

Abstract
Selenoproteins contain the amino acid selenocysteine (Sec), co-translationally inserted at a predefined UGA opal codon by means of Sec-specific translation machineries. In Escherichia coli, this process is dependent upon binding of the Sec-dedicated elongation factor SelB to a Sec insertion sequence (SECIS) element in the selenoprotein-encoding mRNA and competes with UGA-directed translational termination. Here, we found that Sec can also be efficiently incorporated at a predefined UAG amber codon, thereby competing with RF1 rather than RF2. Subsequently, utilizing the RF1-depleted E. coli strain C321.ΔA, we could produce mammalian selenoprotein thioredoxin reductases with unsurpassed purity and yield. We also found that a SECIS element was no longer absolutely required in such a system. Human glutathione peroxidase 1 could thereby also be produced, and we could confirm a previously proposed catalytic tetrad in this selenoprotein. We believe that the versatility of this new UAG-directed production methodology should enable many further studies of diverse selenoproteins.
Keywords: anticodon, Escherichia coli (E. coli), glutathione peroxidase, recombinant protein expression, selenocysteine, selenocysteine insertion sequence (SECIS), selenoprotein, thioredoxin reductase, transfer RNA (tRNA), translation release factor
Introduction
Selenocysteine (Sec; U)2 is referred to as the 21st naturally occurring amino acid. As its name suggests, it is structurally similar to cysteine (Cys), but with an atom of selenium taking the place of sulfur. Sec has unique biochemical properties, including a low pKa, a high reactivity with many electrophilic agents, and resistance to excessive oxidation (1, 2). Selenoproteins, containing one or several Sec residues, are found in all three domains of life (3–6). There are 25 selenoprotein-encoding genes in human and 24 in rodents (3), at least 15 different classes of selenoproteins described in different bacteria and archaea, and only a single selenoprotein (thioredoxin reductase) in Caenorhabditis elegans (4, 7). In addition, more than 300 distinct selenoprotein genes were found in samples from the Sargasso Sea (8), and more than 3600 selenoprotein genes could be derived from 58 selenoprotein families in data from the Global Ocean Sampling expedition (9). Escherichia coli has three selenoproteins, all of which are formate dehydrogenases expressed under different growth conditions (10). Mammalian selenoproteins can be categorized into two major groups, one group with Sec located close to the C terminus, such as in thioredoxin reductases (TrxRs), and the other group having Sec located in an N-terminal domain, often within a thioredoxin fold, such as in glutathione peroxidases (GPxs) (3, 6). Most, if not all, selenoproteins have enzymatic redox activities that employ the unique chemical reactivity of Sec during catalysis, and importantly, several mammalian selenoproteins are believed to be either essential or related to disease (11, 12). Synthetic selenoproteins can also be utilized for a number of novel applications (13–16). It is thus of both medical and biotechnological importance to characterize and understand selenoprotein function. Detailed studies of most selenoproteins have, however, remained hampered by major hurdles in their recombinant production, due to the unique features of Sec insertion during selenoprotein translation.
The position of Sec insertion into a selenoprotein during translation is encoded by a predefined opal termination codon UGA, which is redefined from a translational termination to a sense codon by highly intricate mechanisms involving many dedicated factors. This expansion of the genetic code requires a stem-loop structure in the mRNA, called a SECIS (Sec insertion sequence) element (17, 18). The selenoprotein synthesis machineries that accomplish Sec insertion can generally be divided into two main categories. Eukaryotes and archaea have similar machineries, whereas bacteria have a divergent pathway, among other aspects differing in the location and structure of the SECIS element in the selenoprotein mRNA. In bacterial selenoprotein mRNAs, a SECIS element is situated within the open reading frame next to the Sec-encoding UGA codon (19–21). In eukaryotes and archaea, however, other SECIS element structures are found in the untranslated region of the mRNAs (22–25). This difference makes direct selenoprotein expression from mammalian selenoprotein-encoding genes in E. coli impossible (26). Alternative approaches for production of selenoproteins include chemical methods (14, 15, 27) or the utilization of novel tRNA species compatible with elongation factor Tu that thereby bypass requirements for SelB and SECIS elements in E. coli (28–32). Yields and specificities in Sec insertion using these methods, however, still require improvements. Because of the fact that the Sec residue of mammalian TrxR is penultimately positioned close to the C-terminal end of the protein, it was found that a variant bacterium-type SECIS element could be engineered next to the UGA codon, without interfering with the coding region and thus enabling heterologous expression of Sec-containing TrxR1 in E. coli (33). Using that method for recombinant selenoprotein production, expression conditions were further optimized to yield an enzyme product with about half Sec contents and half being the result of UGA-mediated translational termination (34). Different attempts have been made to further increase the Sec contents of such produced enzyme but have thus far proven too expensive, laborious, or resulting in low yields (35–37). A second major limitation with the methodology is that only a few types of selenoproteins, such as TrxRs having a penultimate Sec residue, can be expressed with that approach of engineered SECIS elements, at least without introduction of mutations in the protein-encoding parts of the gene to be expressed (38). The inherent barrier of the bacterial selenoprotein biosynthesis machinery has thus remained difficult to breach for production of selenoproteins containing internal Sec residues.
In this study, we provide a novel strategy for recombinant selenoprotein production, employing SelB-mediated Sec insertion at a predefined UAG codon instead of the native UGA. We show that this approach significantly lessens translational termination as a competing event when utilized in the E. coli strain C321.ΔA having all its endogenous 321 UAG stop codons replaced by UAA and its UAG-specific release factor 1 (RF1) deleted (39). We furthermore found that SECIS dependence for Sec incorporation is thereby lessened, allowing recombinant expression of human GPx1 with native sequence as an enzymatically active selenoprotein. We trust that this new selenoprotein production methodology shall facilitate further progress in characterization of natural as well as synthetic selenoproteins, having major importance in several lines of research (14–16).
Results and Discussion
Amber UAG Codon Can Be Efficiently Utilized for Sec Insertion
Considering that the RF1-depleted E. coli strain C321.ΔA was recently developed as the first genomically recoded organism made to relieve the UAG amber stop codon for alternative usage (39), we wished to ask whether this strain could be employed for improved selenoprotein synthesis. First, we analyzed whether the UAG codon can be redefined as a Sec-defining codon using the endogenous E. coli selenoprotein synthesis machinery. In a first pilot study, we utilized a combination of four plasmids (Table 1), as follows: (i) the pET-TRSTER originally developed for rat TrxR1 expression using a natural Sec codon of UGA and a SECIS element compatible with the bacterial selenoprotein synthesis machinery (33); (ii) a novel pET-TRSUAG derivative having the Sec codon changed to UAG; (iii) the pSUABC plasmid providing additional Sec synthase (SelA), the Sec specialized elongation factor (SelB), and tRNASer[Sec] (SelC), shown earlier to result in improved Sec incorporation (33); and (iv) a novel pCDF-SelC2 plasmid providing a mutated SelC species with its anticodon changed for pairing with UAG. In combined use of these plasmids together with 75Se labeling, we could easily probe the efficiency of Sec insertion into TrxR1. It should be noted that the Sec residue in TrxR1 is the penultimate amino acid of a 55-kDa protein, which results in identical migration in SDS-PAGE of Sec-deficient as well as Sec-containing TrxR1 species (33). As shown in Fig. 1, TrxR1 could be produced as a 75Se-labeled selenoprotein in BL21(DE3) cells using a construct with a UGA codon defining Sec (lane 1) but not with UAG (lane 5), with efficiency in Sec insertion improved upon co-transformation with pSUABC (cf. lanes 1 and 2), also as shown earlier (33). Interestingly, upon co-transformation with pSUABC, there was also increased Sec insertion using a UAG codon (Fig. 1, lane 6), but the yield was further increased when the cells were also transformed with the pCDF-SelC2 plasmid carrying tRNASer[Sec](CUA) tailored to be fully compatible with a UAG codon (Fig. 1, lane 8). This mutated SelC product was also functional in the absence of pSUABC (Fig. 1, lane 7) but not in translation from a transcript carrying the native UGA codon for Sec (lane 3). Considering that all conditions had about the same expression level of the recombinant protein (see Ponceau S staining in Fig. 1, bottom panel), a similar level of 75Se-labeling in UGA decoding (lane 2) and UAG targeting (lane 8) revealed that UAG can efficiently be utilized as a Sec codon by a correspondingly tailored E. coli selenoprotein synthesis machinery. Therefore, we next analyzed this production approach using the C321.ΔA strain as a host cell, which is reported to lack translational termination at UAG codons due to its lack of RF1 (39).
TABLE 1.
Plasmids and E. coli strains used in this study
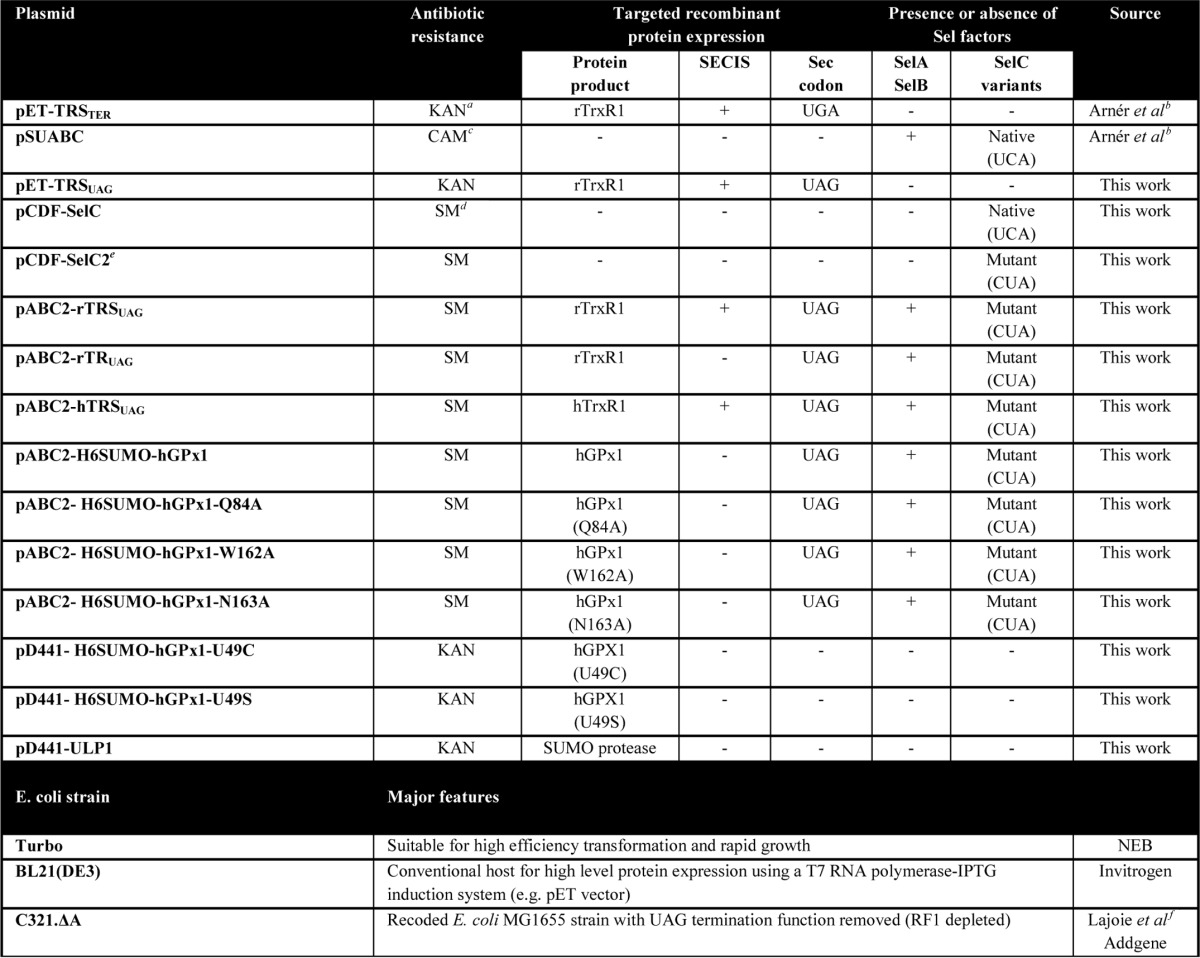
FIGURE 1.
Sec insertion into recombinant TrxR1 species using either UGA or UAG codons in combination with variants of the Sel gene products. Sec insertion is visualized here using autoradiography with SDS-PAGE (top) upon 75Se-radiolabeling of selenoproteins, with total protein loading visualized using Ponceau S staining (bottom). Lanes 1–4 show analyses of crude lysates of BL21(DE3) cells transformed with pET-TRSTER, having a UGA codon for Sec, and lanes 5–8 utilized pET-TRSUAG, having a UAG codon for Sec, in combination with pSUABC and/or pCDF-SelC2, as indicated in the figure. For further descriptions of plasmids, see the text and Table 1. Migration of recombinant TrxR1 is indicated to the right, as is that of chloramphenicol transferase (CAT) introduced by pSUABC plasmid. Size markers are indicated to the left and shown in kDa.
High Efficiency and High Fidelity TrxR1 Production Using UAG for Sec in an RF1-depleted Host
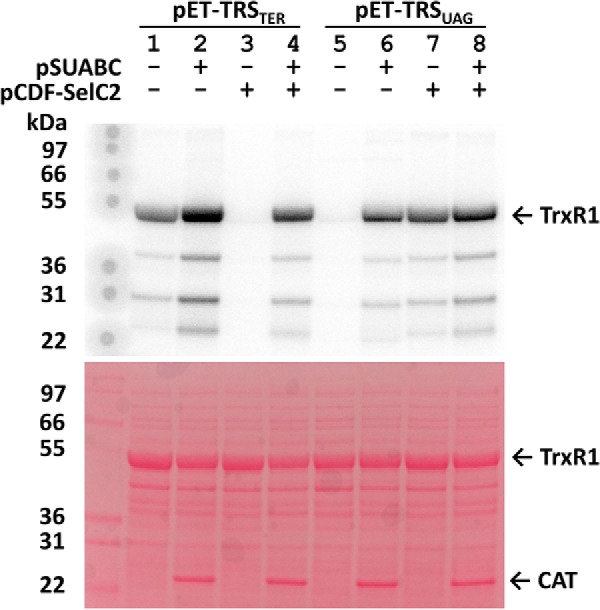
To avoid co-transformation with different plasmids, some of them incompatible with novel host strains, we first constructed a plasmid named pABC2-rTRSUAG. This plasmid contains the genes of selA, selB, and a mutated selC providing tRNASer[Sec](CUA), as well as an IPTG-inducible rat TrxR1 expression cassette containing a UAG codon for Sec and a SECIS element compatible with SelB (Table 1). Using pABC2-rTRSUAG for expression of rat TrxR1 in the RF1-depleted E. coli strain C321.ΔA, we found that this indeed resulted in high yields of TrxR1 with a very high enzymatic specific activity. Using our previously described methodology for recombinant TrxR1 production in E. coli and the classic purification scheme for the enzyme, we routinely obtain about 40 mg of purified rat TrxR1 from 1 liter of bacterial culture, with a specific activity of about 10–20 units/mg (33). This enzyme typically contains significant amounts of a two-amino acid truncated and Sec-deficient product explaining its lower activity compared with native enzyme (33, 40). With the novel pABC2-rTRSUAG plasmid used in C321.ΔA, we obtained about 20 mg of enzyme purified from 1 liter of bacterial culture, having 39.8 units/mg in specific activity. The specific activity of native TrxR1 purified from rat liver has about 35 units/mg (41), suggesting that our novel production of recombinant TrxR1 gave about the same, if not higher, activity as native enzyme. Producing recombinant human TrxR1 using the same strategy, we again obtained 20 mg of protein purified per liter of culture, and the specific activity of that enzyme was 29.8 units/mg. To assess Sec incorporation by a method other than activity measurements, which only infers the Sec content from the notion that fully active enzyme requires Sec, we analyzed the intact enzyme preparations with mass spectrometry. This revealed that the recombinant TrxR1 preparations displayed one dominant peak representing a full-length Sec-containing rat TrxR1 monomer (54,671 Da) as well as a full-length human variant (54,753 Da), with very little presence of truncated or other enzyme species (Fig. 2, green and red arrows). These findings showed that Sec insertion into recombinant TrxR1 is efficient also when tailored for decoding of an amber UAG codon, and that this approach when used in the RF1-depleted C321.ΔA host strain essentially obliterates translational termination, thereby resulting in nearly full Sec content in the final product. We next asked whether the SECIS element is still required for Sec insertion in this system.
FIGURE 2.
Production of Sec-containing TrxR1 using UAG for Sec validated with mass spectrometry. Utilizing expression with UAG targeting in C321.ΔA host cells, the purified enzymes were analyzed with electrospray mass spectrometry. These analyses revealed that rat TrxR1 (top) as well as human TrxR1 (bottom) were purified as dominant forms of intact Sec-containing enzymes (green arrows) and with very little UAG-truncated protein detected (red arrows). The x axes indicate the mass range analyzed.
SECIS-independent Sec Incorporation at UAG in the Absence of RF1
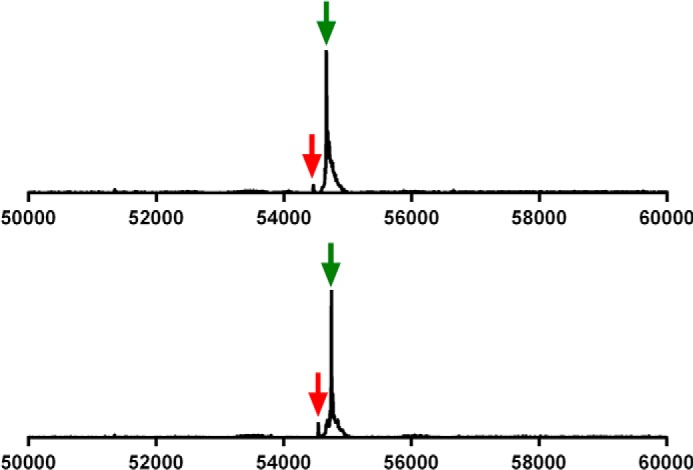
With the C321.ΔA cells lacking RF1, we hypothesized that SelB might facilitate incorporation of Sec at a UAG codon even in the absence of a SECIS element. If this would occur, even with lower efficiency, it would constitute a significant achievement in the field of recombinant selenoprotein production, because no SECIS element constructs would then be required in the mRNA. This would enable production also of recombinant selenoproteins with internal Sec residues. To test whether a selenoprotein could be produced using UAG in the C321.ΔA strain in the absence of a SECIS element, we first removed the SECIS element from the pABC2-rTRSUAG plasmid, making pABC2-rTRUAG. We compared the result with production of either a Sec-to-Cys mutant of TrxR1 (U498C), shown earlier to have about 1–10% activity of wild type enzyme depending upon choice of substrate (42), or a Sec-to-Ser mutant (U498S) expected to be essentially devoid of activity for most substrates (42). The activity of the new preparation of TrxR1 was about 15-fold higher than that achieved with the U498C mutant and at least 100-fold higher than that of the U498S mutant in a thioredoxin-linked insulin reduction assay (Fig. 3). The specific activity of TrxR1 produced with pABC2-rTRUAG was 4 units/mg, suggesting that Sec-containing TrxR1 species were indeed produced, as with pABC2-rTRSUAG, but at lower efficiency than upon inclusion of a SECIS element. A mass spectrometry analysis confirmed that this was the case, revealing the presence of two other unexpected dominant enzyme species in addition to the Sec-containing enzyme (54.671 Da, Fig. 4, green arrow). One of these forms corresponded, surprisingly, to UAG-truncated TrxR1 (54,464 Da, Fig. 4, red arrow), hence produced although RF1 was not present. Perhaps this truncation was RF2-mediated, especially as stoichiometry between ribosomal protein L11, RF1 and RF2, known to affect termination efficiency (43), is considerably distorted in these cells. The other peak corresponded in mass to a Sec-to-Gln and/or Sec-to-Lys variant (54,649 Da, Fig. 4, blue arrow). Although these findings showed that the SECIS element is required to obtain nearly complete Sec incorporation at high yields (Fig. 2), it also showed that significant amounts of Sec-containing enzyme can indeed be produced in the absence of both a SECIS element and RF1. As this fact could open the possibility to produce selenoproteins with internal Sec residues, we next attempted production of the human selenoprotein GPx1.
FIGURE 3.
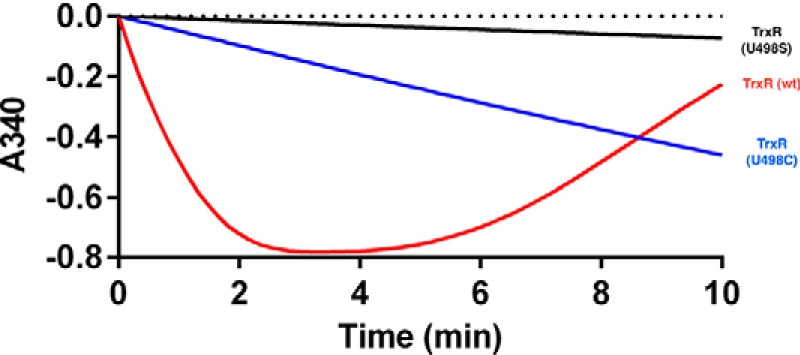
Enzymatically active TrxR1 can be produced in E. coli without use of a SECIS element. Rat TrxR1 was purified from C321.ΔA host cells harboring the pABC2-rTRUAG plasmid with a UAG codon for Sec but lacking an engineered SECIS element. Using 45 nm TrxR1 produced with this approach, its activity was measured with a thioredoxin-linked insulin reduction assay, in which consumption of NADPH was followed as a decrease in absorbance at 340 nm (red curve). As reduced insulin is accumulated it precipitates, explaining increased A340 at later time points. As comparison, activities were also determined using equal concentrations (45 nm) of purified TrxR1 variants having a Sec-to-Cys substitution (U498C, blue curve) or Sec-to-Ser substitution (U498S, black curve). Calculated turnover numbers were 241.3, 16.8, and 2.6 min−1 for the three TrxR1 species, respectively. This should be compared with turnover of fully Sec-containing rat TrxR1 (Fig. 2, top panel) calculated to be 3568 min−1.
FIGURE 4.
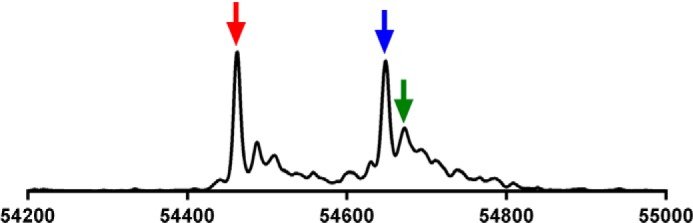
TrxR1 produced using UAG for Sec in absence of both RF1 and a SECIS element resulted in a mixture of Sec-containing enzyme, UAG-truncated variant, and additional forms presumed to be Sec-to-Gln or Sec-to-Lys substituted variants. Shown is an electrospray mass spectrometry analysis of rat TrxR1 produced using the pABC2-rTRUAG plasmid in C321.ΔA host cells. The three major species of enzyme detected corresponded in size to a native Sec-containing variant (green arrow), UAG-truncated enzyme (red arrow), and a variant likely corresponding to Sec-to-Gln- or Sec-to-Lys-substituted forms (blue arrow). See text for further discussion.
Production of Sec-containing Enzymatically Active Human GPx1
Enzymatic activity of the hereby produced and purified recombinant human GPx1 was determined using a GR-coupled assay with H2O2 as GPx1 substrate, which revealed significant GPx1 activity (Fig. 5A). Because of the peculiar kinetics of GPx that do not reach substrate saturation (44), regular Michaelis-Menten kinetic parameters cannot be determined. Under our assay conditions the specific activity was 10 units/mg. Electrospray mass spectrometry analysis of the intact enzyme preparation (Fig. 5B) revealed clear peaks representing a Sec-containing wild type GPx1 (22,088 Da) as well as Sec-to-Gln and/or Sec-to-Lys species of GPx1 (22,066 Da). There were also peaks corresponding in mass to oxidized species of the wild type enzyme. In-gel tryptic digestion followed by peptide mapping confirmed that both Sec-to-Gln and Sec-to-Lys species were present, as Lys introduced a new cleavage site for trypsin producing a unique shorter peptide, whereas the Sec-to-Gln containing peptide was also found (Fig. 5C). Both of these mass spectrometry analyses agreed with an estimation from the enzymatic activity assay that the Sec-containing GPx1 enzyme constituted about 20–30% of the purified enzyme species. This preparation is thus not of the same homogeneous quality as TrxR1 produced above in the presence of a SECIS element or as expected of native GPx1 purified from mammalian tissue. UAG-directed Gln suppression has also been reported earlier (45–47). The hereby enabled recombinant production methodology nonetheless gives the possibilities for new types of studies, such as evaluating the effects of point mutations within a selenoprotein scaffold. We next determined to use this technique to probe the role of potential key residues, apart from the Sec, that can be important for maintained activity of human GPx1.
FIGURE 5.
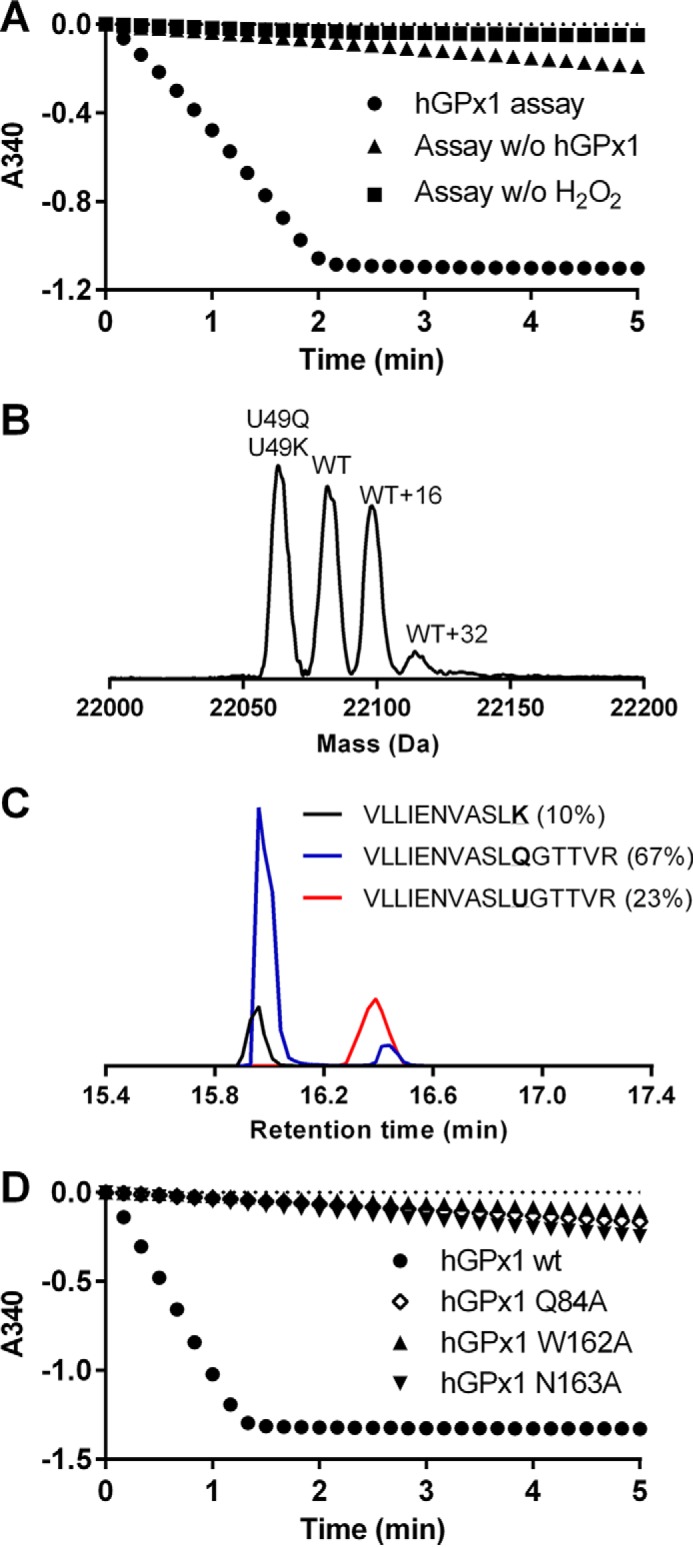
Production of enzymatically active human GPx1 and validation of its active site tetrad. A, enzymatic activity of recombinant human GPx1 (1 μm, circles) was assessed through the decrease of absorbance at 340 nm over time upon addition of H2O2 (0.25 mm) to a cuvette containing GR (1 unit/ml), GSH (1 mm), and NADPH (0.2 mm). Control reactions were either without hGPx1 (triangles) or without H2O2 (squares). B, electrospray mass spectrometry analysis of human GPx1 produced in C321.ΔA host cells was performed. The major peaks are annotated according to the enzyme species as inferred from their molecular weights, including a combination of Sec-to-Gln- and Sec-to-Lys-substituted variants (U49Q, U49K), a Sec-containing wild type GPx1 form (WT), as well as potentially oxidized forms of wild type GPx1 (WT+16 and WT+32). C, results of LC/MS mass spectrometry analyses of tryptic digests of the GPx1 preparation used in B are shown, confirming the existence of a Sec-containing peptide (red curve), a Sec-to-Gln peptide (blue curve), and a Sec-to-Lys peptide (black curve; Lys serves as a cleavage site for trypsin, hence resulting in a shorter peptide in this analysis). The sequences of the peptides are also indicated, and their ratios in percentages as indicated in parentheses were calculated from the area under the curve. D, similar assay was performed as shown in A, using either the wild type GPx1 preparation (circles) or the Q84A, W162A, and N163A variants (each at 1 μm), as indicated.
Catalytic Tetrad of Human GPx1 Validated Using Recombinant Selenoprotein Variants
It was previously suggested that the enzyme activities of different GPxs depend upon an active site tetrad involving Gln-84, Trp-162, and Asn-163 in addition to Sec49, with this numbering referring to the sequence of human GPx1 (44, 48). Such tetrad configuration has thus far only been possible to experimentally address for GPx from Drosophila melanogaster (DmGPx), because the active site of DmGPx naturally has a Cys residue in place of Sec, thus allowing for introduction of mutations of other residues in the tetrad while keeping the native catalytic Cys residue intact (48). It could well be, however, that the tetrad would only be required in Cys-dependent GPxs, whereas the presence of a more reactive Sec residue in the active site might obliterate the need for such a tetrad. This notion has so far not been tested because Sec-containing GPxs have remained difficult to produce in recombinant form. We therefore here expressed human GPx1 as a selenoprotein, using replacement with an alanine for each of the three amino acids supposed to make up the tetrad together with Sec49, thereby producing Q84A, W162A, and N163A variants in a human Sec49-containing GPx1 scaffold. When determining GPx activities with H2O2 as substrate, all of these variants displayed very little activity above background compared with the native enzyme, thereby strongly suggesting that the previously proposed active site tetrad of GPxs is essential for activity also in Sec-containing GPx variants (Fig. 5D).
Summary of Yield and Specificity in Sec Insertion
The results obtained here reveal that Sec insertion can be efficient at UAG codons, provided that the SelC anticodon is altered to become compatible with UAG. If expression with such system is performed in the RF1-depleted C321.ΔA host strain, efficiency is greatly increased and results in production of TrxR1 species with nearly full Sec contents. We also found that the SECIS element is no longer absolutely required for SelB-mediated Sec insertion in this system, which enabled production of human Sec-containing GPx1. However, in this case the specific activity was lower due to a combination of UAG-directed translational termination and suppression with Gln and Lys. Assessing yields, specific activities, and mass spectrometric analyses of the recombinant purified enzyme products, we can estimate the efficiency of the different production systems, as illustrated in Fig. 6. With production of TrxR1 using the previously developed method of a tailored SECIS element and expression in regular BL21(DE3) hosts, our yield of recombinant protein is ∼40 mg/liter bacterial culture with an estimated 30–50% Sec content, 50–70% UGA-directed truncation, and little suppression of the UGA with other amino acids (Fig. 6A). Here, we found that the UGA could be changed to UAG, provided that the Sec anticodon was correspondingly changed to CUA, resulting in a similar result (Fig. 6B). Additional codon-anticodon variations have been reported, but have not yet been analyzed at depth regarding yields and efficiencies in final products of recombinant selenoproteins (29, 49). Importantly, here we found that if UAG-directed TrxR1 production is performed in the RF1-depleted C321.ΔA host strain with a maintained variant SECIS element, almost full Sec content is achieved due to the fact that termination no longer occurs (Fig. 6C). As we found that the SECIS element was no longer required under such conditions, TrxR1 and, importantly, GPx1 could thereby also be produced, albeit with lower yields. Under such conditions the purified enzymes become mixtures of the native selenoprotein with a truncated form as well as UAG suppression with Gln/Lys for TrxR1, or only Sec insertion or Gln/Lys suppression in the case of GPx1, purified at lower but still useful yields for subsequent kinetics determinations (Fig. 6D). To conclude, here we could produce human and rat TrxR1 as selenoproteins with nearly full Sec contents at a yield of about 20 mg/liter bacterial culture (Fig. 6C), and human GPx1 with about 15–30% Sec contents at a yield of about 1–2 mg/liter bacterial culture (Fig. 6D). In Table 2 we compare the features of our method as developed here with the other recently published methods for expression of heterologous selenoproteins in E. coli that were mentioned in the Introduction.
FIGURE 6.
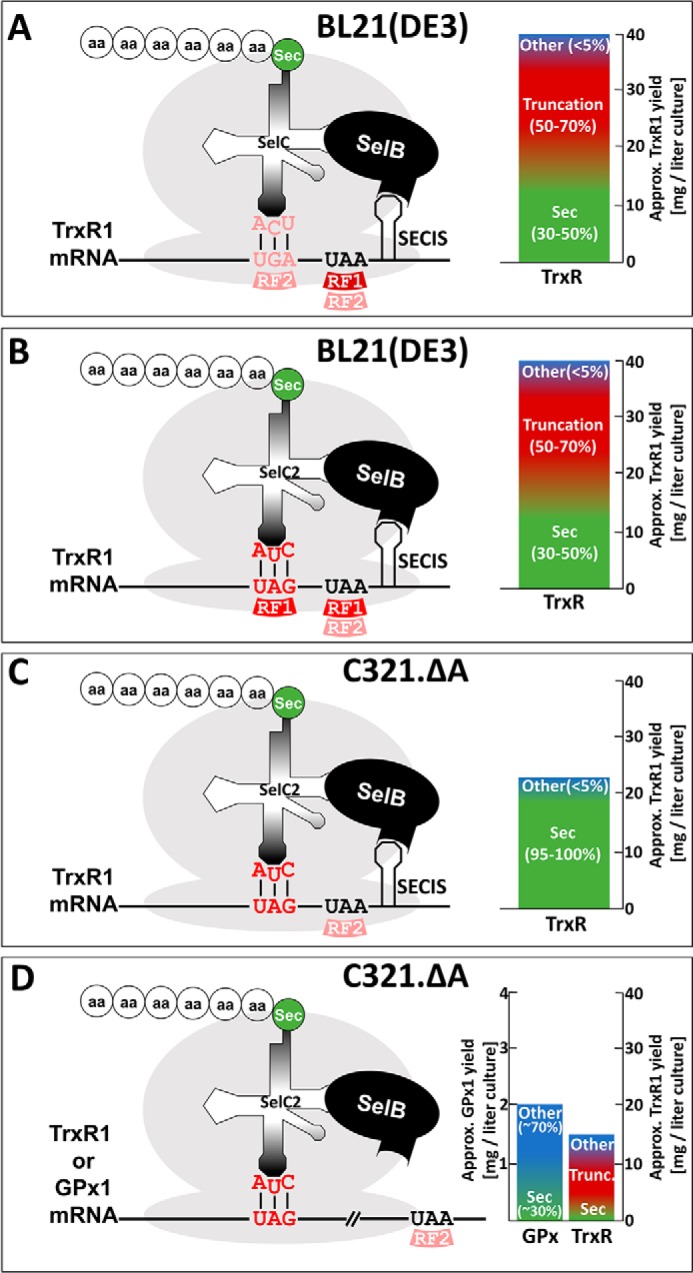
Schematic illustration of constructs, efficiencies, and yields from different systems of recombinant selenoprotein production as studied here. The schemes graphically summarize the principles for recombinant selenoprotein production using different E. coli host strains with alternative codon-anticodon combinations with or without RF1 and RF2, in the presence or absence of an engineered SECIS element, as discussed in the text and indicated in the figures. Shown are also the approximate final yields of selenoproteins purified from the different conditions, with Sec-containing variants shown in green, truncated in red, and suppression of the Sec codon with other amino acids such as Gln or Lys indicated in blue. A, expression of TrxR1 in BL21(DE3) with an engineered SECIS element and UGA for Sec is shown. B, results of the same expression of TrxR1 as in A is shown, but using UAG as the Sec codon together with a corresponding mutation of the SelC anticodon. C, results are shown using the same principle as in B, but changing from BL21(DE3) to the RF1-depleted C321.ΔA host strain. D, results producing TrxR1 or GPx1 are summarized, as obtained using the principle given in C, but upon removal of the SECIS element. See text for further details.
TABLE 2.
Comparison of the method developed here with other approaches for expression of heterologous selenoproteins in E. coli
FDH is formate dehydrogenase; EF-Tu is elongation factor Tu.
| Approach | Selenoproteins expressed | Proof of Sec incorporation | Specificity in Sec incorporation | Total yield of selenoprotein | Competing events reported | Refs. |
|---|---|---|---|---|---|---|
| 16S rRNA mutations for improved Sec insertion efficiency at the ribosome | Luciferase reporter downstream of SECIS element, endogenous FDH | Luciferase activity of reporter, benzyl viologen assay, for endogenous FDH activities | Best mutants, ≈30% | Unclear, not quantified | UGA truncation, Trp-mediated UGA suppression | Thyer et al. (31) |
| Synthetic tRNAUtu species for EF-Tu-mediated Sec insertion at either UGA or UAG codons | Endogenous FDH, GPx1 or Sec-substituted Grx1 | Benzyl viologen assay for endogenous FDH, mass spectrometry of Grx1, enzyme activities of Grx1 and GPx1 | ≈50–65% (Sec-Grx1 vs. Ser-Grx1 or Sec-GPx1 vs. Ser-GPx1), double activity of Sec-Grx1 vs. Cys-Grx1, GPx1 ≈6 units/mg | Unclear, not quantified | Ser-mediated suppression | Aldag et al. (28) |
| SelB-mediated suppression of alternative codons using tRNASec variants with altered anticodon sequences | Endogenous FDH, human TrxR1 | Benzyl viologen assay for endogenous activities of FDH and specific activities of purified FDH, mass spectrometry, specific activity of TrxR | FDH, 12–100% depending upon codon usage; TrxR ≈95–98% | FDH, up to 12-fold levels of endogenous wild type FDH; TrxR unclear, not quantified | Unclear, codon-dependent | Bröcker et al. (29) |
| Engineered tRNASec for EF-Tu-mediated Sec insertion at UAG, also tested in the RF1- depleted C321.ΔA host | Sec-substituted NMC-A β-lactamase, endogenous FDH, Sec-substituted DHFR, Sec-substituted azurin, human GPx1 | Antibiotic resistance due to functional β-lactamase, benzyl viologen assay for endogenous FDH, mass spectrometry | Sec-DHFR, ≈100%, other selenoproteins produced unclear specificity | Unclear, not quantified | Ser-mediated suppression | Thyer et al. (32) |
| Further engineered tRNASec for EF-Tu-mediated Sec insertion at UAG with lower Ser-mediated suppression | Endogenous FDH, Sec-substituted Grx1 | Benzyl viologen assay for endogenous FDH, specific activities of FDG and Sec-substituted Grx1, mass spectrometry | FDH, ≈70%; Grx1, ≈100% | Unclear, not quantified using expression in vivo; in cell-free systems ≈2–80 ng obtained per reaction depending upon construct | Ser-mediated suppression | Miller et al. (30) |
| UAG-directed Sec insertion in RF1-depleted host, with or without bacterial SECIS element | Rat and human TrxR isoenzymes, human GPx1 | Enzyme activities and mass spectrometry | With SECIS, ≥95%. No SECIS, ≈30%. TrxR1 ≈35–40 units/mg. GPx1 ≈10 units/mg | With SECIS, ≈20 mg/liter. No SECIS, 1–3 mg/liter | UAG-directed truncation, Gln- and Lys-mediated suppression | This work |
We believe that our method for production of TrxR1 with full Sec contents at high yields can now be widely employed for production of the wide variety of selenoproteins in nature that carry a penultimate or ultimate Sec residue (3, 4, 8, 24). Our method for recombinant GPx1 production, or of other selenoproteins with internal Sec residues, should preferably be further improved in terms of Sec insertion efficiency using measures to minimize Gln or Lys suppression of the UAG. Still, we conclude that our new methodology should constitute a powerful resource enabling many forthcoming studies of recombinant selenoproteins, considering its versatility and comparable ease of use.
Experimental Procedures
Plasmids and E. coli Strains
The E. coli strains and plasmids developed and used in this study are listed in Table 1. The RF1-depleted E. coli strain C321.ΔA strain developed by others (39) and used here for selenoprotein production was obtained from Addgene (catalogue no. 488998). The E. coli strain Turbo used for routine cloning and non-selenoprotein production was obtained from New England Biolabs. The pD441 plasmid providing IPTG-inducible protein expression cassette as well as a high copy number replication origin was obtained from DNA2.0. The pCDF plasmid used to provide antibiotic resistance cassette was obtained from Novagen. Rat thioredoxin reductase ORF was obtained from pET-TRSTER (33) with its Sec codon mutated to TAG. The selA, selB, and selC genes, including their native E. coli promoters, were obtained from plasmid pSUABC (33). A series of cloning procedures gave the final construct named pABC2-rTRSUAG (Fig. 7) for production of rat thioredoxin reductase. XbaI and EagI restriction sites were constructed in this plasmid to be used for insert exchange to make expression vectors for other selenoprotein variants. Utilized primers are given in Table 3. The gene for human TrxR1 was synthesized (GeneArt, Table 4) with codons optimized for expression in E. coli and cloned into the pABC2 vector, which resulted in plasmid pABC2-hTRSUAG. The gene for human GPx1 was synthesized (DNA2.0, Table 4) with codons optimized for expression in E. coli and, in addition, introducing a His-tagged SUMO tag (H6SUMO) to the N-terminal end of hGPx1, with this fragment subsequently cloned into the pABC2 vector, resulting in plasmid pABC2-H6SUMO-hGPx1. Several mutants of these constructs were constructed using conventional cloning methods (Table 1). The plasmids for expressing wild type thioredoxin reductases, glutathione peroxidase, and its Sec-containing mutants were transformed into the C321.ΔA strain. All other plasmids were transformed into the New England Biolabs Turbo strain.
FIGURE 7.
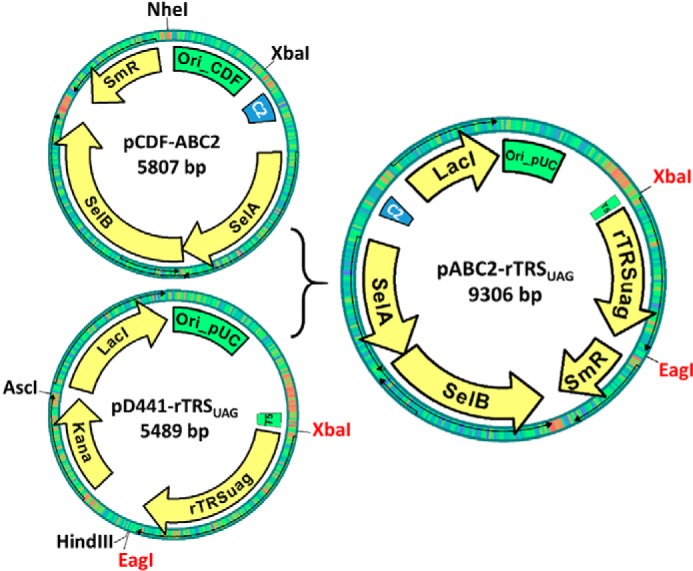
Simplified schemes of key plasmids used in this study. The pABC2-rTRSUAG plasmid was made from pCDF-ABC2 and pD44a-rTRSUAG as indicated in the figure, with key functional elements indicated. See description of experimental procedures for further details.
TABLE 3.
Primer sequences
| Primers | Sequence (5′–3′) |
|---|---|
| P1 | CTAGGGCTAATAATCGGTTGCAG |
| P2 | GATTATTAGCCCTAGCATCCGGACTGGAGG |
| P3 | CTCTAAATCCAGTTGGGGCCGCC |
| P4 | CAACTGGATTTAGAGTCCAGCCGCCTCACCG |
| P5 | CATAGGCTAACGATTGATTATTAGCCCTAGCATC |
| P6 | AATCGTTAGCCTATGCGGCC |
| P7 | GAAAACGCCAAAAACGAAGAAATTCTG |
| P8 | GTTTTTGGCGTTTTCTGCATGGCCAAACTGATTACACGG |
| P9 | TTTGAGAAATTTCTGGTTGGTCC |
| P10 | CAGAAATTTCTCAAAGTTTGCTGCAACATCATTACGACAA |
| P11 | CAGAAATTTCTCAAATGCCCATGCAACATCATTACGACAA |
| P12 | CAGGCTTGCAACATTTTCAATCAGC |
| P13 | AATGTTGCAAGCCTGTGCGGCACCACCGTTCGTGATTATA |
| P14 | AATGTTGCAAGCCTGAGCGGCACCACCGTTCGTGATTATA |
| P15 | CTCGAAAATAATAAAGGGAAAATCAG |
| P16 | CACGGCCGAAGCTTAGACTCGAGCTCGGATCCTTATTATTTTAAAGCGTCGGTTAAAATC |
TABLE 4.
Sequences of synthetic genes
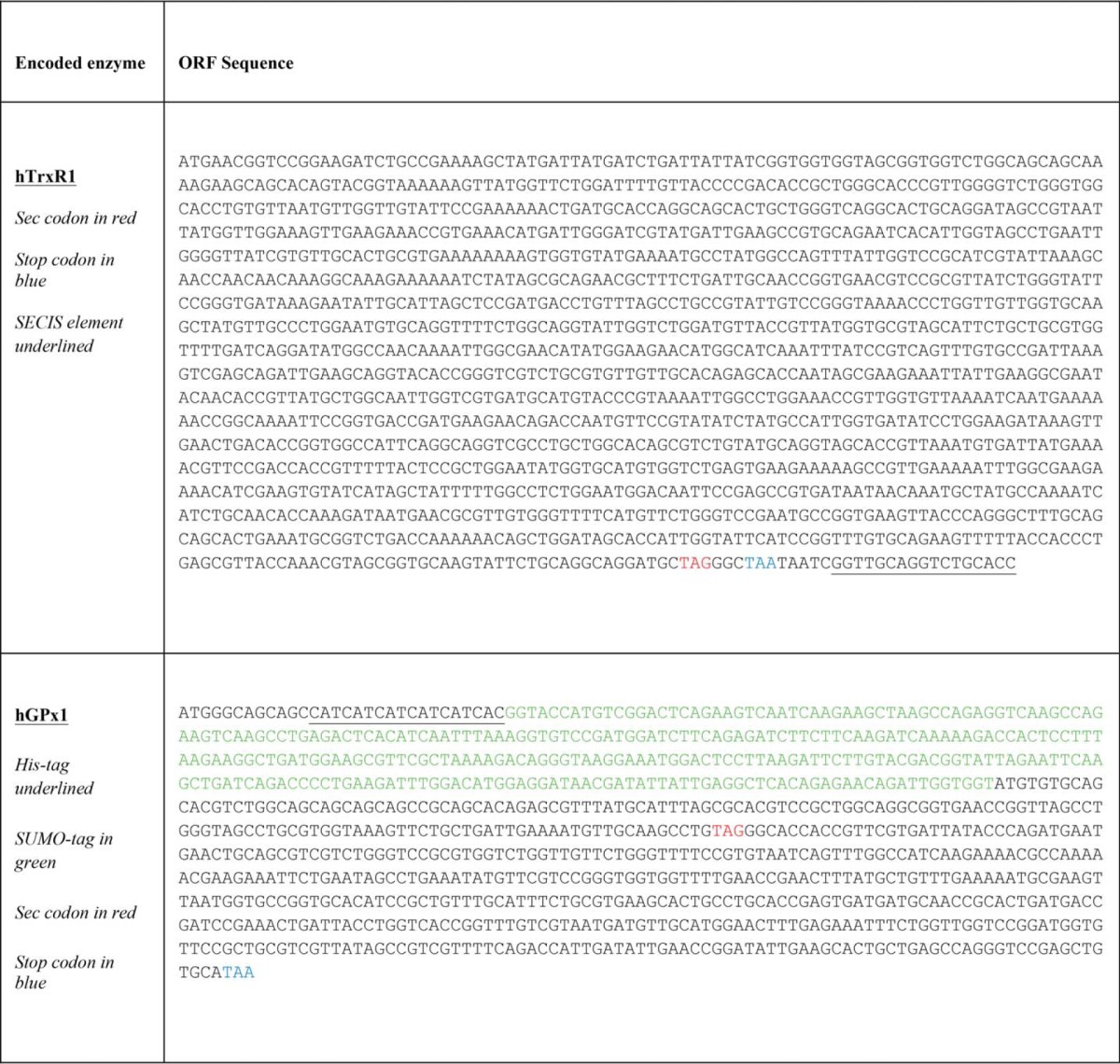
Cloning Procedures
All enzymes used in molecular cloning were obtained from Thermo Fisher Scientific if not specified otherwise.
The Sec codon in pET-TRSTER was point mutated to TAG using primer pair P1 and P2 to generate pET-TRSUAG. The insert was then transferred into pD441-NC (DNA2.0) to generate pD441-rTRSUAG. The selA, selB, and selC genes, including their native E. coli promoters, were obtained from plasmid pSUABC. Briefly, the selC gene was first obtained from pSUABC by XbaI/EcoRI digestion and transferred into plasmid pUC18 (Thermo Fisher Scientific) at the same restriction site. The resulting plasmid pUC-SelC was digested again with XbaI and Acc65I, and the selC containing fragment was ligated into plasmid pCDF-1b (Novagen) at the same restriction site to generate pCDF-SelC. The Sec anticodon (UCA) in this plasmid was changed to CUA using primer pairs P3 and P4, which yielded the plasmid pCDF-SelC2. The selA- and selB-encoding operon was isolated from pSUABC by HindIII/NdeI digestion and ligated into plasmid pET-20b (Novagen) at the same sites. The resulting plasmid pET20b-SelAB was first digested with XbaI and blunted by DNA polymerase I, large (Klenow) fragment, after which the linearized plasmid was digested with HindIII to generate a selA- and selB-containing fragment with one blunt end (from XbaI) and one sticky end (from HindIII). This fragment was subsequently ligated into pCDF-SelC2 with similar restriction enzyme treatment between its XhoI (blunted) and HindIII (remained sticky) sites to generate the pCDF-ABC2 vector. The sequence of this plasmid was verified by primer-walking sequencing (GATC Biotech).
Next, the pCDF-ABC2 was digested with NheI and XbaI, and the large fragment was blunted using DNA polymerase I, large (Klenow) fragment. Similarly, pD441-rTRSUAG was digested with HindIII and AscI with the large fragment recovered and blunted. The two fragments were then ligated to generate pABC2-rTRSUAG (Fig. 7). The orientation and exact sequence of the new plasmid were confirmed by primer-walking sequencing (GATC Biotech). The remaining XbaI and EagI sites in the vector (Fig. 7, highlighted in red) were used for insert exchange to make expression vectors for other selenoprotein variants. The SECIS element sequence was removed from pABC2-rTRSUAG using primer pair P5 and P6, which resulted in plasmid pABC2-rTRUAG. The gene for human TrxR1 was synthesized by a commercial service (GeneArt, Table 4) with codons optimized for expression in E. coli and cloned into the pABC2 vector between XbaI and EagI, which resulted in plasmid pABC2-hTRSUAG.
The gene for human GPx1 was synthesized by a commercial service (DNA2.0, Table 4) with codons optimized for expression in E. coli and, in addition, introducing a His-tagged SUMO tag (H6SUMO) to the N-terminal end of hGPx1, with this fragment subsequently cloned into the pABC2 vector between XbaI and EagI, resulting in plasmid pABC2-H6SUMO-hGPx1. The three hGPx1 active site mutants with intact Sec residues were constructed using primer pair P7 and P8 for pABC2-H6SUMO-hGPx1-Q84A, primer pair P9 and P10 for pABC2-H6SUMO-hGPx1-W162A, and primer pair P9 and P11 for pABC2-H6SUMO-hGPx1-Q84A, respectively. The H6SUMO-hGPx1 insert was also cloned into plasmid pD441-NC between XbaI and EagI, and the Sec residue was mutated to either Cys using primer pair P12 and P13 or Ser using primer pair P12 and P14, which resulted in plasmids pD441-H6SUMO-hGPx1-U49C and pD441-H6SUMO-hGPx1-U49S, respectively. The cDNA for His-tagged ULP1 (SUMO protease) was kindly provided by Dr. David L. Williams (Rush University Medical Center) and transferred into the pD441 vector using primer pair P15 and P16, resulting in plasmid pD441-ULP1.
Recombinant Protein Expression and Purification
The E. coli C321.ΔA strain is ampicillin-resistant (Table 1), and its growth is impaired above 34 °C (39), so all cultures using this strain were grown at 30 °C or lower, with supplementation of ampicillin (100 μg/ml) in addition to other antibiotics as dictated by the use of plasmids (Table 1).
The plasmids for expressing wild type thioredoxin reductases, glutathione peroxidase, and its Sec containing mutants, including pABC2-rTRSUAG, pABC2-rTRUAG, pABC2-hTRSUAG, pABC2-H6SUMO-hGPx1, pABC2-H6SUMO-hGPx1-Q84A, pABC2-H6SUMO-hGPx1-W162A, and pABC2-H6SUMO-hGPx1-N163A, were transformed into the C321.ΔA strain. The plasmids for expression of two selenocysteine mutants of GPx1, pD441-H6SUMO-hGPx1-U49C and pD441-H6SUMO-hGPx1-U49S, were transformed into the New England Biolabs Turbo strain.
For recombinant protein production, a single colony of the corresponding transformant was inoculated into 10 ml of modified LB media (10 g of tryptone, 10 g of yeast extract, and 10 g of NaCl per liter) and incubated at 30 °C overnight with shaking (250 rpm). The overnight culture was then transferred into 2 liters of LB media. Incubation was then continued with agitation at 30 °C until A600 reached 0.7. At that point, 5 μm selenite, 5 mm l-Cys, and 0.5 mm IPTG were added into the culture, and incubation was then continued at 24 °C overnight.
The bacteria were harvested by centrifugation at 5000 × g for 15 min and suspended in binding buffer (TE buffer: 50 mm Tris-HCl, 2 mm EDTA (pH 7.5) for TrxR and its variants; 50 mm Tris-HCl, 250 mm NaCl, and 20 mm imidazole for GPx1 and its variants). Subsequently, the bacteria were lysed using a French press cell disruptor (Thermo Fisher Scientific). Lysates were then centrifuged at 18,000 rpm at 4 °C for 30 min, and the supernatants were subjected to affinity chromatography purification.
Thioredoxin reductase variants were purified over 2′,5′-ADP-Sepharose (GE Healthcare) followed by size exclusion chromatography on an ÄKTAExplorer 10 FPLC system equipped with a HiLoad 16/60 Superdex 200-pg gel filtration column (GE Healthcare) as described previously (35). Purified protein was concentrated and stored in TE buffer containing 50% glycerol for long term storage at −20 °C. The protein concentration for TrxR1 was determined spectrophotometrically by FAD absorption at 463 nm (ϵ = 13,600 m−1 cm−1, one FAD presumed to correspond to one TrxR subunit).
All GPxs were made with an N-terminal His-tagged SUMO tag (H6SUMO), whereby the H6SUMO-GPx variants were first purified over IMAC (ÄKTAExplorer 10 FPLC equipped with a 5-ml HisTrap FF column, GE Healthcare). To the subsequently eluted fractions containing H6SUMO-GPx, 15 μg/ml His-tagged ULP1 (SUMO protease) was added, and the protein mixture was kept at 4 °C overnight with gentle shaking to cleave the H6SUMO tag. The excessive imidazole in the digestion mixture was subsequently removed using a NAP-25 desalting column (GE Healthcare), and the protein mixture was subjected to IMAC for a second time. The non-cleaved H6SUMO-GPx, cleaved H6SUMO tag, His-tagged ULP1, as well as any impurity obtained from the first IMAC thereby bound to the nickel column, whereas cleaved non-tagged GPx was collected from the flow through. This purified protein was concentrated and stored in TE buffer containing 50% glycerol for long term storage at −20 °C. The protein concentration was determined by Bradford Protein Assay (Bio-Rad).
Expression and purification of His-tagged ULP1 were done in a manner similar to the purification of H6SUMO-GPx. After the first IMAC procedure, the His-tagged ULP1 was concentrated and stored in 50 mm Tris-HCl (pH 7.5), 100 mm NaCl, 1 mm DTT, and 50% glycerol that was kept at −20 °C. The protease was fully active in the presence of 150 mm imidazole so it was added directly into the eluted H6SUMO-tagged protein without changing buffers. It should be noted that ULP1 does not leave any residues in the cleaved target protein, which was thus purified as a non-tagged protein without any mutation at the N terminus.
Enzyme Activity Measurements
Different protocols for enzyme activity assays were used as described previously (51, 52). Briefly, direct NADPH-dependent reduction of 5,5′-dithiobis(2-nitrobenzoic) acid (DTNB) was used to determine specific activities of different TrxR1 preparations. This assay is based upon the reduction of one DTNB molecule into two 5-thio-2-nitrobenzoic acid anions (TNB−) by TrxR under consumption of NADPH. TNB− has a strong absorbance at 412 nm, and the reaction velocity is calculated as micromoles of DTNB reduced per min (U) from the extinction coefficient for TNB− (13,600 m−1 cm−1). The specific activity of the enzyme is given as units/mg taking into consideration the enzyme amount used in the assay (53). The alternative insulin-coupled Trx reduction assay is more specific and was used to further verify Sec-dependent activities of TrxR1, following consumption of NADPH through decreased absorbance at 340 nm, also resulting in insulin precipitation as the assay progresses (53). A GR-coupled assay was used to measure GPx activities. In this assay, hydrogen peroxide (H2O2) was used as substrate for GPx, which oxidizes reduced glutathione (GSH) to its disulfide form (GSSG) that is subsequently recycled by GR using NADPH. The assay is therefore based upon measurement of decreased absorbance at 340 nm due to NADPH consumption. Units for GPx are defined as micromoles of NADPH consumed per mg of GPx under our assay conditions using NADPH′s extinction coefficient of 6200 m−1 cm−1, with GPx concentration determined using Protein Assay from Bio-Rad.
Mass Spectrometry
The mass spectrometry analyses were performed at the Science for Life Laboratory Mass Spectrometry-based Proteomics Facility in Uppsala, Sweden. Briefly, aliquots with purified proteins (TrxR, GPx, and their mutants) were prepared in 20% acetonitrile and 1% formic acid (v/v). The samples were subsequently diluted 10-fold and injected on a C4 column, separated in reversed phase using an 8-min-long gradient, and electrosprayed on line to a LC/MSD TOF MS (Agilent Technologies). The detected mass-to-charge-ratios were deconvoluted into intact masses applying the MassHunter software (Agilent). The purified wild type human GPx1 was also digested in-gel by trypsin according to a standard operating procedure. Thereafter the resulting peptides were dried in a SpeedVac system and resolved in 15 μl of 0.1% formic acid (v/v). The peptides were separated in reversed phase on a C18 column and electrosprayed on line to an LTQ-Orbitrap Velos Pro ETD MS (Thermo Finnigan). Tandem mass spectrometry was performed applying collision-induced dissociation. The data were searched using the Sequest algorithm embedded into Proteome Discoverer 2.1 (Thermo Fisher Scientific) against the protein sequence for human glutathione peroxidase 1 as well as its mutant sequences. The search criteria for protein identification were set to at least two matching peptides of 95% confidence level per protein.
Selenium-75 (75Se) Labeling to Visualize Recombinant Selenoproteins
Selenium-75 labeling was used to visualize selenium incorporation under different conditions of the recombinant selenoprotein expression systems. Briefly, [75Se] selenite (University of Missouri Research Reactor Center) was added together with the addition of IPTG to the bacterial cultures to enable visualization of de novo synthesis of [75Se]Sec using [75Se]selenite as the selenium source. Consequently, [75Se]Sec is incorporated into the nascent selenoprotein with the intensity in radiolabeling reflecting the efficiency of selenoprotein production. In addition to [75Se]selenite (0.5–1 μCi/ml bacterial culture), 5 μm non-reactive sodium selenite was also included to ensure a sufficient selenium supply, together with 100 μg/ml l-cysteine to prevent non-specific selenium incorporation through pathways of sulfur metabolism (50). After IPTG induction, aliquots of the bacterial cultures were in this case lysed directly in SDS loading buffer, and proteins were separated with SDS-PAGE, followed by transfer to a nitrocellulose membrane (iBlot system, Thermo Fisher Scientific), with the membrane subsequently stained with Ponceau S (Sigma) to visualize total protein loading. Next, the membrane was air-dried and secured against a phosphorimaging screen overnight, whereupon the 75Se autoradiography imaging was obtained using a Typhoon FLA 7000 system (GE Healthcare).
Author Contributions
Q. C. and E. S. J. A. designed the experiments. Q. C. carried out the experiments. Q. C. and E. S. J. A. analyzed the data and drafted and edited the manuscript to its final version.
Acknowledgments
We are grateful to Malin Petersson at Karolinska Institutet for experimental assistance; Dr. Margareta Ramström and team at the Science for Life Laboratory Mass Spectrometry-based Proteomics Facility in Uppsala, Sweden, for mass spectrometry support and analysis; Dr. Gottfried Otting at Australian National University for discussions; and Dr. David L Williams at Rush University Medical Center for providing the ULP1 coding plasmid.
This work was supported by the Swedish Research Council, the Swedish Cancer Society, The Knut and Alice Wallenberg Foundations, and Karolinska Institutet. The authors declare that they have no conflicts of interest with the contents of this article.
- Sec (U)
- selenocysteine
- TrxR
- thioredoxin reductase
- SECIS
- Sec insertion sequence
- GPx
- glutathione peroxidase
- GR
- glutathione reductase
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- TNB−
- 5-thio-2-nitrobenzoic acid anion
- IMAC
- immobilized metal affinity chromatography
- DTNB
- 5,5′-dithiobis(2-nitrobenzoic) acid.
References
- 1. Arnér E. S. (2010) Selenoproteins–what unique properties can arise with selenocysteine in place of cysteine? Exp. Cell Res. 316, 1296–1303 [DOI] [PubMed] [Google Scholar]
- 2. Reich H. J., and Hondal R. J. (2016) Why nature chose selenium. ACS Chem. Biol. 11, 821–841 [DOI] [PubMed] [Google Scholar]
- 3. Kryukov G. V., Castellano S., Novoselov S. V., Lobanov A. V., Zehtab O., Guigó R., and Gladyshev V. N. (2003) Characterization of mammalian selenoproteomes. Science 300, 1439–1443 [DOI] [PubMed] [Google Scholar]
- 4. Kryukov G. V., and Gladyshev V. N. (2004) The prokaryotic selenoproteome. EMBO Rep. 5, 538–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lobanov A. V., Fomenko D. E., Zhang Y., Sengupta A., Hatfield D. L., and Gladyshev V. N. (2007) Evolutionary dynamics of eukaryotic selenoproteomes: large selenoproteomes may associate with aquatic life and small with terrestrial life. Genome Biol. 8, R198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lobanov A. V., Hatfield D. L., and Gladyshev V. N. (2009) Eukaryotic selenoproteins and selenoproteomes. Biochim. Biophys. Acta 1790, 1424–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taskov K., Chapple C., Kryukov G. V., Castellano S., Lobanov A. V., Korotkov K. V., Guigó R., and Gladyshev V. N. (2005) Nematode selenoproteome: the use of the selenocysteine insertion system to decode one codon in an animal genome? Nucleic Acids Res. 33, 2227–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y., Fomenko D. E., and Gladyshev V. N. (2005) The microbial selenoproteome of the Sargasso Sea. Genome Biol. 6, R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Y., and Gladyshev V. N. (2008) Trends in selenium utilization in marine microbial world revealed through the analysis of the global ocean sampling (GOS) project. PLoS Genet. 4, e1000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sawers G., Heider J., Zehelein E., and Böck A. (1991) Expression and operon structure of the sel genes of Escherichia coli and identification of a third selenium-containing formate dehydrogenase isoenzyme. J. Bacteriol. 173, 4983–4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lei X. G., Zhu J. H., Cheng W. H., Bao Y., Ho Y. S., Reddi A. R., Holmgren A., and Arnér E. S. (2016) Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol. Rev. 96, 307–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rayman M. P. (2009) Selenoproteins and human health: insights from epidemiological data. Biochim. Biophys. Acta 1790, 1533–1540 [DOI] [PubMed] [Google Scholar]
- 13. Cheng Q., Stone-Elander S., and Arnér E. S. (2006) Tagging recombinant proteins with a Sel-tag for purification, labeling with electrophilic compounds or radiolabeling with 11C. Nat. Protoc. 1, 604–613 [DOI] [PubMed] [Google Scholar]
- 14. Metanis N., and Hilvert D. (2014) Natural and synthetic selenoproteins. Curr. Opin. Chem. Biol. 22, 27–34 [DOI] [PubMed] [Google Scholar]
- 15. Hondal R. J. (2009) Using chemical approaches to study selenoproteins–focus on thioredoxin reductases. Biochim. Biophys. Acta 1790, 1501–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johansson L., Gafvelin G., and Arnér E. S. (2005) Selenocysteine in proteins-properties and biotechnological use. Biochim. Biophys. Acta 1726, 1–13 [DOI] [PubMed] [Google Scholar]
- 17. Berry M. J., Banu L., Harney J. W., and Larsen P. R. (1993) Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons. EMBO J. 12, 3315–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thanbichler M., and Böck A. (2002) The function of SECIS RNA in translational control of gene expression in Escherichia coli. EMBO J. 21, 6925–6934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zinoni F., Heider J., and Böck A. (1990) Features of the formate dehydrogenase mRNA necessary for decoding of the UGA codon as selenocysteine. Proc. Natl. Acad. Sci. U.S.A. 87, 4660–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suppmann S., Persson B. C., and Böck A. (1999) Dynamics and efficiency in vivo of UGA-directed selenocysteine insertion at the ribosome. EMBO J. 18, 2284–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Böck A., Forchhammer K., Heider J., Leinfelder W., Sawers G., Veprek B., and Zinoni F. (1991) Selenocysteine: the 21st amino acid. Mol. Microbiol. 5, 515–520 [DOI] [PubMed] [Google Scholar]
- 22. Fletcher J. E., Copeland P. R., Driscoll D. M., and Krol A. (2001) The selenocysteine incorporation machinery: interactions between the SECIS RNA and the SECIS-binding protein SBP2. RNA 7, 1442–1453 [PMC free article] [PubMed] [Google Scholar]
- 23. Novoselov S. V., Lobanov A. V., Hua D., Kasaikina M. V., Hatfield D. L., and Gladyshev V. N. (2007) A highly efficient form of the selenocysteine insertion sequence element in protozoan parasites and its use in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 104, 7857–7862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Castellano S., Gladyshev V. N., Guigó R., and Berry M. J. (2008) SelenoDB 1.0: a database of selenoprotein genes, proteins and SECIS elements. Nucleic Acids Res. 36, D332–D338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rother M., Resch A., Gardner W. L., Whitman W. B., and Böck A. (2001) Heterologous expression of archaeal selenoprotein genes directed by the SECIS element located in the 3′ non-translated region. Mol. Microbiol. 40, 900–908 [DOI] [PubMed] [Google Scholar]
- 26. Arnér E. S. (2002) Recombinant expression of mammalian selenocysteine-containing thioredoxin reductase and other selenoproteins in Escherichia coli. Methods Enzymol. 347, 226–235 [DOI] [PubMed] [Google Scholar]
- 27. Casi G., Roelfes G., and Hilvert D. (2008) Selenoglutaredoxin as a glutathione peroxidase mimic. ChemBioChem 9, 1623–1631 [DOI] [PubMed] [Google Scholar]
- 28. Aldag C., Bröcker M. J., Hohn M. J., Prat L., Hammond G., Plummer A., and Söll D. (2013) Rewiring translation for elongation factor Tu-dependent selenocysteine incorporation. Angew. Chem. Int. Ed. Engl. 52, 1441–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bröcker M. J., Ho J. M., Church G. M., Söll D., and O'Donoghue P. (2014) Recoding the genetic code with selenocysteine. Angew. Chem. Int. Ed. Engl. 53, 319–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miller C., Bröcker M. J., Prat L., Ip K., Chirathivat N., Feiock A., Veszprémi M., and Söll D. (2015) A synthetic tRNA for EF-Tu mediated selenocysteine incorporation in vivo and in vitro. FEBS Lett. 589, 2194–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thyer R., Filipovska A., and Rackham O. (2013) Engineered rRNA enhances the efficiency of selenocysteine incorporation during translation. J. Am. Chem. Soc. 135, 2–5 [DOI] [PubMed] [Google Scholar]
- 32. Thyer R., Robotham S. A., Brodbelt J. S., and Ellington A. D. (2015) Evolving tRNA(Sec) for efficient canonical incorporation of selenocysteine. J. Am. Chem. Soc. 137, 46–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arnér E. S., Sarioglu H., Lottspeich F., Holmgren A., and Böck A. (1999) High-level expression in Escherichia coli of selenocysteine-containing rat thioredoxin reductase utilizing gene fusions with engineered bacterial-type SECIS elements and co-expression with the selA, selB and selC genes. J. Mol. Biol. 292, 1003–1016 [DOI] [PubMed] [Google Scholar]
- 34. Rengby O., Johansson L., Carlson L. A., Serini E., Vlamis-Gardikas A., Kårsnäs P., and Arnér E. S. (2004) Assessment of production conditions for efficient use of Escherichia coli in high-yield heterologous recombinant selenoprotein synthesis. Appl. Environ. Microbiol. 70, 5159–5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cheng Q., Sandalova T., Lindqvist Y., and Arnér E. S. (2009) Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J. Biol. Chem. 284, 3998–4008 [DOI] [PubMed] [Google Scholar]
- 36. Rengby O., Cheng Q., Vahter M., Jörnvall H., and Arnér E. S. (2009) Highly active dimeric and low-activity tetrameric forms of selenium-containing rat thioredoxin reductase 1. Free Radic. Biol. Med. 46, 893–904 [DOI] [PubMed] [Google Scholar]
- 37. Rengby O., and Arnér E. S. (2007) Titration and conditional knockdown of the prfB gene in Escherichia coli: effects on growth and overproduction of the recombinant mammalian selenoprotein thioredoxin reductase. Appl. Environ. Microbiol. 73, 432–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang Z., Arnér E. S., Mu Y., Johansson L., Shi J., Zhao S., Liu S., Wang R., Zhang T., Yan G., Liu J., Shen J., and Luo G. (2004) Expression of selenocysteine-containing glutathione S-transferase in Escherichia coli. Biochem. Biophys. Res. Commun. 321, 94–101 [DOI] [PubMed] [Google Scholar]
- 39. Lajoie M. J., Rovner A. J., Goodman D. B., Aerni H. R., Haimovich A. D., Kuznetsov G., Mercer J. A., Wang H. H., Carr P. A., Mosberg J. A., Rohland N., Schultz P. G., Jacobson J. M., Rinehart J., Church G. M., and Isaacs F. J. (2013) Genomically recoded organisms expand biological functions. Science 342, 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu J., Croitoru V., Rutishauser D., Cheng Q., and Arnér E. S. (2013) Wobble decoding by the Escherichia coli selenocysteine insertion machinery. Nucleic Acids Res. 41, 9800–9811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luthman M., and Holmgren A. (1982) Rat liver thioredoxin and thioredoxin reductase: purification and characterization. Biochemistry 21, 6628–6633 [DOI] [PubMed] [Google Scholar]
- 42. Zhong L., and Holmgren A. (2000) Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 275, 18121–18128 [DOI] [PubMed] [Google Scholar]
- 43. Bouakaz L., Bouakaz E., Murgola E. J., Ehrenberg M., and Sanyal S. (2006) The role of ribosomal protein L11 in class I release factor-mediated translation termination and translational accuracy. J. Biol. Chem. 281, 4548–4556 [DOI] [PubMed] [Google Scholar]
- 44. Toppo S., Flohé L., Ursini F., Vanin S., and Maiorino M. (2009) Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim. Biophys. Acta 1790, 1486–1500 [DOI] [PubMed] [Google Scholar]
- 45. Mukai T., Hayashi A., Iraha F., Sato A., Ohtake K., Yokoyama S., and Sakamoto K. (2010) Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 38, 8188–8195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nilsson M., and Rydén-Aulin M. (2003) Glutamine is incorporated at the nonsense codons UAG and UAA in a suppressor-free Escherichia coli strain. Biochim. Biophys. Acta 1627, 1–6 [DOI] [PubMed] [Google Scholar]
- 47. George S., Aguirre J. D., Spratt D. E., Bi Y., Jeffery M., Shaw G. S., and O'Donoghue P. (2016) Generation of phospho-ubiquitin variants by orthogonal translation reveals codon skipping. FEBS Lett. 590, 1530–1542 [DOI] [PubMed] [Google Scholar]
- 48. Tosatto S. C., Bosello V., Fogolari F., Mauri P., Roveri A., Toppo S., Flohé L., Ursini F., and Maiorino M. (2008) The catalytic site of glutathione peroxidases. Antioxid. Redox Signal. 10, 1515–1526 [DOI] [PubMed] [Google Scholar]
- 49. Mukai T., Englert M., Tripp H. J., Miller C., Ivanova N. N., Rubin E. M., Kyrpides N. C., and Söll D. (2016) Facile recoding of selenocysteine in nature. Angew. Chem. Int. Ed. Engl. 55, 5337–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Müller S., Heider J., and Böck A. (1997) The path of unspecific incorporation of selenium in Escherichia coli. Arch. Microbiol. 168, 421–427 [DOI] [PubMed] [Google Scholar]
- 51. Arnér E. S., and Holmgren A. (2001) Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol. 2002, Chapter 7, Unit 7. [DOI] [PubMed] [Google Scholar]
- 52. Esworthy R. S., Chu F. F., and Doroshow J. H. (2001) Analysis of glutathione-related enzymes. Curr. Protoc. Toxicol. 2001, Chapter 7, Unit 7.1 [DOI] [PubMed] [Google Scholar]
- 53. Arnér E. S., and Holmgren A. (2000) in Current Protocols in Toxicology (Maines M., Costa L., Reed D., and Sassa S., eds) pp. 7.4.1–7.4.14, John Wiley & Sons, Inc., New York: 5 [Google Scholar]