

Abstract
Reactive oxygen species generate potentially cytotoxic and mutagenic lesions in DNA, both between and within the nucleosomes that package DNA in chromatin. The vast majority of these lesions are subject to base excision repair (BER). Enzymes that catalyze the first three steps in BER can act at many sites in nucleosomes without the aid of chromatin-remodeling agents and without irreversibly disrupting the host nucleosome. Here we show that the same is true for a protein complex comprising DNA ligase IIIα and the scaffolding protein X-ray repair cross-complementing protein 1 (XRCC1), which completes the fourth and final step in (short-patch) BER. Using in vitro assembled nucleosomes containing discretely positioned DNA nicks, our evidence indicates that the ligase IIIα-XRCC1 complex binds to DNA nicks in nucleosomes only when they are exposed by periodic, spontaneous partial unwrapping of DNA from the histone octamer; that the scaffolding protein XRCC1 enhances the ligation; that the ligation occurs within a complex that ligase IIIα-XRCC1 forms with the host nucleosome; and that the ligase IIIα-XRCC1-nucleosome complex decays when ligation is complete, allowing the host nucleosome to return to its native configuration. Taken together, our results illustrate ways in which dynamic properties intrinsic to nucleosomes may contribute to the discovery and efficient repair of base damage in chromatin.
Keywords: base excision repair (BER), chromatin, DNA enzyme, DNA repair, histone
Introduction
Reactive oxygen species, produced during normal oxidative metabolism and by exposure to ionizing radiation, generate some ∼30,000 oxidized bases and abasic sites in the DNA of every nucleated human cell every day (Refs. 1 and 2) and reviewed in Ref. 3). The vast majority of these oxidative lesions are subject to base excision repair (BER,2 reviewed in Refs. 4–9). Left unrepaired or misrepaired, oxidative lesions can lead to polymerase stalling, DNA strand breaks, base mutations, and cell death. Germline mutations in one BER enzyme were recently found to promote adenomatous polyposis and colorectal cancer (10), illustrating the critical importance of this repair pathway. The BER of oxidized bases begins with their discovery and excision by bifunctional DNA glycosylases. The DNA backbone at the resulting abasic site is then cleaved by either a lyase activity associated with the DNA glycosylase or apurinic endonuclease (APE1). This leaves a polymerase-blocking moiety on the 3′ side that is removed by either APE1 or polynucleotide kinase phosphatase. This leaves a single-strand deoxynucleotide gap flanked by a 3′ hydroxyl group and, in some cases, a 5′-2-deoxyribose phosphate moiety. In short-patch BER, DNA polymerase β (pol β) fills the DNA gap and removes the blocking 5′-2-deoxyribose phosphate group, leaving a nick that is sealed by DNA ligase IIIα (LigIIIα) in association with the scaffolding protein X-ray repair cross-complementing protein 1 (XRCC1). If pol β fails to remove the 5′ moiety, then long-patch BER ensues. This entails recruitment of a replicative polymerase that displaces DNA linked to the 5′ moiety. The resulting DNA flap is removed by flap endonuclease 1, leaving a DNA nick that is sealed by DNA ligase I.
In eukaryotes, BER occurs in chromatin, where most of the DNA is wrapped around histone octamers to form nucleosomes (11). The close association between DNA and histone octamers and the changes in major and minor groove geometry associated with DNA bending about the octamer impede the binding of many regulatory factors and enzymes. These impediments are sometimes relieved by chromatin remodeling agents (reviewed in Ref. 12). The nucleosome remodeling complexes Switch/Sucrose Non-Fermentable (SWI/SNF) and imitation-switch 1 & 2 (ISW1 and ISW2) enhance the activity of certain BER enzymes on nucleosome substrates in vitro (13, 14), and, in budding yeast, depletion of an essential subunit in the chromatin remodeling complex RSC (remodeling the structure of chromatin) produces numerous DNA and chromatin-related defects, among them defects in BER (15). However, it is not yet clear that chromatin-remodeling agents act at sites of oxidative damage in cells. To identify chromatin-associated, rate-limiting steps in BER and possible “pioneer factors” that may recruit chromatin-remodeling agents, we have investigated interactions between BER factors and nucleosomes (16–20). The DNA glycosylases, APE1, and pol β, which catalyze the first three steps in BER, proved able to process their substrates without irreversibly disrupting the host nucleosome (17). DNA ligase IIIα differs from the enzymes that catalyze early steps BER in that it fully encircles its DNA substrate (as is also the case for DNA ligase I, which completes long-patch BER and DNA replication (21, 22)). This suggested that binding to, and sealing of, DNA nicks by either ligase I or IIIα would require a more extensive disruption of the host nucleosome. Even so, both enzymes proved able to detect and process DNA nicks in nucleosomes (17, 23), and LigIIIα-XRCC1, which can bind single base gaps as well as DNA nicks, enhanced the activity of pol β on nucleosome substrates. This enhancement of pol β was evident even in the presence of mutations that weaken its binding to XRCC1, which suggested that LigIIIα-XRCC1 renders substrates more readily accessible by altering nucleosome structure (17). However, the exact nature of these alterations, and indeed whether host nucleosomes survive ligation by either ligase, remained to be determined. Here we report that the structural perturbations in nick-containing nucleosomes, which enable ligase IIIα to function, are reversible. More specifically, DNA ligation takes place within a complex that LigIIIα-XRCC1 forms with nick-containing nucleosomes; when ligation is complete, LigIIIα-XRCC1 dissociates, allowing the nucleosome to return to its native configuration. We relate these findings to discrete steps in the folding of DNA ligase IIIα into its active configuration and ways in which LigIIIα-XRCC1 may function in cells.
Results
Evidence That Spontaneous Partial Unwrapping of DNA from the Histone Octamer Enables LigIIIα-XRCC1 to Discover Nicks in Nucleosomes
We showed previously that the bifunctional DNA glycosylases human endonuclease III-like protein 1 (NTHL1) and human endonuclease VIII-like protein (NEIL1) are able to bind and remove oxidized thymine residues from nucleosomes in vitro and. thereby, initiate BER (16, 18). These reactions occur within ternary complexes that can be visualized in gel mobility shift assays and proceed without irreversibly disrupting the host nucleosome or shifting its translational position. We subsequently demonstrated the handoff of NTHL1 products in nucleosomes to APE1 and, from there, to pol β and LigIIIα-XRCC1 (17). The efficiency with which the first three of these BER enzymes (i.e. DNA glycosylases, APE1, and pol β) act in nucleosomes varies considerably with the helical orientation of substrates with respect to the underlying histone octamer (reviewed in Ref. 24). In large part, this variability reflects two different modes of substrate binding (16, 19). The first is direct binding of BER enzymes to substrates that are located at accessible sites in the nucleosome. As DNA glycosylases, APE1, and pol β all bend DNA, their binding to substrates at sterically accessible sites may require local changes in DNA configuration. The ease with which this occurs may be influenced by local variation in, e.g., DNA twist and bending angles; this may account for variation in the rate of excision even among similarly oriented lesions (25). The second, much less efficient binding mode occurs when substrates are transiently exposed to solvent by spontaneous, partial unwrapping of DNA from the histone octamer. The extent of this unwrapping is variable, and the duration of the unwrapped state is on the order of milliseconds (26–30). As a result, the rate of processing of additional nicks in the nucleosome population is governed by local enzyme concentration and the frequency with which unwrapping events expose additional substrate (19).
Because the first three enzymes that act in BER demonstrated a strong preference for the outward-facing lesions, we do not believe that local variation in nucleosome architecture is playing a strong role at these particular nucleosome locations. That is, the lesion orientation (inward versus outward) mattered more than any DNA twist or specific histone contacts that may be present. However, in the final step, the orientation of DNA nicks in nucleosomes did not affect the efficiency of ligation by LigIIIα-XRCC1 (17). Given that DNA ligases must fully encircle their substrates, the more likely explanation is that LigIIIα-XRCC1 can bind DNA nicks only when they are transiently exposed to solvent by partial unwrapping of nucleosomal DNA. If true, the efficiency of ligation of a DNA nick located 46 nucleotides from the dyad axis of the nucleosome would exceed that for a nick located only 26 nucleotides from the dyad. To test this prediction, we prepared 32P end-labeled DNA fragments that contained the sea urchin 5S ribosomal DNA (rDNA) nucleosome-positioning element and a single, discretely positioned DNA nick, as depicted in Fig. 1A. We positioned these nicks so that they would face into the histone octamer, making them difficult or impossible to detect except when exposed by DNA unwrapping. We assembled these fragments into nucleosomes and used non-denaturing gels to visualize and quantify the efficiency of nucleosome assembly (Fig. 1B). Fig. 1, C and D, demonstrates that a 15-fold molar excess of LigIIIα-XRCC1 sealed nicks in most of the naked (i.e. non-nucleosomal) DNA fragments in a minute or less; the same amount of enzyme sealed a smaller but still significant fraction of DNA nicks at the −46 site in nucleosomes. As Fig. 1D indicates, the fraction of ligated nucleosomal DNA reached 30–40% of that in the naked DNA reactions. In contrast, the ligation efficiency for a similarly oriented nick 20 base pairs closer to the dyad axis was another ∼3-fold lower (Fig. 1E). This result supports the unwrapping hypothesis.
FIGURE 1.

Evidence that spontaneous partial unwrapping of DNA from the histone octamer enables LigIIIα-XRCC1 to discover nicks in nucleosomes. A, schematic depicting features of the nucleosomes used in the experiments shown in Figs. 1–6. The gray oval represents the predominant translational position of the 5S rDNA nucleosome; minor translational variants, in which the octamer is positioned ∼10 bp to either the left or right, also occurred (16, 19, 58) but did not affect the interpretation of the results. Ligatable, single-strand DNA nicks were positioned either −46 or −26 nt away from the nucleosome center (the dyad axis). Also shown are restriction endonuclease cleavage sites used in Fig. 6 to monitor nucleosome integrity during ligation. B, representative nucleosome (Nuc) preparation examined by native PAGE. Nucleosomes were assembled with 2.5 nm labeled, nicked DNA in the presence of 200 nm of nonspecific cold carrier (to ensure nucleosome stability) and then further diluted for subsequent experiments. The presence of this nonspecific DNA had no effect on ligation efficiency (refer to Fig. 4 for competition assays). C, representative 8% denaturing gel used to monitor ligation of nicks in nucleosomes and naked DNA controls as detailed under “Experimental Procedures.” Ligation products migrate as 183-nt bands. The minor band denoted by an asterisk corresponds to unligated DNA located 3′ to the nick and did not interfere with the quantification of ligation products. D, LigIIIα-XRCC1 ligated a significant fraction of the DNA nicks at position −46 even though these nicks faced into the histone octamer, making them accessible only during episodes of spontaneous partial unwrapping of DNA from the histone octamer (see text for further discussion). E, ligation efficiency dropped a further ∼3-fold for nicks positioned 20 nt further in from the nucleosome edge (position −26). This supports the hypothesis that nucleosome unwrapping is rate-limiting LigIIIα-XRCC1. Error bars represent standard deviations.
The kinetics of the ligation reactions shown in Fig. 1 also are consistent with the unwrapping hypothesis. Specifically, the naked DNA reaction exhibited monophasic kinetics. (The small fraction of non-ligated, naked DNA that remained at the end of the reaction can be attributed to a combination of factors, including substrate depletion and possible accumulation of abortive ligation intermediates.) Ligation products from the reactions with nick-containing, 5S rDNA-based nucleosomes accumulated with biphasic kinetics. The early, rapid accumulation of product can be attributed to that fraction of nicks exposed to solvent at the time of enzyme addition or sometime prior to the first (1-min) time point (by spontaneous partial unwrapping of DNA from the histone octamer). The second, much slower kinetic phase is limited by the rate of unwrapping-dependent exposure of additional substrate.
Binding of LigIIIα-XRCC1 Does Not Irreversibly Disrupt DNA Nick- or Gap-containing Nucleosomes
As noted earlier, we expected that LigIIIα-XRCC1 binding to gaps or nicks would require substantial disruption of host nucleosomes, and previous gel mobility shift assays suggested that this disruption might be irreversible (17). The mobility shift assays in Fig. 2A, lanes 3–6, are consistent with this inference. These lanes show loss of the canonical nucleosome band with addition of increasing amounts of LigIIIα-XRCC1 to −46 nick-containing nucleosomes and the concurrent accumulation of a slower-migrating complex. The reaction mixtures in these experiments were quenched after 30 s by addition of an EDTA-containing, native gel loading buffer and fractionated through native gels. However, when identical reaction mixtures were quenched with buffer containing excess unlabeled DNA and electrophoresed as before (Fig. 2A, lanes 7–10), the additional DNA competed for binding of LigIIIα-XRCC1. This resulted in nucleosome bands reappearing; their mobility was identical to that of the input nucleosomes. Analysis of DNA from a third identical set of reaction mixtures confirmed that ligation had occurred in these experiments (Fig. 2B). Thus, LigIIIα-XRCC1 appears able to seal DNA nicks without irreversibly disrupting host nucleosomes. As noted earlier, LigIIIα-XRCC1 can also bind to DNA gap-containing nucleosomes and enhance the activity of pol β. Fig. 2C shows that addition of LigIIIα-XRCC1 to gap-containing nucleosomes results in formation of a slowly migrating complex indistinguishable from the DNA nick-containing complex and that addition of excess naked DNA restores gap-containing nucleosomes to their original mobility. Thus, the LigIIIα-XRCC1-dependent structural changes that promote the activity of pol β in nucleosomes also are reversible.
FIGURE 2.
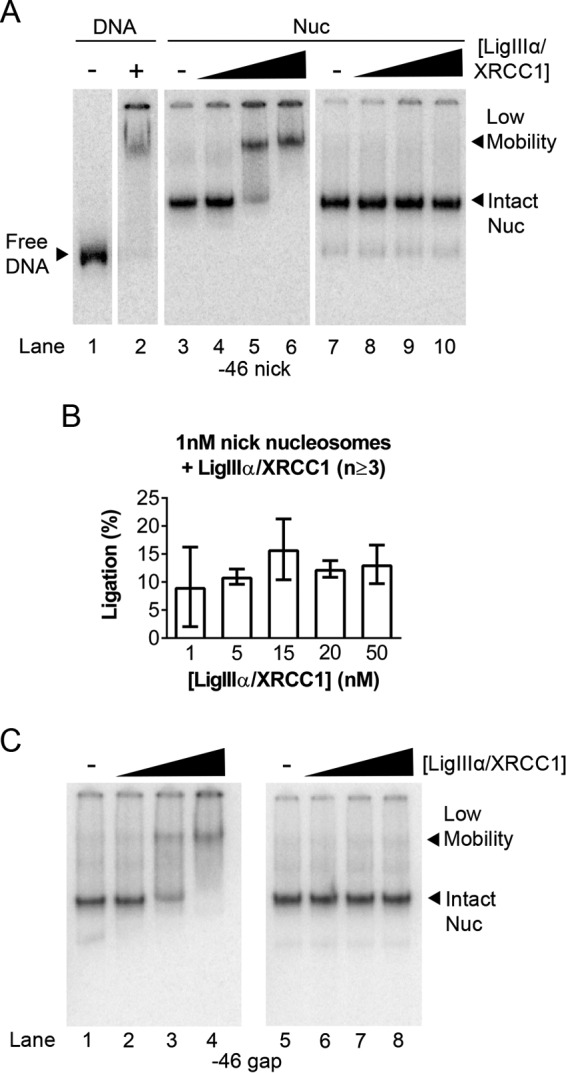
Binding of LigIIIα-XRCC1 does not irreversibly disrupt DNA nick- or gap-containing nucleosomes. A, lanes 1 and 2 show migration in native (mobility shift) gels of an end-labeled, 183-bp DNA fragment containing a single DNA nick in the absence and presence of LigIIIα-XRCC1. Lanes 3–6 show migration of nucleosomes (Nuc) assembled with the same DNA and then incubated in the absence and presence of 1, 15, or 50 nm LigIIIα-XRCC1. In all samples containing LigIIIα-XRCC1, substrate concentrations were 1 nm, and incubation times were 30 s. Ligation reactions were halted by addition of an EDTA-containing gel loading buffer (lanes 3–6) and then run on mobility shift gels. The samples in lanes 7–10 were treated identically to those in lanes 3–6, except for the addition of 1 μg of nonspecific carrier DNA to the gel loading buffer; this DNA competed for LigIIIα-XRCC1 binding and enabled nucleosomes to reform, indicating that all nucleosome components are present in the low-mobility complexes evident in lanes 5–6. B, DNA from LigIIIα-XRCC1 reactions was fractionated through denaturing gels to quantify ligation products generated in the 30-s reactions. Error bars represent standard deviations. C, mobility shift gels of 1 nm −46 gap-containing nucleosomes incubated with varying amounts of LigIIIα-XRCC1 as in A.
The experiments in Fig. 2 suggest that ligation occurs within a slow mobility complex that forms between LigIIIα-XRCC1 and nick-containing nucleosomes and that this complex formation can be reversed by addition of cold competitor DNA. If the slow mobility complex is a true BER intermediate, it ought to contain both substrate and product DNAs. To test this prediction, we incubated −46 nick nucleosomes with 15 nm LigIIIα-XRCC1 for 3 and 30 min and then separated intact nucleosomes from LigIIIα-XRCC1-containing ternary complexes as before, producing the first-dimension gel shown in Fig. 3A. Inspection of the low mobility band in Fig. 3A shows that its intensity diminishes with reaction time, whereas the intensity of the band corresponding to the intact nucleosome increased (as quantified in Fig. 3B). This suggests that the LigIIIα-XRCC1-containing ternary complex resolves following ligation. We next excised lanes from a gel identical to that shown in Fig. 3A and fractionated DNA from both the nucleosome and ternary complex by electrophoresis on a second-dimension sequencing gel (see “Experimental Procedures”). Fig. 3C shows that the ligated DNA products (boxed) were present in both the ternary complex and the intact nucleosome, as predicted. The aforementioned decrease in the ternary complex with reaction time is also evident in the two-dimensional gel (as quantified in Fig. 3D). This result is further evidence that the structural perturbations that enable LigIIIα-XRCC1 to discover and seal nicks in nucleosomes are reversible and that, after nick closure, LigIIIα-XRCC1 dissociates, enabling nucleosomes to reform.
FIGURE 3.

Ligation occurs within a complex that LigIIIα-XRCC1 forms with nick-containing nucleosomes. A, 1 nm −46 nick nucleosomes (Nuc) incubated without or with 15 nm LigIIIα-XRCC1 for 3 or 30 min were fractionated through mobility shift gels. (Note the longer incubation times compared with Fig. 2). B, quantification of bands in gels such as those in A shows a decline in the nucleosome-LigIIIα-XRCC1 ternary complex and a corresponding increase in intact nucleosome with time. This suggests that the ternary complex decays when ligation is complete. C, lanes were excised from a mobility shift gel identical to A, and DNA in each of the complexes was fractionated through a second-dimension sequencing gel (see “Experimental Procedures”). D, DNA; N = nucleosome; S, super-shifted complex. Boxes indicate nicked and ligated DNAs that are present within both the nucleosome band and the ternary complex. This result indicates that productive ligation occurs within a low-mobility ternary complex. D, the fraction of total ligation product in the low-mobility ternary complex declines with time, supporting the inference that, when ligation is complete, the ternary complex decays, allowing the host nucleosome to return to its native configuration. Error bars represent standard deviations (n = 3); * = p < 0.05.
The Efficiency with Which LigIIIα-XRCC1 Discovers Nicks in Both Naked DNA and Nucleosomes Is Enhanced by Its Capacity to Bind DNA Ends
The capacity of any DNA structure or sequence-specific binding protein to locate its target in DNA depends in part on its relative specific versus nonspecific DNA binding affinities (31). Previous gel mobility shift assays indicated that, at relatively high concentrations, LigIIIα-XRCC1 bound to nucleosomes containing undamaged DNA almost as well as it did to nucleosomes containing a DNA nick or gap (Ref. 17 and Fig. 5). It is highly unlikely that this apparent nonspecific binding is due to interactions between LigIIIα-XRCC1 and one or more histones. This is because all experiments in this study included ∼80 nm unlabeled nucleosomes preassembled with mono- and oligonucleosome-sized DNA (“cold carrier”); this ensured that our end-labeled test nucleosomes would remain stable (32). Clearly, these excess nucleosomes did not abolish ligation in the reactions in Figs. 1–3.
FIGURE 5.
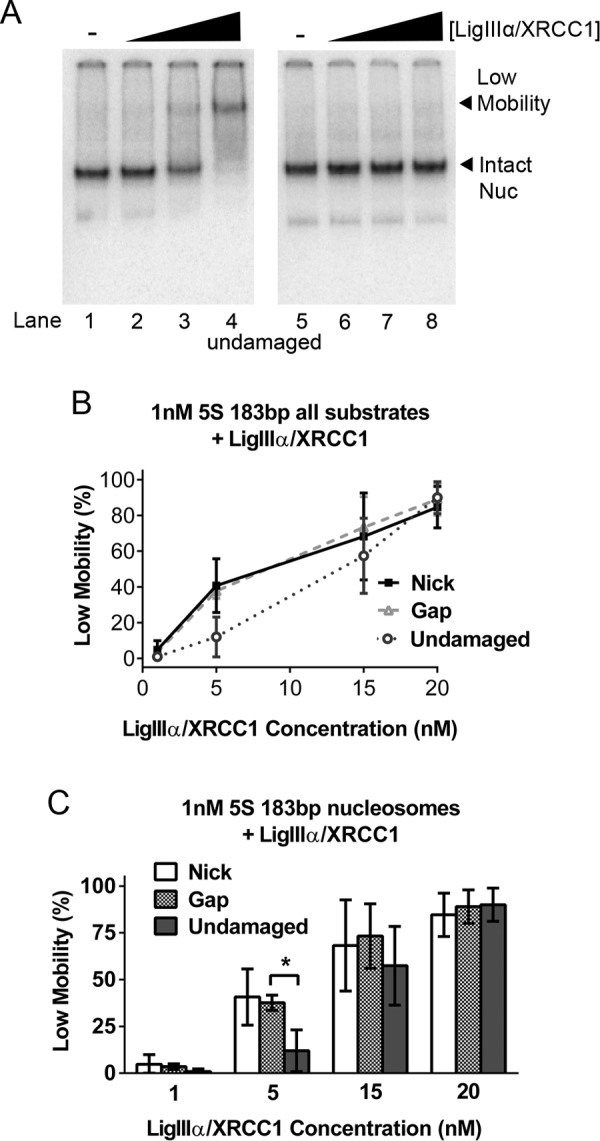
Interactions between LigIIIα-XRCC1 and undamaged nucleosomes. A, gel mobility shift assay of 1 nm 5S rDNA-based, undamaged nucleosomes (Nuc) in the absence (lanes 1 and 5) and presence of increasing concentrations of LigIIIα-XRCC1 (lanes 2–4 and 6–8). As in Fig. 2, A and C, the samples in lanes 5–8 were identical to those in lanes 1–4, expect for the addition of unlabeled competitor DNA just prior to loading samples on to gels. The low-mobility complex evident in lanes 3–4 is indistinguishable from the complexes formed by addition of LigIIIα-XRCC1 to nick- and gap-containing nucleosomes. B, the fraction of nucleosomes shifted into low-mobility complexes in all gel shift experiments conducted (see A and Fig. 2, A and C, respectively, for representative gels) was quantified and plotted as function of nucleosome type and LigIIIα-XRCC1 concentration. For ease of comparison, the same data were plotted in the histograms in C. The somewhat lower affinity of LigIIIα-XRCC1 for nucleosomes with undamaged DNA reached statistical significance only when enzyme concentrations were reduced to 5 nm. Error bars represent standard deviations; * = p < 0.05.
Ligase III binds DNA ends (33) and catalyzes intermolecular end joining (34–36). We did not expect this to appreciably affect this study because all DNA substrates were blunt-ended and used at concentrations well below those needed for efficient blunt end ligation. Moreover, we never detected intermolecular ligation products in our experiments. Nevertheless, the control experiments summarized below indicated that the apparent nonspecific binding of LigIIIα-XRCC1 to intact nucleosomes could be explained by its binding to free DNA ends. Fig. 4A shows that the addition of a single 32P residue to one 5′ end of a DNA fragment enhanced the nick-sealing activity of LigIIIα-XRCC1. 5′-phosphate residues similarly enhanced the nick-sealing activity of T4 DNA ligase, indicating that this stimulatory mechanism is not unique to LigIIIα-XRCC1. Comparisons between nucleosomes that differ only in the presence (Fig. 1D) or absence of 5′-phosphate residues (Fig. 4B) indicate that 5′-phosphate residues also stimulated the sealing of nicks in our model nucleosomes. The 5′-phosphate residues increased the extent of ligation by ∼50% (from ∼20% to 30% within the time frame of the reaction). Collectively, these results indicate that DNA end binding requires a 5′-phosphate and also explain why DNA ends associated with excess cold carrier had little effect on ligation efficiency. Specifically, we prepared carrier DNA fragments by incubating purified chromatin with micrococcal nuclease, which generates DNA ends with 3′-phosphate and 5′-hydroxyl groups. Fig. 4C shows that even a 400-fold molar excess of DNA lacking 5′-phosphates had little effect on ligation efficiency.
FIGURE 4.
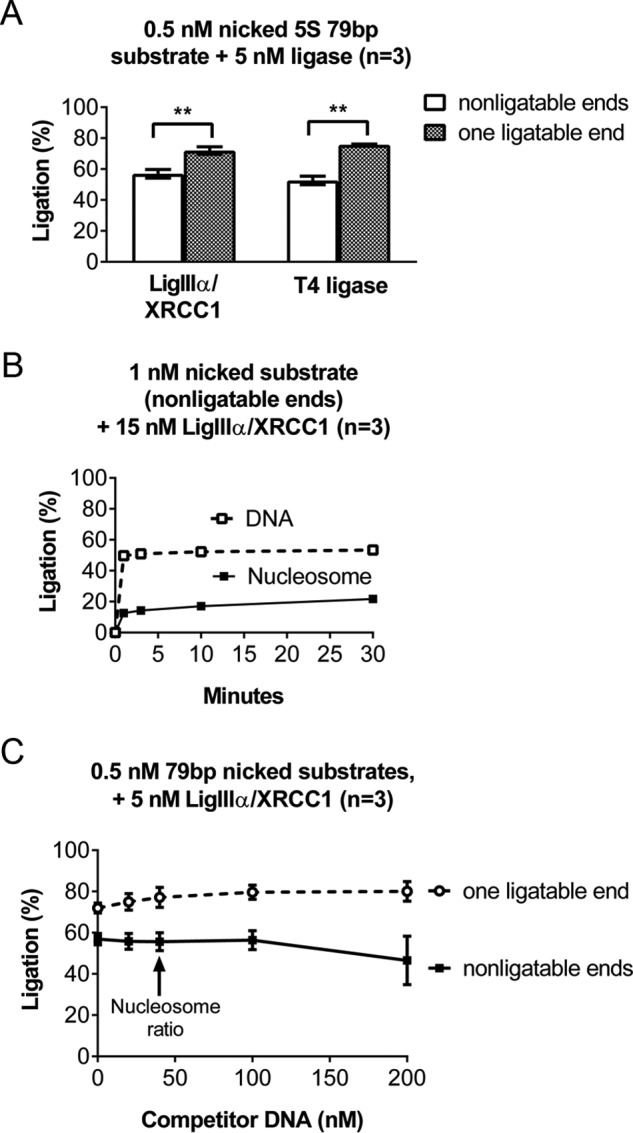
The efficiency with which LigIIIα-XRCC1 discovers nicks in both naked DNA and nucleosomes is enhanced by its capacity to bind DNA ends. A, to examine the impact of DNA end binding by LigIIIα-XRCC1, we prepared a 79-bp DNA fragment containing a single nick and OH groups at both the 5′ and 3′ ends. (The DNA was a truncated version of that used to assemble 5S rDNA-based nucleosomes for many of the experiments reported in this paper.) We labeled this DNA by the addition of 32P to an internal site, producing DNA that is not competent for end binding or blunt end ligation. We also labeled a second aliquot of DNA, identical except for one end containing a 5′-phosphate group. We incubated these differently ended DNAs (0.5 nm) with 5 nm LigIIIα-XRCC1 for 30 min at 37 °C, separated substrate and products on denaturing gels such as those shown in Fig. 1C, and quantified the fraction of substrate ligated using phosphorimaging. The presence of a phosphate group at the 5′ end significantly enhanced ligation. This enhancement was also evident in control reactions using T4 DNA ligase. B, the effect of 5′-phosphate-dependent, DNA end binding on ligation of nicks in nucleosomes. The conditions in these reactions were identical to those in Fig. 1D, except for use of an internally labeled substrate that lacked 5′-phosphate residues and thus would not support DNA end binding. As with the naked DNA substrates, ligation efficiency was significantly reduced (from ∼30% to ∼20%) in the absence of 5′-phosphates. C, to ligation reactions containing the same substrates used in A, we added 0–200 nm naked, unlabeled carrier DNA lacking 5′-phosphate residues. The results indicate that this carrier DNA did not suppress ligation, even at concentrations well above the 80-fold excess carrier used in all other assays. Importantly, we did not detect any dimeric, blunt end ligation products in any of our experiments. Error bars represent standard deviations; ** = p < 0.01.
We also investigated the binding of LigIIIα-XRCC1 to nucleosomes under various conditions. Fig. 5A shows that, at the concentrations tested, LigIIIα-XRCC1 bound to nucleosomes containing undamaged DNA nearly as well as they did to gap- or nick-containing nucleosomes (Fig. 5B); only the 5 nm enzyme point yielded any significant difference between undamaged and damaged nucleosomes (Fig. 5C). It is likely that DNA end binding by LigIIIα-XRCC1 enhanced ligation of nicks in both naked DNA and nucleosomal templates by increasing the local concentration of DNA ligase; however, it is unlikely that end binding had any direct impact on changes in nucleosomes that render them ligation-permissive, as we have no evidence that end binding contributes to either nucleosome disruption or faster unwrapping rates.
LigIIIα-XRCC1 Disrupts Only Histone-DNA Contacts Close to the Site of the Nick
Collectively, the gel mobility shift assays in Figs. 2, 3, and 5 indicate that the slowly migrating complexes that LigIIIα-XRCC1 forms with nucleosomes may, in one instance, reflect either DNA end binding and, in another instance, the complex that the enzyme forms when it binds a DNA nick and folds into its active configuration. Although these complexes must differ from one another, they could not be distinguished from one another in gel mobility shift assays. Therefore, to further investigate interactions between LigIIIα-XRCC1 and nick-containing nucleosomes, we conducted restriction site accessibility assays. Fig. 1A depicts an AgeI restriction site in the DNA we used for most of the experiments in this study; it is well separated from the DNA nicks used to assay LigIIIα-XRCC1 function. Thus, in naked DNA, AgeI had no impact on the efficiency of nick ligation, nor did we observe ligation of AgeI-generated, sticky DNA ends (data not shown). In nucleosomes, however, the −46 nick and AgeI are relatively close to one another (Fig. 6A). If nick binding by LigIIIα-XRCC1 is accompanied by extensive disruption of the host nucleosome, we might observe increased access to the AgeI site, which ordinarily is almost completely resistant to cleavage (16). Instead, as shown in Fig. 6B, the extent of AgeI cleavage actually declined with the addition of increasing amounts of LigIIIα-XRCC1 (sufficient to ligate ∼25% of the molecules), possibly because LigIIIα-XRCC1 blocked access to the AgeI restriction site. The addition of AgeI did not affect ligation efficiency in nucleosomes (Fig. 6C).
FIGURE 6.

LigIIIα-XRCC1 disrupts only histone-DNA contacts close to the site of the nick. A, schematic depicting the relative position in DNA nick-containing nucleosomes of restriction sites used to probe the extent of nucleosome disruption during ligation. B, ligation of a DNA nick located 46 nt from the dyad axis does not interfere with nucleosome-mediated protection from cleavage at an AgeI site. 1 nm −46 nick nucleosomes were incubated with 25 units of AgeI together with 1–15 nm LigIIIα-XRCC1, and ligation and cleavage amounts were determined as described under “Experimental Procedures.” AgeI cleavage was calculated as a fraction of naked DNA, whereas nucleosome ligation was reported as an absolute fraction. C, the presence of AgeI in the ligation reactions reported in Fig. 2B had no significant (ns) impact on the efficiency of ligation of the −46 nicks in nucleosomes or in naked −46 nick DNA controls (where ligation reached ∼77%, data not shown). D, the left axis shows that 15 nm LigIIIα-XRCC1 did not appreciably affect cleavage at a PsiI site located approximately midway between the −46 DNA nick and the edge of the nucleosome (A and Fig. 1A). The right axis shows efficient ligation of −46 nick nucleosomes in the absence PsiI. Ligation also occurred in the presence of PsiI but could not be quantified, as the Psi I site lies between the DNA nick and the 5′ 32P label. Error bars represent standard deviations. ns = not significant; p < 0.05.
The AgeI experiments suggested that LigIIIα-XRCC1 binding does not disrupt a large enough number of histone-DNA contacts to destabilize the entire host nucleosome. To test for possible changes closer to the DNA nicks, we asked whether LigIIIα-XRCC1 affected access to a PsiI site located 11–16 nt from the nick at position −46 (see schematics in Figs. 1A and 6A) and largely occluded in the intact nucleosome (16). Again, control experiments indicated that LigIIIα-XRCC1 did not block access to the PsiI site in naked DNA (data not shown). Thus, if LigIIIα-XRCC1 binding freed the PsiI site from its association with the histone octamer, then we should have seen increased PsiI cleavage. Instead, as in the AgeI experiment, LigIIIα-XRCC1 had little or no effect on PsiI cleavage in the nucleosome (Fig. 6D). This result suggests that histone-DNA contacts disrupted by the binding of ligase IIIα are highly localized and do not extend much beyond the DNA residues that interact directly with the enzyme.
Highly Stable 601 Nucleosomes Are Only Marginally Permissive for DNA Ligation by LigIIIα-XRCC1
Nucleosomes assembled with synthetic 601 DNA are more stable than even 5S rDNA-containing nucleosomes (37, 38). Also, 601 nucleosomes occupy a single translational position, whereas most 5S rDNA nucleosomes are distributed among three translational positions, separated from one another by 10-bp intervals (16, 19). Their exceptional stability makes 601 nucleosomes relatively tractable for in vitro studies, although they may not as faithfully reflect chromatin dynamics in cells (39). Despite this caveat, we used nick-containing 601 nucleosomes to determine whether nucleosome stability or positional uniformity influences the efficiency of nick discovery or ligation. Fig. 7A shows that the overall ligation efficiency was substantially lower than for 5S rDNA-containing nucleosomes. To eliminate possible variation in ligation efficiency because of differences in sequence context, Fig. 7B presents the same ligation efficiency data but expressed as a fraction of the ligation efficiency measured for their respective DNA controls. It is possible that the ∼3-fold difference in ligation extent was due to the short segment of linker DNA in the (159-bp) 5S rDNA nucleosome. However, as reported in Fig. 7C, addition of linker DNA to 601 DNA nucleosomes had little or no impact on ternary complex formation or ligation efficiency.
FIGURE 7.

Highly stable, 601 nucleosomes are only marginally permissive for DNA ligation by LigIIIα-XRCC1. A, ligation of nicks in 159-bp, 5S rDNA-containing nucleosomes compared with ligation of nicks in 147-bp, 601 DNA-containing nucleosomes. Nucleosomes were incubated with 15 nm LigIIIα-XRCC1 at 37 °C for 30 min, and the extent of ligation was measured as before. As indicated, very little ligation occurred in the 601 nucleosomes. B, ligation data from A after normalizing to the extent of ligation obtained in reactions with the corresponding naked DNAs. The extent of ligation of nicks in 5S rDNA nucleosomes was 3- to 4-fold higher than in the 601 nucleosomes. C, the efficiency of ligation of nicks in 601 nucleosomes is not limited by the absence of linker DNA. 601 nucleosomes containing intact DNA and either no linker DNA (Short) or 26-bp linker DNA added to the nick-proximal side of the nucleosome (Left Overhang) were incubated with LigIIIα-XRCC1 and analyzed in gel shift assays as in Fig. 3. The bar graph shows that 26 bp of linker DNA had virtually no impact on ligation or the extent of LigIIIα-XRCC1 binding to undamaged nucleosome in the gel shift assay. Error bars represent standard deviations.
XRCC1 Promotes Ligation of Nicks by LigIIIα
XRCC1 not only serves as a scaffold for the binding of downstream BER enzymes but also binds preferentially to DNA nicks and single base gaps, with reported KD values of ∼65 nm and ∼34 nm, respectively (40). Reported KD values for the binding of ligase III to a ligatable nick, at physiological salt concentrations, range between 5 and 200 nm, depending on assay methods (33, 41). The relatively robust activity that we observed in reactions containing 5–15 nm LigIIIα-XRCC1 is consistent with a KD value at the lower end of this range, but it is also possible that the overall DNA binding affinity for LigIIIα-XRCC1 exceeds that of either protein alone, as appears to be the case for pol β-XRCC1 complexes (42). To determine whether XRCC1 facilitates the discovery of DNA nicks in nucleosomes, we first compared the activity of recombinant human LigIIIα (from Escherichia coli) with that of recombinant human LigIIIα-XRCC1 (from Sf9 insect cells). Fig. 8, A and B, shows significantly less ligation in the absence of XRCC1. However, the addition of E. coli-expressed XRCC1 failed to restore normal levels of ligation (data not shown). This result could have been due to the absence of posttranslational modifications that potentially stabilize interactions between LigIIIα and XRCC1. A further concern was whether any comparison involving LigIIIα alone is biologically meaningful. LigIIIα lacks a nuclear localization sequence, and, in cells, its import into nuclei depends on its association with XRCC1 or other proteins (reviewed in Ref. 43).
FIGURE 8.

XRCC1 promotes ligation of nicks by LigIIIα. A, DNA nick-containing, 5S rDNA-based nucleosomes were incubated at 37 °C for 30 min with either 15 nm Sf9-expressed LigIIIα-XRCC1 or an equivalent amount of E. coli-expressed LigIIIα (using concentrations specified by the supplier), and the extent of ligation was measured as before. B, comparison of nucleosome ligation efficiency between LigIIIα-XRCC1 and LigIIIα. The values shown are based on data from A after normalizing to the extent of ligation obtained in reactions with the corresponding naked DNAs. In the absence of XRCC1, the extent of ligation was ∼⅔ that observed with LigIIIα-XRCC1. C, time course of 15 nm Sf9-expressed LigIIIα-TDP1 and LigIIIα-TDP1-XRCC1 incubated with 1 nm nick-containing DNA or nucleosomes (Nuc) under conditions identical to Figs. 1D and A. Data from Fig. 1D are included to facilitate comparisons. LT, LigIIIα-TDP1; LX, LigIIIα-XRCC1; LTX, LigIIIα-TDP1-XRCC1. D, ligation products from reactions with nucleosomes in C were recalculated as a fraction of naked DNA ligation to adjust for differing naked DNA ligation efficiencies. Note that ligation efficiencies were markedly higher in complexes containing XRCC1. E, in gel shift experiments conducted as in Fig. 3, E. coli-purified recombinant XRCC1 did not detectably alter the mobility of nucleosomes containing a single nucleotide gap DNA/nucleosomes. This result suggests that nucleosome binding by LigIIIα-XRCC1 is driven primarily by DNA ligase IIIα. Error bars represent standard deviations; ** = p < 0.01.
To circumvent the limitations associated with the above-described comparisons, we took advantage of the fact that LigIIIα interacts not only with XRCC1 but also with the DNA nick-associated repair factor topoisomerase I-associated tyrosine phosphatase (TDP1). The fact that TDP1 and XRCC1 interact with different regions in LigIIIα enabled us to compare the nick-sealing activities of LigIIIα-TDP1 with that of LigIIIα-TDP1-XRCC1. To ensure the presence of posttranslational modifications, both complexes were expressed in and purified from Sf9 cells. Fig. 8C shows that LigIIIα-TDP1 was less active on nicks in naked DNA and only marginally active on nicks in nucleosomes. In contrast, the LigIIIα-TDP1-XRCC1 complex exhibited a level of activity on nicks in both nucleosomal and naked DNA substrates closer to that seen with LigIIIα-XRCC1. To ensure that differences in the activity of the different enzyme complexes on nucleosome substrates could be attributed to the presence or absence of XRCC1, the data from Fig. 8C were normalized against the naked DNA results and replotted in Fig. 8D. These results suggest that XRCC1 promotes DNA ligation. Fig. 8E shows that XRCC1 alone did not detectably interact with either naked DNA or the nucleosome in a gel mobility shift assay. Thus, XRCC1 appears to facilitate the nick discovery step only when complexed with LigIIIα.
Discussion
Prior to this study, we had reconstituted the entire four-step BER reaction with nucleosomes containing oxidized bases and determined that the first three steps in BER can occur without irreversibly disrupting or shifting the position of the host nucleosome. Instead, each BER enzyme in turn formed a ternary complex with the host nucleosome (17). The results in this study indicate that the same is true for LigIIIα-XRCC1, which catalyzes the fourth and final step in short-patch BER. A key difference, however, is that enzymes that act early in BER can bind directly to substrates that are optimally oriented with respect to the underlying histone octamer. Binding to substrates at sterically occluded sites appears to occur only when substrates are exposed by spontaneous, transient partial unwrapping of DNA from the histone octamer regardless of nick orientation. This makes structural sense in that LigIIIα is the only short-patch BER enzyme that fully encircles DNA in its active configuration.
LigIIIα-XRCC1 proved less efficient at ligating nicks in 601 than in 5S rDNA nucleosomes. The octamer structure within 601 nucleosomes is virtually identical to that in other nucleosomes (44). The reduced ligation efficiency might instead reflect the unusual curvature or rigidity of 601 DNA. This could manifest itself at the level of lower, spontaneous nucleosome unwrapping rates (19) or in the ease with which DNA untwists to accompany the folding of ligase IIIα into its active configuration. It is also possible that substrates ligated in the 5S rDNA nucleosomes were disproportionately associated with translational variants in which the nick was within the nucleosome but still relatively close (∼17 bp) to the nucleosome edge, where unwrapping events are most frequent.
We reported previously that LigIIIα-XRCC1 enhanced the gap-filling activity of DNA pol β in nucleosomes (17), and we show here that XRCC1 enhances ligation of nicks in nucleosomes (Fig. 8). DNA pol β interacts with an N-terminal segment of XRCC1, whereas LigIIIα interacts with a BRCT (BRCA1 C terminus) domain in the C-terminal region of XRCC1 in the C-terminal region of XRCC1. Thus, it is possible that, during BER, XRCC1 first facilitates gap discovery and binding of pol β and subsequently facilitates nick discovery and binding of LigIIIα. This mechanism is consistent with the likely role of XRCC1 as a scaffold. In the context of BER in nucleosomes, this mechanism would mean that LigIIIα binding would not depend on an independent, de novo DNA unwrapping event. There is some uncertainty as to when the pol β-XRCC1-LigIIIα complex is recruited during BER in vivo and whether the complex includes BER factors that act at earlier steps in repair (e.g. Ref. 45). In situ localization studies show rapid, poly(ADP-ribose) polymerase 1 (PARP1)-dependent recruitment of XRCC1 to sites of single-strand DNA break repair but delayed, PARP1-independent recruitment of XRCC1 to sites of BER (46). This observation suggests that pol β-XRCC1-LigIIIα may be recruited after the initial discovery and excision of oxidized bases.
As with the DNA ligases, RNA polymerase II encircles DNA, and its transit through a nucleosome requires that it (or DNA) rotate relative to the histone octamer, thus disrupting DNA-histone contacts along the way (47). This transit, which likely requires ATP-dependent chromatin remodelers (48), does not invariably alter the translational position of the nucleosome but appears to entail loss of a single H2A/H2B dimer from the host nucleosome (49). Based on our gel mobility shift assay, ligation at the nick positions tested did not result in histone loss. However, it is possible that the integrity of the octamer (and the capacity of the host nucleosome to refold following ligation) varies with the site of nick repair.
Summary and Perspectives
We have shown that dynamic properties intrinsic to nucleosomes facilitate the ligation of nicked DNA ligation by human LigIIIα-XRCC1, and help explain the survival of host nucleosomes. That the final ligation step of BER by ligase IIIα can occur on nucleosomes raises interesting parallels with the replicative ligase, DNA ligase I, which also is able to seal nicks in nucleosomes in vitro (23). The packaging of newly replicated DNA into nucleosomes is coordinated with movement of the replication fork through physical interactions between replication machinery and nucleosome assembly factors (Ref. 50 and reviewed in Ref. 51). Thus, the ligase I-mediated sealing of DNA nicks during the processing of Okazaki fragments may occur during or after DNA has been incorporated into nucleosomes (52, 53). That both ligases I and IIIα can act on nucleosome substrates is consistent with reports that ligase I can complete BER in the absence of ligase III (54, 55) and that ligase III can function as a replicative ligase (56). Replication-coupled nucleosome assembly requires specific histone secondary modifications and histone chaperones that might be present during the replicative process and assist with ligation of Okazaki fragments. It is possible as well that chromatin-remodeling agents associated with newly replicated chromatin promote or amplify the unwrapping events that enable ligase I to load onto nucleosomal DNA. These many considerations suggest that we are far from being able to account for how BER occurs in chromatin in vivo. Further progress will require investigating the possible contributions to BER of histone modifiers, histone chaperones, and chromatin-remodeling agents as well as the posttranslational modifications that regulate BER enzymes.
Experimental Procedures
Preparation of DNA Substrates
We assembled undamaged nucleosomes and DNA nick- or gap-containing nucleosomes with either the synthetic 601 nucleosome positioning sequence (57) or the Lytechinus variegatus 5S rDNA (5S) nucleosome positioning sequence (58). 601 DNA adopts a single helical orientation and translational position with respect to the underlying histone octamer, allowing us to predict the location and orientation of each base in the sequence. 5S rDNA also adopts a single helical orientation but, depending on reconstitution conditions, may adopt one or a few translational positions separated by ∼10-bp intervals (16, 59, 60). To prepare DNA for these nucleosomes, we annealed equimolar amounts of a top-strand DNA oligomer containing sequences 3′ to the desired nick or gap to a full-length (147- to 183-nt) bottom strand oligomer. (All oligomers were purchased from Integrated DNA Technologies.) The top strand oligomer contained a 5′-phosphate moiety to permit ligation and was lightly end-labeled with [γ-32P]ATP (PerkinElmer Life Sciences) and polynucleotide kinase (New England Biolabs) to help track DNA through subsequent steps. The resulting duplex was mixed with the thermostable DeepVent exopolymerase (New England Biolabs) and extended in a linear PCR amplification reaction (30 cycles × 30 s at 95 °C, 30 s at 46 °C, and 30 s at 68 °C). To verify quantitative extension, reaction products were examined by denaturing PAGE and then annealed to a [γ-32P]ATP end-labeled oligomer containing sequences 5′ to the desired nick or gap. We were able to use this second 32P-labeled oligomer to monitor ligation because it migrated differently from the first on gels and was labeled to a higher specific activity. To assemble undamaged DNA substrates, we omitted the 3′ oligomer annealing and extension step and instead annealed and extended the 5′ oligomer.
Nucleosome Reconstitutions
The 32P-labeled DNAs were combined with an 80-fold molar excess of unlabeled carrier DNA purified from chicken chromatin that had been digested with micrococcal nuclease to mono-, di-, and trinucleosome lengths as described in Ref. 16. This nuclease cleaves 3′ of the phosphodiester backbone in DNA, producing DNA fragments with 5′-hydroxyl and 3′-phosphate groups. Hence, this DNA did not interfere with our ligation assays (Fig. 2C). To this DNA mixture, we added a slight molar excess of histone octamer in 25 mm HEPES (pH 8.0), 1 mm EDTA, 1 mm dithiothreitol, and 2 m sodium chloride (reconstituted from E. coli-expressed recombinant human or Xenopus histones and purified as described in Ref. 61, 62). Samples were then slowly dialyzed, in a stepwise fashion, into the same buffer lacking sodium chloride, beginning at 37 °C for the first step and ending with an overnight dialysis at 4 °C (note that nucleosomes are stable in low salt whereas the histone octamer is not). Reconstitution efficiencies were assessed using 5% native PAGE in ½ × Tris borate-EDTA (gel shift assay, or EMSA gels) and were virtually 100% for the 601 nucleosomes and typically >90% for 5S rDNA nucleosomes. The end-labeled nucleosomes were adjusted to 2.5 nm (∼200 nm total nucleosome concentration) for storage at 4 °C and diluted to 1 nm (80 nm total nucleosome concentration) just prior to use in ligation assays. This is well above the concentration where one can see dilution-driven nucleosome dissociation (32).
Ligation Assays
Human DNA LigIIIα-XRCC1, LigIIIα-TDP1, and LigIIIα-TDP1-XRCC1 were expressed in and purified from baculovirus-infected Sf9 insect cells. E. coli-expressed human LigIIIα was from Enzymax, LLC, and E. coli-expressed XRCC1 was a gift from Michael Weinfeld. Unless otherwise noted, ligation reactions contained 1 nm of 32P end-labeled nucleosomes or naked DNA lacking or containing a single DNA nick or gap and ∼80 nm unlabeled nucleosomes or naked DNA in our standard assay buffer (25 mm HEPES (pH 8.0), 100 mm sodium chloride, 5 mm magnesium chloride, 1 mm dithiothreitol, 1 mm ATP, and 0.1 mg/ml bovine serum albumin) in a final volume of 5–10 μl. Reaction mixtures were incubated at 37 °C with the indicated enzyme(s) at the concentrations and times specified in the text or figure legends. To monitor ligation, reactions were quenched by adding ¼ volume of 0.5% sodium dodecyl sulfate, 50 mm EDTA, and 0.5 mg/ml of freshly added proteinase K. Samples were incubated for 30 min at 37 °C, mixed with an equal volume of formamide containing 20 mm EDTA and 2 mg/ml each of bromphenol blue and xylene cyanol, and fractionated on 8% denaturing gels. Gels were dried and exposed to phosphorimaging screens (Bio-Rad), and bands of interest were quantified using a PharosFX Plus PhosphorImager with Quantity One software (Bio-Rad). We calculated ligation as a percentage of the total DNA substrate after adjusting for background (using the global subtraction method). The impact of small amounts of contaminating naked DNA in the nucleosome reactions was computationally eliminated as described in Ref. 16.
Restriction Enzyme Assays for Nucleosome Integrity
To assess the fate of nucleosomes during ligation, we monitored the availability of restriction endonuclease sites in 5S rDNA that are normally inaccessible (16, 18). We added 25 units of AgeI (New England Biolabs) to 5-μl reaction mixtures containing 0–15 nm LigIIIα-XRCC1, 1 nm −46 nicked DNA or nucleosomes, and 80 nm unlabeled naked DNA or nucleosomes. After 45 min at 37 °C, we measured the extent of ligation by quenching a portion of each reaction and processing samples as described above. To measure the extent of AgeI cleavage in the presence or absence of LigIIIα-XRCC1, we added to a second portion of each reaction an equal volume of 100 mm Tris base, 25 mm EDTA, 0.2% sodium dodecyl sulfate, 2 mg/ml bromphenol blue, and 12% glycerol and fractionated samples through native 8% gels. We monitored access to a PsiI site closer to the DNA nick in an identical manner using 15 nm LigIIIα-XRCC1 and 2.5 units of PsiI.
Ternary Complex Analyses
To visualize the impact of adding LigIIIα-XRCC1 to intact or nick- or gap-containing nucleosomes, portions of the above-described reactions were mixed with equal volumes of 10% glycerol, 25 nm HEPES (pH 8.0), and 20 mm EDTA, immediately fractionated through 5% EMSA gels, and visualized as before. To determine whether DNA ligation occurs within the ternary complex that LigIIIα-XRCC1 forms with nick-containing nucleosomes, ternary complexes were separated from nucleosomes using EMSA gels. Gel lanes were then excised and incubated first in 0.1% sodium dodecyl sulfate, 10 mm EDTA (pH 8.0), and 0.1 mg/ml proteinase K for 30 min at 50 °C to degrade protein and then in 0.1 m sodium hydroxide, 5% glycerol, and 0.0125% each of bromphenol blue and xylene cyanol for 10 min to denature DNA. DNA associated with individual particles was then fractionated through an 8% sequencing gel.
Author Contributions
W. J. C. conducted the experiments, analyzed the results, prepared all figures, and wrote the paper. D. S. P. conceived the project, helped analyze the results, and wrote the paper with W. J. C. I. R. expressed and purified the LigIIIα-TDP1 and LigIIIα-TDP1-XRCC1 complexes for the studies in Fig. 8. S. S. W. and A. E. T. helped to write the paper.
Acknowledgments
We thank Drs. Joyce Heckman and Robyn Maher for critical comments on the manuscript, Dr. Michael Weinfeld for purified XRCC1, Dr. Amalthiya Prasad for purified chicken nuclei, and April Averill for the purification of histones.
This work was supported by NCI, National Institutes of Health Grants P01-CA098993 (to S. S. W.) and CA92584 (to A. E. T.) and NIEHS, National Institutes of Health Grant ES012512 (to A. E. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- BER
- base excision repair
- APE1
- apurinic endonuclease
- LigIIIα
- DNA ligase IIIα
- nt
- nucleotide
- pol β
- DNA polymerase β
- TDP1
- topoisomerase I-associated tyrosine phosphatase
- XRCC1
- X-ray repair cross-complementing protein 1
- rDNA
- ribosomal DNA.
References
- 1. Lindahl T., and Nyberg B. (1972) Rate of depurination of native deoxyribonucleic acid. Biochemistry 11, 3610–3618 [DOI] [PubMed] [Google Scholar]
- 2. Fraga C. G., Shigenaga M. K., Park J. W., Degan P., and Ames B. N. (1990) Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. U.S.A. 87, 4533–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Friedberg E. C., Walker G. C., Siede W., and Wood R. D. (2005) DNA Repair and Mutagenesis, pp. 9–49, American Society for Microbiology Press, Washington, D. C. [Google Scholar]
- 4. David S. S., O'Shea V. L., and Kundu S. (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hegde M. L., Hazra T. K., and Mitra S. (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res 18, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robertson A. B., Klungland A., Rognes T., and Leiros I. (2009) DNA repair in mammalian cells: base excision repair: the long and short of it. Cell Mol. Life Sci. 66, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duclos S., Doublié S., and Wallace S. S. (2012) in The Cellular Response to the Genotoxic Insult: The Question of Threshold for Genotoxic Carcinogens (Greim H., and Albertini R., eds), First Ed., pp. 115–159, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 8. Krokan H. E., and Bjørås M. (2013) Base excision repair. Cold Spring Harb. Perspect. Biol. 5, a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wallace S. S. (2014) Base excision repair: a critical player in many games. DNA Repair 19, 14–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weren R. D., Ligtenberg M. J., Kets C. M., de Voer R. M., Verwiel E. T., Spruijt L., van Zelst-Stams W. A., Jongmans M. C., Gilissen C., Hehir-Kwa J. Y., Hoischen A., Shendure J., Boyle E. A., Kamping E. J., Nagtegaal I. D., et al. (2015) A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 47, 668–671 [DOI] [PubMed] [Google Scholar]
- 11. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., and Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 12. Clapier C. R., and Cairns B. R. (2009) The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 78, 273–304 [DOI] [PubMed] [Google Scholar]
- 13. Menoni H., Gasparutto D., Hamiche A., Cadet J., Dimitrov S., Bouvet P., and Angelov D. (2007) ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A.Bbd nucleosomes. Mol. Cell. Biol. 27, 5949–5956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakanishi S., Prasad R., Wilson S. H., and Smerdon M. (2007) Different structural states in oligonucleosomes are required for early versus late steps of base excision repair. Nucleic Acids Res. 35, 4313–4321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Czaja W., Mao P., and Smerdon M. J. (2014) Chromatin remodelling complex RSC promotes base excision repair in chromatin of Saccharomyces cerevisiae. DNA Repair 16, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prasad A., Wallace S. S., and Pederson D. S. (2007) Initiation of base excision repair of oxidative lesions in nucleosomes by the human, bifunctional DNA glycosylase NTH1. Mol. Cell. Biol. 27, 8442–8453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Odell I. D., Barbour J. E., Murphy D. L., Della-Maria J. A., Sweasy J. B., Tomkinson A. E., Wallace S. S., and Pederson D. S. (2011) Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell. Biol. 31, 4623–4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Odell I. D., Newick K., Heintz N. H., Wallace S. S., and Pederson D. S. (2010) Non-specific DNA binding interferes with the efficient excision of oxidative lesions from chromatin by the human DNA glycosylase, NEIL1. DNA Repair 9, 134–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maher R. L., Prasad A., Rizvanova O., Wallace S. S., and Pederson D. S. (2013) Contribution of DNA unwrapping from histone octamers to the repair of oxidatively damaged DNA in nucleosomes. DNA Repair 12, 964–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cannan W. J., Tsang B. P., Wallace S. S., and Pederson D. S. (2014) Nucleosomes suppress the formation of double-strand DNA breaks during attempted base excision repair of clustered oxidative damages. J. Biol. Chem. 289, 19881–19893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pascal J. M., O'Brien P. J., Tomkinson A. E., and Ellenberger T. (2004) Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature 432, 473–478 [DOI] [PubMed] [Google Scholar]
- 22. Cotner-Gohara E., Kim I. K., Hammel M., Tainer J. A., Tomkinson A. E., and Ellenberger T. (2010) Human DNA ligase III recognizes DNA ends by dynamic switching between two DNA-bound states. Biochemistry 49, 6165–6176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chafin D. R., Vitolo J. M., Henricksen L. A., Bambara R. A., and Hayes J. J. (2000) Human DNA ligase I efficiently seals nicks in nucleosomes. EMBO J. 19, 5492–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Odell I. D., Wallace S. S., and Pederson D. S. (2013) Rules of engagement for base excision repair in chromatin. J. Cell. Physiol. 228, 258–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ye Y., Stahley M. R., Xu J., Friedman J. I., Sun Y., McKnight J. N., Gray J. J., Bowman G. D., and Stivers J. T. (2012) Enzymatic excision of uracil residues in nucleosomes depends on the local DNA structure and dynamics. Biochemistry 51, 6028–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Polach K. J., and Widom J. (1995) Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol. 254, 130–149 [DOI] [PubMed] [Google Scholar]
- 27. Anderson J. D., and Widom J. (2000) Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 296, 979–987 [DOI] [PubMed] [Google Scholar]
- 28. Li G., and Widom J. (2004) Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 11, 763–769 [DOI] [PubMed] [Google Scholar]
- 29. Li G., Levitus M., Bustamante C., and Widom J. (2005) Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 12, 46–53 [DOI] [PubMed] [Google Scholar]
- 30. Tims H. S., Gurunathan K., Levitus M., and Widom J. (2011) Dynamics of nucleosome invasion by DNA binding proteins. J. Mol. Biol. 411, 430–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. von Hippel P. H., and Berg O. G. (1989) Facilitated target location in biological systems. J. Biol. Chem. 264, 675–678 [PubMed] [Google Scholar]
- 32. Pederson D. S., Venkatesan M., Thoma F., and Simpson R. T. (1986) Isolation of an episomal yeast gene and replication origin as chromatin. Proc. Natl. Acad. Sci. U.S.A. 83, 7206–7210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cotner-Gohara E., Kim I. K., Tomkinson A. E., and Ellenberger T. (2008) Two DNA-binding and nick recognition modules in human DNA ligase III. J. Biol. Chem. 283, 10764–10772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taylor R. M., Whitehouse C. J., and Caldecott K. W. (2000) The DNA ligase III zinc finger stimulates binding to DNA secondary structure and promotes end joining. Nucleic Acids Res. 28, 3558–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen X., Ballin J. D., Della-Maria J., Tsai M. S., White E. J., Tomkinson A. E., and Wilson G. M. (2009) Distinct kinetics of human DNA ligases I, IIIα, IIIβ, and IV reveal direct DNA sensing ability and differential physiological functions in DNA repair. DNA Repair 8, 961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kukshal V., Kim I. K., Hura G. L., Tomkinson A. E., Tainer J. A., and Ellenberger T. (2015) Human DNA ligase III bridges two DNA ends to promote specific intermolecular DNA end joining. Nucleic Acids Res. 43, 7021–7031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thåström A., Lowary P. T., and Widom J. (2004) Measurement of histone-DNA interaction free energy in nucleosomes. Methods 33, 33–44 [DOI] [PubMed] [Google Scholar]
- 38. Maskell D. P., Renault L., Serrao E., Lesbats P., Matadeen R., Hare S., Lindemann D., Engelman A. N., Costa A., and Cherepanov P. (2015) Structural basis for retroviral integration into nucleosomes. Nature 523, 366–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thåström A., Lowary P. T., Widlund H. R., Cao H., Kubista M., and Widom J. (1999) Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 288, 213–229 [DOI] [PubMed] [Google Scholar]
- 40. Mani R. S., Karimi-Busheri F., Fanta M., Caldecott K. W., Cass C. E., and Weinfeld M. (2004) Biophysical characterization of human XRCC1 and its binding to damaged and undamaged DNA. Biochemistry 43, 16505–16514 [DOI] [PubMed] [Google Scholar]
- 41. Leppard J. B., Dong Z., Mackey Z. B., and Tomkinson A. E. (2003) Physical and functional interaction between DNA ligase IIIα and poly(ADP-ribose) polymerase 1 in DNA single-strand break repair. Mol. Cell. Biol. 23, 5919–5927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marintchev A., Mullen M. A., Maciejewski M. W., Pan B., Gryk M. R., and Mullen G. P. (1999) Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nat. Struct. Biol. 6, 884–893 [DOI] [PubMed] [Google Scholar]
- 43. Tomkinson A. E., and Sallmyr A. (2013) Structure and function of the DNA ligases encoded by the mammalian LIG3 gene. Gene 531, 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vasudevan D., Chua E. Y., and Davey C. A. (2010) Crystal structures of nucleosome core particles containing the “601” strong positioning sequence. J. Mol. Biol. 403, 1–10 [DOI] [PubMed] [Google Scholar]
- 45. Prasad R., Williams J. G., Hou E. W., and Wilson S. H. (2012) Pol β associated complex and base excision repair factors in mouse fibroblasts. Nucleic Acids Res. 40, 11571–11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Campalans A., Kortulewski T., Amouroux R., Menoni H., Vermeulen W., and Radicella J. P. (2013) Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Res. 41, 3115–3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kulaeva O. I., Hsieh F. K., Chang H. W., Luse D. S., and Studitsky V. M. (2013) Mechanism of transcription through a nucleosome by RNA polymerase II. Biochim. Biophys. Acta 1829, 76–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hodges C., Bintu L., Lubkowska L., Kashlev M., and Bustamante C. (2009) Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science 325, 626–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kireeva M. L., Walter W., Tchernajenko V., Bondarenko V., Kashlev M., and Studitsky V. M. (2002) Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol. Cell 9, 541–552 [DOI] [PubMed] [Google Scholar]
- 50. Smith S., and Stillman B. (1991) Stepwise assembly of chromatin during DNA replication in vitro. EMBO J. 10, 971–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Krude T. (1999) Chromatin assembly during DNA replication in somatic cells. Eur. J. Biochem. 263, 1–5 [DOI] [PubMed] [Google Scholar]
- 52. Smith D. J., and Whitehouse I. (2012) Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 483, 434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bielinsky A. K., and Gerbi S. A. (1999) Chromosomal ARS1 has a single leading strand start site. Mol. Cell 3, 477–486 [DOI] [PubMed] [Google Scholar]
- 54. Gao Y., Katyal S., Lee Y., Zhao J., Rehg J. E., Russell H. R., and McKinnon P. J. (2011) DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 471, 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Simsek D., Furda A., Gao Y., Artus J., Brunet E., Hadjantonakis A. K., Van Houten B., Shuman S., McKinnon P. J., and Jasin M. (2011) Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 471, 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Arakawa H., and Iliakis G. (2015) Alternative Okazaki fragment ligation pathway by DNA ligase III. Genes 6, 385–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lowary P. T., and Widom J. (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42 [DOI] [PubMed] [Google Scholar]
- 58. Simpson R. T., and Stafford D. W. (1983) Structural features of a phased nucleosome core particle. Proc. Natl. Acad. Sci. U.S.A. 80, 51–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Flaus A., Luger K., Tan S., and Richmond T. J. (1996) Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc. Natl. Acad. Sci. U.S.A. 93, 1370–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dong F., Hansen J. C., and van Holde K. E. (1990) DNA and protein determinants of nucleosome positioning on sea urchin 5S rRNA gene sequences in vitro. Proc. Natl. Acad. Sci. U.S.A. 87, 5724–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dyer P. N., Edayathumangalam R. S., White C. L., Bao Y., Chakravarthy S., Muthurajan U. M., and Luger K. (2004) Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 [DOI] [PubMed] [Google Scholar]
- 62. Luger K., Rechsteiner T. J., and Richmond T. J. (1999) Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304, 3–19 [DOI] [PubMed] [Google Scholar]