

Abstract
Peptide antigen presentation by major histocompatibility complex (MHC) class I proteins initiates CD8+ T cell-mediated immunity against pathogens and cancers. MHC I molecules typically bind peptides with 9 amino acids in length with both ends tucked inside the major A and F binding pockets. It has been known for a while that longer peptides can also bind by either bulging out of the groove in the middle of the peptide or by binding in a zigzag fashion inside the groove. In a recent study, we identified an alternative binding conformation of naturally occurring peptides from Toxoplasma gondii bound by HLA-A*02:01. These peptides were extended at the C terminus (PΩ) and contained charged amino acids not more than 3 residues after the anchor amino acid at PΩ, which enabled them to open the F pocket and expose their C-terminal extension into the solvent. Here, we show that the mechanism of F pocket opening is dictated by the charge of the first charged amino acid found within the extension. Although positively charged amino acids result in the Tyr-84 swing, amino acids that are negatively charged induce a not previously described Lys-146 lift. Furthermore, we demonstrate that the peptides with alternative binding modes have properties that fit very poorly to the conventional MHC class I pathway and suggest they are presented via alternative means, potentially including cross-presentation via the MHC class II pathway.
Keywords: antigen presentation, major histocompatibility complex (MHC), natural killer cells (NK cells), peptide interaction, protein crystallization, protein structure, T-cell receptor (TCR), Toxoplasma gondii
Introduction
Peptide presentation by MHC class I molecules regulates which fragments of a pathogen or cancer antigen are displayed to cytotoxic T cells for immune recognition. Understanding the mechanism of antigen presentation by MHC I is crucial in an attempt to design therapeutic strategies aimed at modulating subsequent immune responses to control disease.
Toxoplasmosis is a parasitic disease caused by infection with the large intracellular protozoan Toxoplasma gondii (1, 2). Although generally asymptomatic in healthy adults, T. gondii infection can cause congenital toxoplasmosis during pregnancy and result in abortion or neonatal disease (1, 2). T cell-mediated immunity against T. gondii-derived peptide antigens provides strong protection against T. gondii and involves both peptide presentation by major histocompatibility complex class I (MHC I) and class II (MHC II) proteins (3–5). Although T. gondii can interfere with CD4 T cell responses by down-regulating MHC II expression in IFN-γ-activated macrophages, immunization with T. gondii MHC II peptide ligands can elicit a potent CD4 T cell response that can lower parasite burden in the brain (6, 7). Immunocompromised individuals and patients with T cell deficiencies are highly susceptible to T. gondii infections (8, 9).
CD8+ T cell responses have been studies more widely than CD4+, and peptide ligands for MHC I have been identified to be derived from surface proteins or proteins of specialized secretory organelles (rhoptry proteins) that can be secreted into either the parasite cytosol or the parasitophorous vacuole (8–14).
HLA-A*02:01 has been the focus of studies aimed at identifying MHC I-restricted peptide ligands that confer protection against T. gondii in HLA transgenic mice (15) and as such is a suitable MHC class I allele to study the basic rules of peptide presentation. Generally, most canonical peptide ligands for MHC I are 9–10 amino acids in length. However, peptide ligands with more than 11 amino acids have been identified as ligands for MHC I in general and form the non-canonical ligand group (13, 16, 17). These long peptides have been shown to interact with the residues of the binding groove of HLA class I heavy (α) chain much like the canonical binders with some changes. The second (P2) and C-terminal (PΩ) residues of the antigen peptide anchor into the A and F pockets of the binding groove, respectively, whereas the middle portion of these oversized peptides either “bulge out” or “zigzag” in the binding groove to be accommodated (18, 19).
In contrast to these “bulged” peptides, we recently identified longer T. gondii peptides eluted from HLA-A*02:01 molecules that had a conserved N-terminal start but differed in their residue composition at the C terminus (20). We showed through crystallographic studies that in the HLA-A*02:01 complex with one 12-mer peptide residue Tyr-84 of the MHC heavy chain swung out and opened the F pocket, allowing the C-terminal amino acid of the peptide to protrude into the solvent, whereas the nested 11-mer N-terminal core peptide bound in a conventional zigzag orientation tucked with both peptide ends inside the peptide binding groove.
To further investigate whether the opening of the binding groove could be achieved with other peptides presented on T. gondii-infected cells and to understand what the structural requirements are to enable such unconventional modes of binding, we crystallized complexes of HLA-A*02:01 with several pairs of core (nested) and C-terminally extended peptides. Surprisingly, we found that that there are at least two distinct modes of opening the F pocket of HLA-A*02:01 involving the residues Tyr-84 and Lys-146. We suggest that these unconventional modes of binding will help better understand targets of MHC class I-restricted epitope recognition.
Results
Crystal Structures of HLA-A*02:01 in Complex with Conventional and Extended Peptides
To identify additional peptides with likely unconventional binding motifs, we scanned the set of peptides eluted from HLA-A*02:01 for those that had poor predicted binding affinity of the full-length peptide (percentile rank >10%) but contained a nested N-terminal peptide with high predicted affinity (percentile rank <2%). In our previous study, we had examined one such peptide (FVLELEPEWTVK), which had a single lysine added to the C terminus of the core peptide (FVLELEPEWTV) and induced a structural change in Tyr-84 of HLA-A*02:01 (20). In contrast, in the current study, we examined three sets of peptides that had C-terminal amino acid additions that contained negatively charged amino acids or both negatively and positively charged residues (Fig. 1). To investigate whether the F pocket of HLA-A*02:01 could also be opened by these extending peptides, we refolded HLA-A*02:01 with several nested and extending peptides and determined the crystal structures of these complexes. We obtained crystal structures for all complexes at resolutions between 1.85 and 2.75 Å (Table 1). Electron densities for all the peptides were well defined over the entire peptide length that is bound within the binding groove, whereas C-terminally extending residues that did not contact HLA-A*02:01 were disordered (Fig. 1). When all the different peptides are compared, slight structural changes in HLA-A*02:01 are observed in the A pocket. Peptides with an N-terminal tyrosine (YLSPIASPL, YLSPIASPLL, and YLSPIASPLLDGKSLR) open the A pocket slightly for the bulky side chain to be accommodated, whereas peptides that begin with glycine (GLKEGIPAL, GLKEGIPALDN, GLLPELPAV, and GLLPELPAVGGNE) are more buried inside the A pocket because they lack any side chain (Fig. 1). In addition, subtle structural changes are observed throughout the binding groove to allow optimal binding of the different amino acid side chains. However, when structures of the core peptides are compared with their respective extended peptides, the position of Tyr-84 of HLA-A*02:01 was unchanged. Surprisingly, however, Lys-146 of the F pocket, which is located close to Tyr-84 and forms a “lid” to bury the PΩ amino acid in the core peptides, moved upward to open the F pocket when the extending peptides were bound (Fig. 1). Although Lys-146 adopts slightly different positions when all the extending peptide structures are compared, in each structure the Lys-146 lid was opened for the C-terminal extensions to protrude from the F pocket.
FIGURE 1.

HLA-A*02:01·peptide structures. a, overall structure of HLA-A*02:01 with heavy chain in gray and light chain (β2m) in blue. Peptide backbones are superimposed within the binding groove. b–d, molecular surface representation of the binding groove of HLA-A*02:01 with the individual peptides bound. Peptide sequences are labeled, and charged amino acids are colored in blue (positive) and red (negative). 2FoFc electron density for the peptide (shown in blue mesh) is contoured at 1σ.
TABLE 1.
Data collection and refinement statistics
r.m.s.d., root mean square deviation.
| HLA-A*02:01·peptide (Protein Data Bank code) |
|||||||
|---|---|---|---|---|---|---|---|
| G9L (5ENW) | G11N (5F7D) | G9V (5FA3) | G13E (5EOT) | Y9L (5F9J) | Y10L (5FDW) | Y16R (5FA4) | |
| Data collection | |||||||
| Resolution range (Å)a | 50.0–1.85 (1.89–1.85) | 50.0–2.30 (2.38–2.3) | 51.6–1.86 (1.89–1.85) | 40–2.10 (2.18–2.10) | 50.0–2.5 (2.59–2.5) | 50–2.7 2.8–2.7 | 50.0–2.4 (2.44–2.4) |
| Completeness (%)a | 93.2 (96.1) | 96.6 (81.1) | 98.8 (97.5) | 99.6 (97.9) | 100 (100) | 93.4 (95.4) | 97.9 (85.2) |
| Number of unique reflections | 35,425 | 19,954 | 37,513 | 26,570 | 15,320 | 11,732 | 17,776 |
| Redundancy | 2.7 | 3.4 | 3.7 | 3.6 | 3.7 | 2.8 | 3.4 |
| Rsym (%) | 8.6 (53.1) | 7.1 (33.5) | 7.8 (31.1) | 12.8 (57.0) | 16.1 (71.1) | 19.0 (66.4) | 13.5 (66.3) |
| Rpim (%) | 6.1 (38.5) | 4.5 (21.7) | 4.7 (18.8) | 7.8 (37.7) | 9.7 (42.9) | 13.1 (45.4) | 8.4 (42.6) |
| I/σa | 21.5 (3.1) | 21.2 (3.3) | 20.3 (4.6) | 13.3 (2.1) | 7.9 (2.1) | 8.3 (2.4) | 11.3 (1.7) |
| Refinement statistics | |||||||
| Number of reflections (F > 0) | 33,590 | 18,963 | 35,290 | 25,169 | 14,464 | 11,305 | 16,851 |
| Maximum resolution (Å) | 1.85 | 2.3 | 1.86 | 2.1 | 2.51 | 2.7 | 2.4 |
| Rcryst (%) | 20.8 (25.1) | 20.9 (36.9) | 20.9 (23.9) | 20.9 (30.8) | 19.9 (26.3) | 21.3 (24.3) | 20.4 (35.5) |
| Rfree (%) | 24.4 (29.4) | 25.7 (34.1) | 23.4 (29.4) | 23.7 (31.3) | 27.5 (33.3) | 28.5 (32.9) | 25.8 (36.3) |
| Number of atoms | 3,371 | 3,151 | 3,397 | 3,227 | 3,170 | 3,142 | 3,210 |
| Protein | 3,047 | 3,011 | 3,060 | 3,015 | 3,012 | 2,996 | 3,032 |
| Peptide | 62 | 70 | 59 | 66 | 67 | 75 | 77 |
| Glycerol | 3 | 2 | 3 | 0 | 3 | 1 | 2 |
| Solvent molecules (waters) | 241 | 57 | 280 | 143 | 71 | 62 | 83 |
| Ramachandran statistics | |||||||
| Favored | 98.7 | 97.6 | 98.7 | 98.9 | 97.6 | 96.0 | 97.9 |
| Outliers | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| r.m.s.d. from ideal geometry | |||||||
| Bond length (Å) | 0.0064 | 0.0075 | 0.0061 | 0.0076 | 0.01 | 0.01 | 0.009 |
| Bond angles (°) | 1.12 | 1.22 | 1.11 | 1.22 | 1.45 | 1.46 | 1.33 |
| Average B values (Å2) | |||||||
| Protein | 29.6 | 48.5 | 15.1 | 27.5 | 26.3 | 20.8 | 36.9 |
| Peptide | 17.8 | 48.2 | 13.0 | 26.2 | 25.8 | 26.3 | 28.8 |
| Water molecules | 29.4 | 41.7 | 24.8 | 26.7 | 21.0 | 15.8 | 35.2 |
a Numbers in parentheses refer to highest resolution shell.
Hydrogen Bond Network for Nested and Longer Peptide Pairs
Next, we looked at the detailed interactions between HLA-A*02:01 and the individual peptides. In the case of peptide GLKEGIPAL, an extensive hydrogen bond network is seen involving the PΩ leucine residue and residues of the heavy chain that line the binding groove including Asp-76, Thr-80, Tyr-84, Thr-143, Lys-146, and Trp-147 (Fig. 2a, upper panel). In contrast, for the extended peptide GLKEGIPALDN, the hydrogen bond between Lys-146 and terminal carboxyl group of PΩ leucine is replaced with one between Lys-146 and the PΩ+1 aspartate side chain because Lys-146 adopts a different orientation (Fig. 2a, lower panel). In the case of the peptide pair GLLPELPAV and GLLPELPAVGGNE, a similar hydrogen bond network is observed for both peptides with only a minor difference in the crystal structure of the longer peptide. The hydrogen bond interaction between the terminal carboxylate of PΩ valine and Lys-146 is missing in the crystal structure with the longer peptide (Fig. 2b). The same is true for the peptide pair YLSPIASPL and YLSPIASPLLDGKSLR (Fig. 2c). As a result, the change in the orientation of Lys-146 leads to the loss of hydrogen bond formation with the carboxylate of the PΩ amino acid. However, the hydrogen bond interaction between HLA-A*02:01 residue Trp-147 and the backbone oxygen of the P8 amino acid remains conserved. Depending on the amino acid following residue P8, a novel hydrogen bond can be formed with the side chain of a compatible amino acid at PΩ+1 (here Asp-10). Because the C-terminally extending amino acids project away from the peptide binding groove, electron density becomes increasingly disordered as the peptide exits the F pocket (Fig. 1).
FIGURE 2.
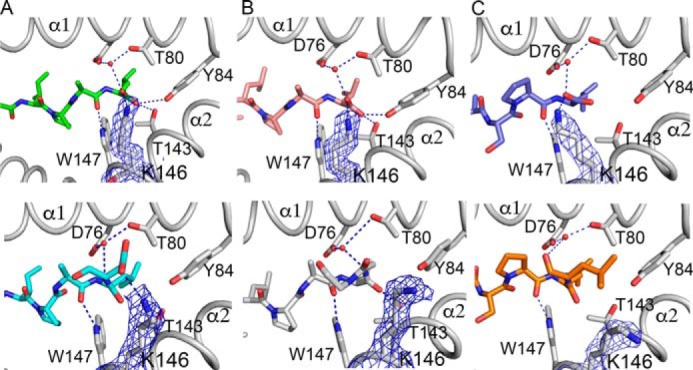
Detailed hydrogen bond interactions around the F pocket. HLA-A*02:01 is shown in gray, peptides are shown in color, and electron density for Lys-146 is shown as a blue mesh contoured at 1σ. Hydrogen bonds between 2.5 and 3.65 Å are shown as blue dashed lines. Structures of nested peptides are shown in the top panel, and the corresponding extending peptides are shown below. a, peptide pair GLKEGIPAL (green) and GLKEGIPALD (cyan). b, GLLPELPAV (pink) and GLLPELPAVGGNE (gray). c, peptide pair YLSPIASPL (violet) and YLSPIASPLLDGKSLR (orange).
Extending Peptides Do Not Significantly Destabilize HLA-A*02:01
To determine the relative stability of the individual HLA-A*02:01·peptide complexes, we followed their thermal denaturation by differential scanning fluorimetry. The melting temperatures (Tm) obtained from the melt curves allowed us to compare the stability of the different complexes (Fig. 3). We observed that complexes of HLA-A*02:01 with extended peptides had similar stability to those with their equivalent nested peptides with not more than 8 °C difference between them. For example, the Tm for HLA-A*02:01 complex with GLKEGIPAL is 63 °C, whereas that with its longer peptide counterpart is 61 °C. Addition of 6 extra residues to peptide YLSPIASPLL also only changes the Tm of the complex with peptide YLSPIASPLLDGKSLR by about 7 °C (Fig. 3 and Ref. 20). Interestingly, some of the complexes with nested peptides are as stable as those with longer peptides (compare GLKEGIPAL with GLLPEPPVGGNE; Fig. 3). It is also worth noting that there are some variations in the stability of complexes with different nested peptides. For instance, peptide GLKEGIPAL forms a less stable complex as compared with peptide GLLPELPAV (Fig. 3). Although most interactions between HLA-A*02:01 and the peptides are conserved, there is a significant difference in the hydrogen bond interaction between Tyr-84 and their terminal carboxylate (3.65 Å for G9L and 2.9 Å for G9V; Fig. 2). The lack of an intimate hydrogen bond interaction of the terminal amino acid with Tyr-84 in G9L peptide is likely a major contributor to the reduced melting temperature. Compared with our previous study, we noticed that a single positively charged amino acid addition (compare FVLELEPEWTV and FVLELEPEWTVK; Ref. 20) destabilizes the protein·peptide complex more than extending peptides that follow a short (2-amino acid) negatively charged residue addition (compare GLKEGIPAL with GLKEGIPALDN; Fig. 3). However, longer peptide additions (4–6 amino acids) that contain a negatively charged amino acid (YLSPIASPLL versus YLSPIASPLLDGKSLR and GLLPELPAV versus GLLPELPAVGGNE) reduce the protein·peptide complex to the same extend as the single “Lys” addition found in peptide FVLELEPEWTVK (20). This highlights that the structural change involving Tyr-84 of HLA-A*02:01 is more destabilizing than that of Lys-146 when the negatively charged peptide extension is very short.
FIGURE 3.
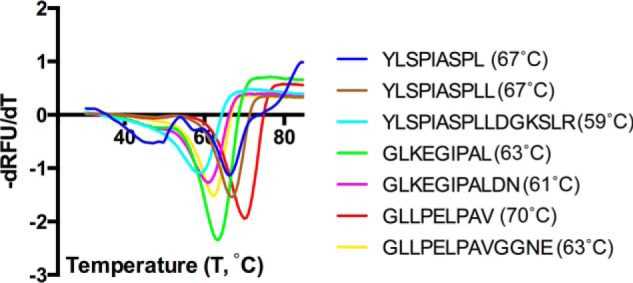
Thermal denaturation assay. First derivative of the melt curve for an individual HLA-A*02:01·peptide complex. Calculated melting temperatures for each complex are provided in parentheses. RFU, relative fluorescence units.
Lysine 146 Lift
The different orientations of Lys-146 upon binding of the longer peptides open the F pocket of HLA-A*02:01 and are required for the C-terminally extending amino acids to project into the solvent because Lys-146 forms a partial lid above the F pocket held in position by the hydrogen bond interaction of the carboxylate of the C-terminal amino acid (PΩ) of any nested peptide (Figs. 2 and 4). Although the position of Lys-146 is not precisely conserved between the different structures of HLA-A*02:01 bound to the extending peptide, the lift of the residue to accommodate the peptide extension seems to be consistent. In the case of the crystal structure of HLA-A*02:01 with YLSPIASPL, YLSPIASPLL, and YLSPIASPLLDGKSLR, there are variations in the way that the two nested peptides are accommodated in the binding groove. With YLSPIASPL, the binding of the residues is quite conventional with the P2 and PΩ anchor residues binding to the A and F pockets. Surprisingly, however, YLSPIASPLL, the nested peptide with one extra leucine residue at the C-terminal end of the peptide, undergoes a certain extent of bulging to accommodate the terminal leucine residue (PΩ+1 instead of PΩ) as the anchor residue in the F pocket (Fig. 4d). The difference in the binding of these two nested peptides to HLA-A*02:01 underscores the requirement for a sequence motif or particular amino acid features within the bound peptide to induce movement of Lys-146 to open the F pocket. Because a mere increase in length of the peptide does not cause the change in orientation of Lys-146, it is likely that the addition of charged residues within the C-terminal extension is the contributing factor to open the F pocket. Thus, in addition to the previously identified “Tyr-84 swing” to accommodate the peptide FVLELEPEWTVK (UFP(16–27)) (20), we observed a “Lys-146 lift” in HLA-A*02:01 as a second mechanism of opening the F pocket induced by the extending peptides YLSPIASPLLDGKSLR, GLKEGIPALDN, and GLLPELPAVGGNE (Fig. 5).
FIGURE 4.

Binding comparison between nested and extending peptides. a, binding groove of HLA-A*02:01 with the nested peptide GLKEGIPAL (green) superimposed with extended peptide GLKEGIPALD (cyan). b, superimposition of nested peptide GLLPELPAV (pink) with extended peptide GLLPELPAVGGNE (gray). c, superimposition of nested peptide YLSPIASPL (violet) with extended peptide YLSPIASPLLDGKSLR (orange). d, superimposition of nested peptide YLSPIASPL (violet) with nested peptide YLSPIASPLL (yellow).
FIGURE 5.

Mechanisms of F pocket opening and peptide sequences. a, “tyrosine swing.” Tyr-84 adopts a different rotamer to open the F pocket and accommodate the longer peptide FVLELEPEWTVK (UFP(16–27)) (15). b, “lysine lift.” Residue Lys-146 is lifted to accommodate longer peptides including GLKEGIPALD, GLLPELPAVGGNE, and YLSPIASPLLDGKSLR. c, list of nested and longer T. gondii peptides. Positively charged residues (blue) open the F pocket via the tyrosine swing, whereas negatively charged residues (red) open the binding groove via the lysine lift. aa, amino acids.
It's a Game of Charge
The negatively charged amino acids in the extended peptides do not always immediately follow the nested conventional PΩ anchor residue but can be several residues downstream (Fig. 5c). The previously reported longer peptide contains a C-terminal addition of a positively charged residue (FVLELEPEWTVK) that opens the binding groove using the Tyr-84 swing mechanism. Here, we observed that all extended peptides with a negatively charged residue only open the binding groove using the Lys-146 lift mechanism. This included the peptide YLSPIASPLLDGKSLR, which contains a lysine (positive charge) residue following the aspartate (negative charge), but no Tyr-84 swing was observed. This suggested that the first charged residue determined which of the two distinct structural modes of binding an extended peptide will induce in HLA-A*02:01.
Extensions with Negative Charges Are Longer and More Frequent than Extensions with Positive Charges
Given the discovery of several ligands binding in an unconventional mode, we aimed to assess the generality of these findings. In the T. gondii peptide elution data set, a total of 134 peptides were predicted to bind with conventional P2-PΩ anchors with predicted rank ≤10%. Of the remaining 150 peptides with poor predicted binding, 108 contained a nested strong binder (rank ≤2%) at the N terminus with 1 or more residues extending beyond PΩ. These 108 peptides are expected to be enriched for examples with a similar unconventional mode of binding as those in our structural studies.
We classified the ligands into having “negative” or “positive” extension based on the first charged residue found after the nested binding peptide. Nearly half of the extended ligands contained a negatively charged residue as the first charged amino acid within the first 3 residues of the extension, whereas positively charged extensions were less frequent (Fig. 6a). Positive extensions were short (less than 3 residues on average), whereas negative extensions had an average length of 8.7 residues (Fig. 6b). Notably, 66% of the positive extensions consisted of a single or 2 residues compared with only 8% of negative extensions being shorter than 3 amino acids. Considering only the longest version of ligands with extensions of multiple sizes, the average length of negative extensions was of 11.0 residues.
FIGURE 6.

Properties of ligands with C-terminal extensions. a, negatively charged extensions (Asp and Glu) in the T. gondii eluted set were more frequent than expected compared with a background distribution of resampled control data sets, whereas positively charged extensions (Arg and Lys) were less frequent. b, positive extensions were on average shorter than 3 residues, and negative extensions had a mean length larger than 8 residues. c, TAP transport scores for extended ligands were significantly lower than for canonical ligands and not significantly different from random. d, proteasome cleavage scores for extended ligands were significantly lower than for canonical ligands and not significantly higher than random. e, the C-terminal composition of T. gondii ligands correlates with the C-terminal enrichment of class II ligands with positively charged residues in the first quadrant and hydrophobic amino acids dominating the third quadrant. IEDB, Immune Epitope Database. Error bars represent S.D.
Extended Peptide MHC I Ligands Show a Putative Processing Motif That Is More Similar to MHC Class II Ligands than Conventional MHC I Ligands
Given the unconventional length and mode of binding of the observed extended peptides and given that T. gondii has an unusual compartmentalized life cycle in the cells it infects, we wanted to examine whether the unconventional ligands found had the typical motifs of peptides derived from the conventional MHC class I processing and presentation pathway. As shown in Fig. 6, this was not the case. Extended ligands had significantly lower scores for TAP2 transport (Fig. 6c) compared with canonical T. gondii ligands (p = 1.2 × 10−5, Wilcoxon rank sum test) but were not significantly different from those of random peptides (p = 0.34). Similarly, proteasome cleavage scores (Fig. 6d) for extended ligands were significantly lower compared with canonical ligands (p = 5 × 10−16) but were not significantly higher than the random natural peptides (p = 0.24). In other words, long ligands with terminal extensions were predicted to be poor substrates both for proteasome cleavage and TAP transport, suggesting an alternative mechanism for the generation and translocation to MHC class I of these extended ligands.
Given the life cycle of T. gondii, it is possible that the unconventional MHC I peptide ligands derived from it are processed and (cross-)presented through the same pathway as MHC II ligands. If that is the case, we would not only expect that these ligands have different amino acid motifs as those found for MHC I ligands (as was shown above) but also that they have a pattern congruent with what is found for MHC II ligands. Accordingly, we examined the amino acid patterns of the C-terminal residues in extended ligands. Remarkably, despite being predominantly negatively charged or uncharged in the first 3 residues of the extension, a large fraction of ligands presented either an arginine or lysine at the very C-terminal residue (42 of 108). If these ligands were cross-presented, we would expect similar trimming motifs in class II ligands, and we examined published data sets of MHC II ligands for evidence of such a motif (see “Experimental Procedures”). Indeed, we found that the C-terminal composition of the extended T. gondii class I ligands strongly correlated to the residue distribution at the C terminus of eluted MHC class II ligands where positively charged amino acids were also enriched (Fig. 6e). Taken together, we found that the unconventional MHC I ligands presented by T. gondii have sequence motifs much more consistent with cross-presentation than with generation through proteasomal cleavage and TAP transport.
Discussion
αβ T cell receptor (TCR) recognition of MHC-presented microbial peptides initiates T cell-mediated immunity against infection. Generally, the TCR binds with both TCR α and β chains in a diagonal orientation above the MHC molecule (28). Although the germ line-encoded complementarity-determining region 1 and 2 loops bind to MHC, the hypervariable loops complementarity-determining regions 3α and 3β specifically bind and recognize the peptide and provide antigen specificity (21). MHC I has a closed binding groove, and peptides bind with both ends tucked inside the binding pocket, whereas MHC II has an open binding pocket, and peptide ligands (typically 15–20 amino acids) bind with both N and C termini hanging over the end of the groove. Because MHC I presents peptides in a more confined space compared with MHC II, the TCR of CD8+ T cells often contacts and discriminates their entire peptide sequence (21). In our present study, we have focused on a panel of C-terminally extended T. gondii peptides that contain a negatively charged amino acid within their C-terminal extensions. Analysis of a large data set of HLA-A*02:01-eluted T. gondii peptides from our previous study (20) demonstrated that C-terminally extending peptides are very common in T. gondii and that additions that contain negatively charged amino acids are more represented than those that contain only positively charged residues.
These peptides contain a canonical HLA-A2*02:01 binding motif at their N termini, but addition of the C-terminal extensions render predictions about their binding to MHC I difficult. Although the N termini of these peptides bind like canonical peptides to the MHC I allele HLA-A*02:01, the C-terminal extensions induce a structural change at the F pocket to allow their extension into the solvent. As such, algorithms aimed at predicting peptide binding to MHC I need to capture rules that allow identification of a canonical N-terminal MHC I binding motif, whereas adding descriptors, such as charged residues in the C-terminal extension, is necessary to predict binding of C-terminally extending peptides. In principle, these rules could be learned directly from peptide binding data using machine learning techniques, such as the recently described extended NNalign method (22, 23). However, this is complicated by the very limited amount of quantitative data characterized by non-canonical binding available. We would envision this situation to change as more binding data become available and as MHC ligand data are included in the training data of MHC class I binding prediction algorithms. We showed that only charged residues that follow the P9 amino acid of a canonical peptide within 3 or fewer amino acids induce the structural change that allows them to bind to MHC I and stabilize the complex. These rules can now be incorporated into existing algorithms to predict MHC I binding peptides at least for HLA-A*02:01.
Because the residues that are involved in the F pocket opening (Tyr-84 and Lys-146) are conserved across all HLA-A, -B, and -C alleles (and found in many non-classical MHC I molecules, such as HLA-E, HLA-G, and Qa-1 as well as viral MHC I mimics such as UL18), we postulate that the ability to open the F pocket is a universal characteristic found across many MHC I molecules. Although it is not clear to what extent this structural change affects CD8+ T cell recognition, killer immunoglobulin receptors (KIRs), a family of activating and inhibitory receptors expressed on natural killer cells, natural killer T cells, and many CD4+ and CD8+ T cells, bind directly above the F pocket. As a consequence, any structural change around the F pocket would likely affect KIR binding and directly modulate host immune responses (24, 25). In particular, the inability of inhibitory KIRs to engage MHC I would lower the threshold of activation for many more immune cells to combat infection or cancers. Future studies will have to address the origin and potential function of these longer peptides in host immunity.
Experimental Procedures
HLA-A*02:01 Expression and Purification
HLA-A*02:01 class I heavy chain ectodomain (residues 21–274) and human β2-microglobulin (β2m) (residues 1–99) were expressed as inclusion bodies and refolded as reported previously (20) with modifications reported here. Briefly, both the heavy chain and light chain were expressed in Escherichia coli BL21 DE3 cells and induced at A600 of 0.6 with 1 mm isopropyl 1-thio-d-galactopyranoside, and cells were harvested after 4 h by centrifugation (5000 × g for 20 min). Cells were resuspended separately in lysis buffer (100 mm Tris-HCl, pH 7.0, 5 mm EDTA, 5 mm DTT, 0.5 mm PMSF), and the cells were broken with four to five passes through a microfluidizer (20 kilopascals) (Microfluidics). Cell lysate was centrifuged (50,000 × g for 30 min at 4 °C) to collect inclusion bodies. Inclusion bodies were further resuspended in wash buffer A (100 mm Tris-HCl, pH 7.0, 5 mm EDTA, 5 mm DTT, 2 m urea, 2% (w/v) Triton X-100), centrifuged again, and washed in wash buffer B (100 mm Tris-HCl, pH 7.0, 5 mm EDTA, 2 mm DTT). Finally, the inclusion bodies were denatured in extraction buffer (50 mm Tris-HCl, pH 7.0, 5 mm EDTA, 2 mm DTT, 6 m guanidine HCl) for subsequent refolding. 3 mg of β2m was added dropwise to 250 ml of refolding buffer (0.1 m Tris-HCl, pH 8.0, 2 mm EDTA, 400 mm l-arginine, 5 mm oxidized glutathione, 5 mm reduced glutathione) and stirred for 1–2 h. Between 11 and 15 mg of HLA-A heavy chain mixed with 2–3 mg of individual peptide (GenScript) was then added to the refolding mixture and further stirred at 4 °C for 72 h. Final heavy chain:light chain:peptide ratios were in the range of 2:1:12 and 2.5:1:12 for the different peptides. Following refolding, the refolding mixture was centrifuged at 50,000 × g to remove any precipitated protein, and the supernatant was concentrated to about 3 ml for size exclusion chromatography using a Superdex S200 HR16/60 gel filtration column pre-equilibrated with size exclusion chromatography buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl). Fractions containing refolded HLA-A*02:01·β2m·peptide complexes were pooled, concentrated to 5–12 mg/ml, and used for subsequent crystallization experiments.
Crystallization and Data Collection
Initial attempts to obtain crystals for the HLA-A*02:01·peptide complexes using factorial screens were not successful. Thin needle-shaped sea urchin crystals of HLA-A*02:01·UFP(16–27) complex obtained in 1.2 m sodium citrate were used to cross-seed the other complexes. The complexes were equilibrated in 30% PEG 4000, 0.1 m Tris-HCl, pH 8.0, 0.2 m lithium sulfate for 1–2 h by mixing 0.15 μl of complex and 0.15 μl of precipitant at 20 °C before seeding. Thin platelike crystals were obtained by sitting drop vapor diffusion at 20 °C after 2–4 days. The crystals were flash frozen in cryoprotectant (crystallization solution, 100% glycerol; 3:1) using liquid nitrogen.
Diffraction data for HLA-A*02:01 complex with peptides G9V, G11N, G13E, and Y16R were collected remotely at beamline 7.1 at the Stanford Synchrotron Radiation Lightsource and processed to 1.86-, 2.3-, 2.1-, and 2.4-Å resolution, respectively, using HKL2000. Diffraction data for HLA-A*02:01 complex with peptides G9L, Y9L, and Y10L were collected remotely at beamline 12.3.1 at the Advanced Light Source and processed to 1.85-, 2.5-, and 2.75-Å resolution, respectively, using HKL2000. Phases were obtained by molecular replacement with Phaser MR (26) in ccp4i (27, 28) using the protein coordinates for HLA-A*02:01 (Protein Data Bank code 3MRE) and resulted in unambiguous electron density for all the peptides. Model building was carried out using Coot (29, 30). Structures were refined using Refmac (31). Data collection and structure refinement parameters are provided in Table 1.
Thermal Denaturation Assay
HLA-A*02:01·β2m·peptide complexes with the various peptides were analyzed for thermal denaturation by differential scanning fluorimetry using a LightCycler 480 (Roche Applied Science). HLA-A*02:01·β2m·peptide complexes with different peptides at 100 μm in reaction buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl) were used as stock solutions. Each reaction mixture constituted 1–2 μl of protein complex stock solution and 2 μl of SYPRO Orange dye (100×; Invitrogen) made up to 20 μl in reaction buffer in a 96-well white plate compatible with the instrument. A temperature gradient from 20 to 85 °C at steps of 0.06 °C/s and 10 acquisitions/°C was used for the experiment. Each experiment with an individual protein·peptide complex was repeated thrice. A melt curve of the total fluorescence of the run was plotted against temperature. The minima of the first derivative of the melt curve from raw fluorescence data (temperature differential of absolute fluorescence versus temperature) provided the Tm for individual HLA-A*02:01·β2m·peptide complexes (inflection point of the melt curve) (32).
Binding Affinity Predictions and Analysis of Extensions
Binding affinities for all 284 eluted ligands to HLA-A*02:01 in the data set of McMurtrey et al. (20) were predicted using NetMHCpan-3.0 (21). Peptides with conventional P2-PΩ anchors and predicted rank of up to 10% were considered as canonical binders. A rank score lower than 10% indicates that a peptide is among the 10% strongest binders for HLA-A*02:01 in a large pool of random natural peptides. Peptides that were not predicted to bind canonically but contained a nested 8–11-mer subsequence with predicted high affinity (rank within top 2%) at the N terminus were classified as extended peptides. Extensions following the PΩ of unconventional binders were categorized based on the first charged residue of the extension found within the first 3 residues of the extension. Extensions where the first charged residue was an Arg or Lys were classified as positive, whereas extensions where the first residue was a Asp or Glu were considered negative. As controls, we randomly picked amino acid sequences from the same set of source proteins with the same length distribution as the observed extensions. We repeated the sampling for random extensions a thousand times to be able to calculate distributions and compared them with the observed extensions.
Derivation of a MHC II Processing Motif Based on Published Data
A large set of 16,868 unique eluted HLA class II ligands was downloaded from the Immune Epitope Database (33) and inspected for amino acid enrichment at the C terminus. We compared the frequency of the very last residue at the C terminus in the MHC II ligands and in the T. gondii ligands, applying a pseudocount correction (34) with β = 50. Pseudocounts exploit information about amino acid similarity to smooth the observed amino acid frequencies of small sequence data sets. This correction has a negligible effect on the large set of MHC II ligands, but it is important in the T. gondii data set because some amino acids were never observed at the C terminus. Enrichment scores were then calculated as SA = log2(fA/qA) where fA is the pseudocount corrected frequency for amino acid A and qA is the background frequency of A in natural proteins.
Prediction of Proteasomal Cleavage and TAP Transport
Predictions of proteasomal cleavage and TAP transport were obtained using the MHC class I processing tools of the Immune Epitope Database (35, 36) for ligands predicted to bind both canonically and in the extended mode. For the processing predictions, precursors for all ligands were obtained by elongating them by 1 residue at the N terminus (to allow for transport of elongated precursors by TAP) and 5 residues at the C terminus (to cover the residues thought to impact cleavage by the proteasome) using the context of their source protein. As a control, we also produced proteasome and TAP scores for 100 random natural sequences extracted from UniProt.
Author Contributions
S. G. M. conducted most of the experiments including protein refolding, crystallization, and structure determination. G. Y. assisted in protein refolding. M. A. performed bioinformatics analysis of the T. gondii peptide data set with early contributions from T. K. C. M. and W. H. identified the T. gondii peptides. S. G. M., M. N., B. P., and D. M. Z. wrote the paper.
Acknowledgments
We thank the support staff at the Advanced Light Source and Stanford Synchrotron Radiation Lightsource for access to remote data collection. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the United States Department of Energy Office of Biological and Environmental Research and by the National Institute of General Medical Sciences (including Grant P41GM103393) and National Center for Research Resources (Grant P41RR001209), National Institutes of Health. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract DE-AC02-05CH11231.
This work was supported by National Institutes of Health Grants AI128609 (to D. M. Z.) and AI062629 (subcontract to W. H.) and Immune Epitope Database National Institutes of Health Contract HHSN272201200010C (to B. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TAP
- transporter associated with antigen processing
- TCR
- T cell receptor
- KIR
- killer immunoglobulin receptor
- β2m
- human β2-microglobulin.
References
- 1. Montoya J. G., and Liesenfeld O. (2004) Toxoplasmosis. Lancet 363, 1965–1976 [DOI] [PubMed] [Google Scholar]
- 2. Webster J. P. (2010) Review of “Toxoplasmosis of Animals and Humans (Second Edition)” by J.P. Dubey. Parasit. Vectors 3, 112 [Google Scholar]
- 3. Cong H., Mui E. J., Witola W. H., Sidney J., Alexander J., Sette A., Maewal A., El Bissati K., Zhou Y., Suzuki Y., Lee D., Woods S., Sommerville C., Henriquez F. L., Roberts C. W., et al. (2012) Toxoplasma gondii HLA-B*0702-restricted GRA7(20–28) peptide with adjuvants and a universal helper T cell epitope elicits CD8+ T cells producing interferon-gamma and reduces parasite burden in HLA-B*0702 mice. Hum. Immunol. 73, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blanchard N., and Shastri N. (2010) Topological journey of parasite-derived antigens for presentation by MHC class I molecules. Trends Immunol. 31, 414–421 [DOI] [PubMed] [Google Scholar]
- 5. Lüder C. G., and Seeber F. (2001) Toxoplasma gondii and MHC-restricted antigen presentation: on degradation, transport and modulation. Int. J. Parasitol. 31, 1355–1369 [DOI] [PubMed] [Google Scholar]
- 6. Leroux L. P., Dasanayake D., Rommereim L. M., Fox B. A., Bzik D. J., Jardim A., and Dzierszinski F. S. (2015) Secreted Toxoplasma gondii molecules interfere with expression of MHC-II in interferon γ-activated macrophages. Int. J. Parasitol. 45, 319–332 [DOI] [PubMed] [Google Scholar]
- 7. Grover H. S., Blanchard N., Gonzalez F., Chan S., Robey E. A., and Shastri N. (2012) The Toxoplasma gondii peptide AS15 elicits CD4 T cells that can control parasite burden. Infect. Immun. 80, 3279–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Israelski D. M., and Remington J. S. (1993) Toxoplasmosis in patients with cancer. Clin. Infect. Dis. 16, Suppl. 2, S423–S435 [DOI] [PubMed] [Google Scholar]
- 9. Luft B. J., and Remington J. S. (1992) Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis. 15, 211–222 [DOI] [PubMed] [Google Scholar]
- 10. Deckert-Schlüter M., Schlüter D., Schmidt D., Schwendemann G., Wiestler O. D., and Hof H. (1994) Toxoplasma encephalitis in congenic B10 and BALB mice: impact of genetic factors on the immune response. Infect. Immun. 62, 221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hakim F. T., Gazzinelli R. T., Denkers E., Hieny S., Shearer G. M., and Sher A. (1991) CD8+ T cells from mice vaccinated against Toxoplasma gondii are cytotoxic for parasite-infected or antigen-pulsed host cells. J. Immunol. 147, 2310–2316 [PubMed] [Google Scholar]
- 12. Denkers E. Y. (1999) T lymphocyte-dependent effector mechanisms of immunity to Toxoplasma gondii. Microbes Infect. 1, 699–708 [DOI] [PubMed] [Google Scholar]
- 13. Hassan C., Chabrol E., Jahn L., Kester M. G., de Ru A. H., Drijfhout J. W., Rossjohn J., Falkenburg J. H., Heemskerk M. H., Gras S., and van Veelen P. A. (2015) Naturally processed non-canonical HLA-A*02:01 presented peptides. J. Biol. Chem. 290, 2593–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blanchard N., Gonzalez F., Schaeffer M., Joncker N. T., Cheng T., Shastri A. J., Robey E. A., and Shastri N. (2008) Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat. Immunol. 9, 937–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cong H., Mui E. J., Witola W. H., Sidney J., Alexander J., Sette A., Maewal A., and McLeod R. (2011) Towards an immunosense vaccine to prevent toxoplasmosis: protective Toxoplasma gondii epitopes restricted by HLA-A*0201. Vaccine 29, 754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schittenhelm R. B., Sian T. C., Wilmann P. G., Dudek N. L., and Purcell A. W. (2015) Revisiting the arthritogenic peptide theory: quantitative not qualitative changes in the peptide repertoire of HLA-B27 allotypes. Arthritis Rheumatol. 67, 702–713 [DOI] [PubMed] [Google Scholar]
- 17. Burrows S. R., Rossjohn J., and McCluskey J. (2006) Have we cut ourselves too short in mapping CTL epitopes? Trends Immunol. 27, 11–16 [DOI] [PubMed] [Google Scholar]
- 18. Schaible U. E., Hagens K., Fischer K., Collins H. L., and Kaufmann S. H. (2000) Intersection of group I CD1 molecules and mycobacteria in different intracellular compartments of dendritic cells. J. Immunol. 164, 4843–4852 [DOI] [PubMed] [Google Scholar]
- 19. Tynan F. E., Borg N. A., Miles J. J., Beddoe T., El-Hassen D., Silins S. L., van Zuylen W. J., Purcell A. W., Kjer-Nielsen L., McCluskey J., Burrows S. R., and Rossjohn J. (2005) High resolution structures of highly bulged viral epitopes bound to major histocompatibility complex class I. Implications for T-cell receptor engagement and T-cell immunodominance. J. Biol. Chem. 280, 23900–23909 [DOI] [PubMed] [Google Scholar]
- 20. McMurtrey C., Trolle T., Sansom T., Remesh S. G., Kaever T., Bardet W., Jackson K., McLeod R., Sette A., Nielsen M., Zajonc D. M., Blader I. J., Peters B., and Hildebrand W. (2016) Toxoplasma gondii peptide ligands open the gate of the HLA class I binding groove. Elife 5, e12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rossjohn J., Gras S., Miles J. J., Turner S. J., Godfrey D. I., and McCluskey J. (2015) T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 33, 169–200 [DOI] [PubMed] [Google Scholar]
- 22. Andreatta M., and Nielsen M. (2016) Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 511–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nielsen M., and Andreatta M. (2016) NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 8, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saunders P. M., Vivian J. P., O'Connor G. M., Sullivan L. C., Pymm P., Rossjohn J., and Brooks A. G. (2015) A bird's eye view of NK cell receptor interactions with their MHC class I ligands. Immunol. Rev. 267, 148–166 [DOI] [PubMed] [Google Scholar]
- 25. Vivier E., and Anfossi N. (2004) Inhibitory NK-cell receptors on T cells: witness of the past, actors of the future. Nat. Rev. Immunol. 4, 190–198 [DOI] [PubMed] [Google Scholar]
- 26. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 28. Potterton E., Briggs P., Turkenburg M., and Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 29. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 30. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 32. Tanford C. (1968) Protein denaturation. Adv. Protein Chem. 23, 121–282 [DOI] [PubMed] [Google Scholar]
- 33. Vita R., Overton J. A., Greenbaum J. A., Ponomarenko J., Clark J. D., Cantrell J. R., Wheeler D. K., Gabbard J. L., Hix D., Sette A., and Peters B. (2015) The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 43, D405–D412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peters B., Bulik S., Tampe R., Van Endert P. M., and Holzhütter H. G. (2003) Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol. 171, 1741–1749 [DOI] [PubMed] [Google Scholar]
- 36. Tenzer S., Peters B., Bulik S., Schoor O., Lemmel C., Schatz M. M., Kloetzel P. M., Rammensee H. G., Schild H., and Holzhütter H. G. (2005) Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell. Mol. Life Sci. 62, 1025–1037 [DOI] [PubMed] [Google Scholar]