

Abstract
Previous studies have shown that glucagon cooperatively interacts with insulin to stimulate hepatic FGF21 gene expression. Here we investigated the mechanism by which glucagon and insulin increased FGF21 gene transcription in primary hepatocyte cultures. Transfection analyses demonstrated that glucagon plus insulin induction of FGF21 transcription was conferred by two activating transcription factor 4 (ATF4) binding sites in the FGF21 gene. Glucagon plus insulin stimulated a 5-fold increase in ATF4 protein abundance, and knockdown of ATF4 expression suppressed the ability of glucagon plus insulin to increase FGF21 expression. In hepatocytes incubated in the presence of insulin, treatment with a PKA-selective agonist mimicked the ability of glucagon to stimulate ATF4 and FGF21 expression. Inhibition of PKA, PI3K, Akt, and mammalian target of rapamycin complex 1 (mTORC1) suppressed the ability of glucagon plus insulin to stimulate ATF4 and FGF21 expression. Additional analyses demonstrated that chenodeoxycholic acid (CDCA) induced a 6-fold increase in ATF4 expression and that knockdown of ATF4 expression suppressed the ability of CDCA to increase FGF21 gene expression. CDCA increased the phosphorylation of eIF2α, and inhibition of eIF2α signaling activity suppressed CDCA regulation of ATF4 and FGF21 expression. These results demonstrate that glucagon plus insulin increases FGF21 transcription by stimulating ATF4 expression and that activation of cAMP/PKA and PI3K/Akt/mTORC1 mediates the effect of glucagon plus insulin on ATF4 expression. These results also demonstrate that CDCA regulation of FGF21 transcription is mediated at least partially by an eIF2α-dependent increase in ATF4 expression.
Keywords: bile acid, eIF2, glucagon, insulin, liver, mammalian target of rapamycin (mTOR), PKA, transcription regulation
Introduction
FGF21 is an atypical member of the fibroblast growth factor family that lacks the ability to bind to heparin sulfate proteoglycans, allowing it to escape the extracellular matrix and function in an endocrine manner (1–3). Studies investigating the biological action of FGF21 have shown that starvation and other nutritional stresses (e.g. dietary protein restriction, consumption of a high-fat, low-carbohydrate ketogenic diet, and consumption of a high-fat obesogenic diet) stimulate an increase in the expression and secretion of FGF21 by the liver, the predominant site of FGF21 production in the body (4–10). FGF21 signals through FGF receptor 1c (FGFR1c) linked to the co-receptor β-klotho to increase food intake, energy expenditure, gluconeogenesis, and insulin sensitivity and inhibit growth and female fertility in response to nutritional stress (1–6, 11).
Several signaling pathways have been identified that mediate the effects of nutritional stress on FGF21 expression. One such pathway involves the activation of the nuclear receptor peroxisome proliferator-activated receptor α (PPARα).2 A PPARα response element (PPRE) has been identified in the 5′-flanking region of the murine and human FGF21 genes (8, 12). Ablation of the PPARα gene suppresses the ability of starvation and ketogenic diet consumption to increase hepatic FGF21 mRNA abundance and serum FGF21 concentration (7, 8).
Another pathway mediating the nutritional regulation of FGF21 expression is activated by the glucagon receptor. This has been deduced from studies in mice showing that ablation of the glucagon receptor suppresses the ability of starvation to increase hepatic FGF21 mRNA abundance and serum FGF21 concentration (1). In studies examining the mechanism by which glucagon increases FGF21 production, we have shown that incubating rat and human hepatocyte cultures with glucagon causes a 3-fold increase in FGF21 secretion into the culture medium (14). Interestingly, the glucagon-induced increase in FGF21 secretion in hepatocytes is associated with a transient decrease in FGF21 mRNA abundance, suggesting that glucagon acts at a translational/posttranslational step to increase hepatic FGF21 secretion.
The inability of glucagon to induce FGF21 mRNA abundance in hepatocyte cultures (14) contrasts with the results of glucagon receptor ablation studies (1) demonstrating that the starvation-induced increase in FGF21 mRNA abundance is mediated at least partially by glucagon receptor activation. One possible explanation for the discrepant findings is that glucagon stimulation of FGF21 mRNA abundance requires the presence of another hormone or signaling factor. Results of studies with intact mice containing defects in the insulin signaling pathway suggest that insulin is one such factor that potentiates the ability of glucagon to increase FGF21 mRNA abundance. Dong et al. (15) have shown that liver-specific ablation of insulin receptor substrate 1 (IRS-1) and IRS-2 causes a decrease in hepatic FGF21 mRNA abundance during both the fed state and the starved state. In addition, Haeusler et al. (16) have reported that streptozotocin-induced diabetes suppresses the stimulatory effect of starvation on hepatic FGF21 mRNA abundance. Although insulin is generally regarded as a hormone signaling the fed state, these observations suggest that basal insulin levels during the starved state play a role in mediating the increase in FGF21 mRNA abundance caused by starvation. In support of this possibility, we have shown that insulin potentiates the ability of glucagon to stimulate FGF21 mRNA abundance in primary rat hepatocyte cultures (14). Treatment with insulin alone stimulates a 3.5-fold increase in FGF21 mRNA abundance, and the addition of glucagon in the presence of insulin causes a further 2.5-fold elevation in FGF21 mRNA abundance. This cooperative interaction between glucagon and insulin in the regulation of FGF21 mRNA abundance is associated with a substantially greater induction of FGF21 secretion (i.e. 28-fold) relative to that by glucagon or insulin alone. Dose-response experiments have shown that insulin is effective in unmasking glucagon regulation of FGF21 mRNA abundance at a concentration observed in the portal circulation during fasted conditions (i.e. 1 nm) (14). We have postulated that insulin maintains a basal level of hepatic FGF21 mRNA abundance during the carbohydrate-fed state and synergistically interacts with elevated glucagon levels during the starved state to stimulate a further increase in FGF21 mRNA abundance. The mechanism by which glucagon and insulin cooperatively increase FGF21 mRNA abundance is presently not known.
In addition to PPARα, glucagon, and insulin, the farnesoid X receptor (FXR) plays a role in the regulation of hepatic FGF21 production. We have shown that natural (i.e. bile acids) and synthetic activating ligands (i.e. GW4064) of FXR stimulate an increase in hepatic FGF21 mRNA abundance and secretion in rodent and human hepatocyte cultures (17). The effect of GW4064 on FGF21 gene transcription is mediated, at least in part, by a conserved FXR response element (FXRE) that binds heterodimers comprised of FXR and retinoid X receptor (RXR). Ablation of the FXR gene suppresses the ability of ketogenic diet consumption to induce hepatic FGF21 mRNA abundance and serum FGF21 concentration.
In our studies analyzing the bile acid regulation of FGF21 expression, chenodeoxycholic acid (CDCA) was substantially more effective in inducing FGF21 gene expression in hepatocytes than a selective FXR-activating ligand (i.e. GW4064) that bound to FXR with a >100-fold higher affinity than that of CDCA (17). Incubating rodent and human hepatocyte cultures with an optimal concentration of CDCA (100 μm) stimulated a 25-fold increase in FGF21 mRNA abundance, whereas incubating hepatocytes with an optimal concentration of GW4064 (3 μm) stimulated a 3.2-fold increase in FGF21 mRNA abundance. These observations suggested that (an)other mechanism(s) besides ligand activation of FXR is/are involved in mediating the stimulatory effect of bile acids on FGF21 gene transcription. The nature of this mechanism has not yet been defined.
The objective of this study is to characterize the mechanisms by which glucagon plus insulin and CDCA increase hepatic FGF21 gene expression. We show that glucagon plus insulin and CDCA increase the expression and DNA binding activity of the stress-associated transcription factor activating transcription factor 4 (ATF4) and that this protein plays a key role in mediating the stimulatory effect of these signaling factors on FGF21 gene transcription. We have also characterized the proximal signaling pathways mediating the effects of glucagon plus insulin and CDCA on ATF4 and FGF21 expression.
Results
Identification of Cis-acting Sequences That Mediate the Stimulatory Effect of Glucagon Plus Insulin on FGF21 Gene Transcription
To identify cis-acting sequences mediating the stimulatory effect of glucagon plus insulin on FGF21 gene expression, transient transfection experiments were performed in primary rat hepatocyte cultures using reporter constructs containing 5′ deletions of the rat FGF21 promoter linked to the luciferase gene. In cells transfected with the longest FGF21 fragment (−2940 to +68 bp), treatment with glucagon plus insulin stimulated a 10.8-fold increase in luciferase activity (Fig. 1). 5′ deletion of FGF21 sequences to −1656 bp and −1316 bp had no effect on glucagon plus insulin responsiveness, whereas 5′ deletion of sequences to −1241 bp caused a 55% decrease in glucagon plus insulin responsiveness. Further 5′ deletion of FGF21 sequences to −1164 bp had no effect on remaining glucagon plus insulin responsiveness, whereas deletion of sequences from −1164 to −103 caused a 46% decrease in glucagon plus insulin responsiveness. These results indicate that there are two regions (−1316 to −1241 bp and −1164 to −103 bp) that confer the stimulatory effect of glucagon plus insulin on FGF21 gene transcription. Because of the small effect of insulin alone on FGF21 gene expression and the variability of the transient transfection assay, we were unable to identify FGF21 sequences that conferred regulation of transcription by insulin alone. Insulin treatment in the absence of glucagon did not stimulate a significant increase in the activity of any of the FGF21 reporter constructs (supplemental Fig. 1).
FIGURE 1.

Effects of deletions of the 5-flanking region of the rat FGF21 gene on transcriptional activity in the absence and presence of glucagon plus insulin. Primary rat hepatocytes were transiently transfected with a series of plasmids containing fragments of the rat FGF21 gene linked to the luciferase (Luc) gene as described under “Experimental Procedures.” After transfection, cells were treated with or without 25 nm glucagon (Gln) and 50 nm insulin (Ins) for 24 h. Cells were harvested, extracts were prepared, and luciferase assays were performed. Left, the constructs used in these experiments. The number at the left of each construct is the 5′ end of FGF21 DNA in nucleotides relative to the transcription initiation site. The 3′ end of each construct is +68 bp. The location of a previously identified FXRE (−1222 to −1210 bp), PPRE (−1215 to −1203 bp), ChoRE (−72 to −56 bp), and AARE (−1282 to −1274 bp and −140 to −132 bp) are indicated by boxes with different fills or patterns. Right, the luciferase activity of cells transfected with the −2940 to +68 bp FGF21 construct and treated with vehicle was set at 1, and all other activities were adjusted proportionately. The -fold stimulation by glucagon plus insulin was calculated by dividing the luciferase activity for cells treated with glucagon plus insulin by that for cells treated with vehicle. The -fold responses were calculated for individual experiments and then averaged. The results are the means ± S.E. of six experiments. Different superscript letters indicate that the means are significantly (p ≤ 0.05) different.
FGF21 sequences between −1316 to −1241 bp and between −1164 to −103 bp each contain a previously characterized sequence element that binds the stress-associated transcription factor ATF4 (18). Both of these elements play a role in mediating the stimulatory effect of essential amino acid deficiency on FGF21 gene transcription. They are designated as amino acid response element (AARE) 1 and AARE2 (Fig. 2A). To determine whether AARE1 and AARE2 mediate the stimulatory effect of glucagon plus insulin on FGF21 gene transcription, FGF21 reporter constructs were developed that contained site-specific mutations that abolished ATF4 binding to AARE1, AARE2, or both AARE1 and AARE2 in the context of the −1316 to +68 bp FGF21 reporter construct. Mutation of the upstream AARE1 (−1282 to −1274 bp) or the downstream AARE2 (−140 to −132 bp) caused a 54–56% decrease in glucagon plus insulin responsiveness (Fig. 2B). Mutation of both AARE1 and AARE2 caused an 87% decrease in glucagon plus insulin responsiveness. In contrast to the effect of mutation of AARE1 and AARE2 on glucagon plus insulin regulation of FGF21 promoter activity, mutations that abolished FXR and PPARα binding to the FGF21 FXRE/PPRE (−1222 to −1203 bp) and carbohydrate response element binding protein binding to the FGF21 carbohydrate response element (ChoRE, −72 to −56 bp) had no effect on the ability of glucagon plus insulin to stimulate FGF21 promoter activity (Fig. 2B). These data indicate that glucagon and insulin act selectively through both of the AAREs in the FGF21 5-flanking DNA to induce FGF21 gene transcription.
FIGURE 2.

Two AAREs in the FGF21 gene confer the stimulatory effect of glucagon plus insulin on FGF21 transcription. Reporter plasmids containing mutations (Mut) of AARE1, AARE2, FXRE/PPRE, and/or ChoRE in the context of the −1316 to +68 bp FGF21 promoter fragment were transiently transfected into hepatocytes as described in the legend for Fig. 1 and under “Experimental Procedures.” A, native and mutant sequences of AARE1, AARE2, FXRE/PPRE, and ChoRE in the rat FGF21 gene. The native sequence of each regulatory element is indicated in bold letters, and the mutated sequence is shown underneath. The hexameric half-sites comprising the FXRE and PPRE are indicated by arrows. B, luciferase activity of hepatocytes transfected with wild-type and mutant reporter plasmids. Mutation of AARE1, AARE2, FXRE/PPRE, and ChoRE is indicated by an X through the box designated for that element. The results are the means ± S.E. of six experiments. Different superscript letters indicate that the means are significantly (p ≤ 0.05) different. Gln, glucagon; Ins, insulin.
Role of ATF4 in Mediating the Stimulatory Effect of Glucagon Plus Insulin on FGF21 Gene Transcription
Previous studies have shown that ATF4 binds to AARE1 and AARE2 in hepatocytes and that expression of exogenous ATF4 in hepatocytes induces FGF21 gene transcription (18). These observations plus the results of this study demonstrating that glucagon and insulin signal through AARE1 and AARE2 to induce FGF21 gene transcription led us to investigate whether glucagon and insulin modulated ATF4 expression in hepatocyte cultures. Incubating hepatocytes with insulin or glucagon alone for 12 h had no effect on ATF4 protein concentration or ATF4 mRNA abundance, whereas incubating hepatocytes with glucagon plus insulin stimulated a 5.3-fold increase in ATF4 protein concentration and a 1.6-fold increase in ATF4 mRNA abundance (Fig. 3A). This cooperative interaction between glucagon and insulin in the regulation of ATF4 expression mirrored the regulation of FGF21 mRNA abundance by these hormones. In contrast, glucagon antagonized the ability of insulin to induce the expression of the lipogenic transcription factor SREBP-1c (Fig. 3A). These findings demonstrate that glucagon and insulin cooperatively interact to increase ATF4 expression and that this effect is selective for ATF4 and its downstream targets.
FIGURE 3.

Glucagon and insulin cooperatively induce ATF4 expression and ATF4 binding to AARE1 and AARE2. A, primary rat hepatocytes were incubated with or without glucagon, insulin, or glucagon plus insulin. The abundance of ATF4 mRNA, FGF21 mRNA, and SREBP-1c mRNA in total RNA was measured after 6 h of treatment. The abundance of ATF4 protein in cell extracts was measured after 12 h of treatment. The level of ATF4 protein, ATF4 mRNA, FGF21 mRNA, and SREBP-1c mRNA in cells incubated with vehicle was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. of seven experiments. An asterisk indicates that the mean is significantly (*, p ≤ 0.05) higher than any other mean. B, gel mobility shift analyses were performed using nuclear extracts (N.E.) prepared from hepatocytes treated with or without glucagon (Gln) plus insulin (Ins) for 12 h. The sequences of 32P-labeled probes containing AARE1 or AARE2 are shown in Fig. 2A. The binding reactions were performed as described under “Experimental Procedures.” Nuclear extracts were incubated with antibodies against ATF4 prior to addition of the probe. Competition analyses were performed by mixing the labeled probe with a 5- and 50-fold molar excess of unlabeled probe (Self Comp.) or a competitor DNA containing a mutation of AARE1 or AARE2 (AARE Mut Comp.). Positions of specific DNA-protein complexes (brackets) and supershifted complexes (SS) are indicated.
Gel mobility shift assays were performed to determine whether the stimulatory effect of glucagon plus insulin on ATF4 expression was associated with an elevation in the binding of ATF4 to AARE1 and AARE2. Nuclear extracts were prepared from hepatocytes incubated with or without glucagon plus insulin for 12 h. Incubation of nuclear extracts with 32P-labeled DNA probes containing the AARE1 or AARE2 resulted in the formation of multiple protein-DNA complexes (Fig. 3B). Competition analyses indicated that the binding of several of these DNA-protein complexes was specific. The abundance of these specific protein-DNA complexes was increased in nuclear extracts from hepatocytes treated with glucagon plus insulin. Preincubation of nuclear extracts with ATF4 antibody partially disrupted the formation of these protein-DNA complexes and caused the formation of new supershifted complexes. The abundance of these supershifted complexes was elevated in nuclear extracts from hepatocytes treated with glucagon plus insulin. These observations indicate that glucagon plus insulin increases the ATF4 binding to AARE1 and AARE2.
To further establish a role of ATF4 in mediating the stimulatory effect of glucagon plus insulin on FGF21 expression, we investigated whether knockdown of ATF4 expression modulated the ability of glucagon plus insulin to increase FGF21 expression. Primary rat hepatocytes were transfected with siRNAs targeting ATF4 (ATF4 siRNA 1 and ATF4 siRNA 2) or a non-targeting control siRNA and were treated with or without glucagon plus insulin. Transfection of hepatocytes with ATF4 siRNA 1 or ATF4 siRNA 2 inhibited both basal ATF4 protein expression and glucagon plus insulin induction of ATF4 protein expression relative to untransfected cells or cells transfected with control siRNA (Fig. 4). These effects on ATF4 expression were associated with a reduction in basal FGF21 mRNA abundance and the ability of glucagon plus insulin to increase FGF21 mRNA abundance. These results demonstrate that glucagon plus insulin regulation of FGF21 gene expression is dependent on ATF4.
FIGURE 4.
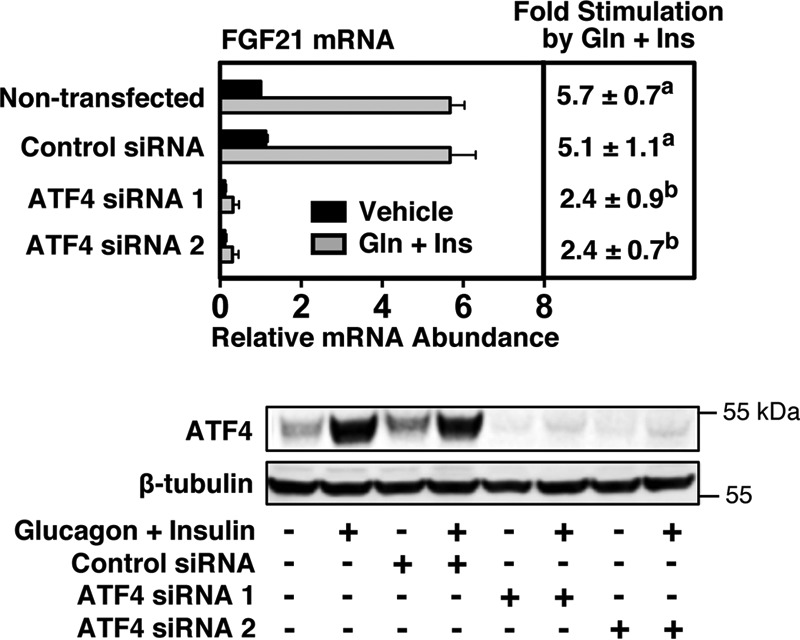
Knockdown of ATF4 expression suppresses the ability of glucagon plus insulin to increase FGF21 mRNA abundance. Primary rat hepatocytes were transfected with control siRNA or siRNA targeting ATF4 as described under “Experimental Procedures.” After transfection, cells were incubated with glucagon (Gln) and insulin (Ins) for 12 h. Cells were then harvested, total RNA was isolated, and cellular extracts were prepared. Top panel, FGF21 mRNA abundance was measured in total RNA. The level of FGF21 mRNA in non-transfected cells incubated with vehicle was set at 1, and all other values were adjusted proportionately. The -fold stimulation by glucagon plus insulin was calculated by dividing the FGF21 mRNA abundance of cells treated with glucagon plus insulin by that of cells treated with vehicle. The -fold responses were calculated for individual experiments and then averaged. Values are the means ± S.E. of five experiments. Different superscript letters indicate that the means are significantly (p ≤ 0.05) different. Bottom panel, the abundance ATF4 protein and β-tubulin in cell lysates was measured by Western blotting analysis. These data are representative of five experiments.
PI3K/Akt/mTORC1 and PKA Mediate the Stimulatory Effect of Insulin and Glucagon on FGF21 and ATF4 Expression
We next investigated the proximal signaling pathways mediating the synergistic effect of glucagon and insulin on FGF21 and ATF4 expression. Insulin activation of the insulin receptor stimulates PI3K activity, which, in turn, increases the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3) (19). Elevated phosphatidylinositol 3,4,5-triphosphate levels lead to an increase in the phosphorylation and activation of Akt (also known as protein kinase B). We performed studies with specific inhibitors of PI3K (i.e. LY294002) and Akt (i.e. Akti-1/2) to investigate the role of the PI3K/Akt pathway in mediating the cooperative interaction of insulin with glucagon in inducing FGF21 gene expression. Incubating hepatocytes with LY294002 or Akti-1/2 suppressed the ability of glucagon plus insulin to stimulate FGF21 mRNA abundance and ATF4 protein concentration in a dose-dependent manner (Fig. 5, A and B). In agreement with previous studies (20), Western blotting analyses showed that insulin increased the abundance of the phosphorylated, active form of Akt (Ser473) and that the presence of glucagon amplified this effect (Fig. 5A). The ability of LY294002 and Akti-1/2 to suppress the stimulatory effect of glucagon plus insulin on FGF21 and ATF4 expression was associated with a decrease in phosphorylated Akt (Fig. 5, A and B). These data demonstrate that insulin interacts with glucagon in a cooperative manner to stimulate Akt activity and that Akt is required for the stimulatory effect of glucagon plus insulin on FGF21 and ATF4 expression.
FIGURE 5.

Inhibition of PI3K, Akt, or mTORC1 suppresses the ability of glucagon plus insulin to induce FGF21 mRNA abundance and ATF4 protein expression. A–C, primary rat hepatocytes were isolated and incubated in serum-free Medium 199. At 47 h of incubation, the medium was replaced with one of the same composition containing vehicle or the indicated concentrations of LY294002 (A), Akti-1/2 (B), or rapamycin (C). At 48 h of incubation, glucagon (Gln) and/or insulin (Ins) was added to the medium, and the incubation was continued for 12 h. Cells were harvested, total RNA was isolated, and cellular extracts were prepared. Left panels, the abundance of FGF21 mRNA in total RNA and the level of ATF4 protein in total cell lysates were measured as described under “Experimental Procedures.” Values for cells incubated in the absence of inhibitor and hormones were set at 1, and the other values were adjusted proportionately. Values are means ± S.E. of four experiments. *, p ≤ 0.05. Right panels, the abundance of phosphorylated Akt (Ser473, P-Akt), phosphorylated ribosomal protein S6 (Ser235/236, P-RPS6), total Akt, and total RPS6 in total cell lysates was measured by Western blotting analysis. These data are representative of four experiments.
The hepatic insulin signaling pathway bifurcates at a step distal to Akt. Akt phosphorylates FoxO1, leading to inactivation of gluconeogenic enzyme gene transcription (21, 22). Alternatively, Akt phosphorylates tuberous sclerosis complex 2 (TSC2), leading to activation of mammalian target of rapamycin complex 1 (mTORC1) and an increase in lipogenic gene expression (22, 23). To evaluate the role of mTORC1 in mediating the induction of FGF21 gene expression by glucagon plus insulin, we conducted experiments employing the specific mTORC1 inhibitor rapamycin. Incubating hepatocytes with 1 nm rapamycin suppressed the ability of glucagon plus insulin to increase FGF21 mRNA abundance and ATF4 protein concentration by 65% and 44%, respectively (Fig. 5C). The rapamycin-mediated reduction in glucagon plus insulin regulation of FGF21 mRNA abundance and ATF4 protein concentration was associated with a decrease in the phosphorylated active form of ribosomal protein S6, an effector protein downstream of mTORC1. In contrast, treatment with rapamycin had no effect on the abundance of the phosphorylated, active form of Akt. These data suggest that insulin signals through the mTORC1 branch of the insulin pathway to potentiate the stimulatory effect of glucagon on FGF21 and ATF4 expression.
We next characterized the glucagon signaling pathway mediating the induction of FGF21 and ATF4 expression by glucagon plus insulin. Glucagon regulates hepatic metabolic processes by binding to the glucagon receptor, stimulating an increase in adenylyl cyclase activity, resulting in an elevation in cAMP production (24). Increased intracellular cAMP levels, in turn, activate PKA and exchange protein directly activated by cAMP (EPAC), a guanine nucleotide exchange factor that activates the small GTPase Rap1 (24, 25). To assess the role of the cAMP/PKA pathway and the cAMP/EPAC pathway in mediating the stimulatory effect of glucagon plus insulin on FGF21 gene expression, we tested the ability of a membrane-permeable form of cAMP (i.e. dibutyryl cAMP), a PKA-selective agonist (i.e. N6-benzoyladenosine-3′,5′-cyclic monophosphate (6-Bnz-cAMP)) (26), and an EPAC-selective agonist (i.e. 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (cpTOME)) (27) to mimic the glucagon induction of FGF21 mRNA abundance in the presence of insulin. In hepatocytes incubated in culture medium containing insulin, addition of dibutyryl cAMP or 6-Bnz-cAMP stimulated a 10- to 11-fold increase in FGF21 mRNA abundance (Fig. 6A). This increase in FGF21 mRNA abundance was similar in magnitude to that observed when glucagon was added to the culture medium. Dibutyryl cAMP and 6-Bnz-cAMP also mimicked the ability of glucagon to stimulate ATF4 protein concentration, ATF4 mRNA abundance, Akt Ser473 phosphorylation, and ribosomal protein S6 Ser235/236 phosphorylation in the presence of insulin (Fig. 6, A and B). In contrast, addition of cpTOME to the culture medium had no effect on FGF21 mRNA abundance, ATF4 protein concentration, ATF4 mRNA abundance, and Akt Ser473 phosphorylation in hepatocytes incubated with insulin. cpTOME treatment was effective in stimulating ribosomal protein S6 Ser235/236 phosphorylation, suggesting that EPAC activation in the presence of insulin induces ribosomal protein S6 phosphorylation via a mechanism that is independent of changes in Akt activity. This observation is concordant with previous work showing that EPAC activation of Rap1 stimulates ribosomal protein S6 phosphorylation via a PI3K/Akt-independent mechanism (28). These data suggest that glucagon signals through cAMP and PKA, but not through EPAC, to induce FGF21 and ATF4 expression in the presence of insulin.
FIGURE 6.

Dibutyryl cAMP and 6-Bnz-cAMP mimic the ability of glucagon to stimulate FGF21 and ATF4 expression in the presence of insulin. Primary rat hepatocytes were isolated and incubated in serum-free Medium 199. At 48 h of incubation, the medium was replaced with one of the same composition containing glucagon (25 nm), dibutyryl cAMP (100 μm), 6-Bnz-cAMP (100 μm), cpTOME (5 μm), or vehicle in the absence or presence of insulin (50 nm). After 12 h of incubation, cells were harvested, total RNA was isolated, and cellular extracts were prepared. A, the abundance of FGF21 mRNA and ATF4 mRNA in total RNA and the level of ATF4 protein in total cell lysates were measured as described under “Experimental Procedures.” Values for cells incubated with vehicle alone were set at 1, and the other values were adjusted proportionately. Values are means ± S.E. of three experiments. An asterisk indicates that the mean is significantly (*, p ≤ 0.05) higher compared with that of cells incubated with insulin alone. B, the abundance of ATF4, β-tubulin, phosphorylated Akt (Ser473), phosphorylated RPS6 (Ser235/236), total Akt, and total RPS6 in total cell lysates was measured by Western blotting analysis. These data are representative of three experiments. Quantitation of the signals for ATF4 protein is shown in A. C, signaling diagram showing the proteins that are activated by dibutyryl cAMP, 6-Bnz-cAMP, and cpTOME.
To obtain further evidence that PKA plays a role in mediating the stimulatory effect of glucagon and insulin on FGF21 gene expression, experiments were conducted using the PKA-selective inhibitor H89 (29). Incubating hepatocytes with H89 suppressed the ability of glucagon plus insulin to induce FGF21 mRNA abundance and ATF4 protein concentration (Fig. 7A). As a positive control for inhibition of PKA, H89 was effective in suppressing the stimulatory effect of glucagon on the abundance of mRNA encoding PEPCK, a PKA target (Fig. 7B) (30). These results provide support for a role of PKA in mediating the increase in FGF21 gene expression caused by glucagon plus insulin.
FIGURE 7.
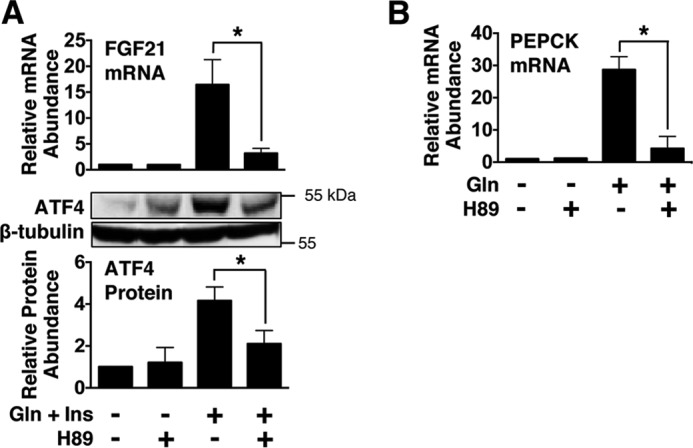
Inhibition of PKA suppresses the ability of glucagon plus insulin to induce FGF21 mRNA abundance and ATF4 protein expression. Primary rat hepatocytes were isolated and incubated in serum-free Medium 199. At 47 h of incubation, the medium was replaced with one of the same composition containing H89 (20 μm) or vehicle. A and B, at 48 h of incubation, glucagon (Gln) plus insulin (Ins) (A) or glucagon alone (B) was added to the medium, and the incubation was continued for 12 h. Cells were harvested, total RNA was isolated, and cellular extracts were prepared. The abundance of FGF21 mRNA and PEPCK mRNA in total RNA and the level of ATF4 protein in total cell lysates was measured as described under “Experimental Procedures.” Values for cells incubated in the absence of H89 and hormones were set at 1, and the other values were adjusted proportionately. Values are means ± S.E. of three experiments. *, p ≤ 0.05).
CDCA Stimulates FGF21 Gene Expression via an ATF4-dependent Mechanism
CDCA induces FGF21 gene expression not only by ligand activation of FXR but also by an undefined mechanism that is independent of ligand activation of FXR (17). Other work has shown that elevated levels of hepatic bile acids stimulate the accumulation of aberrant proteins in the endoplasmic reticulum (ER) and induce markers of ER stress (i.e. Grp78 and Chop) (31). These observations in combination with the current findings demonstrating that the stress-associated protein ATF4 mediates the stimulatory effect of glucagon plus insulin on FGF21 expression prompted us to investigate whether ATF4 plays a role in mediating the stimulatory effect of CDCA on FGF21 expression. We first examined whether CDCA modulated hepatic ATF4 expression. Incubating hepatocyte cultures with 100 μm CDCA for 2 h stimulated a 6-fold increase in ATF4 protein concentration and a 2.7-fold increase in ATF4 mRNA abundance (Fig. 8A). Results from gel mobility shift analyses demonstrated that this CDCA-induced increase in ATF4 expression was associated with an elevation in ATF4 binding to AARE1 and AARE2 (Fig. 8B). The specific binding of proteins to AARE1 and AARE2 was elevated in nuclear extracts from hepatocytes treated with CDCA. Preincubation of nuclear extracts with ATF4 antibody disrupted the formation of these DNA-protein complexes and caused the formation of new supershifted complexes. The abundance of these supershifted complexes was elevated in nuclear extracts from hepatocytes treated with CDCA.
FIGURE 8.

CDCA induces ATF4 expression and ATF4 binding to AARE1 and AARE2, and knockdown of ATF4 expression suppresses the ability of CDCA to increase FGF21 mRNA abundance. A, effect of CDCA on ATF4 expression in primary rat hepatocytes. The abundance of ATF4 protein in total cell lysates and levels of ATF4 mRNA and FGF21 mRNA in total RNA were measured after 2 h of treatment with CDCA (100 μm) or vehicle. The level of ATF4 protein, ATF4 mRNA, and FGF21 mRNA in cells incubated with vehicle was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. of seven experiments. An asterisk indicates that the mean is significantly (*, p ≤ 0.05) higher compared with that of cells treated with vehicle. B, gel mobility shift analyses were performed as described under “Experimental Procedures” using nuclear extracts (N.E.) prepared from hepatocytes treated with or without CDCA for 2 h. Positions of specific DNA-protein complexes (brackets) and supershifted complexes (SS) are indicated. C, effect of knockdown of ATF4 expression on CDCA regulation of FGF21 gene expression. Hepatocytes were transfected with control siRNA or siRNA targeting ATF4 as described under “Experimental Procedures.” After transfection, cells were incubated with CDCA for 2 h. Cells were then harvested, total RNA was isolated, and cellular extracts were prepared. Top panel, FGF21 mRNA abundance was measured in total RNA. The level of FGF21 mRNA in non-transfected cells incubated with vehicle was set at 1, and all other values were adjusted proportionately. The -fold stimulation by CDCA was calculated by dividing the FGF21 mRNA abundance of cells treated with CDCA by that of cells treated with vehicle. The -fold responses were calculated for individual experiments and then averaged. The results are the means ± S.E. of five experiments. Different superscript letters indicate that the means are significantly (p ≤ 0.05) different. Bottom panel, the abundance ATF4 protein and β-tubulin in cell lysates was measured by Western blotting analysis. These data are representative of five experiments.
We next investigated whether knockdown of ATF4 expression modulated the ability of CDCA to induce FGF21 expression. Transfection of hepatocytes with ATF4 siRNA 1 or ATF4 siRNA 2 suppressed the ability of CDCA to increase FGF21 mRNA abundance by 53–59% relative to cells transfected with control siRNA (Fig. 8C). Diminished CDCA regulation of FGF21 expression in cells transfected with ATF4 siRNA 1 and ATF4 siRNA 2 was associated with a marked reduction in ATF4 protein expression. These results indicate that CDCA increases ATF4 expression and ATF4 binding to the FGF21 gene and that ATF4 is required for CDCA induction of FGF21 gene expression.
Akt and Phosphorylated eIF2α Mediate the Stimulatory Effect of CDCA on FGF21 Gene Expression
Treatment of hepatocytes with CDCA stimulates Akt activity via mechanisms involving increased production of mitochondrial reactive oxygen species (32) and increased production of phosphatidic acid, a cellular metabolite that promotes Akt phosphorylation (33). These observations led us to investigate whether Akt plays a role in mediating the stimulatory effect of CDCA on FGF21 and ATF4 expression. Treatment of hepatocytes with Akti-1/2 suppressed the ability of CDCA to stimulate FGF21 mRNA abundance but had no effect on the ability of CDCA to increase ATF4 protein concentration (Fig. 9A). This finding suggests that Akt mediates the stimulatory effect of CDCA on FGF21 expression but not through a mechanism involving changes in ATF4 expression. The inability of Akt inhibition to suppress the stimulatory effect of CDCA on ATF4 expression raised the possibility that another signaling pathway besides Akt is involved in mediating CDCA regulation of FGF21 expression. ER stress increases the phosphorylation (Ser51) of eIF2α (34). Phosphorylated eIF2α (P-eIF2α) represses global protein translation while selectively increasing the translation of ATF4 mRNA (35). To investigate the role of P-eIF2α in mediating the stimulatory effect of CDCA on FGF21 and ATF4 expression, experiments were performed using integrated stress response inhibitor B (ISRIB), a cell-permeable small molecule that inhibits the downstream actions of P-eIF2α without affecting the eIF2α phosphorylation state (36). Treatment of hepatocyte cultures with CDCA stimulated a 3.5-fold increase in the abundance of P-eIF2α but had no effect on the abundance of total eIF2α (Fig. 9B). Incubating cells with ISRIB suppressed the ability of CDCA to increase FGF21 mRNA abundance and ATF4 protein concentration by 86% and 55%, respectively. These results suggest that CDCA signals through P-eIF2α to induce FGF21 and ATF4 expression.
FIGURE 9.

Inhibition of Akt and eIF2α signaling activity suppresses the ability of CDCA to induce FGF21 mRNA abundance. A and B, primary rat hepatocytes were isolated and incubated in serum-free Medium 199. At 47 h of incubation, the medium was replaced with one of the same composition containing 0.3 or 1 mm Akti-1/2 (A), 10 nm ISRIB (B), or vehicle. At 48 h of incubation, CDCA was added to the medium, and the incubation was continued for 2 h. Cells were then harvested, total RNA was isolated, and cellular extracts were prepared. The abundance of FGF21 mRNA and ATF4 mRNA in total RNA and the level of ATF4 protein, β-tubulin, phosphorylated Akt (Ser473), phosphorylated eIF2α (Ser51), total Akt, and total eIF2α in total cell lysates was measured as described under “Experimental Procedures.” Values for cells incubated in the absence of inhibitor and hormones were set at 1, and the other values were adjusted proportionately. Values are means ± S.E. of four experiments. *, p ≤ 0.05.
Discussion
This study identifies two new signaling pathways controlling FGF21 gene transcription. In the first pathway, glucagon activation of PKA in the presence of insulin stimulates the activity of mTORC1, a signaling complex that promotes an increase in the expression of the transcription factor ATF4. Elevated ATF4 expression, in turn, stimulates FGF21 gene transcription (Fig. 10). In the second pathway, CDCA stimulates eIF2α phosphorylation, causing an elevation in ATF4 expression and an increase in FGF21 gene transcription. To our knowledge, this is the first report demonstrating a role for ATF4 in mediating the effects of glucagon, insulin, and bile acids on hepatic gene expression. An elevation in ATF4 expression has been shown to mediate the stimulatory effect of essential amino acid deficiency on FGF21 gene transcription (18). This finding, together with the results of this study, indicate that ATF4 is a distal regulatory factor that integrates a wide range of nutritional and hormonal signals controlling FGF21 gene transcription.
FIGURE 10.

Proposed model for how glucagon, insulin, and CDCA increase hepatic FGF21 gene expression and secretion. Glucagon binding to the glucagon receptor stimulates cAMP production, resulting in an activation of PKA and EPAC. Activation of PKA enhances the ability of insulin to stimulate the activity of mTORC1, a signaling complex that promotes an increase in ATF4 expression. ATF4 binds to the FGF21 gene, triggering an increase in FGF21 transcription and FGF21 secretion. In combination with EPAC, PKA also stimulates FGF21 secretion via a translational and/or posttranslational mechanism that remains to be defined. In addition to glucagon and insulin, CDCA induces FGF21 transcription by increasing ATF4 expression. Here the elevation in ATF4 expression is mediated by phosphorylated eIF2α. CDCA also induces FGF21 transcription by ligand activation of FXR and by an undefined mechanism requiring Akt activity. Results from genetic ablation studies and correlative analyses suggest that glucagon, insulin, and bile acid signaling activity plays a role in mediating the induction of hepatic FGF21 expression during starvation and conditions related to metabolic syndrome (1, 15, 16, 47–49). Dose-response studies performed in hepatocyte cultures have shown that insulin is effective in stimulating FGF21 mRNA abundance at a concentration observed in the portal vein under fasted conditions (i.e. 1 nm) (14).
Previous studies have shown that ectopic activation of mTORC1 via knockdown of TSC1 causes an increase in FGF21 gene expression (37). Expression of exogenous ATF4 or a constitutively active form of Akt also induces FGF21 gene expression (18, 38). These findings provide support for our model that an elevation in mTORC1 signaling activity and ATF4 expression is involved in mediating the stimulatory effect of glucagon plus insulin on FGF21 gene transcription.
Interactions between insulin and glucagon play an important role in regulating hepatic metabolic processes. For example, insulin acts in a dominant manner to suppress the stimulatory effect of glucagon on the transcription of the gluconeogenic genes PEPCK and glucose 6-phosphatase (30). This effect of insulin on gluconeogenic enzyme expression is mediated by the Akt/FoxO1 branch of the insulin signaling pathway (39, 40). Other studies have shown that glucagon acts in a dominant manner to suppress the stimulatory effect of insulin on the expression of the lipogenic transcription factor SREBP-1c and its downstream target genes acetyl-CoA carboxylase and fatty acid synthase (41, 42). This effect of glucagon on SREBP-1c expression is mediated at least partially through the mTORC1 branch of the insulin signaling pathway (22, 42). In addition to these two types of antagonistic interactions, our studies investigating the regulation of FGF21 expression describe a third type of interaction in which glucagon cooperatively interacts with insulin to stimulate ATF4 and FGF21 expression. Glucagon also cooperatively interacts with insulin to stimulate hepatic DNA synthesis and cell proliferation (43, 44) and to increase the activity of Akt and mTORC1 (Figs. 5A and 6B) (20), signaling proteins that are required for glucagon and insulin regulation of ATF4 and FGF21 expression. The observation that both antagonistic interactions (i.e. SREBP-1c) and cooperative interactions (i.e. FGF21) require the presence of mTORC1 suggests that a bifurcation of the insulin signaling pathway exists downstream of mTORC1.
Previous studies by our laboratory have shown that glucagon stimulates hepatic FGF21 secretion not only by a transcriptional mechanism but also by a posttranscriptional mechanism (14). In the absence of insulin, glucagon regulation of FGF21 expression is solely posttranscriptional, as glucagon treatment under this condition stimulates a 3-fold increase in FGF21 secretion without a corresponding elevation in FGF21 mRNA abundance. Results of experiments employing selective agonists and/or inhibitors of PKA and EPAC indicate that glucagon regulation of FGF21 secretion in the absence of insulin is mediated by both the PKA and EPAC branch of the cAMP pathway. In contrast, the results of this study demonstrate that glucagon regulation of FGF21 gene transcription in the presence of insulin is mediated only by the PKA branch of the cAMP pathway (Figs. 6 and 7). Together, these findings indicate that the PKA branch of the cAMP pathway acts at both a transcriptional step and a posttranscriptional step to control FGF21 secretion, whereas the EPAC branch of the cAMP pathway acts only at a posttranscriptional step to control FGF21 secretion. These findings also indicate that cooperative interactions with insulin are restricted to the PKA branch of the cAMP pathway.
In contrast to the mechanism mediating the regulation of FGF21 transcription by glucagon plus insulin, CDCA and other bile acids induce FGF21 transcription in part through ligand activation of FXR (17). The results of this study indicate that two additional pathways also contribute to the CDCA induction of FGF21 gene transcription. One pathway involves CDCA stimulation of ATF4 expression via a P-eIF2α-dependent mechanism (Fig. 9B). The other pathway requires Akt activity and is independent of changes in ATF4 expression (Fig. 9A). We postulate that that the latter pathway involves the transcription factor nuclear factor E2-related factor 2 (Nrf2), as previous studies have shown that bile acids stimulate Nrf2 activity via a PI3K/Akt-dependent mechanism and that Nrf2 induction stimulates hepatic FGF21 expression in diabetic mice (45, 46). The ability of CDCA to act through multiple signaling pathways to induce FGF21 gene expression provides a means through which CDCA can finely control FGF21 production during different conditions.
Nutritional stress (e.g. starvation) and diseases associated with metabolic syndrome (e.g. obesity, type 2 diabetes, nonalcoholic fatty liver disease) stimulate an increase in hepatic FGF21 expression and serum FGF21 levels (5, 7–10). Elevated FGF21 production, in turn, mediates adaptive changes in insulin sensitivity, growth, circadian behavior, and energy metabolism under these conditions (1–7, 11). What is the role of the CDCA/eIF2α/ATF4 pathway and the glucagon/insulin/PKA/mTORC1/ATF4 pathway in mediating changes in FGF21 expression caused by nutritional stress and metabolic syndrome? Previous studies have shown that hepatic levels of bile acids and serum levels of bile acids, glucagon, and insulin are elevated in obesity, type 2 diabetes, and nonalcoholic fatty liver disease (47–49). Hepatic ATF4 expression, eIFα phosphorylation, PKA activity, and mTORC1 activity are also elevated in obesity, type 2 diabetes, and nonalcoholic fatty liver disease (50–53). Recent studies have shown that insulin induction of mTORC1 activity under conditions of insulin resistance requires signaling through Akt1/2 (54). These observations provide support for a role of both the CDCA/eIF2α/ATF4 pathway and the glucagon/insulin/PKA/mTORC1/ATF4 pathway in mediating changes in FGF21 expression caused by metabolic syndrome.
There is evidence that the glucagon/insulin/PKA/mTORC1/ATF4 pathway also plays a role in mediating the increase in FGF21 expression caused by starvation. First, results of experiments employing mice lacking hepatic insulin signaling activity (i.e. liver-specific deletion of IRS-1 and IRS-2) or glucagon signaling activity (i.e. deletion of the glucagon receptor) have shown that both of these pathways are required for the starvation-induced increase in hepatic FGF21 expression (1, 15, 16). Second, fasting for 48 h causes an increase in hepatic PKA and mTORC1 signaling activity and ATF4 protein expression (30, 55, 56). Previous studies have shown that mTORC1 signaling activity induces both cap-dependent and cap-independent translation of selective internal ribosome entry site-containing mRNAs (57). We hypothesize that glucagon plus insulin stimulates ATF4 protein expression by enhancing mTORC1-dependent internal ribosome entry site translation of ATF4 mRNA (58).
In conclusion, the results of this study demonstrate that glucagon and insulin act through PKA and mTORC1 to induce the expression of ATF4, a transcription factor that binds the FGF21 gene and activates transcription. This finding in combination with the observation that PKA activity, mTORC1 activity, and ATF4 expression are elevated after 48 h of starvation supports previous studies demonstrating that both insulin signaling activity and glucagon signaling activity are required for the stimulatory effect of starvation on hepatic FGF21 gene expression (1, 15, 16). The results of this study also demonstrate that alterations in ATF4 expression play a role in mediating the stimulatory effect of CDCA on FGF21 gene expression and that eIF2α mediates the increase in ATF4 expression caused by CDCA. Previous studies have shown that the eIF2α/ATF4 pathway mediates the induction of hepatic FGF21 expression caused by essential amino acid deficiency (5, 18). Knockout studies performed in mice suggest that the eIF2α kinase general control nonderepressible 2 (GCN2) plays a role a mediating the induction of ATF4 and FGF21 expression caused by acute dietary protein restriction (59). The role of GCN2 and other eIF2α kinases in mediating the stimulatory effect of bile acids on FGF21 expression is the subject of future investigations.
Experimental Procedures
Cell Culture
Primary hepatocytes were isolated from 24-h-starved male Sprague-Dawley rats (∼175–200 g) as described by Stabile et al. (60). Cells were plated on 35-mm or 60-mm collagen-coated dishes (1.4 × 105cells/cm2) containing Waymouth medium MD752/1 supplemented with 20 mm HEPES (pH 7.4), 0.5 mm serine, 0.5 mm alanine, 100 μg/ml penicillin, 100 μg/ml streptomycin, 50 mg/ml gentamicin, and 5% newborn calf serum. At 4 h of incubation, the medium was replaced with one of the same composition lacking newborn calf serum. A Matrigel overlay (0.3 mg/ml) and insulin (50 nm) were added at this time. At 24 h of incubation, the cells were washed in serum-free Medium 199 lacking insulin, and the incubation was continued in serum-free Medium 199. At 48 h of incubation, the medium was replaced with one of the same composition containing the treatments indicated in the figure legends. Hepatocyte cultures were maintained in a humidified chamber at 37 °C in 5% CO2 and 95% air. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes Health and was approved by the Institutional Animal Care and Use Committee of West Virginia University (protocol approval no. 15-0904). Chenodeoxycholic acid, dibutyryl cAMP, and Akti-1/2 (1,3-dihydro-1-(1-((4-(6-phenyl-1H-imidazo[4,5-g]quinoxalin-7-yl)phenyl)methyl)-4-piperidinyl)-2H-benzimidazol-2-one) were obtained from Sigma-Aldrich. Rapamycin and LY294002 were purchased from LC Laboratories. H89 and ISRIB were obtained from Cayman. Bovine insulin was a gift from Lilly. N6-benzoyladenosine-3′,5′-cyclic monophosphate (Biolog) and 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (R&D Systems) were obtained from the indicated sources.
Transient Transfection
The construction of reporter plasmids containing fragments of the rat FGF21 promoter/regulatory region linked to the luciferase gene has been described previously (17). Site-directed mutations were introduced into the −1316 to +68 bp FGF21 reporter plasmid using the Agilent QuikChange II XL site-directed mutagenesis kit. Hepatocytes were plated on 35-mm dishes and transfected with 1 μg of the −2940 to +68 FGF21 reporter plasmid or an equimolar amount of another reporter plasmid using Lipofectin reagent (Invitrogen). At 18 h of incubation, the transfection medium was replaced with fresh medium, and a Matrigel overlay (0.3 mg/ml) was added. At 48 h of incubation, the medium was replaced with fresh medium with or without glucagon plus insulin. At 72 h of incubation, cells were harvested, and cell extracts were prepared in 1× cell culture lysis buffer (Promega). Cell extracts were centrifuged at 12,000 × g for 2 min, and the supernatants were assayed for protein concentration and luciferase activity. Luciferase assay reagent was obtained from Promega.
siRNA Knockdown Experiments
Primary hepatocytes were plated on 35-mm dishes and transfected with 30 pmol of siRNA targeting ATF4 (Silencer Select siRNA IDs s135172 and s135173, Ambion) or 30 pmol of negative control #1 siRNA (Ambion) using Lipofectamine RNAiMAX reagent (Invitrogen). At 18 h of incubation, the transfection medium was replaced with Medium 199, and a Matrigel overlay (0.3 mg/ml) was added. The medium was replaced with fresh medium at 48 h and 66 h of incubation. After the medium change at 66 h of incubation, cells were treated with or without CDCA for 2 h or glucagon plus insulin for 12 h. Cells were then harvested, cell extracts were prepared, and total RNA was isolated.
Western Blotting Analysis
Total cell extracts were prepared from hepatocytes as described by Hansmannel et al. (61), except that the lysis buffer contained 25 mm Tris-HCl (pH 7.6), 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, and a mixture of protease inhibitors and phosphatase inhibitors (Halt, Thermo Scientific). Equal amounts of denatured protein were subjected to electrophoresis in SDS-polyacrylamide gels and then transferred to polyvinylidene difluoride membranes (Immobilon-FL, Millipore) using an electroblotting apparatus (Bio-Rad). The blots were blocked in TBST (10 mm Tris-HCl (pH 8.0), 150 mm NaCl, and 0.1% Tween) containing 5% nonfat dry milk for 1 h at room temperature and then incubated with primary antibody diluted 1:1000 in TBST containing 5% bovine serum albumin. After incubation with primary antibody for 12 h at 4 °C, the blots were washed in TBST. Next, the blots were incubated with secondary antibody conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories) diluted 1:5000 in TBST for 1 h at room temperature. After washing with TBST, antibody-protein complexes on blots were detected using enhanced chemiluminescence (Amersham Biosciences). Fluorescence on the blots was visualized using a Typhoon 9410 imager, and signals were quantified using ImageQuant software. Antibodies against ATF4, phosphorylated Akt (Ser473), phosphorylated eIF2α (Ser51), phosphorylated ribosomal protein S6 (Ser235/236), total Akt, total eIF2α, total ribosomal protein S6, and β-tubulin were obtained from Cell Signaling Technology.
Isolation of RNA and Quantitation of mRNA Levels
Total RNA was extracted from cell cultures by the guanidinium thiocyanate/phenol/chloroform method (62). The abundance of mRNA encoding FGF21, ATF4, sterol regulatory element-binding protein 1c (SREBP-1c), and phosphoenolpyruvate carboxykinase (PEPCK) was measured by quantitative real-time PCR analysis using the Qiagen Quantitect SYBR Green RT-PCR system. Samples of DNase I-treated RNA (100 ng) were analyzed in triplicate according to the instructions of the manufacturer. PCR was performed in 96-well plates using a Bio-Rad iCycler iQ. The relative amount of mRNA was calculated using the comparative Ct method. Rat cyclophilin and glucuronidase β were used as reference genes. Amplification of specific transcripts was confirmed by analyzing the melting curve profile performed at the end of each run and by determining the size of the PCR products using agarose electrophoresis and ethidium bromide staining. The sequences of the primer sets are available upon request.
Gel Mobility Shift Analysis
Nuclear extracts were prepared from primary rat hepatocytes as described previously (63). Double-stranded oligonucleotides were labeled by filling in overhanging 5′ ends using the Klenow fragment of Escherichia coli DNA polymerase in the presence of [α-32P]dGTP. The binding reactions were carried out in 30 μl containing 15 mm HEPES (pH 7.9), 150 mm NaCl, 0.5 mm MgCl2, 0.35 mm EDTA, 0.35 mm dithiothreitol, 15% glycerol (v/v), 0.2 mg/ml bovine serum albumin, 0.25% Triton X-100 (v/v), and 0.5 μg poly(dI-dC). The reactions contained 50,000 cpm of labeled DNA and 20 μg of nuclear extract. The reactions were incubated on ice for 60 min. DNA and DNA-protein complexes were resolved on 5% nondenaturing polyacrylamide gels at 4 °C in 0.5 m Tris (pH 8.8) and 4 m glycine. Following electrophoresis, the gels were dried and subjected to storage phosphor autoradiography. For gel supershift experiments, nuclear extracts were incubated with antibodies for 1 h at 0 °C prior to addition of the oligonucleotide probe.
Statistical Methods
Data were subjected to analysis of variance, and statistical comparisons were made with the Student's t test.
Author Contributions
F. B. H. and K. M. A. designed the experiments and analyzed the data. K. M. A. performed the experiments. G. P. M. provided intellectual and technical assistance for the siRNA knockdown experiments. F. B. H. and K. M. A. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported by American Heart Association Grant-in-aid 17GRNT33210002 (to F. B. H.), American Diabetes Association Basic Science Award 1-12-BS-75 (to F. B. H.), and a West Virginia University Foundation distinguished doctoral scholarship (to K. M. A.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Fig. 1.
- PPARα
- peroxisome proliferator-activated receptor α
- PPRE
- peroxisome proliferator-activated receptor α response element
- FXR
- farnesoid X receptor
- FXRE
- farnesoid X receptor response element
- CDCA
- chenodeoxycholic acid
- AARE
- amino acid response element
- ChoRE
- carbohydrate response element
- mTORC
- mammalian target of rapamycin complex
- EPAC
- exchange protein directly activated by cAMP
- cpTOME
- 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate
- 6-Bnz-cAMP
- N6-benzoyladenosine-3′,5′-cyclic monophosphate; phosphoenolpyruvate carboxykinase
- ER
- endoplasmic reticulum
- ISRIB
- internal stress response inhibitor B
- ATF4
- activating transcription factor
- IRS
- insulin receptor substrate.
References
- 1. Potthoff M. J., Kliewer S. A., and Mangelsdorf D. J. (2012) Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 26, 312–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kharitonenkov A., and DiMarchi R. (2015) FGF21 revolutions: recent advances illuminating FGF21 biology and medicinal properties. Trends Endocrinol. Metab. 26, 608–617 [DOI] [PubMed] [Google Scholar]
- 3. Fisher F. M., and Maratos-Flier E. (2016) Understanding the physiology of FGF21. Annu. Rev. Physiol. 78, 223–241 [DOI] [PubMed] [Google Scholar]
- 4. De Sousa-Coelho A. L., Relat J., Hondares E., Pérez-Martí A., Ribas F., Villarroya F., Marrero P. F., and Haro D. (2013) FGF21 mediates the lipid metabolism response to amino acid starvation. J. Lipid Res. 54, 1786–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laeger T., Henagan T. M., Albarado D. C., Redman L. M., Bray G. A., Noland R. C., Münzberg H., Hutson S. M., Gettys T. W., Schwartz M. W., and Morrison C. D. (2014) FGF21 is an endocrine signal of protein restriction. J. Clin. Invest. 124, 3913–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Markan K. R., Naber M. C., Ameka M. K., Anderegg M. D., Mangelsdorf D. J., Kliewer S. A., Mohammadi M., and Potthoff M. J. (2014) Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 63, 4057–4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Badman M. K., Pissios P., Kennedy A. R., Koukos G., Flier J. S., and Maratos-Flier E. (2007) Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 [DOI] [PubMed] [Google Scholar]
- 8. Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V., Elmquist J. K., Gerard R. D., Burgess S. C., Hammer R. E., Mangelsdorf D. J., and Kliewer S. A. (2007) Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 [DOI] [PubMed] [Google Scholar]
- 9. Dushay J., Chui P. C., Gopalakrishnan G. S., Varela-Rey M., Crawley M., Fisher F. M., Badman M. K., Martinez-Chantar M. L., and Maratos-Flier E. (2010) Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 139, 456–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang X., Yeung D. C., Karpisek M., Stejskal D., Zhou Z. G., Liu F., Wong R. L., Chow W. S., Tso A. W., Lam K. S., and Xu A. (2008) Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 57, 1246–1253 [DOI] [PubMed] [Google Scholar]
- 11. Owen B. M., Mangelsdorf D. J., and Kliewer S. A. (2015) Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 26, 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lundåsen T., Hunt M. C., Nilsson L. M., Sanyal S., Angelin B., Alexson S. E., and Rudling M. (2007) PPARα is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 360, 437–440 [DOI] [PubMed] [Google Scholar]
- 13. Deleted in proof
- 14. Cyphert H. A., Alonge K. M., Ippagunta S. M., and Hillgartner F. B. (2014) Glucagon stimulates hepatic FGF21 secretion through a PKA- and EPAC-dependent posttranscriptional mechanism. PLoS ONE 9, e94996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dong X. C., Copps K. D., Guo S., Li Y., Kollipara R., DePinho R. A., and White M. F. (2008) Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 8, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haeusler R. A., Han S., and Accili D. (2010) Hepatic FoxO1 ablation exacerbates lipid abnormalities during hyperglycemia. J. Biol. Chem. 285, 26861–26868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cyphert H. A., Ge X., Kohan A. B., Salati L. M., Zhang Y., and Hillgartner F. B. (2012) Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J. Biol. Chem. 287, 25123–25138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Sousa-Coelho A. L., Marrero P. F., and Haro D. (2012) Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem. J. 443, 165–171 [DOI] [PubMed] [Google Scholar]
- 19. Taniguchi C. M., Emanuelli B., and Kahn C. R. (2006) Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96 [DOI] [PubMed] [Google Scholar]
- 20. Zhao A. Z., Shinohara M. M., Huang D., Shimizu M., Eldar-Finkelman H., Krebs E. G., Beavo J. A., and Bornfeldt K. E. (2000) Leptin induces insulin-like signaling that antagonizes cAMP elevation by glucagon in hepatocytes. J. Biol. Chem. 275, 11348–11354 [DOI] [PubMed] [Google Scholar]
- 21. Gross D. N., Wan M., and Birnbaum M. J. (2009) The role of FOXO in the regulation of metabolism. Curr. Diab. Rep. 9, 208–214 [DOI] [PubMed] [Google Scholar]
- 22. Li S., Brown M. S., and Goldstein J. L. (2010) Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 3441–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yecies J. L., Zhang H. H., Menon S., Liu S., Yecies D., Lipovsky A. I., Gorgun C., Kwiatkowski D. J., Hotamisligil G. S., Lee C. H., and Manning B. D. (2011) Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 14, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Habegger K. M., Heppner K. M., Geary N., Bartness T. J., DiMarchi R., and Tschöp M. H. (2010) The metabolic actions of glucagon revisited. Nat. Rev. Endocrinol. 6, 689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Almahariq M., Mei F. C., and Cheng X. (2014) Cyclic AMP sensor EPAC proteins and energy homeostasis. Trends Endocrinol. Metab. 25, 60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beebe S. J., Blackmore P. F., Chrisman T. D., and Corbin J. D. (1988) Use of synergistic pairs of site-selective cAMP analogs in intact cells. Methods Enzymol. 159, 118–139 [DOI] [PubMed] [Google Scholar]
- 27. Enserink J. M., Christensen A. E., de Rooij J., van Triest M., Schwede F., Genieser H. G., Døskeland S. O., Blank J. L., and Bos J. L. (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 4, 901–906 [DOI] [PubMed] [Google Scholar]
- 28. Kelly P., Bailey C. L., Fueger P. T., Newgard C. B., Casey P. J., and Kimple M. E. (2010) Rap1 promotes multiple pancreatic islet cell functions and signals through mammalian target of rapamycin complex 1 to enhance proliferation. J. Biol. Chem. 285, 15777–15785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hidaka H., Watanabe M., and Kobayashi R. (1991) Properties and use of H-series compounds as protein kinase inhibitors. Methods Enzymol. 201, 328–339 [DOI] [PubMed] [Google Scholar]
- 30. Pilkis S. J., and Granner D. K. (1992) Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu. Rev. Physiol. 54, 885–909 [DOI] [PubMed] [Google Scholar]
- 31. Bochkis I. M., Rubins N. E., White P., Furth E. E., Friedman J. R., and Kaestner K. H. (2008) Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat. Med. 14, 828–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dent P., Fang Y., Gupta S., Studer E., Mitchell C., Spiegel S., and Hylemon P. B. (2005) Conjugated bile acids promote ERK1/2 and AKT activation via a pertussis toxin-sensitive mechanism in murine and human hepatocytes. Hepatology 42, 1291–1299 [DOI] [PubMed] [Google Scholar]
- 33. Cai K., and Sewer M. B. (2013) Diacylglycerol kinase theta couples farnesoid X receptor-dependent bile acid signalling to Akt activation and glucose homoeostasis in hepatocytes. Biochem. J. 454, 267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wek R. C., Jiang H. Y., and Anthony T. G. (2006) Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34, 7–11 [DOI] [PubMed] [Google Scholar]
- 35. Vattem K. M., and Wek R. C. (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 101, 11269–11274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sidrauski C., McGeachy A. M., Ingolia N. T., and Walter P. (2015) The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. eLife 4, e05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cornu M., Oppliger W., Albert V., Robitaille A. M., Trapani F., Quagliata L., Fuhrer T., Sauer U., Terracciano L., and Hall M. N. (2014) Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc. Natl. Acad. Sci. U.S.A. 111, 11592–11599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Izumiya Y., Bina H. A., Ouchi N., Akasaki Y., Kharitonenkov A., and Walsh K. (2008) FGF21 is an Akt-regulated myokine. FEBS Lett. 582, 3805–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Puigserver P., Rhee J., Donovan J., Walkey C. J., Yoon J. C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., and Spiegelman B. M. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423, 550–555 [DOI] [PubMed] [Google Scholar]
- 40. Liao J., Barthel A., Nakatani K., and Roth R. A. (1998) Activation of protein kinase B/Akt is sufficient to repress the glucocorticoid and cAMP induction of phosphoenolpyruvate carboxykinase gene. J. Biol. Chem. 273, 27320–27324 [DOI] [PubMed] [Google Scholar]
- 41. Foretz M., Pacot C., Dugail I., Lemarchand P., Guichard C., Le Lièpvre X., Berthelier-Lubrano C., Spiegelman B., Kim J. B., Ferré P., and Foufelle F. (1999) ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol. Cell Biol. 19, 3760–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Owen J. L., Zhang Y., Bae S.-H., Farooqi M. S., Liang G., Hammer R. E., Goldstein J. L., and Brown M. S. (2012) Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. U.S.A. 109, 16184–16189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kimura M., and Ogihara M. (1997) Proliferation of adult rat hepatocytes in primary culture induced by insulin is potentiated by cAMP-elevating agents. Eur. J. Pharmacol. 327, 87–95 [DOI] [PubMed] [Google Scholar]
- 44. Bucher M. L., and Swaffield M. N. (1975) Regulation of hepatic regeneration in rats by synergistic action of insulin and glucagon. Proc. Natl. Acad. Sci. U.S.A. 72, 1157–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arisawa S., Ishida K., Kameyama N., Ueyama J., Hattori A., Tatsumi Y., Hayashi H., Yano M., Hayashi K., Katano Y., Goto H., Takagi K., and Wakusawa S. (2009) Ursodeoxycholic acid induces glutathione synthesis through activation of PI3K/Akt pathway in HepG2 cells. Biochem. Pharmacol. 77, 858–866 [DOI] [PubMed] [Google Scholar]
- 46. Furusawa Y., Uruno A., Yagishita Y., Higashi C., and Yamamoto M. (2014) Nrf2 induces fibroblast growth factor 21 in diabetic mice. Genes Cells 19, 864–878 [DOI] [PubMed] [Google Scholar]
- 47. Li T., Francl J. M., Boehme S., Ochoa A., Zhang Y., Klaassen C. D., Erickson S. K., and Chiang J. Y. (2012) Glucose and insulin induction of bile acid synthesis: mechanisms and implication in diabetes and obesity. J. Biol. Chem. 287, 1861–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reaven G. M. (2005) The insulin resistance syndrome: definition and dietary approaches to treatment. Annu. Rev. Nutr. 25, 391–406 [DOI] [PubMed] [Google Scholar]
- 49. Haeusler R. A., Astiarraga B., Camastra S., Accili D., and Ferrannini E. (2013) Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes 62, 4184–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jiang S., Yan C., Fang Q. C., Shao M. L., Zhang Y. L., Liu Y., Deng Y. P., Shan B., Liu J. Q., Li H. T., Yang L., Zhou J., Dai Z., Liu Y., and Jia W. P. (2014) Fibroblast growth factor 21 is regulated by the IRE1α-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J. Biol. Chem. 289, 29751–29765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Khamzina L., Veilleux A., Bergeron S., and Marette A. (2005) Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 146, 1473–1481 [DOI] [PubMed] [Google Scholar]
- 52. Ozcan U., Cao Q., Yilmaz E., Lee A.-H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., and Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 53. Yang H., and Yang L. (2016) Targeting cAMP/PKA pathway for glycemic control and type 2 diabetes therapy. J. Mol. Endocrinol. 57, R93-R108 [DOI] [PubMed] [Google Scholar]
- 54. Cook J. R., Langlet F., Kido Y., and Accili D. (2015) Pathogenesis of selective insulin resistance in isolated hepatocytes. J. Biol. Chem. 290, 13972–13980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anand P., and Gruppuso P. A. (2005) The regulation of hepatic protein synthesis during fasting in the rat. J. Biol. Chem. 280, 16427–16436 [DOI] [PubMed] [Google Scholar]
- 56. Sokolović M., Sokolović A., Wehkamp D., Ver Loren van Themaat E., de Waart D. R., Gilhuijs-Pederson L. A., Nikolsky Y., van Kampen A. H., Hakvoort T. B., and Lamers W. H. (2008) The transcriptomic signature of fasting murine liver. BMC Genomics 9, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tsai B. P., Jimenez J., Lim S., Fitzgerald K. D., Zhang M., Chuah C. T., Axelrod H., Wilson L., Ong S. T., Semler B. L., and Waterman M. L. (2014) A novel Bcr-Abl-mTOR-eIF4A axis regulates IRES-mediated translation of LEF-1. Open Biol. 4, 140180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chan C. P., Kok K. H., Tang H. M., Wong C. M., and Jin D. Y. (2013) Internal ribosome entry site-mediated translational regulation of ATF4 splice variant in mammalian unfolded protein response. Biochim. Biophys. Acta 1833, 2165–2175 [DOI] [PubMed] [Google Scholar]
- 59. Laeger T., Albarado D. C., Burke S. J., Trosclair L., Hedgepeth J. W., Berthoud H.-R., Gettys T. W., Collier J. J., Münzberg H., and Morrison C. D. (2016) Metabolic responses to dietary protein restriction require an increase in FGF21 that is delayed by the absence of GCN2. Cell Rep. 16, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stabile L. P., Klautky S. A., Minor S. M., and Salati L. M. (1998) Polyunsaturated fatty acids inhibit the expression of the glucose-6-phosphate dehydrogenase gene in primary rat hepatocytes by a nuclear posttranscriptional mechanism. J. Lipid Res. 39, 1951–1963 [PubMed] [Google Scholar]
- 61. Hansmannel F., Mordier S., and Iynedjian P. B. (2006) Insulin induction of glucokinase and fatty acid synthase in hepatocytes: analysis of the roles of sterol-regulatory-element-binding protein-1c and liver X receptor. Biochem. J. 399, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chomczynski P., and Sacchi N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159 [DOI] [PubMed] [Google Scholar]
- 63. Carey M. F., Peterson C. L., and Smale S. T. (2009) Dignam and Roeder nuclear extract preparation. Cold Spring Harb. Protoc. 2009, pdb.prot5330 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.