensity distribution of the population has been plotted. The box plot represents data from more than 200 beads analyzed from two different experiments (*, p value <0.05). E, THP-1-derived macrophages were pretreated with EGTA (3 mm), TMB8 (100 μm), and EGTA + TMB8 for 30 min before addition of FITC latex beads (1 μm). At respective time points, the cells were fixed and analyzed by flow cytometry. Data shown are average from three different experiments and represented as % uptake in treated cells with respect to the untreated cells (values ± S.D.). F, THP-1-derived macrophages were pretreated with EGTA (3 mm), TMB8 (100 μm), and EGTA + TMB8 for 30 min before addition of PKH-labeled H37Rv. At respective time points, the cells were fixed and analyzed by flow cytometry. Data shown are average from three different experiments and represented as % uptake in treated cells with respect to the untreated cells (values ± S.D.).
Extracellular Calcium (Ca2+) Is Needed for Phagocytosis and Focal Exocytosis of Golgi-derived Vesicles at the Site of Phagocytosis
We next wanted to understand the immediate early mediators of focal exocytosis during phagocytosis. Focal exocytosis of vesicles derived from the recycling endosomes as well as lysosomes depends on the functioning of key Ca2+ sensors in these compartments (10, 11, 13). It was therefore imperative to test the role of Ca2+ in the focal movement of Golgi-derived vesicles. Release of Ca2+ from intracellular stores is one of the key signaling events during the course of phagocytosis and downstream maturation of the phagosomes (34). As expected, phagocytosis of both latex beads and H37Rv in THP-1 macrophages was severely hampered in the presence of TMB-8 (Fig. 7, E and F), an inhibitor of the IP3 receptor that serves as the Ca2+ release channel from intracellular stores like ER upon binding with IP3 (35). Intriguingly, the presence of the Ca2+ chelator EGTA in the extracellular media also inhibited phagocytic uptake of both latex beads and H37Rv in THP-1 macrophages (Fig. 7, E and F), thereby suggesting the involvement of extracellular Ca2+ in the phagocytic uptake. However, it could simply imply the effect of capacitative Ca2+ influx, which typically requires the CRAC channels and happens immediately after the intracellular stores are exhausted of Ca2+ (35). Treatment with both TMB-8 and EGTA had an additive effect on the uptake of latex beads but not on the uptake of M. tuberculosis by THP-1 macrophages (Fig. 7, E and F). We also noted a much more pronounced and sustained effect of TMB-8 and EGTA + TMB8 (∼70 and ∼85% inhibition respectively, Fig. 7E) on the uptake of latex beads as against that of M. tuberculosis (∼25 and ∼45% inhibition respectively, Fig. 7F). As observed in the case of wortmannin treatment, inhibition of Ca2+ signaling also resulted in a marked decline in the recruitment of mannosidase-II at the nascent phagosomes (Fig. 7, C and D).
Entry of Extracellular Ca2+ through Voltage-gated Ca2+ Channels Helps Create the Foci for the Recruitment of Vesicles during Phagocytosis
The role and mechanism of recruitment of extracellular Ca2+ during phagocytosis are limited to the capacitative influx (36) or through passive accumulation of Ca2+ in the phagosomes during phagocytosis (37). An increase in the local Ca2+ concentration around phagosomes has been reported to facilitate phagocytosis (38). Interestingly, the role of extracellular Ca2+ during the membrane repair process and focal exocytosis of vesicles to the damaged site have been extensively reported (39–41). Similarly, during neurotransmitter release in the neuronal cells, activation of voltage-gated Ca2+ channels during action potential helps focal exocytosis of secretory vesicles (42). We hypothesized that one of the earliest signals during phagocytic uptake could consist of Ca2+ entry into the cells through the voltage-gated Ca2+ channel. In THP-1 macrophages treated with loperamide hydrochloride or amlodipine besylate (inhibitors of L/P-type and L-type voltage-gated Ca2+ channels respectively) (43, 44), phagocytosis of E. coli declined considerably (Fig. 8A). These treatments were also effective against phagocytosis of latex beads and M. tuberculosis in THP-1 macrophages (Fig. 8, B and C, respectively). Treatment with any of these two inhibitors also blocked mannosidase-II recruitment at the nascent phagosomes during uptake of latex beads in THP-1 macrophages (Fig. 8D). Thus, voltage-gated channel appeared to play an important role during phagocytosis of diverse targets.
FIGURE 8.

Ca2+ entry through voltage-gated Ca2+ channel helps phagocytosis. A, THP-1-derived macrophages were pretreated with amlodipine (100 μm) or loperamide (100 μm) for 30 min before addition of GFP expressing E. coli. At respective time points, the cells were fixed and analyzed by flow cytometry. Data shown are average from three different experiments and represented as % uptake in the treated cells with respect to the untreated cells (values ± S.D.). B, THP-1-derived macrophages were pretreated with loperamide hydrochloride (100 μm) and amlodipine besylate (100 μm) for 30 min and incubated with FITC-labeled latex beads. The cells were fixed at respective points and analyzed by flow cytometry (values ± S.D.). C, THP-1-derived macrophages were pretreated with loperamide hydrochloride (100 μm) and amlodipine besylate (100 μm) for 30 min and infected with PKH67-labeled H37RV. The cells were fixed at respective points and analyzed by flow cytometry (values ± S.D.). D, RAW264.7 macrophages expressing mCherry-MAN2A (red) were pretreated with amlodipine (100 μm) or loperamide (100 μm) for 30 min and subsequently incubated with mouse serum-coated latex beads for 30 min and 1 h. The total intensity of mCherry-MAN2A puncta on the bead surface was determined using 3D spot creation module in Imaris 7.2, and the intensity distribution of the population has been plotted. The box plot represents data from more than 200 beads analyzed from two different experiments (*, p value <0.05; **, p value <0.01; and ***, p value <0.005).
It now seemed plausible that extracellular Ca2+ helped decide the foci for the recruitment of Golgi-derived vesicles during phagosome formation (42). To test that, we investigated the accumulation of PIP3 at the site of engulfment. Inhibition of PIP3 by wortmannin treatment abrogates the uptake as observed in this study (Fig. 7) as well as reported earlier (14). PIP3 is known to interact with the pleckstrin homology (PH) domain of proteins. In cells expressing AKT-PH domain fused with mCherry (AKT-PH-mCherry), at 5 min post-addition of E. coli, we observed, as expected, a significant accumulation of PIP3 at the site of bacterial entry (Fig. 9A). Also there was a gradual decline in the PIP3 levels as we move further into the cells from the site of phagocytosis (Fig. 9A). However, in cells pretreated with the Ca2+ chelator (EGTA, 3 mm), the selective accumulation of PIP3 at the site of phagocytosis was lost (Fig. 9A), and PIP3 was almost uniformly distributed across the cell irrespective of the site of engagement with the bacteria (Fig. 9A). Surprisingly, similar effect on PIP3 distribution was observed when these cells were pretreated with either loperamide or amlodipine (Fig. 9A). In both of these cases, the distribution of PIP3 in the cells was not influenced by the site of engagement with the bacteria (Fig. 9A). In cells treated with the PI3K inhibitor wortmannin, AKT-PH-mCherry was, as expected, more uniformly distributed (Fig. 9A). Quantitatively, it was evident from the signal intensity plots (see “Materials and Methods”) in Fig. 9B that a gradient of PIP3 is established during phagocytosis, with the highest concentration at the site of entry. Moreover, because blocking the entry of extracellular Ca2+ either through chelators or inhibitors of voltage-gated channels resulted in a loss of PIP3 gradient in a similar fashion as in the case of wortmannin treatment, it was evident that the formation of the PIP3 gradient was dependent on Ca2+, and more likely on the entry of Ca2+ from the extracellular milieu through the voltage-gated Ca2+ channels (Fig. 9B). Together, these results imply that the extracellular Ca2+, entering through a voltage-gated Ca2+ channel, helps form the foci for the recruitment of PI3K and initiate the signaling cascade for phagocytic uptake in the macrophages.
FIGURE 9.

Role of voltage-gated Ca2+ channels in establishing the PIP3 gradient during phagocytosis. A, RAW264.7 macrophages were transfected with AKT-PH-mCherry. At 24 h, cells were incubated with GFP expressing E. coli for 5 min. In parallel, we also had AKT-PH-mCherry-expressing cells that were pretreated with EGTA (3 mm), loperamide (100 μm), amlodipine (100 μm), or wortmannin (5 μm) followed by incubation with GFP expressing E. coli for 5 min. Samples were fixed at 5 min and analyzed by confocal microscopy. The arrows in the far right image in each of the panels highlight the fluorescence intensity measurements for the analysis presented in B (scale bar, 4 μm). DIC, differential interference contrast. B, images in A (arrows) were analyzed using intensity profile line tool in the NIS elements software (see “Materials and Methods”). The data represent median from more than 20 fields for each case.
MAN2A-positive Vesicles Represent Golgi-derived Secretory Vesicles
During the membrane repair and neurotransmitter release, secretory vesicles derived from the Golgi are rushed to the site of repair or release (40). Fusion of secretory vesicles with the membrane results in closure of membrane damage or release of the vesicular content (40). The fusion involves interaction between v-SNARE VAMP2 on the secretory vesicles and t-SNARE syntaxin 1 at the plasma membrane (45). We did observe VAMP2 in the phagosome proteome thereby providing a clue that the mannosidase-II vesicles could most likely be secretory vesicles derived from the Golgi. In macrophages, we confirmed that endogenous VAMP2 or overexpressed VAMP2 showed nearly similar distribution (Fig. 10A), as also reported earlier (46). With respect to VAMP3, VAMP2 largely targets distinct compartments, albeit with some overlaps (Fig. 10A). Furthermore, it was evident that in macrophages the VAMP2 pathways were distinct from the VAMP3 pathway because unlike VAMP3, which was insensitive to BfA treatment (Fig. 5D), VAMP2 recruitment to the phagosomes was sensitive to BfA treatment (Fig. 10B). Functional distinction between VAMP2 and VAMP3 has also been shown previously on multiple occasions in different cellular systems (46–50). We next tested the uptake of E. coli in cells where VAMP2 was knocked down using siRNA. We could achieve more than 55% knockdown of VAMP2 in THP-1 macrophages (Fig. 10C). In VAMP2 knockdown cells, E. coli uptake was reduced by ∼50% at 10 min and ∼35% by 20 min (Fig. 10C). In U937 cells expressing mCherry-MAN2A, we observed upon siRNA treatment recruitment of mannosidase-2 at the phagosomes declined by nearly 50% at 10 min to ∼75% at 30 min (Fig. 10D). Interestingly, there was no decline in the recruitment of VAMP3 to the E. coli phagosomes in VAMP2 knockdown cells (Fig. 10E). The progressive loss of mannosidase-II-positive E. coli phagosomes in the absence of VAMP2 indicates a compensatory mechanism whereby in the absence of secretory vesicles, membrane from other sources, like recycling endosomes, ER, etc., gets recruited to help phagocytosis. Because VAMP2 is required for the fusion of secretory vesicles with the target membrane, loss of mannosidase-II at the phagosomes in the absence of VAMP2 suggests inhibition of fusion of Golgi-derived vesicles with the phagosomes. However, we could not rule out whether VAMP2 knockdown also influenced vesicle release from the GA.
FIGURE 10.

Recruitment of mannosidase-II at phagosomes require secretory vesicle-specific v-SNARE VAMP2. A, upper panel, U937 cells expressing EGFP-VAMP2 were fixed and stained with anti-VAMP3 antibody followed by Alexa Fluor 568-labeled secondary antibody (scale bar, 4 μm). White box in the merge panel identifies the area that was magnified for the zoom panel. Lower panel, U937 cells were fixed and stained with anti-VAMP3 (rabbit) antibody and anti-VAMP2 (mouse) antibody followed by anti-rabbit Alexa Fluor 647 secondary antibody (pseudocolor blue) and labeled anti-mouse Alexa Fluor 488-labeled secondary antibody (scale bar, 15 μm). White box in the merge panel identifies the area that was magnified for the zoom panel. Percent co-localization ± S.D. is marked on the merged images. DIC, differential interference contrast. B, EGFP-VAMP2-expressing U937 cells were incubated with serum-coated latex beads in the presence or absence of BfA for 30 min. Samples were fixed and stained with anti-VAMP3 antibody followed by Alexa Fluor 568-labeled secondary antibody. Images were analyzed using 3D module in Imaris 7.2, and VAMP2 intensity distribution in the latex beads were calculated (***, p value <0.001). C, THP-1-derived macrophages were transfected with VAMP2 siRNA or scrambled siRNA control for 48 h; cells were lysed and probed with anti-VAMP2 antibody in the Western blotting. D, THP-1-derived macrophages were transfected with VAMP2 siRNA and at 48 h post-transfection; uptake of E. coli was assayed in comparison with the control cells that received scrambled siRNA. Uptake of PKH67-labeled E. coli was monitored at 10, 20, and 30 min. Data represent average from three replicates, and error bars are S.D. E, in mCherry-MAN2A-expressing cells, VAMP2 was knocked down and incubated with GFP expressing E. coli for different time points as mentioned. Samples were then fixed and analyzed through a confocal microscope. Data represent average of more than 200 bacteria from two different experiments (values ± S.D.; scale bar). F, in the experiment mentioned in B, samples were also stained with anti-VAMP3 antibody followed by Alexa Fluor 405-labeled secondary antibody and analyzed through a confocal microscope. Data represent average of more than 200 bacteria from two different experiments (values ± S.D.).
Mannosidase-II-positive Early Phagosomes Are Also NCS1-positive
Having established the role of Ca2+ in regulating the movement of mannosidase-II vesicles during phagocytosis, we next wanted to understand how the focal movement of Golgi-derived vesicles is triggered in phagocytosing macrophages. NCS1 is an EF-hand motif containing protein, originally believed to exclusively express in the neuronal cells (51). Subsequently, it was shown to be expressed in a variety of cell lines as a TGN resident Ca2+-interacting protein (51). NCS1 was reported to be involved in the recruitment of Golgi-derived vesicles during plasma membrane repair in macrophages and was integral to these vesicles (23, 52). In cells expressing mCherry-MAN2A, we stained for NCS1 during uptake of latex beads or M. tuberculosis. In both the cases, NCS1 co-localized with the cargo (Fig. 11, A and B). In the case of M. tuberculosis, nearly all M. tuberculosis-containing phagosomes were also positive for NCS1, whereas some NCS1-positive phagosomes did not contain mannosidase-II (Fig. 11B). Similarly for beads, NCS1 intensity was much higher compared with mannosidase-II intensity (Fig. 11B). Moreover, we observed NCS1 to co-localize with mannosidase-II in vesicles that were not part of the phagosome (Fig. 11C), corroborating the fact that both mannosidase-II and NCS1 are components of the secretory vesicles derived from the GA (23, 52).
FIGURE 11.

Neuronal calcium sensor (NCS1) in the Golgi apparatus recognizes Ca2+ signal for focal release of mannosidase-II vesicles. A, mCherrry-MAN2A-expressing U937 macrophages were incubated with latex beads (30 min) or infected with PKH67-labeled H37Rv (1 h). Cells were fixed and stained with anti-NCS1 antibody followed by Alexa Fluor 405-labeled secondary antibody. For the upper panel, latex beads were given green pseudo-color using Imaris (scale bar, 4 μm). B, upper panel, in U937 macrophages, incubated with beads for 30 min or 1 h, samples were stained with anti-NCS1 antibody. Presence of NCS1 at the bead surface was calculated using the 3D spot creation module in Imaris 7.2 software. The box plot at right shows data from more than 100 beads from two independent experiments. For lower panel, mCherry-MAN2A (red)-expressing U937-derived macrophages were infected with PKH67-labeled H37Rv (green) for 1 and 2 h. At the respective time points, samples were fixed and stained with anti-NCS1 antibody followed by Alexa Fluor 405-tagged secondary antibody. The images are representative of the 1-h time point. Percent co-localization of H37Rv with mannosidase-II, NCS1, or both mannosidase-II and NCS1 was calculated using Imaris 7.2. The data represent an average of more than 150 bacteria from three different experiments (values ± S.D.). DIC, differential interference contrast. C, co-localization of mannosidase-II and NCS1 in U937 macrophages. U937 macrophages expressing mCherry-MAN2A were stained with anti-NCS1 antibody followed by secondary antibody (pseudo-colored green) (scale bar, 15 μm). White box in the merge panel identifies the area that was magnified for the zoom panel. D, siRNA-mediated knockdown of NCS1 was confirmed by Western blottings on the whole cell lysates from the transfected cells. Knockdown was monitored at 48 h post-transfection. E, THP-1-derived macrophages treated with NCS1 siRNA were incubated with GFP expressing E. coli for 5 and 15 min. Cells were stained with anti-mannosidase-II antibody to assess the recruitment of mannosidase-II at the phagosomes in NCS1-depleted cells. Percent co-localization of E. coli with mannosidase-II was calculated using Imaris 7.2. The data represent an average of more than 150 bacteria from three different experiments (values ± S.E., *, p value <0.05). F, in the similar experiment as mentioned in E, samples were stained with anti-VAMP3 antibody to assess the recruitment of VAMP3 at the phagosomes in NCS1-depleted cells. Percent co-localization of E. coli with mannosidase-II was calculated using Imaris 7.2. The data represent an average of more than 100 bacteria from three different experiments (value ± S.D.). G, THP-1 macrophages were treated with siRNA against NCS1 or scrambled control. At 48 h post-siRNA treatment, cells were monitored to uptake latex beads (1 μm), E. coli, S. typhimurium, or H37Rv for indicated time points. Data are shown as % uptake in the siRNA-treated cells with respect to the scrambled siRNA control-treated cells. Data are representative of three independent experiments (values ± S.D.).
Depleting NCS1 Inhibits Focal Exocytosis of MAN2A Vesicle but Not of VAMP3 Vesicles during Phagocytosis
To test whether Ca2+-dependent trigger of vesicular trafficking relied on the ability of NCS1 activation upon Ca2+ binding, we compared the recruitment of mannosidase-II at the nascent phagosome during phagocytosis of E. coli in THP-1 macrophages that were either treated with scrambled siRNA control or NCS1-specific siRNA (Fig. 11D). Knocking down NCS1 resulted in more than a 50% decline in the recruitment of mannosidase-II at the early E. coli phagosomes (Fig. 11E). Requirement of NCS1 was very specific for mannosidase-II recruitment because in NCS1-depleted cells there was no decline in the recruitment of VAMP3 at the phagosomes (Fig. 11F). As expected, knockdown of NCS1 in THP-1 macrophages resulted in a marked decline in the uptake of latex beads, E. coli, Salmonella, and M. tuberculosis (Fig. 11G). True to all other treatments, which inhibited the phagocytic uptake, NCS1 siRNA knockdown also had both quantitative and kinetic effects on the uptake of all of the targets except in Salmonella where the effects were persistent (Fig. 11G). Thus, uptake in the case of latex beads was ∼35, 60, and 85% of the control in the siRNA-treated sets at 30 min and 1 and 2 h, respectively (Fig. 11G). For E. coli, these numbers were 8, 10, and 22% of control at 5, 15, and 30 min (Fig. 11G). In the case of Salmonella, uptake was about 65% of the control set at 15 and 30 min, and in case of M. tuberculosis, the relative uptake in the NCS knockdown cells was 40, 45, and 55% of control at 1, 2, and 4 h, respectively (Fig. 11G).
Discussion
Professional phagocytes like macrophages require continuous supply of membrane to form phagosomes around the phagocytosed particles (53). It is now understood that cellular compartments like recycling endosomes, lysosomes, and ER could supply membrane for the nascent phagosomes (6–8). Adding to the existing pool of membrane sources available for phagosome formation, in this study we show recruitment of Golgi-derived vesicles at the site of phagocytosis in macrophages. However, unlike previous reports, where the studies were mostly restricted to either latex beads or a select organism, we show here a more universal requirement of the Golgi-derived vesicles during phagocytosis by macrophages using inert particles, non-pathogenic bacteria, and two different pathogenic bacterial species as the cargo. It is important to note here that GA serves as the origin for most vesicles in the cell, including recycling endosomes, endocytic machinery, and vesicles destined to the plasma membrane for exocytosis (54). Bacterial pathogens once inside the host cells further subvert the membrane trafficking pathways to ensure prolonged survival and escape from innate defense mechanisms (55). Interestingly, GA serves as the hub for trafficking inside the cells (56). It therefore may make strong sense that GA gets involved and alerted to the incoming pathogen, while the cell has just started to engulf it. This line of investigation seems extremely fascinating at present because it may open a new understanding in the functioning of the innate immune system. However, involvement of Golgi apparatus during phagocytosis was ruled out earlier (16). Coincidentally, all of these reports involved studies on Fcγ receptor-mediated phagocytosis using IgG-coated latex beads (16). A detailed analysis of uptake of latex beads coated with either IgG or serum revealed the selectivity in the involvement of Golgi apparatus during phagocytosis. Although uptake of IgG-coated latex beads was not sensitive to brefeldin A treatment, uptake of serum-opsonized latex beads was sensitive to this treatment. We realized that serum-opsonized latex beads would engage a different set of receptors like complement receptor, mannose receptor, and scavenger receptors on the macrophages unlike IgG-coated beads, which would only engage with Fcγ receptors (3). Thus, although we observed recruitment of Golgi-derived vesicles to most of the cargos studied and that the recruitment of Golgi-derived vesicles was important for their uptake, it was not the case for uptake of IgG-coated beads. Interestingly, brefeldin A treatment also led to a decline in mannosidase-II recruitment at the nascent phagosomes. More importantly, brefeldin A treatment did not have any influence on the recruitment of VAMP3, a recycling endosome vesicle-specific v-SNARE, which is known to be recruited at early phagosomes. Thus, recruitment of mannosidase-II happened through a pathway other than the established recycling endosome pathway of vesicle recruitment. Better understanding of specific signaling events downstream of these receptors could provide answers on how differential receptor engagement ensures recruitment of selective organelles.
Role of Golgi-derived vesicles in supplying the membrane at the plasma membrane is also well known in another context, the membrane repair pathway (23). Using mannosidase-II as specific marker for Golgi-derived vesicles, they showed participation of GA and lysosomes in the repair process through targeted exocytosis (23). The movement of vesicles during phagocytosis from the recycling endosomes and lysosomes was also reported to follow a similarly targeted exocytosis where vesicles are directed toward the phagocytic cup (7, 13, 57). Interestingly, more than 90% of mannosidase-II-positive phagosomes at the site of entry were also found to be positive for VAMP-3 and dynamin-2. Although VAMP-3 gets recruited through focal exocytosis, dynamin-2 is known to regulate post-Golgi transport of vesicles and facilitate focal exocytosis (13). Dynamins have earlier been implicated in regulating the focal exocytosis in phagocytosing macrophages (13, 33, 58). Inhibition of dynamins by dynasore treatment resulted in a reduced phagocytic uptake, which may simply occur due to inhibition of dynamin-mediated scission of phagosomes from the membrane (59). However, dynasore treatment also resulted in reduced mannosidase-II recruitment at the nascent phagosomes, which can only happen if dynasore is inhibiting the generation and focal movement of Golgi-derived vesicles. Moreover, we categorically show that mannosidase-II recruitment is independent of recruitment of markers from endocytic pathways, re-iterating the fact that dynasore mediated effect was mostly due to inhibition of post-Golgi transport of vesicles, consistent with previously reported function of this inhibitor (13). High co-localization of mannosidase-II with TfR further emphasized very early recruitment of Golgi-derived vesicles. Yet another instance where vesicle exocytosis from Golgi has been extensively studied is in the context of neurotransmitter release, which shows remarkable similarity with the focal exocytosis during membrane repair. Thus, it is intriguing to witness the brilliance of cellular economy, where a common mechanism could be utilized to address three entirely independent cellular requirements.
Next, to understand how target recognition at the cell surface for phagocytic uptake could trigger GA to elicit the movement of the vesicles, we took cues from membrane repair pathway where the damage is typically sensed via entry of extracellular Ca2+ into the cells (39, 40). We indeed observed inhibiting either extracellular Ca2+ or the release of Ca2+ from intracellular stores resulted in a loss of phagocytic function in the macrophages. The possibility of a membrane breach during phagocytic uptake in macrophages has never been discussed. Thus the most plausible source for the entry of extracellular Ca2+ was some membrane channels, which are classically involved at a similar step during neurotransmitter release (42, 60). Experiments with loperamide and amlodipine strongly support a critical involvement of voltage-gated Ca2+ channel in the extracellular Ca2+ entry thereby facilitating phagocytosis. Incidentally the role of voltage-gated channel in regulating podosome formation in the macrophages was recently shown (61). Yet another channel TRPV2 was recently shown to be important for phagocytosis in macrophages, and its absence resulted in loss of Ca2+ influx from the extracellular milieu and abrogated phagocytic uptake (62). Curiously, macrophages lacking TRPV2 were also defective in chemoattractant-evoked motility (62). It may not be unusual to assume that some of the inhibitory effects shown in this study were most likely due to inhibition of transient receptor potential cation channel subfamily V at the plasma membrane. Therefore, it seems macrophage membrane depolarization could be a more general mechanism of cellular functioning, including phagocytosis, adherence, and motility.
Phagocytosis was also dependent on the release of Ca2+ from intracellular stores, as TMB8 treatment inhibited phagocytosis. A combination of EGTA and TMB8 had more dramatic effects on phagocytosis, which also showed some sort of selectivity in terms of the cargo. Thus, for latex beads, blocking both intra- and extracellular cargo abolished their uptake by the macrophages; however, in the case of bacteria, the block in uptake was more kinetic in nature. It strongly supports the possibility that Ca2+ may be extremely critical for the uptake of cargos, where cognate receptors are not known/available and thereby rely a lot on the membrane depolarization and associated Ca2+ entry. The graded importance of Ca2+ during phagocytosis of diverse targets needs further exploration for better understanding. Inhibition of Ca2+ also resulted in reduced mannosidase-II recruitment at the phagosomes.
The role of PI3K in phagocytosis is well known, which supposedly helps pseudopod extension while engulfing the target (14). However, it has also been shown that inhibition of PI3K may limit the membrane availability (15). We indeed observed that uptake of M. tuberculosis, Salmonella, or latex beads by macrophages was severely compromised in the presence of wortmannin. Salmonella entry is known to occur via either macropinocytosis or phagocytosis; however, the former is insensitive to PI3K inhibition (63). Thus, the strains used in this study are taken up by the macrophages through phagocytosis. Interestingly, vesicle recruitment has also been reported to be important for membrane ruffle formation, which helps in Salmonella macropinocytosis (64). We also noted significantly reduced recruitment of mannosidase-II vesicles at the nascent phagosomes in the presence of wortmannin. Thus, recruitment of Golgi-derived vesicles at the nascent phagosomes indeed required PIP3. Experiments with AKT-PH-mCherry clearly support the establishment of PIP3 gradient during phagocytosis. Selective PIP3 enrichment has previously been shown regulating cellular polarity, chemotaxis, and pseudopod extension (14, 65, 66). The most dramatic observation was, however, the use of voltage-gated Ca2+ channels by macrophages to set the foci for PIP3 accumulation, which eventually results in all the downstream signaling and recruitments. Interestingly, dynamins have conserved PH domain at their C terminus, making them responsive to the PIP3 levels. It has been shown that mutations in the PH domain of dynamin could result in severe defects in its key functioning like post-Golgi transport and endocytosis (67, 68). Thus PI3K and dynamins seem to work together for the focal recruitment of Golgi-derived vesicles during phagosome biogenesis.
The Ca2+-sensing function in the GA is attributed to the resident molecule NCS1, which was initially discovered in the neuronal cells regulating the synaptic transmission (51). NCS1 was also shown as part of the secretory vesicles (69). In macrophages, ablation of NCS1 resulted in a block in the membrane-resealing pathway (23). We therefore hypothesized that NCS1 could also serve as the Ca2+ sensor in the Golgi apparatus for triggering the exodus of the vesicles during phagocytosis. Interestingly, NCS1 knockdown not only reduced the phagocytic function of macrophages, there was also significantly lower recruitment of mannosidase-II at the phagosomes. Further emphasizing the existence of a separate mechanism for mannosidase-II recruitment, NCS1 knockdown did not influence recruitment of VAMP3 on the early phagosomes.
The results from purified latex bead phagosomes supported the overall observation in this study that phagocytic uptake is a complex process and involves several players. We deliberately focused on the molecules below the 25-kDa cutoff to make sure of the enrichment of three key classes of molecules, including RABs (∼25 kDa), ARFs (∼20 kDa), and VAMPs (∼14 kDa). This was to ensure that the presence of other high abundance and high molecular weight proteins did not mask these low abundant low molecular weight proteins. We identified 17 different RABs on the phagosomes, nine of which were associated with Golgi function. We also identified five of six known ARFs, where ARF1 and ARF3 were particularly important for their known association with Golgi apparatus, phagosome/engulfment, and exocytosis (70–72). We could identify four different VAMPs in the phagosomes, including VAMP2, -3, -4, and -8. Although the role of VAMP3 in phagosome biogenesis is well described, VAMP2 is particularly known for its involvement in exocytosis, neurotransmitter release, and secretory vesicles (27, 28). Previously in a proteomic study of the latex beads containing phagosome, VAMP4 was identified as a phagosome membrane-associated protein (73). VAMP4s are trans-Golgi resident proteins and are also involved in the immature secretory granules (74). We also found SEC22B in the phagosome MS data, an important SNARE known to be important for the recruitment of ER during phagocytic uptake (16). Considering the number of RABs, ARFs, and VAMPs associated with the exocytosis and secretory vesicles, it seems very likely that the true identity of the Golgi-derived vesicles that are recruited during phagosome biogenesis could be secretory vesicles.
At the molecular level it seems that SNARE-mediated fusion may be involved in the phagosome biogenesis. We were able to track the presence of syntaxin 1, the t-SNARE at the plasma membrane involved in SNARE-mediated vesicle fusion, along with mannosidase-II at the early phagosomes (data not shown). Syntaxin-1 is known to facilitate fusion of secretory vesicles (75). Moreover, its presence will also be required for the fusion of VAMP3-positive vesicles, a vSNARE typically located in the recycling endosomes. However, the presence of VAMP2 in the proteomic data, a secretory vesicle-specific v-SNARE, gave us a clue that possibly recruitment and fusion of mannosidase-II vesicles to the phagosomes happen through the secretory pathway. By knocking down VAMP2, we were able to see selective decline in the recruitment for mannosidase-II without influencing VAMP3 recruitment at the phagosomes. The selectivity between VAMP3 and VAMP2 was evident even by their respective sensitivity to BfA treatment. The distinctive aspect of VAMP2 versus VAMP3 regulation has been reported in the past, although in unrelated systems (47, 49, 50). VAMP2 knockdown also resulted in a marked decline in the uptake, reiterating the fact that phagocytosis involves cooperation between multiple sources, which can provide a membrane for membrane biogenesis. Taken together, the results in this study allowed us to reconstruct the process of phagocytosis (Fig. 12). It is important to note that Golgi-derived vesicles are one of the several subcellular vesicular pools available for phagosome biogenesis, including recycling endosomes and lysosomes. It actually explains why in many cases, except for PI3K inhibition, the effects were more kinetic in nature.
FIGURE 12.

Schematic model describing involvement of Golgi-derived vesicles during phagocytic uptake in macrophages. i, initial recognition of an object (bacteria, beads, cell debris, etc.) for phagocytosis results in membrane depolarization possibly due to torsional stress and resulting in the activation of voltage-gated Ca2+ channels, leading to the entry of extracellular Ca2+ into the cells. The entry of Ca2+ through voltage-gated Ca2+ channels sets the focus for quick and efficient recruitment of PI3K and results in the accumulation of PIP3 at the site of phagocytosis. These early events are further aided by signaling through specific pattern recognition receptors and release of Ca2+ from intracellular stores. ii, increased cytosolic Ca2+ is sensed by Golgi-resident NCS1, which triggers the movement of mannosidase-II vesicles toward the site of phagocytosis, guided by a gradient of PIP3, along with dynamin and microtubule. iii, mannosidase-II vesicles fuse with the membrane at the site of phagocytosis and help grow the phagosome around the cargo before final scission and internalization. iv, membrane of the nascent phagosome is contributed in part by the plasma membrane and the rest from the Golgi-derived vesicles (in this model for clarity, we have excluded lysosome and recycling endosomes as additional sources, see text).
In conclusion, we show here an as yet unknown function of Golgi-derived vesicles during phagocytic uptake in macrophages. The targeted exocytosis coupled with phagocytosis has been studied in the past; however, involvement of secretory vesicles from the GA during phagocytosis was unprecedented. Thus, although the utilization of distinct phagocytic pathways by pathogens to escape killing in macrophages is well known, this study highlights the redundancy in the repertoire of membrane sources recruited downstream to distinct phagocytic receptors. How the membrane repertoire utilized during phagocytosis influences the fate of infection poses an attractive direction of investigation. Finally, our finding that the voltage-gated Ca2+ channel could play a role in the process of phagocytosis may be potentially harnessed in the future for developing better therapeutic interventions for various infectious diseases, including tuberculosis.
Materials and Methods
Ethics Statement
The animal experiments were performed upon prior approval from the institutional ethics committee (IAEC) of the International Center for Genetic Engineering and Biotechnology (approval no. ICGEB/AH/2013/03/IMM-38).
Reagents and Antibodies
The following reagents were used in this study: wortmannin (Sigma, W1628); dynasore hydrate (Sigma, D7693); nocodazole (Sigma, M1404); TMB8 (Sigma, T111); EGTA (Amresco, 0732); loperamide (Sigma, L4762); amlodipine besylate (Sigma, A5605); PKH 67 (Sigma, MINI 26); RIPA buffer (Amresco, N653); BSA (Sigma, A2153); saponin (Sigma, 47036); puromycin (Invivogen, ant-pr-1); 4 μm aldehydate-sulfate latex beads (Life Technologies, Inc., A37304); and 1 μm of yellow green aldehyde latex beads (Life Technologies, Inc., F8823). All the siRNA used in this study were siGenome siRNA SmartPool (Dharmacon Inc). The following were used: NCS1 siRNA (M-013024-01-005-siGENOME; sequences: UCUACGACGGGCUGGUAUA, GGGGAACCCUGGAUGAGAA, ACUUGGACAAUGAUGGCUA, and ACAUUGUGGAUGCCAUUUA); VAMP2 siRNA (M-012498-03-0005-siGENOME; sequences: GCGCAAAUACUGGUGGAAA, CCUCCAAACCUCACCAGUA, CAGCAUGUUAAUUAGCGUA, and GUAAAUAGCCAGCUGUUAU); and non-targeting siRNA (D-001206-13-05; sequence: UAGCGACUAAACACAUCAA). The transfection reagent used for siRNA transfections was Dharmafect-2 (Dharmacon Inc.). The primary antibodies used in this study are as follows: mannosidase II (Abcam, ab12277); dynamin (Santa Cruz Biotechnology, sc-6401); VAMP3 (Abcam, ab43080); transferrin (Abcam, ab84036); and VAMP2 (R&D Systems, MAB5136-SP). The secondary antibodies used in this study are Alexa Fluor 405 and Alexa Fluor 568 conjugates from Life Technologies, Inc.
Plasmid Constructs
The plasmid constructs used in this study are mCherry-ManII-N-10 (Addgene plasmid 55073), pcDNA3.1_AktPH-mCherry (Addgene plasmid 67301), pCT-Golgi-GFP (CYTO104-VA-1), and pCT-Mem-GFP (System Biosciences, CYTO100-PA-1), and pEGFP-VAMP2 (Addgene plasmid 42308).
Bacterial Culture Maintenance
Mycobacterial cultures were maintained in 7H9 media (Difco) supplemented with 10% oleic acid/albumin/dextrose/catalase. Single cell suspension of mycobacterial strains were prepared by aspiration of the culture eight times with a 26-gauge needle and six times with a 30-gauge needle. Quantification of this prepared culture was done by taking absorbance at 600 nm wavelength (0.6 OD corresponds to ∼100 × 106 bacteria). The bacteria thus appropriately calculated were added to the cells at the mentioned multiplicity of infection. For microscopy experiments, the desired number of bacteria was stained with PKH67 (Sigma), a lipophilic green fluorescent dye, as per the manufacturer's protocol. The stained bacteria were then passed three times through a 26-gauge needle and used for infection. E. coli and S. typhimurium strains were maintained in LB (Difco). For infection, culture density was determined by absorbance at 600 for both E. coli and S. typhimurium containing GFP, an OD of 1 = 1 × 108 bacteria for both these strains.
Tissue Culture
THP-1 cells (a kind gift from Dr. Dong An, UCLA) and U937 cells (ATCC) were cultured in RPMI 1640 medium (Life Technologies, Inc.), and RAW264.7 (ATCC) murine macrophages were grown in Dulbecco's modified Eagle's media (DMEM, Life Technologies, Inc.) supplemented with 10% fetal bovine serum (FBS, Gibco) and maintained 37 °C in a humidified 5% CO2 atmosphere. THP-1 cells were differentiated with 20 ng/μl PMA for 24 h, washed with plain RPMI 1640 medium, and maintained in 10% FCS-supplemented RPMI 1640 medium for another 24 h. The cells were then infected with respective bacteria/beads. RAW264.7 macrophages were seeded in respective plates and differentiated with 200 ng/ml LPS for 12 h. The cells were washed once with plain DMEM and infected with respective bacteria/beads.
Latex Bead Experiments
Latex beads were coated with anti-human IgG (for THP-1 and U937 macrophages) or anti-mouse IgG (for RAW264.7 macrophages) as described in Ref. 16, 1 mg/ml antibody for 1 h, or incubated with 10 times the volume of mouse/human sera for 1 h and then utilized for further experimentation by confocal or flow cytometry.
Animals and Isolation of BMDMs
BMDMs were isolated from femurs of BALB/c mice (4–6 weeks old, female) obtained from the institutional animal house. BMDMs were obtained by culturing the marrow cells in the presence of macrophage-colony stimulating factor (M-CSF, eBioscience, 14-8983-80) for 7 days. Fully differentiated macrophages were harvested and seeded for infection with H37Rv. The infection protocol was same as described above for THP-1 macrophages.
Experiments with Latex Beads
For microscopy experiments, 4 μm aldehyde sulfate latex beads (Life Technologies, Inc.) were incubated with human/mouse IgG on an agitator overnight. The beads were washed twice with plain DMEM by centrifugation at 2000 rpm for 10 min and added to the cells at a respective m.o.i. For flow cytometry experiments, 1 μm of FITC-labeled aldehyde beads were added at a respective m.o.i. to the differentiated THP-1 cells.
Transfection and Nucleofection Assays
Transfection and transduction was carried out using JetPrime reagent (Himedia) as per the manufacturer's protocol. Nucleofection was carried out with the 4D-NucleofectorTM system Lonza in 20 μl of NucleocuvetteTM strips with the program DS-136, as per the manufacturer's protocol. At 24 h post-nucleofection, these cells expressed the inserted vector as determined by visualization with a fluorescence microscope.
siRNA Transfection and Inhibitor Assays
After 24 h of PMA treatment, the cells were washed once with plain RPMI 1640 medium. The siRNAs were added as per the manufacturer's protocol. After 48 h of incubation, the respective infection/assays were performed. For the inhibitor assays such as dynasore (40 and 80 μm), wortmannin (as specified) was added 4 h before infection. Nocodazole (25 μm) was added 2.5 h before infection, and EGTA (3 mm), TMB8 (100 μm), loperamide hydrochloride (100 μm), and amlodipine besylate (100 μm) were added 30 min before infection.
Creation of Stable Cell Lines
The stable cell lines were created from lentivirus cytotracer plasmids (B4GALT1-mRuby and NEUM-EGFP, System Biosciences). 1 × 106 U937 cells were added to a 24-well plate. The harvested media containing lentivirus (refer to transfection section) was added to them. At 72 h post-infection, the cells were selected on puromycin at 350 ng/ml for 21 days. The population of positive cells was routinely checked by microscopy, and it was found that post-selection for 21 days the population of cells remained stable (in our case 30–40%).
Staining for Confocal Microscopy
At specific time points, the cells were fixed with 4% paraformaldehyde (PFA) for 15 min. This was followed by two washes with 1× PBS. The cells were permeabilized with 0.4% Triton X-100 for 30 min and washed once with 1× PBS. Blocking was performed using 3% (w/v) BSA and 0.5% Tween 20 in 1× PBS for 1 h, followed by one wash with 1× PBS. The cells were now stained with the respective primary antibody made in the blocking solution at specific dilution for 90 min. The coverslips were then washed once with 1× PBST and twice with 1× PBS. This was followed by staining with the respective secondary antibody tagged with the fluorophore of choice for 90 min. The primary and secondary antibodies were diluted in blocking solutions for use. After three washes with 1× PBS, the coverslips were then mounted on glass slides with anti-fade reagent (Life Technologies, Inc.). Images were acquired with a Nikon A1R laser scanning confocal microscope with a ×100/1.4NA Plan Apochromat VC, differential interference contrast N2 objective lens. For image acquisition, the microscope settings (laser power, voltage gain, and offset) were decided with respect to unstained sample for each set to avoid signal from autofluorescence, if any. These settings were kept constant for the entire experiment. Image analysis was exclusively done by Imaris 7.2 using images in ND2 format only. Image processing, viz. 3D reconstruction, co-localization, and intensity measurements were done via Imaris 7.2 (Bitplane) following standard protocols.
Live Cell Imaging
The live cell imaging dish pre-seeded was RAW264.7 macrophages expressing mCherry-MAN2A. The live cell system was set at 5% CO2 and 37 °C. The field was set and imaged for 10 min. This was followed by an on-stage addition of 0.0001% saponin for 5 min. The cells were washed once, supplemented with complete media, and imaging was continued. The entire procedure was performed on stage to analyze the pre- and post-treatment effects on the membrane under our experimental setup.
Flow Cytometry Experiments
At respective time points, the infected cells were washed three times with sterile 1× PBS to remove extracellular bacteria/beads and fixed with 2% PFA. The cells were scraped and run on a FACS Canto or FACS influx cytometer (BD Bioscience).
Western Blotting
Following SDS-PAGE, the proteins were blotted onto a nitrocellulose membrane using a semi-dry transfer system. Following incubation with primary and secondary antibodies, the blots were scanned using Odyssey InfraRed Imaging System (LI-COR BioSciences, Lincoln, NE) at various intensities to obtain a blot scan with a minimum background. All settings were rigorously maintained for all experiments. The scans were quantitatively analyzed using Odyssey InfraRed Imaging System Application Software Version 3.O (commercially available from (LI-COR BioSciences, Lincoln, NE).
Phagosome Isolation
Latex bead phagosome for 1-μm latex beads were isolated as per the protocol described earlier (76). 1 h post addition, the plates were washed four times with cold 1× PBS, ensuring all the extracellular beads were completely washed off. The cells were scraped off with a rubber policeman and centrifuged twice with cold 1× PBS at 1200 rpm for 5 min. The cells were pooled and washed once with homogenization buffer at 1200 rpm for 5 min (3 mm imidazole, 250 mm sucrose, pH 7.4). The cells were then incubated in appropriate volume of homogenization buffer (with protease inhibitor mixture, Amresco) for 30 min. The cells were lysed by Dounce homogenizer until 90% of the cells were disrupted as observed under a light microscope. This lysate was centrifuged at 1200 rpm for 5 min at 4 °C to remove the unbroken cells. This was followed by preparation of sucrose gradient for ultracentrifugation. The homogenate consisted of 40% sucrose by mixing with equal volume of 62% sucrose, and 2.25 ml of the same was layered upon 2.0 ml of 62% sucrose solution. We then added 2.25 ml each of 35%, 25, and 10% sucrose solutions. The tubes were centrifuged at 100,000 × g for 1 h at 4 °C in an SW28 rotor (Beckman Coulter). The LBC band obtained between 10 and 25% sucrose layers was collected and washed once with cold 1× PBS at 40,000 × g at 4 °C in an SW28 rotor (Beckman Coulter).
Mass Spectrometry and Data Analysis
For LC-LTQ Orbitrap MS analysis, samples were re-solubilized in 2% (v/v) acetonitrile, 0.1% (v/v) formic acid in water and injected onto the trap column at a flow rate of 20 μl/min. Subsequently, peptides were separated on Zorbax 300SB-C18 (Agilent, Santa Clara, CA) by a gradient developed from 2% (v/v) acetonitrile, 0.1% (v/v) formic acid to 80% (v/v) acetonitrile, 0.1% (v/v) formic acid in water over 180 min at a flow rate of 300 nl/min onto an Agilent 1200 (Agilent, Santa Clara, CA) nano-flow LC-system that was in-line coupled to the nano-electrospray source of an LTQ-Orbitrap discovery hybrid mass spectrometer (Thermo Scientific, San Jose, CA). Full MS in a mass range between m/z 300 and 2000 was performed in the Orbitrap mass analyzer with a resolution of 30,000 at m/z 400 and an AGC target of 2 × 105. The strongest five signals were selected for collision-induced dissociation (CID)-MS/MS in the LTQ ion trap at a normalized collision energy of 35% using an AGC target of 1 × 105 and two microscans. Dynamic exclusion was enabled with one repeat count during 45 s and an exclusion period of 180 s. Peptide identification was performed by CID-based MS/MS of the selected precursors. For protein/peptide identification, MS/MS data were searched against the Mus musculus amino acid sequence database (downloaded in August, 2015) using an in-house Mascot server (version 2.4) through the Proteome Discoverer 1.4 software. The search was set up for full tryptic peptides with a maximum of three missed cleavage sites, and carbamidomethyl on cysteine and oxidized methionine were included as variable modifications. The precursor mass tolerance threshold was 10 ppm, and the maximum fragment mass error was 0.8 Da. The significance threshold of the ion score was calculated based on a false discovery rate of <1%, estimated by the peptide validator node of the Proteome Discoverer software.
Analysis for Functional Classes
The non-redundant list of proteins identified was matched against a series of gene ontology classes from the AMIGO2 database. The selection of gene ontology was empirically made based on known and perceived classes, which together could represent the set of trafficking proteins identified. The obtained matches were then represented as a network using Cytoscape 3.2.0.
Statistical Analysis
Comparative groups were analyzed using paired two-tailed t test using inbuilt function in MS Excel.
3D Recreation and Analysis
The intensity of a particular fluorophore on the bead was estimated by first creating a three-dimensional bead surface in the captured image (z-stack) using “spot module” in Imaris Version 7.2 (Bitplane), which automatically detects spheres in an image depending upon the dimensions fed. The software then allows one to determine fluorescence intensity on the surface individually for each fluorophore. For each case maximum fluorescence intensity was determined and plotted.
PIP3 Gradient Analysis
The gradient of fluorescence in the AKT-pH mCherry experiments was determined using “intensity profile line tool” in NIS-Elements (NIKON).
Author Contributions
D. K. and N. V. designed the study and wrote the paper. N. V., S. B. A. A., and S. G. performed the experiments. N. V. and D. K. analyzed the data. M. S. provided important reagents for the study. All authors reviewed and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
The work related to M. tuberculosis infections was carried out at Department of Biotechnology-funded Tuberculosis Aerosol Challenge Facility at International Centre for Genetic Engineering and Biotechnology. We thank Prof. Sarman Singh for access to the flow cytometer at All India Institute of Medical Sciences and Purnima Kumar for help in the confocal microscopy. We thank C-CAMP, National Center for Biological Sciences, Bangalore, India, for the mass spectrometry facility.
This work was supported in part by Department of Biotechnology, Government of India, Grant BT/PR14730/BRB/10/874/2010 (to D. K.) and in part by International AIDS Society and Centers for AIDS Research (CFAR)-funded Creative and Novel Ideas in HIV Research (CNIHR) grant from the National Institutes of Health (to D. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1 and S2.
- ER
- endoplasmic reticulum
- PIP3
- phosphatidylinositol 1,4,5-trisphosphate
- PMA
- phorbol 12-myristate 13-acetate
- PH
- pleckstrin homology
- GA
- Golgi apparatus
- Syt
- synaptotagmin
- BMDM
- bone marrow-derived macrophage
- TfR
- transferrin receptor
- ARF
- ADP-ribosylation factor
- BfA
- brefeldin A
- A.U.
- arbitrary unit
- m.o.i.
- multiplicity of infection
- TGN
- trans-Golgi network
- CID
- collision-induced dissociation
- IP3
- inositol 1,4,5-trisphosphate
- PFA
- paraformaldehyde.
References
- 1. Flannagan R. S., Jaumouillé V., and Grinstein S. (2012) The cell biology of phagocytosis. Annu. Rev. Pathol. 7, 61–98 [DOI] [PubMed] [Google Scholar]
- 2. Jutras I., and Desjardins M. (2005) Phagocytosis: at the crossroads of innate and adaptive immunity. Annu. Rev. Cell Dev. Biol. 21, 511–527 [DOI] [PubMed] [Google Scholar]
- 3. Aderem A., and Underhill D. M. (1999) Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17, 593–623 [DOI] [PubMed] [Google Scholar]
- 4. Cannon G. J., and Swanson J. A. (1992) The macrophage capacity for phagocytosis. J. Cell Sci. 101, 907–913 [DOI] [PubMed] [Google Scholar]
- 5. Holevinsky K. O., and Nelson D. J. (1998) Membrane capacitance changes associated with particle uptake during phagocytosis in macrophages. Biophys. J. 75, 2577–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gagnon E., Duclos S., Rondeau C., Chevet E., Cameron P. H., Steele-Mortimer O., Paiement J., Bergeron J. J., and Desjardins M. (2002) Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell 110, 119–131 [DOI] [PubMed] [Google Scholar]
- 7. Bajno L., Peng X. R., Schreiber A. D., Moore H. P., Trimble W. S., and Grinstein S. (2000) Focal exocytosis of VAMP3-containing vesicles at sites of phagosome formation. J. Cell Biol. 149, 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tardieux I., Webster P., Ravesloot J., Boron W., Lunn J. A., Heuser J. E., and Andrews N. W. (1992) Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell 71, 1117–1130 [DOI] [PubMed] [Google Scholar]
- 9. Braun V., and Niedergang F. (2006) Linking exocytosis and endocytosis during phagocytosis. Biol. Cell 98, 195–201 [DOI] [PubMed] [Google Scholar]
- 10. Vinet A. F., Fukuda M., and Descoteaux A. (2008) The exocytosis regulator synaptotagmin V controls phagocytosis in macrophages. J. Immunol. 181, 5289–5295 [DOI] [PubMed] [Google Scholar]
- 11. Czibener C., Sherer N. M., Becker S. M., Pypaert M., Hui E., Chapman E. R., Mothes W., and Andrews N. W. (2006) Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J. Cell Biol. 174, 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reddy A., Caler E. V., and Andrews N. W. (2001) Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 106, 157–169 [DOI] [PubMed] [Google Scholar]
- 13. Samie M., Wang X., Zhang X., Goschka A., Li X., Cheng X., Gregg E., Azar M., Zhuo Y., Garrity A. G., Gao Q., Slaugenhaupt S., Pickel J., Zolov S. N., Weisman L. S., et al. (2013) A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev. Cell 26, 511–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cox D., Tseng C. C., Bjekic G., and Greenberg S. (1999) A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J. Biol. Chem. 274, 1240–1247 [DOI] [PubMed] [Google Scholar]
- 15. Underhill D. M., and Ozinsky A. (2002) Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 20, 825–852 [DOI] [PubMed] [Google Scholar]
- 16. Becker T., Volchuk A., and Rothman J. E. (2005) Differential use of endoplasmic reticulum membrane for phagocytosis in J774 macrophages. Proc. Natl. Acad. Sci. U.S.A. 102, 4022–4026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bainton D. F., Takemura R., Stenberg P. E., and Werb Z. (1989) Rapid fragmentation and reorganization of Golgi membranes during frustrated phagocytosis of immobile immune complexes by macrophages. Am. J. Pathol. 134, 15–26 [PMC free article] [PubMed] [Google Scholar]
- 18. Beemiller P., Hoppe A. D., and Swanson J. A. (2006) A phosphatidylinositol-3-kinase-dependent signal transition regulates ARF1 and ARF6 during Fcγ receptor-mediated phagocytosis. PLoS Biol. 4, e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Braun V., Deschamps C., Raposo G., Benaroch P., Benmerah A., Chavrier P., and Niedergang F. (2007) AP-1 and ARF1 control endosomal dynamics at sites of FcR mediated phagocytosis. Mol. Biol. Cell 18, 4921–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Q., Cox D., Tseng C. C., Donaldson J. G., and Greenberg S. (1998) A requirement for ARF6 in Fcγ receptor-mediated phagocytosis in macrophages. J. Biol. Chem. 273, 19977–19981 [DOI] [PubMed] [Google Scholar]
- 21. Mosser D. M. (1994) Receptors on phagocytic cells involved in microbial recognition. Immunology Series 60, 99–114 [PubMed] [Google Scholar]
- 22. Velasco A., Hendricks L., Moremen K. W., Tulsiani D. R., Touster O., and Farquhar M. G. (1993) Cell type-dependent variations in the subcellular distribution of α-mannosidase I and II. J. Cell Biol. 122, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Divangahi M., Chen M., Gan H., Desjardins D., Hickman T. T., Lee D. M., Fortune S., Behar S. M., and Remold H. G. (2009) Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 10, 899–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Y. A., and Scheller R. H. (2001) SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2, 98–106 [DOI] [PubMed] [Google Scholar]
- 25. Gillingham A. K., and Munro S. (2007) The small G proteins of the Arf family and their regulators. Annu. Rev. Cell Dev. Biol. 23, 579–611 [DOI] [PubMed] [Google Scholar]
- 26. Stenmark H. (2009) Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 [DOI] [PubMed] [Google Scholar]
- 27. Faurschou M., and Borregaard N. (2003) Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 5, 1317–1327 [DOI] [PubMed] [Google Scholar]
- 28. Shen C., Rathore S. S., Yu H., Gulbranson D. R., Hua R., Zhang C., Schoppa N. E., and Shen J. (2015) The trans-SNARE-regulating function of Munc18–1 is essential to synaptic exocytosis. Nat. Commun. 6, 8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kreitzer G., Marmorstein A., Okamoto P., Vallee R., and Rodriguez-Boulan E. (2000) Kinesin and dynamin are required for post-Golgi transport of a plasma-membrane protein. Nat. Cell Biol. 2, 125–127 [DOI] [PubMed] [Google Scholar]
- 30. Henley J. R., and McNiven M. A. (1996) Association of a dynamin-like protein with the Golgi apparatus in mammalian cells. J. Cell Biol. 133, 761–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Warnock D. E., Baba T., and Schmid S. L. (1997) Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol. Biol. Cell 8, 2553–2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Praefcke G. J., and McMahon H. T. (2004) The dynamin superfamily: universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 5, 133–147 [DOI] [PubMed] [Google Scholar]
- 33. Gold E. S., Underhill D. M., Morrissette N. S., Guo J., McNiven M. A., and Aderem A. (1999) Dynamin 2 is required for phagocytosis in macrophages. J. Exp. Med. 190, 1849–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koul A., Herget T., Klebl B., and Ullrich A. (2004) Interplay between mycobacteria and host signalling pathways. Nat. Rev. Microbiol. 2, 189–202 [DOI] [PubMed] [Google Scholar]
- 35. Singh D. K., Kumar D., Siddiqui Z., Basu S. K., Kumar V., and Rao K. V. (2005) The strength of receptor signaling is centrally controlled through a cooperative loop between Ca2+ and an oxidant signal. Cell 121, 281–293 [DOI] [PubMed] [Google Scholar]
- 36. Lee W. L., Harrison R. E., and Grinstein S. (2003) Phagocytosis by neutrophils. Microbes Infect. 5, 1299–1306 [DOI] [PubMed] [Google Scholar]
- 37. Lundqvist-Gustafsson H., Gustafsson M., and Dahlgren C. (2000) Dynamic Ca2+ changes in neutrophil phagosomes. A source for intracellular Ca2+ during phagolysosome formation? Cell Calcium 27, 353–362 [DOI] [PubMed] [Google Scholar]
- 38. Stendahl O., Krause K. H., Krischer J., Jerström P., Theler J. M., Clark R. A., Carpentier J. L., and Lew D. P. (1994) Redistribution of intracellular Ca2+ stores during phagocytosis in human neutrophils. Science 265, 1439–1441 [DOI] [PubMed] [Google Scholar]
- 39. Miyake K., and McNeil P. L. (1995) Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J. Cell Biol. 131, 1737–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Steinhardt R. A., Bi G., and Alderton J. M. (1994) Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 263, 390–393 [DOI] [PubMed] [Google Scholar]
- 41. Togo T., Alderton J. M., Bi G. Q., and Steinhardt R. A. (1999) The mechanism of facilitated cell membrane resealing. J. Cell Sci. 112, 719–731 [DOI] [PubMed] [Google Scholar]
- 42. Südhof T. C. (2012) Calcium control of neurotransmitter release. Cold Spring Harb. Perspect. Biol. 4, a011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Church J., Fletcher E. J., Abdel-Hamid K., and MacDonald J. F. (1994) Loperamide blocks high-voltage-activated calcium channels and N-methyl-d-aspartate-evoked responses in rat and mouse cultured hippocampal pyramidal neurons. Mol. Pharmacol. 45, 747–757 [PubMed] [Google Scholar]
- 44. Kochegarov A. A. (2003) Pharmacological modulators of voltage-gated calcium channels and their therapeutical application. Cell Calcium 33, 145–162 [DOI] [PubMed] [Google Scholar]
- 45. Bennett M. K., Calakos N., and Scheller R. H. (1992) Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257, 255–259 [DOI] [PubMed] [Google Scholar]
- 46. Randhawa V. K., Bilan P. J., Khayat Z. A., Daneman N., Liu Z., Ramlal T., Volchuk A., Peng X. R., Coppola T., Regazzi R., Trimble W. S., and Klip A. (2000) VAMP2, but not VAMP3/cellubrevin, mediates insulin-dependent incorporation of GLUT4 into the plasma membrane of L6 myoblasts. Mol. Biol. Cell 11, 2403–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Caceres P. S., Mendez M., and Ortiz P. A. (2014) Vesicle-associated membrane protein 2 (VAMP2) but not VAMP3 mediates cAMP-stimulated trafficking of the renal Na+-K+-2Cl− co-transporter NKCC2 in thick ascending limbs. J. Biol. Chem. 289, 23951–23962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li S. K., Zhu D., Gaisano H. Y., and Brubaker P. L. (2014) Role of vesicle-associated membrane protein 2 in exocytosis of glucagon-like peptide-1 from the murine intestinal L cell. Diabetologia 57, 809–818 [DOI] [PubMed] [Google Scholar]
- 49. Mendez M., Gross K. W., Glenn S. T., Garvin J. L., and Carretero O. A. (2011) Vesicle-associated membrane protein-2 (VAMP2) mediates cAMP-stimulated renin release in mouse juxtaglomerular cells. J. Biol. Chem. 286, 28608–28618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singh B. B., Lockwich T. P., Bandyopadhyay B. C., Liu X., Bollimuntha S., Brazer S. C., Combs C., Das S., Leenders A. G., Sheng Z. H., Knepper M. A., Ambudkar S. V., and Ambudkar I. S. (2004) VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol. Cell 15, 635–646 [DOI] [PubMed] [Google Scholar]
- 51. Haynes L. P., Thomas G. M., and Burgoyne R. D. (2005) Interaction of neuronal calcium sensor-1 and ADP-ribosylation factor 1 allows bidirectional control of phosphatidylinositol 4-kinase beta and trans-Golgi network-plasma membrane traffic. J. Biol. Chem. 280, 6047–6054 [DOI] [PubMed] [Google Scholar]
- 52. Behar S. M., Divangahi M., and Remold H. G. (2010) Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat. Rev. Microbiol. 8, 668–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vicker M. G. (1977) On the origin of the phagocytic membrane. Exp. Cell Res. 109, 127–138 [DOI] [PubMed] [Google Scholar]
- 54. Bonifacino J. S., and Glick B. S. (2004) The mechanisms of vesicle budding and fusion. Cell 116, 153–166 [DOI] [PubMed] [Google Scholar]
- 55. Alix E., Mukherjee S., and Roy C. R. (2011) Subversion of membrane transport pathways by vacuolar pathogens. J. Cell Biol. 195, 943–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gillingham A. K., and Munro S. (2016) Finding the Golgi: golgin coiled-coil proteins show the way. Trends Cell Biol. 26, 399–408 [DOI] [PubMed] [Google Scholar]
- 57. Niedergang F., and Chavrier P. (2004) Signaling and membrane dynamics during phagocytosis: many roads lead to the phagos(R)ome. Curr. Opin. Cell Biol. 16, 422–428 [DOI] [PubMed] [Google Scholar]
- 58. Di A., Nelson D. J., Bindokas V., Brown M. E., Libunao F., and Palfrey H. C. (2003) Dynamin regulates focal exocytosis in phagocytosing macrophages. Mol. Biol. Cell 14, 2016–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jones S. M., Howell K. E., Henley J. R., Cao H., and McNiven M. A. (1998) Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science 279, 573–577 [DOI] [PubMed] [Google Scholar]
- 60. Augustine G. J. (2001) How does calcium trigger neurotransmitter release? Curr. Opin. Neurobiol. 11, 320–326 [DOI] [PubMed] [Google Scholar]
- 61. Carrithers M. D., Chatterjee G., Carrithers L. M., Offoha R., Iheagwara U., Rahner C., Graham M., and Waxman S. G. (2009) Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A. J. Biol. Chem. 284, 8114–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Link T. M., Park U., Vonakis B. M., Raben D. M., Soloski M. J., and Caterina M. J. (2010) TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat. Immunol. 11, 232–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Drecktrah D., Knodler L. A., Ireland R., and Steele-Mortimer O. (2006) The mechanism of Salmonella entry determines the vacuolar environment and intracellular gene expression. Traffic 7, 39–51 [DOI] [PubMed] [Google Scholar]
- 64. Dai S., Zhang Y., Weimbs T., Yaffe M. B., and Zhou D. (2007) Bacteria-generated PtdIns(3)P recruits VAMP8 to facilitate phagocytosis. Traffic 8, 1365–1374 [DOI] [PubMed] [Google Scholar]
- 65. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., and Horwitz A. R. (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 66. Funamoto S., Meili R., Lee S., Parry L., and Firtel R. A. (2002) Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell 109, 611–623 [DOI] [PubMed] [Google Scholar]
- 67. Achiriloaie M., Barylko B., and Albanesi J. P. (1999) Essential role of the dynamin pleckstrin homology domain in receptor-mediated endocytosis. Mol. Cell. Biol. 19, 1410–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Züchner S., Noureddine M., Kennerson M., Verhoeven K., Claeys K., De Jonghe P., Merory J., Oliveira S. A., Speer M. C., Stenger J. E., Walizada G., Zhu D., Pericak-Vance M. A., Nicholson G., Timmerman V., and Vance J. M. (2005) Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat. Genet. 37, 289–294 [DOI] [PubMed] [Google Scholar]
- 69. Scalettar B. A., Rosa P., Taverna E., Francolini M., Tsuboi T., Terakawa S., Koizumi S., Roder J., and Jeromin A. (2002) Neuronal calcium sensor-1 binds to regulated secretory organelles and functions in basal and stimulated exocytosis in PC12 cells. J. Cell Sci. 115, 2399–2412 [DOI] [PubMed] [Google Scholar]
- 70. Beck R., Sun Z., Adolf F., Rutz C., Bassler J., Wild K., Sinning I., Hurt E., Brügger B., Béthune J., and Wieland F. (2008) Membrane curvature induced by Arf1-GTP is essential for vesicle formation. Proc. Natl. Acad. Sci. U.S.A. 105, 11731–11736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Faúndez V., Horng J. T., and Kelly R. B. (1997) ADP ribosylation factor 1 is required for synaptic vesicle budding in PC12 cells. J. Cell Biol. 138, 505–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Krauss M., Jia J. Y., Roux A., Beck R., Wieland F. T., De Camilli P., and Haucke V. (2008) Arf1-GTP-induced tubule formation suggests a function of Arf family proteins in curvature acquisition at sites of vesicle budding. J. Biol. Chem. 283, 27717–27723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shui W., Sheu L., Liu J., Smart B., Petzold C. J., Hsieh T. Y., Pitcher A., Keasling J. D., and Bertozzi C. R. (2008) Membrane proteomics of phagosomes suggests a connection to autophagy. Proc. Natl. Acad. Sci. U.S.A. 105, 16952–16957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Steegmaier M., Klumperman J., Foletti D. L., Yoo J. S., and Scheller R. H. (1999) Vesicle-associated membrane protein 4 is implicated in trans-Golgi network vesicle trafficking. Mol. Biol. Cell 10, 1957–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Söllner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., and Rothman J. E. (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324 [DOI] [PubMed] [Google Scholar]
- 76. Desjardins M., Huber L. A., Parton R. G., and Griffiths G. (1994) Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 124, 677–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liu Y. W., Lukiyanchuk V., and Schmid S. L. (2011) Common membrane trafficking defects of disease-associated dynamin 2 mutations. Traffic 12, 1620–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.