

Abstract
Cytosolic DNA can elicit beneficial as well as undesirable immune responses. For example, viral or microbial DNA triggers cell-intrinsic immune responses to defend against infections, whereas aberrant cytosolic accumulation of self-DNA results in pathological conditions, such as autoimmunity. Given the importance of these DNA-provoked responses, a better understanding of their molecular mechanisms is needed. Cytosolic DNA engages stimulator of interferon genes (STING) to activate TANK-binding kinase 1 (TBK1), which subsequently phosphorylates the transcription factor interferon regulatory factor 3 (IRF3) to promote interferon expression. Recent studies have reported that additional transcription factors, including nuclear factor κB (NF-κB) and signal transducer and activator of transcription 6 (STAT6), are also activated by cytosolic DNA, suggesting that cytosolic DNA-induced gene expression is orchestrated by multiple factors. Here we show that cytosolic DNA activates STAT3, another member of the STAT family, via an autocrine mechanism involving interferon β (IFNβ) and IL-6. Additionally, we observed a novel cytosolic DNA-induced phosphorylation at serine 754 in the transactivation domain of STAT3. Upon cytosolic DNA stimulation, Ser754 is directly phosphorylated by TBK1 in a STING-dependent manner. Moreover, Ser754 phosphorylation inhibits cytosolic DNA-induced STAT3 transcriptional activity and selectively reduces STAT3 target genes that are up-regulated in response to cytosolic DNA. Taken together, our results suggest that cytosolic DNA-induced STAT3 activation via IFNβ and IL-6 is restrained by Ser754 phosphorylation of STAT3. Our findings reveal a new signaling axis downstream of the cytosolic DNA pathway and suggest potential interactions between innate immune responses and STAT3-driven oncogenic pathways.
Keywords: cell signaling, innate immunity, phosphorylation, STAT transcription factor, STAT3, Cytosolic DNA, STING, TBK1
Introduction
Double-stranded DNA (dsDNA) in the cytosol is a danger-associated molecular pattern that triggers inflammation and immune responses. Cytosolic DNA can be derived from viral or intracellular microbial infections, undigested phagocytosed materials, and activated self-retroelements (1). The presence of cytosolic DNA is detected by several cellular sensors, which in turn initiate signaling cascades to induce inflammatory response and type I interferon production (2). Despite the redundancy between these cytosolic DNA sensors, cyclic GMP-AMP synthase (cGAS)3 is the predominant sensor relaying the presence of cytosolic DNA to downstream signaling cascades (3). Upon binding to dsDNA, cGAS produces cyclic 2′-3′ GMP-AMP (cGAMP), which serves as a second messenger to activate the endoplasmic reticulum adaptor protein stimulator of interferon genes (STING) (3–6). Activation of STING by cGAMP leads to STING oligomerization, followed by recruitment and activation of TANK-binding kinase 1 (TBK1) (7, 8). Subsequently, TBK1 phosphorylates interferon regulatory factor 3 (IRF3) to promote expression of interferons (IFNs), thereby initiating immune responses to establish an antiviral state (7, 9, 10). In addition to IRFs, other transcription factors, including nuclear factor κB (NF-κB) and signal transducer and activator of transcription 6 (STAT6), are also activated by STING and TBK1 downstream of cytosolic DNA (11–14).
The transcription factor STAT3 is activated by tyrosine 705 phosphorylation downstream of a variety of cytokines, such as epidermal growth factor (EGF) and IL-6 (15). Phosphorylation at tyrosine 705 leads to nuclear accumulation of STAT3 homodimers and expression of target genes containing a γ-activated site (GAS) in their promoters (16). STAT3 drives the expression of prosurvival and inflammatory genes, and sustained activation of STAT3 has been shown to promote proliferation, enhance survival of neoplastic cells, and facilitate inflammation-driven tumorigenesis (17–19). In addition to its role in promoting tumorigenesis, STAT3 also represses the anti-tumor activity of hematopoietic cells, making it a key candidate for targeted cancer therapy and immunotherapy (20, 21). STAT1, another STAT family member, predominantly functions downstream of interferons. STAT1 homodimers induced by IFNγ recognize almost identical GAS sites as STAT3 dimers do in vitro, but STAT1 and STAT3 have different, albeit overlapping, target genes in vivo (22). On the other hand, type I IFNs, including IFNα and IFNβ, induce the formation of not only the STAT1 homodimer but also the interferon-stimulated gene factor 3 (ISGF3) complex comprising STAT1, STAT2, and IRF9. The ISGF3 complex promotes the expression of genes containing an IFN-stimulated response element (ISRE) in their promoters (23). Reciprocal antagonizing effects between STAT1 and STAT3 can be observed in certain scenarios (24). For example, STAT3 inhibits STAT1-dependent induction of ISRE genes in response to type I IFN stimulation presumably through STAT1-STAT3 heterodimerization (25).
Whereas dimerization and activity of STAT proteins are controlled by tyrosine phosphorylation, recent studies have demonstrated that the function of STATs can also be modulated by TBK1 and the closely related kinase IκB kinase ϵ (IKKϵ). Phosphorylation of STAT1 at Ser708 by IKKϵ disrupts STAT1 homodimerization and favors ISGF3 formation, thereby shifting the type I IFN-induced gene expression profile from GAS-driven genes to ISRE-driven genes (26). Cytosolic nucleic acids and viral infections engage the STING-TBK1 pathway, leading to TBK1-mediated phosphorylation of STAT6 at Ser407 and STAT6 activation (14). Interestingly, functional loss of cGAS or STING has been observed in colorectal cancer and melanoma and correlates with disease progression and elevated STAT3 activation (27–29), suggesting a role of STING in restricting STAT3 activity and tumor progression. Thus, we sought to determine whether activity of STAT3 can be regulated by the STING-TBK1 pathway downstream of cytosolic DNA. Here we show that STAT3 is activated by cytosolic DNA through an autocrine mechanism involving IFNβ and IL-6. At the same time, cytosolic DNA activates TBK1 in a cGAS- and STING-dependent manner to directly phosphorylate STAT3 at serine 754 in the transactivation domain (TAD). This TBK1-mediated phosphorylation at Ser754 is inhibitory and restrains cytosolic DNA, IL-6, and IFNβ-induced activation of STAT3. Our finding provides a possible explanation for the role of STING in limiting STAT3 activation and further emphasizes the complex signaling cascades and gene expression initiated by cytosolic DNA.
Results
TBK1 Directly Phosphorylates STAT3 at Serine 754
Previous studies demonstrated that TBK1 and IKKϵ, respectively, regulate the function of STAT6 and STAT1 by direct phosphorylation (14, 26). To determine whether TBK1 and IKKϵ also phosphorylate other STATs, we examined the sequences of STATs against the optimal substrate motif for TBK1 and IKKϵ (30, 31) and identified serine 754 in the TAD of STAT3 as a potential TBK1/IKKϵ phosphorylation site (Fig. 1A). To test whether STAT3 is a substrate of TBK1 or IKKϵ, IKKϵ and STAT3 were overexpressed in HEK293T cells, and immunoprecipitated STAT3 was blotted with an IKK family phosphosubstrate motif antibody (see “Experimental Procedures”). Overexpression of wild-type IKKϵ strongly induced phosphorylation of STAT3 at Tyr705 as well as a site recognized by the IKK phosphosubstrate motif antibody (Fig. 1B). We then generated an antibody specific for phospho-Ser754-STAT3 and repeated the experiment with TBK1. Similarly, overexpression of wild-type, but not kinase-dead, TBK1 induced STAT3 phosphorylation at multiple sites, and S754A mutation of STAT3 abolished the signal of Ser(P)754-STAT3-specific antibody (Fig. 1C), suggesting that TBK1 kinase activity is critical for the phosphorylation of STAT3 at Ser754. Overexpression of TBK1 and IKKϵ in HEK293T may lead to activation of other kinases, which in turn phosphorylate STAT3. To determine whether TBK1 is capable of phosphorylating STAT3 directly, we performed an in vitro kinase assay with purified TBK1 and recombinant GST-STAT3 from bacteria. Autoradiography showed that incubation with wild-type but not kinase-dead TBK1 led to strong phosphorylation on wild-type STAT3 and that S754A mutation of STAT3 abolished the phosphorylation (Fig. 1D). This TBK1-mediated phosphorylation on STAT3 was also recognized by the Ser(P)754-STAT3-specific antibody (Fig. 1D). These data show that TBK1 is capable of directly phosphorylating STAT3 at Ser(P)754.
FIGURE 1.

IKKϵ and TBK1 induce STAT3 phosphorylation at Ser754. A, a schematic figure showing the domain structure of STAT3 and the location of Ser754. Sequences of STAT3 from several vertebrates were aligned to compare the homology, with residues critical for TBK/IKKϵ substrate recognition marked in blue and Ser754 marked in red. Also shown are the domain structure of STAT1 and STAT6, with previously identified IKKϵ/TBK1 target serine residues marked in red and the sequence alignments shown on the right. NTD, N-terminal domain. B, FLAG-tagged STAT3 (3 μg) was co-transfected with GST-IKKϵ (3 μg) into HEK293T cells. STAT3 was immunoprecipitated (IP) and blotted with an IKK substrate motif antibody. C, FLAG-tagged wild-type, Y705F, S727A, or S754A STAT3 (3 μg) were co-transfected with wild-type or K38A (kinase-dead; KD) GST-TBK1 (3 μg) into HEK293T cells. STAT3 was immunoprecipitated and blotted with phosphorylation-specific antibodies to detect Ser754, Tyr705, and Ser727 phosphorylation. D, in vitro kinase assay using the GST-tagged C terminus of STAT3 and GST-TBK1 purified from HEK293T as described under “Experimental Procedures.” The mixture was resolved by SDS-PAGE and blotted (WB) with GST and Ser(P)754-STAT3 antibodies. Phosphorylation of STAT3 was also detected by autoradiography. Data in B–D are representative of four, three, and two independent experiments, respectively.
STAT3 Is Phosphorylated at Ser754 in Response to Cytosolic dsDNA
TBK1 is activated downstream of Toll-like receptors (TLRs) and several other TLR-independent pathways (32). To determine whether Ser754 phosphorylation of STAT3 occurs under these conditions, we asked whether these TBK1-activating agonists promote STAT3 phosphorylation at Ser754. L929 cells were treated with lipopolysaccharide (LPS) or transfected with poly(I:C), poly(dA:dT), or a 70-bp-long double-stranded DNA (VACV70mer) (33) to engage TLR4, MDA5/RIG-I, or the cytosolic DNA pathway, respectively. Transfection with poly(I:C) or DNA resulted in varying degrees of TBK1, IRF3, and STAT1 activation, but only transfection with dsDNA, including poly(dA:dT) and VACV70mer, led to strong phosphorylation of STAT3 at Ser754 (Fig. 2A). Induction of STAT3 Ser754 phosphorylation correlated with robust TBK1 activation (as marked by Ser172 phosphorylation) and phosphorylation of IRF3, a major substrate of TBK1 (Fig. 2A). STAT3 was also activated by cytosolic dsDNA transfection, as marked by Tyr705 phosphorylation, which was accompanied by a modest induction of Ser727 phosphorylation (Fig. 2A). This demonstrates that cytosolic dsDNA leads to TBK1 activation, STAT3 activation, and Ser754 phosphorylation of STAT3. To determine whether similar responses can be observed in human cell lines, we tested the human monocytic cell line THP-1 with these stimuli as well as flagellin, which signals through TLR5. Similarly, cytosolic DNA, especially poly(dA:dT), induced the most robust Ser754 and Tyr705 phosphorylation of STAT3. This was accompanied by slight STAT3 Ser727 phosphorylation and robust activation of STAT1 (Fig. 2B). Poly(dA:dT) transfection induced association of STAT3 with TBK1 and IKKϵ, whereas VACV70mer induced association of STAT3 with TBK1 but not IKKϵ (Fig. 2B). These stimuli also induced various degrees of IKKα/IKKβ activation and p65 phosphorylation, indicative of NF-κB activation (Fig. 2B). Finally, a time course experiment revealed that association between TBK1 and STAT3 was induced as early as 1.5 h after dsDNA transfection, coinciding with the timing of Ser754 phosphorylation, whereas Tyr705 phosphorylation of STAT3 was detected at a later time point (Fig. 2C). Taken together, these data show that among different TBK1 activators, cytosolic DNA induces the most robust STAT3 activation and phosphorylation at Ser754.
FIGURE 2.

Cytosolic DNA induces STAT3 activation and phosphorylation at Ser754. A, L929 cells were transfected with poly(I:C), poly(dA:dT), or VACV70mer (hereafter referred to as dsDNA) using Lipofectamine 2000; treated with 1 μg/ml LPS; or serum-starved for 3 h. Lysates were analyzed by Western blotting to determine the levels of TBK1, IRF3, and STAT3 activation. B, THP-1 cells were treated as described in A or treated with 1 μg/ml flagellin for 3 h. Lysates were analyzed by Western blotting, and STAT3 was immunoprecipitated (IP) and blotted for co-precipitated IKKϵ or TBK1. C, a time course analysis of signaling responses to cytosolic DNA. THP-1 cells were mock-transfected or transfected with dsDNA and analyzed by Western blotting at the indicated times. STAT3 was immunoprecipitated and blotted for Ser(P)754 and co-precipitated TBK1. Data in this figure are representative of two independent experiments.
TBK1 Is Required for Cytosolic DNA-induced STAT3 Phosphorylation at Ser754
Given that cytosolic DNA induces robust TBK1 activation, STAT3 phosphorylation, and association between TBK1 and STAT3, we asked whether TBK1 is required for cytosolic DNA-induced Ser754 phosphorylation of STAT3. We found that STAT3 Ser754 phosphorylation and IRF3 phosphorylation were abrogated with genetic ablation of TBK1 or siRNA-mediated TBK1 knockdown (Fig. 3, A and B). On the other hand, IKKϵ knockdown had negligible effects on STAT3 Ser754 or IRF3 phosphorylation (Fig. 3C), indicating that TBK1 but not IKKϵ is required for cytosolic DNA-induced STAT3 phosphorylation. It has been shown that cytosolic DNA also activates IKKα and IKKβ (Fig. 2B) (13) and that the optimal substrate motif of IKKα and IKKβ shares a partial homology to that of TBK1 and IKKϵ (30, 31, 34, 35). To further investigate the role of individual IKKs in promoting STAT3 phosphorylation upon cytosolic DNA challenge, we used an IKKα/IKKβ-specific inhibitor compound A (36) and two TBK1/IKKϵ-specific inhibitors, AZ-5C and AZ-5E (37). The TBK1/IKKϵ inhibitors blocked the induction of phospho-IRF3 and Ser(P)754-STAT3, whereas phospho-p65 was mostly unaffected (Fig. 3D). Tyr705 phosphorylation of STAT3 was also dependent on TBK1 and/or IKKϵ (Fig. 3D). In contrast, the IKKα/IKKβ inhibitor potently blocked the induction of phospho-p65 but had minimal effect on phospho-IRF3, Ser(P)754-STAT3, or Tyr(P)705-STAT3 in THP-1 cells (Fig. 3D). This indicates that phosphorylation of STAT3 Ser754 is mediated by TBK1 and is independent of IKKα and IKKβ, whereas activation of NF-κB p65 is mediated by IKKα and/or IKKβ. Taken together, our data demonstrate that TBK1, rather than IKKα, IKKβ, or IKKϵ, is the principle kinase that mediates Ser754 phosphorylation of STAT3 upon cytosolic DNA challenge.
FIGURE 3.

TBK1 activity is required for cytosolic DNA-induced Ser754 phosphorylation of STAT3. A, wild-type or TBK1 knock-out MEFs were transfected with 5 μg/ml dsDNA and analyzed by Western blotting at the indicated time points. B, L929 cells were transfected with control siRNA or TBK1 siRNA. Forty-eight hours after siRNA transfection, cells were transfected with dsDNA and analyzed by Western blotting. C, L929 cells were transfected with control siRNA or IKKϵ siRNA. Sixty-eight hours after siRNA transfection, cells were transfected with dsDNA and analyzed by Western blotting. D, THP-1 cells were pretreated with DMSO, 5 μg/ml compound A (an IKKα/IKKβ inhibitor), or 2 μm AZ-5C or AZ-5E (IKKϵ/TBK1 inhibitors) for 30 min, followed by transfection with dsDNA for 3 h. Cells were lysed and blotted for phospho-TBK1, phospho-IRF3, phospho-p65, and phospho-STAT3 to determine the activation of corresponding pathways. Data in A–C and D are representative of two and three independent experiments, respectively.
The cGAS-STING-TBK1 Pathway Induces STAT3 Ser754 Phosphorylation in Response to Cytosolic DNA
Because the endoplasmic reticulum membrane protein STING is indispensable for cytosolic DNA-induced TBK1 activation (10, 38), we tested whether STING is also required for STAT3 Ser754 phosphorylation in response to cytosolic DNA. Indeed, knockdown of STING significantly reduced cytosolic DNA-induced Ser(P)754-STAT3 (Fig. 4A). Moreover, Ser(P)754-STAT3 can be induced by ectopic expression of STING in HEK293T cells in a dose-dependent manner, suggesting that Ser754 phosphorylation occurs downstream of STING activation (Fig. 4B). We also tested whether the cytosolic dsDNA sensor cGAS is required in this setting. Knockdown of cGAS led to reduced activation of TBK1 and phosphorylation of IRF3 and a moderate reduction in the levels of Ser(P)754-STAT3 (Fig. 4C). The moderate reduction of Ser(P)754-STAT3 may be due to incomplete knockdown of cGAS or the redundancy of other cytosolic DNA sensors. Activation of cGAS by cytosolic DNA leads to production of cGAMP, which subsequently promotes STING activation and activation of the TBK1-IRF3 signaling axis. We found that transfection of cGAMP is sufficient to induce Ser754 phosphorylation of STAT3 (Fig. 4D), further supporting the model in which Ser754 phosphorylation of STAT3 occurs downstream of STING. These data demonstrate that cytosolic DNA engages cGAS and STING to activate TBK1 and induce STAT3 Ser754 phosphorylation.
FIGURE 4.

Cytosolic DNA induces Ser(P)754-STAT3 via the cGAS/STING axis. A, L929 cells were transfected with control or IKKϵ siRNA. Forty-eight hours post-transfection, cells were transfected with dsDNA and analyzed by Western blotting at the indicated time points. B, increasing amounts of STING expression plasmid were transfected into HEK293T cells with decreasing amounts of pcDNA3 empty vector, such that the same amount of total DNA was used in every transfection. After 24 h, cells were lysed and blotted for Ser(P)754-STAT3. GST-TBK1-transfected cells were used as a positive control. C, L929 cells were transfected with control or cGAS siRNA. After 68 h, cells were harvested for qRT-PCR to measure cGAS expression levels or transfected with dsDNA for 3 h and analyzed by Western blotting. The expression of cGAS was normalized to Gusb. D, L929 cells were mock-transfected or transfected with cGAMP and analyzed by Western blotting. Data in A and B–D are representative of three and two independent experiments, respectively. Error bars, S.D.
Secreted Cytokines Induce Activation of STAT3 in Response to Cytosolic DNA
We next set out to determine the mechanism of STAT3 activation (as marked by Tyr705 phosphorylation) in response to cytosolic DNA. In THP-1 cells, activation of STAT3 was suppressed by TBK1/IKKϵ inhibitors (Fig. 3D), suggesting that TBK1 kinase activity is required for STAT3 activation. Because TBK1 is not a tyrosine kinase, activation of STAT3 is probably mediated by tyrosine kinases that are directly or indirectly activated by TBK1. It is well established that Janus kinases (JAKs) activate STAT3 by phosphorylating Tyr705. We thus asked whether JAKs are involved in STAT3 activation in response to cytosolic DNA. Treating the cells with a pan-JAK inhibitor, pyridine 6 (39), effectively blocked STAT3 phosphorylation at Tyr705, but TBK1 activation and phosphorylation of STAT3 at Ser754 was unaffected (Fig. 5A). These data and the TBK1 inhibitor data (Fig. 3D) suggest that cytosolic DNA-induced STAT3 activation is primarily mediated by JAKs, the activation of which is dependent on TBK1. Because JAKs usually function downstream of cytokine receptors, we hypothesized that cytosolic DNA induces production of cytokines, leading to activation of the JAKs and STAT3 through an autocrine mechanism. An alternative hypothesis would be that TBK1 activates JAKs directly or through a cascade of kinase activation in a cell-autonomous manner. To test these hypotheses, we asked whether conditioned media from dsDNA-transfected cells are able to activate STAT3 in naive recipient cells. We found that conditioned media from dsDNA-transfected THP-1 strongly induced Tyr(P)705-STAT3 but not Ser(P)754-STAT3 in the recipient cells (Fig. 5B, lanes 10–14). When cells were treated with cycloheximide immediately before dsDNA transfection to block protein synthesis, the ability of conditioned media to induce Tyr(P)705-STAT3 in the recipient cells was abrogated (Fig. 5B, lanes 15–18). Although the possibility of cell-autonomous TBK1-mediated JAK activation cannot be completely ruled out because a weak STAT3 activation was still observed in cycloheximide-treated cells (Fig. 5B, lanes 6–9), these results argue that STAT3 activation is primarily mediated by de novo synthesized secreted factors. We noticed that expression of several STAT3-activating cytokines, including IL-6 and IFNβ, was induced by cytosolic DNA, and the induction of IFNβ was blunted by TBK1 inhibitor (supplemental Fig. 1). Therefore, we tested the involvement of IL-6 and IFNβ in STAT3 activation. Indeed, an IFNβ-neutralizing antibody reduced the ability of conditioned media to activate STAT3 in the recipient cells, and an IL-6-neutralizing antibody also had a modest effect (Fig. 5C). Taken together, our data show that cytosolic DNA-induced Ser754 phosphorylation of STAT3 is strictly cell-autonomous, whereas Tyr705 phosphorylation, and thus activation of STAT3, is primarily mediated by secreted factors, such as IFNβ, through an autocrine mechanism.
FIGURE 5.
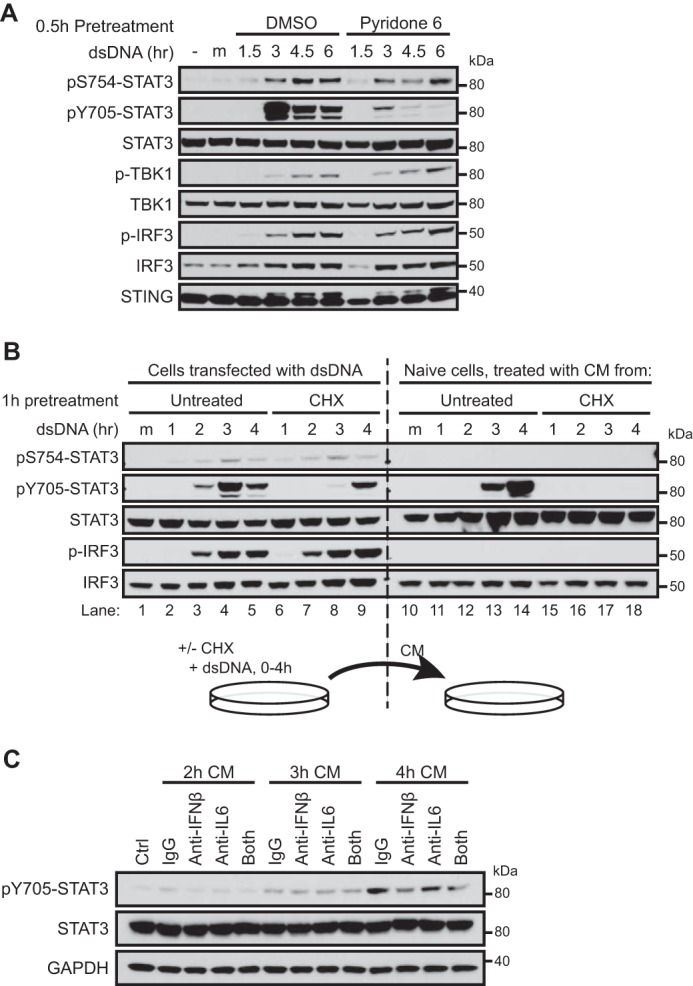
Cytosolic DNA-induced STAT3 activation is mediated by de novo synthesized secreted factors. A, THP-1 cells were pretreated with DMSO or a 100 nm concentration of the pan-JAK inhibitor pyridone 6 for 30 min, followed by transfection of dsDNA. Cells were analyzed by Western blotting at the indicated time points. B, THP-1 cells were left untreated or treated with 30 μg/ml cycloheximide (CHX) to block protein synthesis. After 1 h of treatment, cells were transfected with dsDNA and lysed for Western blotting at the indicated time points, and results are shown in the left half of the blots. Conditioned media (CM) were collected from the dsDNA-transfected cells at the same time points and applied to naive recipient cells for 20 min, and results are shown in the right half of the blots. C, THP-1 cells were transfected with dsDNA, and CM were collected at 2, 3, or 4 h after transfection. Conditioned media were incubated with 20 μg/ml control IgG, 20 μg/ml IL-6-neutralizing antibody, or 40 μg/ml IFNβ-neutralizing antibody for 20 min and applied to naive recipient cells for 20 min before Western blotting. Data in this figure are representative of three independent experiments.
Ser754 Phosphorylation of STAT3 Restricts Cytosolic DNA-induced STAT3 Target Gene Expression
Next, we sought to determine the effect of cytosolic DNA-induced Ser754 phosphorylation on STAT3 activation and target gene expression. THP-1 cells reconstituted with wild-type or mutant STAT3 following CRISPR-mediated knock-out (supplemental Fig. 2) were transfected with dsDNA to induce STAT3 activation and Ser754 phosphorylation. Activation of wild-type and S754D STAT3 by cytosolic DNA was significantly lower than that of the S754A mutant, suggesting that Ser754 phosphorylation inhibits STAT3 activation (Fig. 6A). STAT1 and p65 were also activated by cytosolic DNA, but their activation was not affected by the status of STAT3, and p65 showed constitutive association with STAT3 (Fig. 6A). We also examined the gene expression induced by cytosolic DNA. STAT3 target gene SOCS3 was up-regulated in the presence of STAT3 (Fig. 6B), and its expression was further elevated in the S754A cells, consistent with increased activation of the S754A mutant (Fig. 6A). The NF-κB target gene IL6 was also up-regulated in the presence of STAT3, but Ser754 phosphorylation did not have any measurable effect on its expression (Fig. 6C). This suggests that there may be cooperative binding and transcriptional activation between STAT3 and NF-κB in regulating IL-6 transcription, but Ser754 phosphorylation of STAT3 does not affect the gene expression mediated by this complex. In agreement with this hypothesis, the interaction between p65 and STAT3 was not affected by cytosolic DNA or the status of Ser754 phosphorylation (Fig. 6A). We also asked whether Ser754 phosphorylation of STAT3 modulates the inhibitory effect of STAT3 on ISGF3 target genes (25) and found that CXCL10 was down-regulated by wild-type and mutant STAT3 to similar levels (Fig. 6D), suggesting that Ser754 phosphorylation is not involved in the inhibition of ISGF3 target genes by STAT3. Finally, chromatin immunoprecipitation demonstrated that upon cytosolic DNA challenge, STAT3 was recruited to the predicted binding sites in SOCS3 promoter (Fig. 6E) and that the increased abundance of S754A STAT3 at the SOCS3 promoter correlated with gene expression levels (Fig. 6B). These data demonstrate that cytosolic DNA-induced Ser754 phosphorylation of STAT3 dampens the expression of STAT3 target gene without affecting the expression of NF-κB or ISGF3 genes.
FIGURE 6.

Ser754 phosphorylation restricts STAT3 activation in response to cytosolic DNA. A, ΔSTAT3 THP-1 cells reconstituted with wild-type or mutant STAT3 were transfected with dsDNA for 2 or 4 h. Cells were analyzed by Western blotting to assess the activation of STAT1, STAT3, and NF-κB p65. B–D, ΔSTAT3 and reconstituted THP-1 cells were treated as described A, and RNA was collected for qRT-PCR to determine the expression levels of SOCS3, IL6, and CXCL10. ***, p < 0.001; **, p < 0.01; n.s., not significant; n.d., not determined (below detection threshold). Error bars, S.D. E, diagram of the SOCS3 promoter showing predicted STAT3 binding sites (gray boxes), with numbers indicating locations relative to the transcription start site (TSS). Enrichment of STAT3 at the distal binding site was determined by ChIP as described under “Experimental Procedures.” Immunoprecipitated DNA from control or dsDNA-transfected (3 h) THP-1 cells was quantified by qRT-PCR using primers corresponding to the arrows in the diagram. The quantities of immunoprecipitated DNA are shown as percentages of input DNA. Data in A and B are representative of four independent experiments, and data in C–E are representative of two independent experiments. IP, immunoprecipitation.
Ser754 Phosphorylation Suppresses the Transcriptional Activity of STAT3
Because Ser754 is located in the transactivation domain of STAT3, we hypothesized that phosphorylation at Ser754 modulates the transcriptional activity of STAT3 in response to IL-6 and IFNβ. We asked whether Ser754 phosphorylation of STAT3 affects the expression of a STAT reporter containing tandem GAS sites in response to IFNβ and IL-6 by using a phosphomimetic mutant (S754D), because Ser754 phosphorylation is not induced by IFNβ or IL-6 alone. To avoid interference from endogenous STAT3, the reporter assay was carried out using CRISPR-mediated STAT3 knock-out HEK293T cells (supplemental Fig. 2) or STAT3-null MEFs stably expressing wild-type or mutant STAT3. We found that IFNβ induced a much higher expression of the STAT reporter in the presence of wild-type STAT3, but the transcriptional inactive Y705F mutant inhibited reporter expression (Fig. 7A), indicating that STAT3 contributes to GAS-driven gene expression in response to IFNβ. The reporter expression was reduced with the S754D mutant, suggesting an inhibitory role for Ser754 phosphorylation in the transcriptional activity of STAT3 (Fig. 7A). In contrast, STAT3 did not affect GAS-driven gene expression in response to IFNγ (Fig. 7A), which predominantly activates STAT1 but not STAT3 (15). The S754D mutant also showed decreased activation upon IFNβ stimulation (Fig. 7B), consistent with the results from reporter assays. We then asked whether TBK1-induced Ser754 phosphorylation affects STAT3 activation in response to IFNβ. Because overexpression of TBK1 leads to significant production of cytokines, resulting in elevated basal STAT3 activation (40) (Fig. 1C), here we expressed a moderate amount of TBK1 before treating the cells with IFNβ. We found that TBK1 expression suppressed IFNβ-induced STAT3 activation, but the S754A mutant was more refractory to this TBK1-mediated inhibition (Fig. 7C, lanes 5 and 7). These data show that Ser754 phosphorylation inhibits IFNβ-induced activation of STAT3. We also probed the possibility that Ser754 phosphorylation of STAT3 may affect STAT3-mediated inhibition on ISGF3 target genes. Consistent with CXCL10 expression in THP-1 cells (Fig. 6D), wild-type and mutant STAT3 inhibited IFNβ-induced ISRE reporter expression to comparable levels (supplemental Fig. 3), indicating that Ser754 phosphorylation of STAT3 does not affect ISGF3 activity. Similar to the IFNβ reporter assays, S754D mutant was also less active in response to IL-6 as measured by STAT reporter assays and by the levels of Tyr705 phosphorylation, which inversely correlated with the levels of Ser754 phosphorylation or phosphomimetic mutation (Fig. 7, D and E). Collectively, these data demonstrate that Ser754 phosphorylation suppresses the transcriptional activity of STAT3 induced by IL-6 and IFNβ.
FIGURE 7.

Ser754 phosphorylation inhibits transcriptional activity of STAT3. A, dual luciferase assay was used to determine STAT3 activity as described under “Experimental Procedures.” ΔSTAT3 HEK293T cells in 12-well plates were transfected with 0.5 μg of empty vector (EV) or 4xHA-STAT3 plasmids, 0.5 μg of STAT firefly luciferase plasmid, and 25 ng of TK-Renilla luciferase plasmid, followed by treatment with 25 pg/ml human IFNβ or 200 pg/ml human IFNγ. Cell lysates were used for Western blotting to verify STAT3 expression levels. Data are shown as mean with S.D. ***, p < 0.001. Error bars, S.D. B, ΔSTAT3 HEK293T cells in 6-cm plates were transfected with 3 μg of 4xHA-STAT3 plasmids. Twenty-four hours after transfection, cells were treated with 20 pg/ml of human IFNβ for 30 min and lysed for Western blotting. Densitometric ratios of Tyr(P)705-STAT3 to STAT3 are shown to evaluate the levels of STAT3 activation. C, ΔSTAT3 HEK293T cells in 10-cm plates were transfected with 3 μg of 4xHA-STAT3 plasmids and 1 μg of wild-type or kinase-dead GST-TBK1 plasmids. Twenty-four hours after transfection, cells were treated with 20 pg/ml of human IFNβ and lysed for Western blotting. Densitometric ratios of Tyr(P)705-STAT3 to STAT3 are shown to evaluate the levels of STAT3 activation. D, STAT3-null MEFs reconstituted with wild-type or mutant STAT3 in 12-well plates were transfected with 0.5 μg of STAT firefly luciferase reporter and 33 ng of TK-Renilla luciferase plasmid, followed by treatment of 100 ng/ml mouse IL-6 before Dual-Luciferase assays. Data are shown as mean with S.D. ***, p < 0.001. E, STAT3-null MEFs reconstituted with wild-type or mutant STAT3 were treated with 30 ng/ml mouse IL-6 for 30 min and analyzed by Western blotting to determine the levels of STAT3 activation. Data in A, B and C, and D and E are representative of three, two, and four independent experiments, respectively.
Discussion
In this study, we identified STAT3 as a novel substrate of TBK1 downstream of the cytosolic DNA pathway. In the presence of cytosolic DNA, TBK1 phosphorylates STAT3 at Ser754 to limit STAT3 activity induced by cytokines, such as IL-6 and IFNβ. Previously, it has been shown that IKKϵ regulates STAT1 dimerization and that TBK1 regulates STAT6 activity by direct phosphorylation (14, 26). Our finding places a third STAT member under the control of IKKϵ/TBK1. Interestingly, the IKKϵ/TBK1-mediated phosphorylation sites in STAT1, STAT3, and STAT6 differ in their location within the proteins (Fig. 1A). In the case of STAT1, phosphorylation of Ser708, which resides between the SH2 domain and the TAD, disrupts SH2 domain-mediated STAT1 homodimerization by steric hindrance (26). How TBK1-mediated Ser407 phosphorylation regulates the activity of STAT6 is less clear. Ser407 resides within a highly conserved region of the STAT DNA binding domain, and structural analysis demonstrated that mutations in this region abolish the DNA binding ability of STATs (41). Thus, it is plausible that Ser407 phosphorylation affects the DNA binding affinity of STAT6. It is also worth noting that TBK1 induces a reduced but still significant phosphorylation on STAT6 S407A mutant (14), suggesting the existence of additional TBK1 phosphorylation sites in STAT6. In fact, we identified another IKKϵ/TBK1 substrate motif in STAT6 TAD, in which Ser733 is the residue that corresponds to Ser754 of STAT3. Our preliminary data suggest that TBK1 overexpression also leads to STAT6 phosphorylation at Ser733.4 For future investigations, it would be of interest to determine whether this phosphorylation serves as an additional mechanism by which TBK1 regulates STAT6 activity in a manner similar to what we discovered with STAT3.
The two IKK-related kinases TBK1 and IKKϵ are structurally similar and prefer almost identical substrate sequences in vitro (30, 31). However, they appear to have distinct yet partially overlapping roles in vivo (42). Studies using TBK1 or IKKϵ knock-out cells showed that TBK1 is the principle kinase that phosphorylates IRF3 to initiate interferon production in response to innate immune stimuli and pathogens, whereas IKKϵ has a minor or negligible role in activating IRF3 and interferon production (11, 43, 44). Similarly, in our model, although overexpression of TBK1 and IKKϵ both induced Ser754 phosphorylation of STAT3 (Fig. 1, B and C), endogenous IKKϵ did not have a measurable impact on STAT3 phosphorylation in response to VACV70mer (dsDNA with 33% GC content) transfection (Fig. 3C). However, it is worth noting that whereas VACV70mer only induced interaction between STAT3 and TBK1, poly(dA:dT) transfection induced interaction of STAT3 with TBK1 and IKKϵ (Fig. 2B), suggesting that IKKϵ may contribute to the signaling cascades and STAT3 phosphorylation downstream of AT-rich cytosolic DNA. AT-rich cytosolic DNA not only activates the STING-TBK1 pathway but also engages the cytosolic dsRNA sensor RIG-I by a polymerase III-dependent mechanism (45). Thus, the differential interactions between STAT3 and IKKϵ/TBK1 in response to VACV70mer and poly(dA:dT) may be due to the activation of cytosolic dsRNA pathway specifically downstream of poly(dA:dT). It is also conceivable that IKKϵ will play a more dominant role in scenarios where its expression is highly induced (46). Whether IKKϵ contributes to STAT3 Ser754 phosphorylation and regulation under these conditions remains to be tested.
The NF-κB pathway is also activated by cytosolic DNA, but the roles of different IKKs in this context remain controversial (11–13). Ishii et al. (11) suggested that TBK1 is dispensable for cytosolic DNA-induced NF-κB activation, whereas Abe et al. (12, 13) demonstrated a significant dependence of NF-κB activation on TBK1. In MEFs and THP-1 cells, we observed that IKKα/IKKβ and TBK1 are each responsible for cytosolic DNA-induced p65 NF-κB activation or IRF3 activation (Fig. 3, A and D), indicating that the signaling events dictating NF-κB and IRF3 activation diverge at or above the level of these kinases. Intriguingly, p65 activation in L929 is mostly dependent on TBK1 (Fig. 3B), consistent with the observation made by Abe et al. (13). It is unclear why such differences exist. One possible explanation may be the availability of different signaling molecules and the formation of different complexes. For instance, depending on the cell type, TBK1 may localize to the mitochondria or the endoplasmic reticulum in response to cytosolic DNA (47). Therefore, it is likely that signaling pathways downstream of cytosolic DNA and STING may be influenced by the availability of cell type-specific machinery and platforms as well as the subcellular localization of TBK1.
Although it is unclear why Ser754 phosphorylation dampens the activity of STAT3, studies on a natural occurring STAT3 isoform STAT3β provide a plausible hypothesis. Alternative splicing of the STAT3 transcript results in the truncated STAT3β isoform that is 48 amino acids shorter than the full-length STAT3 (48). STAT3β shows strong Tyr705 phosphorylation and DNA binding activity even in the absence of stimulation, but it lacks intrinsic transcriptional activity due to the lack of TAD (48, 49). Interestingly, STAT3β dimers are more stable than dimers formed by full-length STAT3, and deletion of 19 residues at the C terminus (amino acids 752–770) of full-length STAT3 is sufficient to significantly enhance Tyr705 phosphorylation, DNA binding activity, and dimer stability (50). Because the C terminus of STAT3 is rich in acidic amino acids that are negatively charged, it was therefore proposed that the cluster of negative charges in the TAD interferes with STAT3 dimerization and makes phosphorylated Tyr705 more accessible to phosphatases (50). Given this mechanism, phosphorylation or phosphomimetic mutation of Ser754 will introduce more negative charges to the region and may destabilize STAT3 dimers, thereby reducing its transcriptional activity. Alternatively, it is also possible that Ser754 phosphorylation affects the interaction between STAT3 and its co-activators to modulate STAT3 activity. Future studies will focus on testing these hypotheses.
There is mounting evidence of the critical role of cytosolic DNA in tumorigenesis and anti-tumor immune responses. Functional loss of the cGAS-STING cytosolic DNA pathway has been observed in some commonly used cell lines and high passage immortalized MEFs (5) as well as colorectal cancer and melanoma, in which loss of the cytosolic DNA pathway correlates with tumor progression (27, 28). In a mouse model of colitis-associated colorectal cancer, STING-deficient mice showed elevated NF-κB and STAT3 activity and developed advanced disease, suggesting a role of STING in controlling inflammatory responses and tumorigenesis (29). According to our model, loss of the cytosolic DNA sensing pathway will predict loss of the TBK1-mediated restraint on STAT3 activity, allowing the tumor cells to have elevated STAT3 activation in response to cytokines such as IL-6. Given the well established role of STAT3 in promoting tumorigenesis, our finding suggests a possible mechanism in which tumor cells without the cytosolic DNA pathway may have a survival advantage due to unchecked STAT3 activation. On the other hand, activation of dendritic cells in the tumor microenvironment by engaging the cGAS/STING pathway leads to significant T cell recruitment and anti-tumor immunity (51, 52). Because STAT3 negatively regulates dendritic cell activity and the anti-tumor response of hematopoietic cells (20, 53), cytosolic DNA-mediated restraint on STAT3 activity may serve as an additional mechanism to strengthen the anti-tumor immunity of hematopoietic cells in the tumor microenvironment.
In summary, we identified a novel signaling axis in which TBK1 modulates STAT3 activity in response to cytosolic DNA. Our findings reveal a new mechanism by which the activity of STAT3 can be fine-tuned by a single phosphorylation and shed light on the possible cross-talk between innate immune responses and STAT3-driven oncogenic pathways.
Experimental Procedures
Plasmids and Viruses
Mouse STAT3 was cloned into pBabe-puro (Addgene, catalog no. 1764) (54) using BamHI and EcoRI. N-terminal HA-tagged STAT3 was cloned into 3XHA-pEBB vector using BamHI and NotI. N-terminal FLAG-tagged STAT3 was cloned into pENTR-3C plasmid using BamHI and EcoRI, followed by Gateway recombination into pLenti6/UbC/V5 destination vector (Thermo Fisher). The C terminus of STAT3 (amino acids 700–770) was cloned into pGEX-4T-1 in frame with N-terminal GST using BamHI and EcoRI. STAT3 mutant plasmids were generated by site-directed mutagenesis and confirmed by sequencing. STAT3 CRISPR plasmid was constructed using lentiCRISPRv2 (Addgene, catalog no. 52961) (55) with a single guide RNA targeting the 5′-UTR of human STAT3 (supplemental Fig. 2). The primer sequences for single guide RNA cloning are as follows: 5′-CAC CGT GCC GGA GAA ACA GGT GAA G-3′ and 5′-AAA CCT TCA CCT GTT TCT CCG GCA C-3′.
Protein Purification and in Vitro Kinase Assay
Rosetta cells (Novagen) harboring pGEX-4T-1-STAT3 plasmid were grown to log phase and treated with 0.5 mm isopropyl β-d-1-thiogalactopyranoside at 30 °C for 16 h. The cells were lysed (50 mm Tris, pH 7.6, 150 mm NaCl, 1 mm EDTA, 5 mm DTT, 25 μg/ml lysozyme, protease inhibitor mixture (Promega)) at room temperature, followed by the addition of 1.25 μg/ml sodium deoxycholate, 1.25 μm MgCl2, and 62.5 μg/ml DNase I. Cleared lysates (13,000 rpm, 15 min, 4 °C) were incubated with glutathione-agarose beads (Amersham Biosciences) at 4 °C for 2 h. The beads were washed twice with modified RIPA buffer (150 mm NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 50 mm Tris, pH 7.6, 1 mm β-glycerol phosphate), twice with high salt (500 mm NaCl) modified RIPA, and twice with kinase buffer (1 mm β-glycerol phosphate, 20 mm Tris, pH 7.4, 12 mm MgCl2). Purified protein was eluted by incubating the beads with 30 mm glutathione in kinase buffer with 0.1% Tween 20 for 20 min at room temperature with shaking.
Wild-type and K38A GST-TBK1 were purified from HEK293T transfected with pEBG-GST-TBK1 plasmids as described previously (31). For in vitro kinase assays, 1–2 μl of purified GST-STAT3 was incubated with 2–4 μl of wild-type GST-TBK1 or K38A GST-TBK1 in kinase buffer with 1 mm ATP and 10 μCi of [γ-32P]ATP at room temperature for 2 h. The reaction was resolved by SDS-PAGE, followed by immunoblotting or autoradiography.
Cells
THP-1 cells were maintained in RPMI1640 with 10% FBS, whereas HEK293T, L929, and MEFs were maintained in DMEM with 10% FBS and kept at 37 °C with 5% CO2. STAT3-null MEFs were a gift from Dr. Hua Yu (City of Hope), and TBK1 null MEFs were a gift from Amgen. Retroviruses were produced by co-transfection of pBabe-puro-STAT3 and pCL10A1 plasmids into HEK293T cells, and lentiviruses were produced by co-transfection of pLenti6-STAT3 with psPAX2 and pMD2.G into HEK293T cells. The supernatants containing viruses were 0.45 μm-filtered before being used for transduction. To reconstitute STAT3 expression in STAT3-null MEFs, MEFs were transduced with pBabe-puro-STAT3 retroviruses and selected for puromycin resistance. To generate STAT3 CRISPR knock-out cells, THP-1 or HEK293T cells were transduced with STAT3 CRISPR virus and selected for puromycin resistance, and single clones were analyzed for STAT3 knock-out efficiency (supplemental Fig. 2). To reconstitute STAT3 in THP-1, ΔSTAT3-THP-1 cells were transduced with pLenti6-STAT3 viruses and selected for blasticidin resistance.
To activate TBK1, cells were transfected with poly(I:C), poly(dA:dT), VACV70mer, or 2′-3′ cGAMP using Lipofectamine 2000 (Thermo Fisher) at a 1:1 ratio (μg/μl), and the final concentration of nucleic acids was 2 μg/ml unless otherwise noted. THP-1 cells were treated with 25 ng/ml phorbol 12-myristate 13-acetate (Sigma, P1585) overnight to induce adherence before cytosolic DNA transfection. Poly(I:C) (tlrl-pic) poly(dA:dT) (tlrl-patn), cGAMP (tlrl-cga23), and LPS (tlrl-eblps) were from Invivogen. VACV70mer was prepared by annealing complementary 70-nucleotide primers as described previously (33). For experiments with conditioned media, supernatants from nucleic acid transfected cells were collected and 0.2 μm-filtered; mixed with 20 μg/ml normal mouse IgG (Millipore, 12-371), 20 μg/ml human IL-6-neutralizing antibody (R&D Systems, MAB206), or 40 μg/ml human IFNβ neutralizing antibody (BioLegend, catalog no. 514004); and incubated at room temperature for 20 min before being added to recipient cells.
Transfection of siRNA
Transfection of siRNA was carried out using Lipofectamine 2000 according to the manufacturer's protocol. Mouse Tbk1 (M-063162-01-0005), mouse Ikbke (IKKϵ) (M-040798-01-0005), mouse Tmem173 (STING) (M-055528-01-0005), and control siRNAs (D-001210-03-05) were from Dharmacon. Mouse Mb21d1 (cGAS) siRNA was from Sigma (SASI_Mm01_00129826)
Reporter Assay
The STAT reporter 4xM67 pTATA TK-firefly luciferase plasmid (Addgene, catalog no. 8688) (56) and pRL-TK-Renilla luciferase plasmid (Promega) were co-transfected at 15:1–20:1 ratios into ΔSTAT3 293T cells together with 4xHA-STAT3 plasmids. Twenty-four hours after transfection, cells were treated with 25 pg/ml mouse IFNβ (R&D Systems, 8499-IF-010) or 200 pg/ml mouse IFNγ (R&D Systems, 285-IF-100). Cells were lysed with passive lysis buffer (Promega) for the Dual-Luciferase assay (Promega) at 16–24 h after treatment. The relative luciferase units (RLU) were calculated by normalizing the reading of firefly luciferase to that of Renilla luciferase, and RLU of the control cells was set to 1. For IL-6 reporter assays, STAT3 reconstituted MEFs were transfected with reporters in the same manner and treated with 100 ng/ml mouse IL-6 (BioLegend, catalog no. 575704) for 24 h.
Immunoblotting and Immunoprecipitation
For immunoblotting, cells were lysed in RIPA buffer with protease inhibitor mixture (Promega), phosphatase inhibitor mixture (Sigma), and 1 mm Na3VO4. Cleared lysates were resolved by SDS-PAGE (NuPAGE bis-tris gels, Thermo Fisher), transferred to PVDF membranes (Millipore, IPVH00010), and blocked in 5% nonfat milk in TBST. The membranes were incubated in primary antibodies (1:1000–1:5000) in TBST at 4 °C overnight, washed with TBST, and incubated in appropriate HRP-conjugated secondary antibodies (Promega) (1:10,000). Pierce ECL (Thermo Fisher) was added to the blots, which were then exposed to films and developed or imaged using ChemiDoc (Bio-Rad). For immunoprecipitation, cells were lysed in 0.5% Nonidet P-40 buffer (0.5% Nonidet P-40, 150 mm NaCl, 20 mm Tris, pH 7.6, 1 mm EGTA, 1 mm EDTA, 1 mm β-glycerol phosphate, and 0.5% glycerol) with protease and phosphatase inhibitors and Na3VO4. Cleared lysates were incubated with M2 FLAG (Sigma) or STAT3 antibodies at 4 °C overnight, followed by incubation with Dynabead protein G (Thermo Fisher) for 1 h at 4 °C and washing with 0.5% Nonidet P-40 buffer, and resolved by SDS-PAGE and immunoblotting. The following antibodies were from Cell Signaling Technology: GST (catalog nos. 2624 and 2625), phospho-IKKα/IKKβ (Ser176/Ser180) (catalog no. 2697), IKKβ (catalog no. 2684), IKKϵ (catalog no. 3416), phospho-IRF3 (Ser396) (catalog no. 4947), IRF3 (catalog no. 4302), phospho-TBK1 (Ser172) (catalog no. 5483), TBK1 (catalog no. 3013), phospho-STAT3 (Tyr705) (catalog nos. 9145 and 9132), phospho-STAT3 (Ser727) (catalog no. 9134), phospho-p65 (Ser536) (catalog no. 3033), p65 (catalog no. 8242), STING (catalog no. 13647), phospho-STAT3 (Ser754) (BL14578; catalog no. 5163), and IKK phosphosubstrate motif (G9108). The IKK phosphosubstrate motif antibody was generated against the phosphorylated IKK consensus sequence X(Y/F)XpSLX, where pS is the phosphoserine targeted by IKKs (30, 31, 34, 35). GAPDH (sc-25778) and β-tubulin (sc-9104) antibodies were from Santa Cruz Biotechnology, Inc. Densitometry analyses were carried out using the gel analysis function of ImageJ.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described previously (57). Briefly, cells were fixed by formaldehyde, lysed, and sonicated to yield DNA fragments of 200–500 bp. Lysates were diluted to 0.1% of SDS and precleared by incubating with BSA and salmon sperm DNA-blocked Dynabead magnetic protein G beads (Thermo Fisher). Lysates corresponding to 5 × 106 cells were used for each ChIP with 5 μg of rabbit IgG (Cell Signaling Technology, catalog no. 2729) or rabbit anti-STAT3 antibody (Cell Signaling Technology, catalog no. 12640), followed by capture with Dynabead magnetic protein G beads. DNA-protein-antibody complexes were eluted, and DNA was uncross-linked and purified by phenol-chloroform. The quantity of input DNA was determined by Nanodrop. The quantity of DNA corresponding to the STAT3 binding site in the SOCS3 promoter in immunoprecipitated chromatin was determined by qRT-PCR with three technical repeats using SYBR Green (Thermo Fisher) and the following primers: 5′-TAA GAA GGC TGA TTT CTG GCA GAG G-3′ and 5′-CCA GGT CGG CCT CCT AGA ACT-3′. Data are shown as mean with S.D. and are representative of two independent experiments.
Quantitative Real-time PCR
Total RNA from cells were purified using the Qiagen RNeasy Plus kit according to the manufacturer's protocol. 1–2 μg of RNA was used to synthesize cDNA using Moloney murine leukemia virus reverse transcriptase (Thermo Fisher). Quantitative real-time PCR (qRT-PCR) was carried out using synthesized cDNA and standard TaqMan probes, primers, and reagents (Thermo Fisher). The expression level of target genes was calculated by the ΔΔCt method relative to the level of GUSB. Data shown are the relative quantity, with the relative quantity of the control cells set to 1.
Statistical Analysis
For reporter assays and qRT-PCR, data are shown as mean with S.D. or mean with 95% confidence intervals, respectively. Each data point was from three technical replicates. Analyses were done by Prism (GraphPad Software, Inc., La Jolla, CA) using a t test with false discovery rate controlled at 1%.
Inhibitors and Reagents
The IKKα/IKKβ-specific inhibitor Compound A was a generous gift from Dr. Karl Ziegelbauer (Bayer). The TBK1/IKKϵ-specific inhibitors AZ-5C and AZ-5E were synthesized by Dr. Stephen Frye's group at the University of North Carolina (Chapel Hill, NC). The pan-JAK inhibitor pyridone 6 was from Millipore (catalog no. 420099), and cycloheximide was from Sigma (C7698).
Author Contributions
J. E. H. and A. S. B. conceived the study. J. E. H. designed the experiments in Fig. 1 and performed the experiments in Fig. 1 (A and B). H. C. H. performed the experiments in Fig. 1 (C and D) and all other experiments and analyzed the data. H. C. H. and A. S. B. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank the members of the Baldwin laboratory and Dr. Blossom Damania for constructive feedback. We also thank Dr. Hua Yu (City of Hope) for STAT3-null MEFs, Dr. Jenny P.-Y. Ting (University of North Carolina, Chapel Hill, NC) for FLAG-STING plasmid, and Dr. Stephen Frye (University of North Carolina, Chapel Hill, NC) for TBK1 inhibitors. The IKK phosphosubstrate motif and Ser(P)754-STAT3 antibodies were kindly provided by Cell Signaling Technology, and TBK1-null MEFs were kindly provided by Amgen.
This work was supported by National Institutes of Health (NIH) Grants AI35098 and R35CA197684 (to A. S. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. 1–3.
H.-C. Hsia, unpublished observation.
- cGAS
- cyclic GMP-AMP synthase
- cGAMP
- cyclic 2′-3′ GMP-AMP
- STING
- stimulator of interferon genes
- GAS
- γ-activated site
- ISRE
- IFN-stimulated response element
- IKK
- IκB kinase
- TAD
- transactivation domain
- TLR
- Toll-like receptor
- SH2
- Src homology 2
- RIPA
- radioimmune precipitation assay
- RLU
- relative luciferase unit(s)
- bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- qRT-PCR
- quantitative RT-PCR
- TK
- thymidine kinase.
References
- 1. Paludan S. R., and Bowie A. G. (2013) Immune Sensing of DNA. Immunity 38, 870–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Broz P., and Monack D. M. (2013) Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 13, 551–565 [DOI] [PubMed] [Google Scholar]
- 3. Wu J., Sun L., Chen X., Du F., Shi H., Chen C., and Chen Z. J. (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Diner E. J., Burdette D. L., Wilson S. C., Monroe K. M., Kellenberger C. A., Hyodo M., Hayakawa Y., Hammond M. C., and Vance R. E. (2013) The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 3, 1355–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun L., Wu J., Du F., Chen X., and Chen Z. J. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ablasser A., Goldeck M., Cavlar T., Deimling T., Witte G., Röhl I., Hopfner K.-P., Ludwig J., and Hornung V. (2013) cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tanaka Y., and Chen Z. J. (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu S., Cai X., Wu J., Cong Q., Chen X., Li T., Du F., Ren J., Wu Y.-T., Grishin N. V., and Chen Z. J. (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- 9. Zhong B., Yang Y., Li S., Wang Y.-Y., Li Y., Diao F., Lei C., He X., Zhang L., Tien P., and Shu H.-B. (2008) The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550 [DOI] [PubMed] [Google Scholar]
- 10. Ishikawa H., and Barber G. N. (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ishii K. J., Coban C., Kato H., Takahashi K., Torii Y., Takeshita F., Ludwig H., Sutter G., Suzuki K., Hemmi H., Sato S., Yamamoto M., Uematsu S., Kawai T., Takeuchi O., and Akira S. (2006) A Toll-like receptor: independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 7, 40–48 [DOI] [PubMed] [Google Scholar]
- 12. Abe T., Harashima A., Xia T., Konno H., Konno K., Morales A., Ahn J., Gutman D., and Barber G. N. (2013) STING recognition of cytoplasmic DNA instigates cellular defense. Mol. Cell 50, 5–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abe T., and Barber G. N. (2014) Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 88, 5328–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen H., Sun H., You F., Sun W., Zhou X., Chen L., Yang J., Wang Y., Tang H., Guan Y., Xia W., Gu J., Ishikawa H., Gutman D., Barber G., et al. (2011) Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 147, 436–446 [DOI] [PubMed] [Google Scholar]
- 15. Zhong Z., Wen Z., and Darnell J. E. (1994) Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98 [DOI] [PubMed] [Google Scholar]
- 16. Reich N. C. (2013) STATs get their move on. JAKSTAT 2, e27080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bromberg J. F., Wrzeszczynska M. H., Devgan G., Zhao Y., Pestell R. G., Albanese C., and Darnell J. E. (1999) Stat3 as an oncogene. Cell 98, 295–303 [DOI] [PubMed] [Google Scholar]
- 18. Yu H., Pardoll D., and Jove R. (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grivennikov S. I., and Karin M. (2010) Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 21, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kortylewski M., Kujawski M., Wang T., Wei S., Zhang S., Pilon-Thomas S., Niu G., Kay H., Mulé J., Kerr W. G., Jove R., Pardoll D., and Yu H. (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11, 1314–1321 [DOI] [PubMed] [Google Scholar]
- 21. Kortylewski M., and Yu H. (2008) Role of Stat3 in suppressing anti-tumor immunity. Curr. Opin. Immunol. 20, 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horvath C. M., Wen Z., and Darnell J. E. (1995) A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 9, 984–994 [DOI] [PubMed] [Google Scholar]
- 23. Ivashkiv L. B., and Donlin L. T. (2014) Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Regis G., Pensa S., Boselli D., Novelli F., and Poli V. (2008) Ups and downs: the STAT1:STAT3 seesaw of interferon and gp130 receptor signalling. Semin. Cell Dev. Biol. 19, 351–359 [DOI] [PubMed] [Google Scholar]
- 25. Wang W. B., Levy D. E., and Lee C. K. (2011) STAT3 negatively regulates type I IFN-mediated antiviral response. J. Immunol. 187, 2578–2585 [DOI] [PubMed] [Google Scholar]
- 26. Ng S.-L., Friedman B. A., Schmid S., Gertz J., Myers R. M., Tenoever B. R., and Maniatis T. (2011) IκB kinase epsilon (IKKϵ) regulates the balance between type I and type II interferon responses. Proc. Natl. Acad. Sci. U.S.A. 108, 21170–21175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xia T., Konno H., Ahn J., and Barber G. N. (2016) Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 14, 282–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xia T., Konno H., and Barber G. N. (2016) Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 10.1158/0008-5472.CAN-16-1404 [DOI] [PubMed] [Google Scholar]
- 29. Zhu Q., Man S. M., Gurung P., Liu Z., Vogel P., Lamkanfi M., and Kanneganti T.-D. (2014) Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. J. Immunol. 193, 4779–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hutti J. E., Shen R. R., Abbott D. W., Zhou A. Y., Sprott K. M., Asara J. M., Hahn W. C., and Cantley L. C. (2009) Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKϵ promotes cell transformation. Mol. Cell 34, 461–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hutti J. E., Porter M. A., Cheely A. W., Cantley L. C., Wang X., Kireev D., Baldwin A. S., and Janzen W. P. (2012) Development of a high-throughput assay for identifying inhibitors of TBK1 and IKKϵ. PLoS One 7, e41494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chau T.-L., Gioia R., Gatot J.-S., Patrascu F., Carpentier I., Chapelle J.-P., O'Neill L., Beyaert R., Piette J., and Chariot A. (2008) Are the IKKs and IKK-related kinases TBK1 and IKK-ϵ similarly activated? Trends Biochem. Sci. 33, 171–180 [DOI] [PubMed] [Google Scholar]
- 33. Unterholzner L., Keating S. E., Baran M., Horan K. A., Jensen S. B., Sharma S., Sirois C. M., Jin T., Latz E., Xiao T. S., Fitzgerald K. A., Paludan S. R., and Bowie A. G. (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hutti J. E., Turk B. E., Asara J. M., Ma A., Cantley L. C., and Abbott D. W. (2007) IκB kinase β phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-κB pathway. Mol. Cell Biol. 27, 7451–7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marinis J. M., Hutti J. E., Homer C. R., Cobb B. A., Cantley L. C., McDonald C., and Abbott D. W. (2012) IκB kinase α phosphorylation of TRAF4 downregulates innate immune signaling. Mol. Cell Biol. 32, 2479–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ziegelbauer K., Gantner F., Lukacs N. W., Berlin A., Fuchikami K., Niki T., Sakai K., Inbe H., Takeshita K., Ishimori M., Komura H., Murata T., Lowinger T., and Bacon K. B. (2005) A selective novel low-molecular-weight inhibitor of IkappaB kinase-β (IKK-β) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br. J. Pharmacol. 145, 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang T., Block M. A., Cowen S., Davies A. M., Devereaux E., Gingipalli L., Johannes J., Larsen N. A., Su Q., Tucker J. A., Whitston D., Wu J., Zhang H.-J., Zinda M., and Chuaqui C. (2012) Discovery of azabenzimidazole derivatives as potent, selective inhibitors of TBK1/IKKϵ kinases. Bioorg. Med. Chem. Lett. 22, 2063–2069 [DOI] [PubMed] [Google Scholar]
- 38. Ishikawa H., Ma Z., and Barber G. N. (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pedranzini L., Dechow T., Berishaj M., Comenzo R., Zhou P., Azare J., Bornmann W., and Bromberg J. (2006) Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res. 66, 9714–9721 [DOI] [PubMed] [Google Scholar]
- 40. Korherr C., Gille H., Schäfer R., Koenig-Hoffmann K., Dixelius J., Egland K. A., Pastan I., and Brinkmann U. (2006) Identification of proangiogenic genes and pathways by high-throughput functional genomics: TBK1 and the IRF3 pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 4240–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Becker S., Groner B., and Müller C. W. (1998) Three-dimensional structure of the Stat3β homodimer bound to DNA. Nature 394, 145–151 [DOI] [PubMed] [Google Scholar]
- 42. Clément J.-F., Meloche S., and Servant M. J. (2008) The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 18, 889–899 [DOI] [PubMed] [Google Scholar]
- 43. Hemmi H., Takeuchi O., Sato S., Yamamoto M., Kaisho T., Sanjo H., Kawai T., Hoshino K., Takeda K., and Akira S. (2004) The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199, 1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tenoever B. R., Ng S.-L., Chua M. A., McWhirter S. M., García-Sastre A., and Maniatis T. (2007) Multiple functions of the IKK-related kinase IKKϵ in interferon-mediated antiviral immunity. Science 315, 1274–1278 [DOI] [PubMed] [Google Scholar]
- 45. Chiu Y.-H., Macmillan J. B., and Chen Z. J. (2009) RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shimada T., Kawai T., Takeda K., Matsumoto M., Inoue J., Tatsumi Y., Kanamaru A., and Akira S. (1999) IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IκB kinases. Int. Immunol. 11, 1357–1362 [DOI] [PubMed] [Google Scholar]
- 47. Suzuki T., Oshiumi H., Miyashita M., Aly H. H., Matsumoto M., and Seya T. (2013) Cell type-specific subcellular localization of phospho-TBK1 in response to cytoplasmic viral DNA. PLoS One 8, e83639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Caldenhoven E., van Dijk T. B., Solari R., Armstrong J., Raaijmakers J. A., Lammers J. W., Koenderman L., and de Groot R. P. (1996) STAT3β, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 271, 13221–13227 [DOI] [PubMed] [Google Scholar]
- 49. Schaefer T. S., Sanders L. K., Park O. K., and Nathans D. (1997) Functional differences between Stat3α and Stat3β. Mol. Cell Biol. 17, 5307–5316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park O. K., Schaefer L. K., Wang W., and Schaefer T. S. (2000) Dimer stability as a determinant of differential DNA binding activity of Stat3 isoforms. J. Biol. Chem. 275, 32244–32249 [DOI] [PubMed] [Google Scholar]
- 51. Woo S.-R., Fuertes M. B., Corrales L., Spranger S., Furdyna M. J., Leung M. Y. K., Duggan R., Wang Y., Barber G. N., Fitzgerald K. A., Alegre M.-L., and Gajewski T. F. (2014) STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Corrales L., Glickman L. H., McWhirter S. M., Kanne D. B., Sivick K. E., Katibah G. E., Woo S.-R., Lemmens E., Banda T., Leong J. J., Metchette K., Dubensky T. W. Jr., and Gajewski T. F. (2015) Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 11, 1018–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Melillo J. A., Song L., Bhagat G., Blazquez A. B., Plumlee C. R., Lee C., Berin C., Reizis B., and Schindler C. (2010) Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J. Immunol. 184, 2638–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Morgenstern J. P., and Land H. (1990) Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Besser D., Bromberg J. F., Darnell J. E. Jr., and Hanafusa H. (1999) A single amino acid substitution in the v-Eyk intracellular domain results in activation of Stat3 and enhances cellular transformation. Mol. Cell Biol. 19, 1401–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lawrence C. L., and Baldwin A. S. (2016) Non-canonical EZH2 transcriptionally activates RelB in triple negative breast cancer. PLoS One 11, e0165005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.