

Abstract
Cardiac function requires continuous high levels of energy, and so iron, a critical player in mitochondrial respiration, is an important component of the heart. Hearts from 57Fe-enriched mice were evaluated by Mössbauer spectroscopy. Spectra consisted of a sextet and two quadrupole doublets. One doublet was due to residual blood, whereas the other was due to [Fe4S4]2+ clusters and low-spin FeII hemes, most of which were associated with mitochondrial respiration. The sextet was due to ferritin; there was no evidence of hemosiderin, a ferritin decomposition product. Iron from ferritin was nearly absent in young hearts, but increased steadily with age. EPR spectra exhibited signals similar to those of brain, liver, and human cells. No age-dependent EPR trends were apparent. Hearts from HFE−/− mice with hemochromatosis contained slightly more iron overall than controls, including more ferritin and less mitochondrial iron; these differences typify slightly older hearts, perhaps reflecting the burden due to this disease. HFE−/− livers were overloaded with ferritin but had low mitochondrial iron levels. IRP2−/− hearts contained less ferritin than controls but normal levels of mitochondrial iron. Hearts of young mice born to an iron-deficient mother contained normal levels of mitochondrial iron and no ferritin; the heart from the mother contained low ferritin and normal levels of mitochondrial iron. High-spin FeII ions were nearly undetectable in heart samples; these were evident in brains, livers, and human cells. Previous Mössbauer spectra of unenriched diseased human hearts lacked mitochondrial and blood doublets and included hemosiderin features. This suggests degradation of iron-containing species during sample preparation.
Keywords: cardiac metabolism, electron paramagnetic resonance (EPR), iron, mitochondria, Mossbauer spectroscopy
Introduction
From an early stage of fetal development until the end of life, the heart functions unceasingly to pump blood throughout the body. This requires a high and sustained level of metabolic energy unrivaled by any other organ except perhaps the brain. Mitochondria, the organelle responsible for generating most of the chemical energy in cells, serve a critical role in cardiac function. Improving our understanding of cardiac physiology is important because heart disease is the most common cause of death in the Western world (1). The preponderance of iron-containing centers in respiration-related mitochondrial proteins makes this metal a major player in cardiac physiology.
Much of the iron that enters cardiomyocytes is used to build iron-sulfur clusters (ISCs)3 and heme centers many of which are installed into mitochondrial respiratory complexes and respiration-related proteins. Excess cellular iron is generally stored within the core of cytosolic ferritin, a spherically shaped protein complex composed of variable ratios of H- and L-subunits (2). H-ferritin is especially important in cardiac function (3, 4). Besides storing iron, H-ferritin helps cells manage the metal (5). H-ferritin levels are significantly lower in mice hearts after a myocardial infarct (6). Such hearts contain spotty iron deposits and suffer from excessive oxidative stress. Deleting H-ferritin reduces the viability of cardiomyocytes and increases these deposits (6). Excessive iron and increased oxidative stress damage cardiomyocytes and contribute to heart failure (6). Mitochondrial ferritin (mt-ferritin) is highly expressed in heart mitochondria (7). It functions to sequester FeII in mitochondria and thus protect the organelle from iron-dependent oxidative damage (8).
Cardiomyopathy is common in iron-overload diseases such as hereditary hemochromatosis (HH) (9). HH initially impacts the liver, but eventually affects the heart, causing ROS damage especially to mitochondria (10–12). The most common cause of HH is a mutation in the HFE gene. HFE−/− mutant livers contain 4–6 times more iron than controls, whereas the iron content of mutant hearts is slightly elevated (13, 14). Hemosiderin, a poorly defined iron-loaded breakdown product of ferritin (15), accumulates along with ferritin in mutant livers but not in mutant hearts (13, 14). Hemosiderin is found in some iron-overloaded organs (15–17).
Mössbauer (MB) spectroscopy has been used to investigate the iron content of hearts from patients with β-thalassemia, another iron-overload blood disease (18–20). The average iron concentration in the β-thalassemia heart is nearly 3 times higher than normal (11). MB spectra of β-thalassemia heart tissue exhibit features of ferritin, a high-spin FeIII species, and perhaps hemosiderin (11, 18). The control MB spectrum was too noisy to evaluate because the heart was neither overloaded nor enriched in 57Fe.
Individuals with Friedreich's ataxia (FA) develop cardiomyopathy in conjunction with mitochondrial iron accumulation and ROS damage (21, 22). FA arises from a deficiency in frataxin, a mitochondrial protein involved in ISC assembly (23). Eighty percent of FA patients die of cardiac disease (24).
Mice in which frataxin was deleted in cardiac muscle exhibited the classical progression of FA (20, 24–26). At 7 weeks, mutant mice became deficient in ISCs, and at 9 weeks, the iron concentration in mutant hearts increased and respiratory activity declined (27). At 10 weeks, the concentration of iron in mutant hearts contained nearly 10 times more iron than WT hearts, which contained about 1.3 mm iron (26, 28). The excess iron in the mutant cells flowed into mitochondria where it aggregated and generated ROS (23, 25, 27, 29).
MB spectroscopy was used to characterize the iron aggregates in hearts of 9-week frataxin KO mice (23). The spectrum was noisy because mice were not enriched in 57Fe. It exhibited a single quadrupole doublet due to FeIII oxyhydroxide nanoparticles (23). The low-temperature MB spectrum of the liver from the same frataxin KO mice was dominated by a sextet due to ferritin (11, 23).
Cardiac failure is the most prevalent cause of death in the elderly and is commonly associated with impaired energy homeostasis (30, 31). Aged mitochondria often have respiratory defects (32). Ischemia is associated with a decline in the EPR signal due to the Rieske ISC protein associated with respiratory complex III (32).
Iron-related proteins in mammals are primarily regulated by the IRP1/IRP2 system (33). Under iron-deficient cellular conditions, IRP1/2 bind mRNA transcripts of ferritin and TfR1. Doing so increases TfR1 expression and decrease ferritin levels. IRP2−/− mice accumulate large amounts of iron in the liver (33–36), whereas IRP2−/− brains contain WT levels of iron and WT MB features (34).
MB spectroscopy is the most powerful spectroscopic tool for probing the iron content of biological materials, but it has not been applied extensively to vertebrate animals (37). MB is relatively insensitive and can only detect 57Fe, which accounts for just 2% of naturally occurring iron. As a result, spectra of unenriched mammalian organs are noisy, even when iron overloaded. Enriching mammals in 57Fe is inconvenient and costly. Nevertheless, we enriched mice with 57Fe for this study. In earlier studies, we used MB spectroscopy to characterize the iron content of brain and liver (34, 38, 39). Here we use MB to probe the iron content of healthy mouse hearts during development. We also investigate hearts from HFE−/− mice with hemochromatosis, hearts from IRP2−/− mice, and hearts from an iron-deficient mouse and her offspring.
Results
We wanted to characterize the iron content of healthy mammalian hearts at different developmental stages, and used MB spectroscopy as our primary tool. We enriched mice with 57Fe to improve spectral quality. Analyses were augmented by EPR spectroscopy and ICP-MS analysis. Mice were euthanized at different ages and immediately imported into a refrigerated anaerobic glove box where blood was flushed extensively with Ringer's buffer. Hearts and other organs were removed by dissection, and frozen in MB cups for later analysis. Low-field MB spectra were collected on whole intact hearts from elderly (Fig. 1), young (Fig. 2), and adult (Fig. 3) mice. For each group, multiple spectra are shown to help distinguish sample-to-sample variations from age-dependent changes. Distinguishing this is a challenge for studies like this in which only small numbers of animals can be investigated. After collecting spectra, samples were analyzed for metal concentrations. These results, and the MB parameters used in simulations are compiled in Table 1. EPR spectra were collected (Fig. 4) on different heart samples that had been homogenized and packed into EPR tubes by centrifugation.
FIGURE 1.
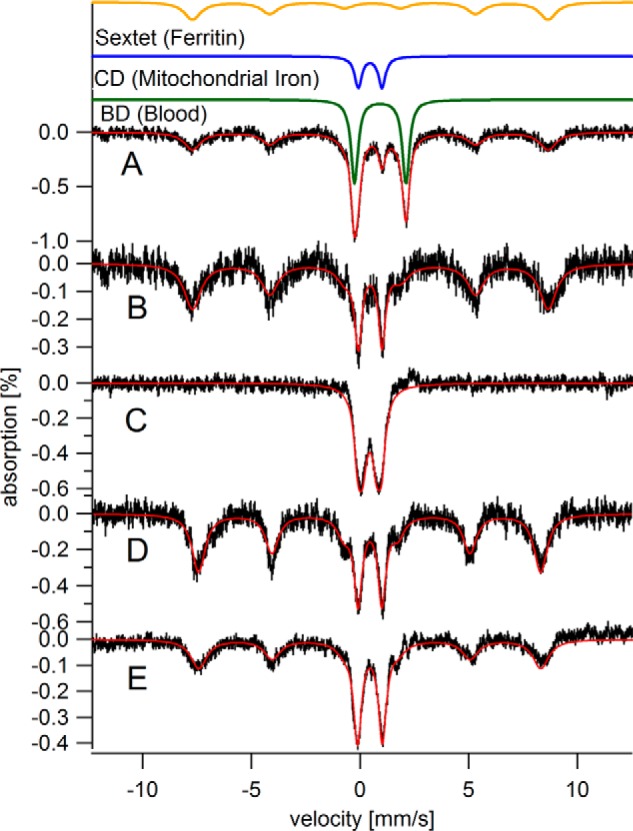
Mössbauer spectra of hearts from elderly mice. All spectra presented in the text were collected at 5 K except for in C, which was collected at 100 K (and in Fig. 5D, which was collected at 70 K). In all MB spectra presented in the text, a 0.05 tesla magnetic field was applied parallel to the gamma rays. The solid red lines overlaying spectra in all MB figures are simulations using parameters listed in Table 1. A, C60, raw spectrum (all other spectra have the blood doublet removed). The solid green, blue, and gold lines above the data are simulations of the blood, CD, and ferritin components, respectively. B, C60 with contribution from blood removed; C, same as B but at 100 K; D, C52; E, I104.
FIGURE 2.
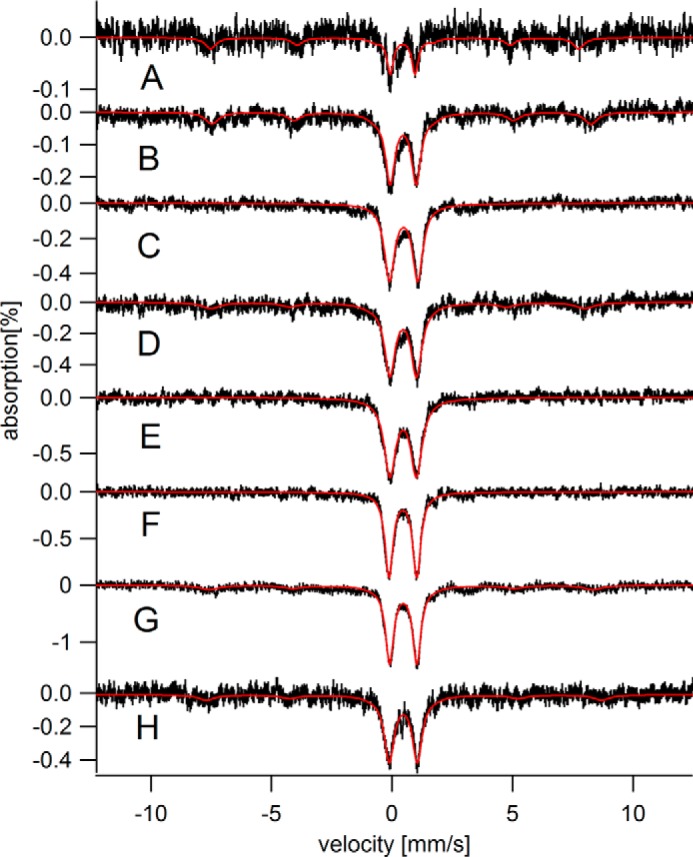
Mössbauer spectra (5 K) of hearts from young mice. A, C00; B, C01; C, C02a; D, C02b; E, C03; F, C03D; G, C04a; H, C04b.
FIGURE 3.
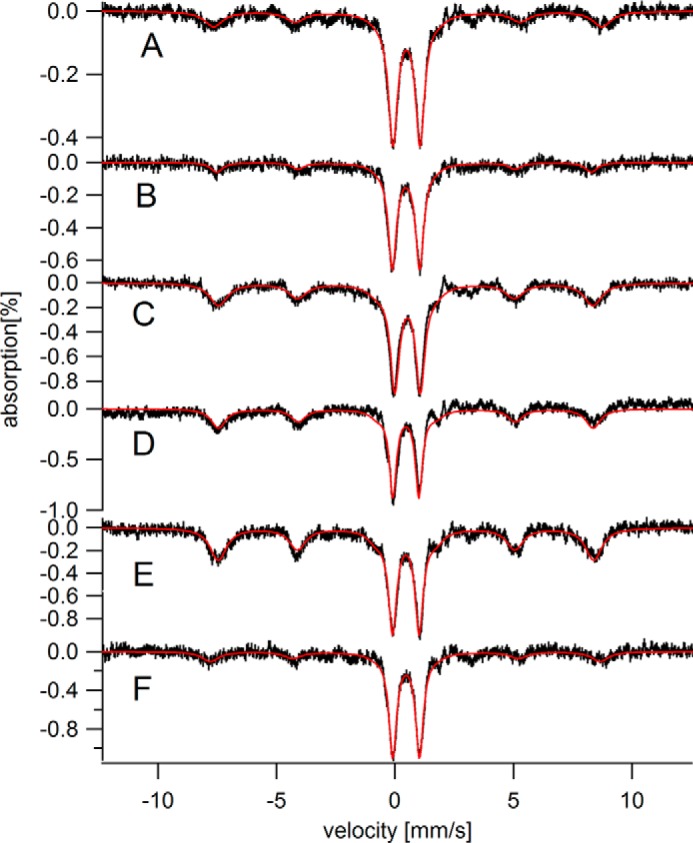
Mössbauer spectra (5 K) of hearts from adult mice. A, C06; B, I08; C, C16; D, C24; E, C28; F, C28D.
TABLE 1.
Metal concentrations in 57Fe-enriched mouse hearts (and livers), and associated Mössbauer parameters
Metal concentrations are in μm, calculated by assuming a tissue density of 1.06 g/ml. Masses of hearts are in mg; uncertainties are ±1 mg. Values in parentheses refer to the number of hearts in the sample. If more than 1 heart was used, the mass refers to the average. Entry indicated with bold and italics is the collective mass of 4–7 hearts that had been combined. This mass, on a per-heart basis, is an outlier relative to the other samples. Sample designations C, H, and I refer to C57BL/6, HFE−/−, and IRP2−/− strains, respectively. The number that follows indicates the age of the mouse in weeks. Samples of the same age are distinguished with a and b. Samples from iron-deficient mice are distinguished by a subscript D. For ICP-MS analysis, each tissue was analyzed in triplicate; reported concentrations are averages; uncertainties are standard deviations. Cobalt and molybdenum concentrations were also determined but no age-dependent trends were apparent; average [Co] and [Mo] (n = 18) were 0.16 ± 0.11 μm and 0.05 ± 0.02 μm, respectively. Isomer shifts δ, quadrupole splittings ΔEQ, and line widths Γ (all in mm/s) used in simulations were: central doublet (0.45 ± 0.01, 1.15 ± 0.02, and 0.44 ± 0.07); low-temperature ferritin (−0.10 ± 0.05, 0.44 ± 0.07, and 0.77 ± 0.14) with Heff = 490 ± 6 kG; blood (0.91 ± 0.06, 2.30 ± 0.04, and 0.43 ± 0.11); high-temperature ferritin (0.44, 0.70, and 0.55). Mössbauer spectra were calibrated against α-iron foil at room temperature. For convenience, the sum of the percent relative intensities for the three major MB components were forced to 100%; however, 10–15% of the actual spectral intensity cannot be accounted for by these three components.
| Sample | Mass of heart | [57Fe] | [Fetot] | [Cu] | [Mn] | [Zn] | Central doublet | Ferritin sextet | Blood doublet |
|---|---|---|---|---|---|---|---|---|---|
| mg | %; μm | %; μm | %; μm | ||||||
| C00 (1 day) | 65 (1) | 240 ± 3 | 300 ± 8 | 10 ± 1 | 1.6 ± 0.2 | 31 ± 7 | 20; 60 | 20; 60 | 60; 180 |
| C01 | 100 | 350 ± 7 | 470 ± 10 | 33 ± 1 | 4.9 ± 0.1 | 43 ± 1 | 38; 180 | 17; 80 | 45; 210 |
| C02a | 180 (1) | 240 ± 9 | 290 ± 10 | 12 ± 0.4 | 2.3 ± 0.1 | 26 ± 1 | 53; 150 | 00; 00 | 47; 140 |
| C02b | 59 (2) | 170 ± 3 | 240 ± 9 | 19 ± 2 | 3.3 ± 0.1 | 26 ± 1 | 46; 110 | 13; 30 | 41; 100 |
| C03 | NDa | ND | ND | ND | ND | ND | 50; ND | 0; ND | 50; ND |
| C04a | 98 (4) | 600 ± 8 | 710 ± 9 | 29 ± 0.3 | 8.9 ± 0.2 | 46 ± 1 | 40; 280 | 10; 70 | 50; 360 |
| C04b | 61 (2) | 570 ± 10 | 610 ± 10 | 48 ± 1 | 11 ± 0.2 | 45 ± 1 | 44; 270 | 12; 70 | 44; 270 |
| C06 | 140 (1) | 620 ± 1 | 670 ± 10 | 42 ± 1 | 8.1 ± 0.1 | 43 ± 1 | 36; 240 | 17; 110 | 47; 320 |
| C12 | 150 (3) | 63 ± 1 | 610 ± 10 | 38 ± 1 | 5.8 ± 0.1 | 37 ± 1 | 31; 190 | 32; 190 | 37; 230 |
| C16 | ND | ND | ND | ND | ND | ND | 33; ND | 28; ND | 39; ND |
| C24 | 240 (2) | 950 ± 8 | 1050 ± 8 | 45 ± 1 | 7.7 ± 0.1 | 47 ± 1 | 30; 320 | 28; 290 | 42; 440 |
| C28 | 140 (2) | 740 ± 10 | 780 ± 10 | 36 ± 0.4 | 5.8 ± 0.1 | 34 ± 1 | 28; 220 | 36; 280 | 36; 280 |
| C52 | 250 (2) | 700 ± 10 | 1100 ± 20 | 39 ± 1 | 8.1 ± 0.1 | 48 ± 1 | 22; 240 | 48; 530 | 30; 330 |
| C60 | 130 (1) | 650 ± 30 | 960 ± 40 | 40 ± 10 | 3.4 ± 0.1 | 34 ± 1 | 16; 150 | 41; 390 | 43; 420 |
| C03D (pups) | 94 (4) | 260 ± 10 | 310 ± 20 | 40 ± 2 | 24 ± 1 | 56 ± 2 | 54; 170 | 00; 000 | 46; 140 |
| C28D (mom) | 390 (1) | 740 ± 5 | 850 ± 5 | 48 ± 0.3 | 8.8 ± 0.1 | 49 ± 1 | 35; 300 | 13; 110 | 52; 440 |
| H12 | 160 (3) | 58 ± 2 | 640 ± 20 | 58 ± 1 | 9.9 ± 0.2 | 60 ± 1 | 27; 170 | 37; 240 | 36; 230 |
| H12 (liver) | 1600 (1) | 1200 ± 200 | 5600 ± 700 | 29 ± 4 | 14 ± 2 | 90 ± 2 | 0.5; 30 | 90; 5040 | 9.5; 530 |
| C12 (liver) | 1300 (1) | 20 ± 90 | 960 ± 500 | 60 ± 30 | 14 ± 6 | 120 ± 70 | 18; 170 | 60; 580 | 22; 210 |
| I08 | 95 (2) | 520 ± 7 | 560 ± 7 | 35 ± 2 | 7.0 ± 0.1 | 32 ± 1 | 38; 210 | 10; 60 | 52; 290 |
| I104 | 200 (1) | 600 ± 20 | 800 ± 30 | 36 ± 1 | 7.7 ± 0.3 | 32 ± 1 | 30; 240 | 34; 270 | 36; 290 |
a ND, not determined.
FIGURE 4.
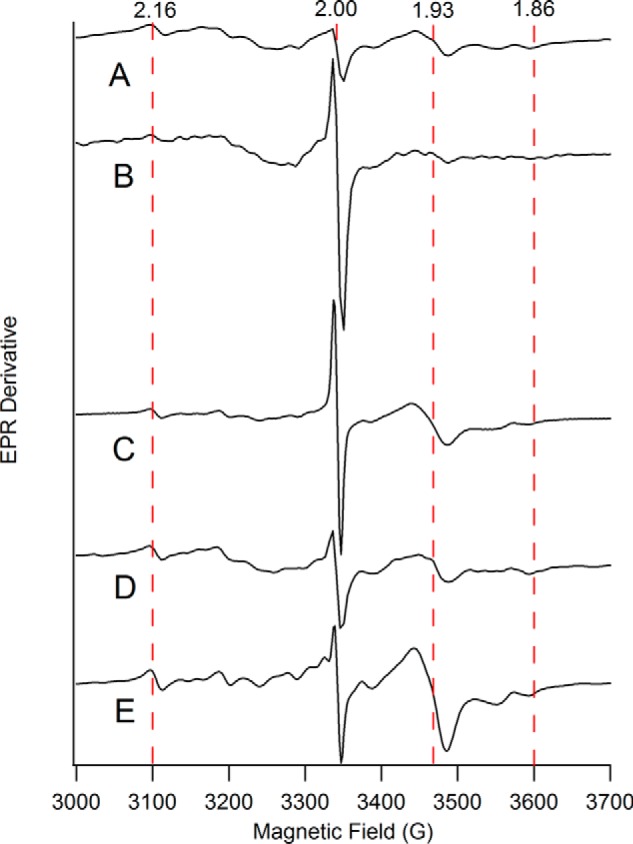
X-band EPR spectra of heart homogenates from mice of different ages. A, 4 weeks; B, 12 weeks; C, 32 weeks; D, 52 weeks; E, 60 weeks. EPR conditions: temperature, 4 K; microwave frequency, 9.37 GHz; microwave power, 0.2 milliwatt (30 dB); modulation amplitude, 10 G. Dashed vertical lines indicate the fields corresponding to g values of 2.16, 2.00, 1.93, and 1.86.
MB spectra were composed of three major features, including a sextet and two quadrupole doublets (Fig. 1A). One doublet had parameters (Table 1) of high-spin FeII hemoglobin. This doublet, simulated in green in Fig. 1, was assigned to residual blood. This so-called “blood doublet” (BD) was present in all raw spectra. The average relative intensity of the BD was 43%, with a standard deviation of just ±5% (n = 18). To view other spectral features better, we have removed the BD contribution from all spectra (except for that in Fig. 1A). The consistent high percentage of iron due to blood in our samples raises doubts that flushing by cardiac puncture is effective in removing blood from the heart itself. Nevertheless, knowing the percent contribution of the BD to the overall intensity of each spectrum and the absolute concentration of iron in each sample allowed us to calculate the concentration of iron within heart cells themselves for the first time. We have already done this for brain and liver; another group has done the same for liver and spleen (40). As such, the iron concentrations listed in Table 1 are probably the most accurate determinations for hearts from iron-sufficient mammals. The average iron concentration in the healthy heart samples (with blood excluded) was 400 ± 200 μm (n = 11), depending on age. The approximate concentration of iron in young, adult, and elderly hearts was 300, 450, and 700 μm, respectively.
We assigned the sextet in heart MB spectra to FeIII aggregates in ferritin cores. The gold line above the data of Fig. 1A simulates the ferritin sextet using parameters in Table 1. To assess whether hemosiderin contributed to this absorption, we collected a 100 K spectrum of the C60 heart. The baseline was devoid of sextet features (Fig. 1C), suggesting the absence of this degradation product.
The other doublet in MB spectra of hearts, called the “central doublet” or CD, was simulated as the blue line in Fig. 1A using parameters in Table 1. The CD is due collectively to [Fe4S4]2+ clusters and low-spin FeII hemes; the two types of centers cannot be resolved by MB spectroscopy because they are diamagnetic and have similar δ and ΔEQ values. The CD dominates the spectrum of mitochondria isolated from brain, liver, and human cells (34, 38, 41), which allows us to assign this spectral feature to respiration-related complexes in that organelle. [Fe4S4]2+ clusters and LS FeII hemes are undoubtedly present in non-mitochondrial regions of the heart, but they are minor contributors to spectral intensity. We have quantified the intensity of the CD in the spectra of elderly hearts (Fig. 1) and find that it corresponds to 110–200 μm iron.
The CD dominated the MB spectra of young hearts (defined as newborn to 4 weeks) with an intensity that corresponded to 60–220 μm (Fig. 2 and Table 1). The intensity of the ferritin sextet was dramatically lower in spectra of young hearts than in those of elderly hearts; indeed the feature was absent in some spectra of young hearts. Adult hearts (Fig. 3, 6–28 weeks) exhibited even higher concentrations of the CD (220–350 μm iron) and intermediate levels of ferritin (55–320 μm iron).
EPR spectra of packed mouse heart homogenates of different ages are shown in Fig. 4. The g = 2 region consisted of numerous overlapping signals including a gave = 1.94 signal due to [Fe2S2]1+ and/or [Fe4S4]1+ clusters, a gave = 1.90 signal due to the [Fe2S2]1+ cluster in the Rieske ISC protein, a radical signal at g = 2.00, and an unassigned resonance at g = 2.16. The same signals were observed in mouse brain and liver homogenates and in mitochondria isolated from these tissues (34, 38). No age-dependent EPR trends were apparent.
MB spectra of HFE−/− hearts and matching controls are shown in Fig. 5, A and B, respectively. Spectra were noisy because the mice were enriched in 57Fe for only 4 weeks. Relative to controls, the HFE−/− heart contained slightly more total iron, including more ferritin and less CD. In a qualitative sense, the HFE−/− heart appeared slightly older. The spectra of the HFE−/− liver and control (Fig. 5, C and E) were strikingly different, with only the HFE−/− liver overloaded with ferritin iron. The 70 K spectrum of the HFE−/− liver (Fig. 5D) was devoid of any sextet features in the baseline, confirming the absence of hemosiderin in this diseased liver. The HFE−/− liver contained significantly less CD than the control liver (notice the absence of spectral intensity between the two inner lines of the sextet of Fig. 5C), implying that the HFE−/− liver contains less mitochondria than WT livers. The MB spectrum of another diseased liver (Fig. 4F of Ref. 34) also showed a deficiency of mitochondrial iron.
FIGURE 5.
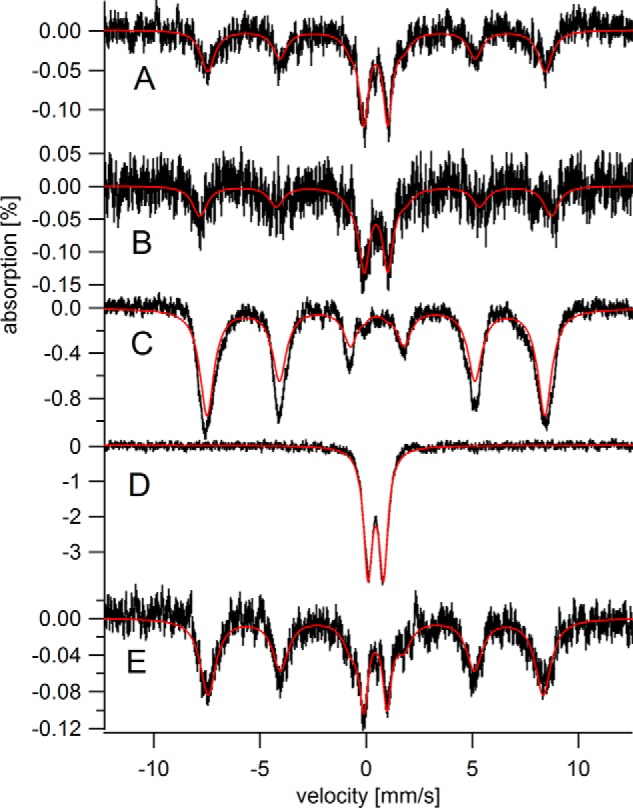
Mössbauer spectra of 12 weeks HFE−/− hearts and livers versus controls. A, HFE−/− hearts; B, control hearts; C and D, HFE−/− livers; E, control livers. All spectra were collected at 5 K except for D, which was collected at 70 K.
Spectra of hearts from IRP2−/− mice (I08 in Fig. 3B and I104 in Fig. 1E) were similar to controls. The intensities of the ferritin sextets were slightly less in mutant spectra but the effect was modest. The intensities of the CD in the mutant spectra were comparable with (if not slightly stronger than) the same feature in the controls. The concentration of iron in I08 and I104 (Table 1) were slightly lower than in the controls. Like HFE−/− livers, IRP2−/− livers accumulated large amounts of ferritin iron; however, they contained more mitochondrial iron (see Fig. 4E of Ref. 34) than did HFE−/− livers.
We fed a pregnant 57Fe-enriched mouse an iron-deficient diet starting 1 week before she gave birth to 4 offspring, and then continued to feed her that diet until the pups were 3 weeks old. Mother and pups were then euthanized and their hearts were examined by MB spectroscopy. The MB spectrum of the pup hearts (Fig. 2F) exhibited standard features of young hearts, i.e. an intense CD and no ferritin sextet. The MB spectrum of the 28-week-old heart from the mother (Fig. 3F) exhibited less than half as much ferritin as a control heart of the same age (110 versus 280 μm). Surprisingly, the CD of the iron-deficient heart was more intense than the control (300 versus 220 μm).
Discussion
This is the first Mössbauer study of the iron content of healthy 57Fe-enriched mammalian hearts. All previous MB studies have investigated diseased human hearts that were overloaded in iron. In those earlier studies, spectra from healthy controls could not be obtained because samples were not 57Fe enriched and so spectral intensities would have been exceedingly weak.
One advantage of obtaining MB spectra of whole hearts is the ability to distinguish iron due to blood in vessels permeating the heart ([Fe]B) from iron in the heart cells themselves ([Fe]H). The total measured iron in whole hearts was [Fe]T = [Fe]B + [Fe]H. Previous determinations of heart iron concentration probably overestimated [Fe]H by nearly a factor of 2 due to the large proportion of blood in the tissue. An exception is an earlier study from our group in which 40% of the iron in newborn to 6-week-old mice hearts was assumed to arise from blood; [Fe]H values between 450 and 720 μm iron were reported (39). Another study reported that hearts from 12-week-old mice contained 1.4–1.6 mm iron (42) (millimolar units calculated from those reported). In another study, heart iron concentrations of 1.1–1.5 mm (28) were reported. If 43% of those concentrations were presumed to be due to blood, [Fe]H would be between 630 and 900 μm, reasonably similar to the concentrations we obtained. Comparing samples more precisely would require matching ages and the concentration of iron in the diet. In other studies, hearts from WT mice reportedly contained 4 (14, 43), 6 (14), 7 (23), and ∼100 mm (36) iron. These values overestimated the actual heart iron concentrations by factors of 10–200.
Apart from blood, the two major forms of iron in heart tissues were ferritin and mitochondrial iron. Iron in hearts of young mice was mostly found in respiration-related ISCs and heme centers within mitochondria. The level of mitochondrial iron increased as young mice matured into adults, and then it stabilized or declined slightly in old age. As mice aged, their hearts accumulated increasing amounts of ferritin iron such that the iron content of elderly mice was dominated by this storage form of iron.
We used the overall concentration of iron in 57Fe-enriched hearts and the percentage of CD and ferritin as determined by MB spectroscopy to calculate the absolute concentration of these two features (Table 1 and Fig. 6). Age-dependent mitochondrial iron concentrations ([CD]age) were assessed by assuming the function in Equation 1.
| (Eq. 1) |
FIGURE 6.

Heart iron concentrations (excluding blood) as a function of age. Black circle, total iron; blue circles, ferritin; red circles, central doublet. Values were obtained from Table 1. The solid red line is the best-fit simulation generated assuming Equation 1. The solid blue line is the equivalent for ferritin. The solid black line is the sum of the red and blue lines. Simulations were optimized against the data by minimizing root mean square deviation. Best-fit parameters (concentrations in μm) were: [CD]newborn, 31; [CD]g, 250; KCD, 2.8 weeks; [FN]newborn, 0; [FN]g, 450; KFN, 14 weeks. Log2 numbers 0, 2, 4, and 6 refer to 1, 4, 16, and 64 weeks, respectively.
A similar function was assumed to assess age-dependent ferritin iron concentrations, [FN]age. In this case, parameters [FN]newborn, [FN]g, and KFN were used. The optimized functions are given as the solid lines in Fig. 6. Total heart iron at any age (ignoring the blood contribution) was calculated as the sum of these two components, i.e. [Heart Fe]age = [CD]age + [FN]age. The best-fit parameters suggest that the heart from a newborn should contain about 60 μm mitochondrial iron and little ferritin, as observed. Mitochondrial iron should increase during the first 6 weeks, maximizing at 280 μm. Ferritin iron levels should increase more slowly over the first 24 weeks, maximizing at 450 μm in adults and elderly.
These trends, illustrated qualitatively in Fig. 7, are easily rationalized given the requirement of the heart to function unceasingly from before birth until death. This high-energy requirement is satisfied by the high levels of mitochondria (and perhaps by not storing much iron). As hearts age, they store increasing amounts of ferritin iron.
FIGURE 7.
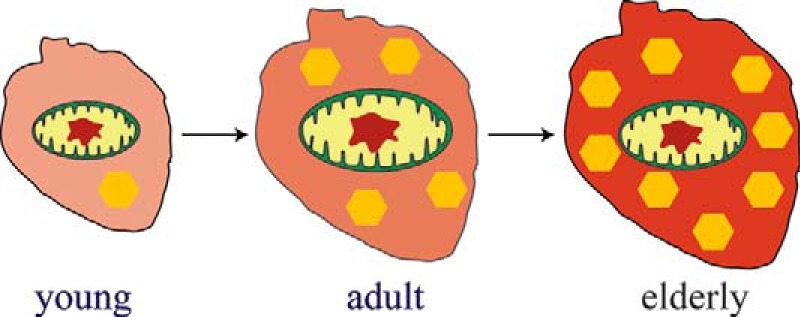
Illustration of how iron content changes with age in healthy mouse hearts. The opacity of the background reflects the overall iron concentration in the heart. The size of the mitochondria reflects the concentration of iron present in that organelle. The number of yellow hexagons represents the concentration of iron present as ferritin.
These age-dependent changes differ from those occurring in the developing brain and liver (38, 34). The iron content of the newborn brain is dominated by ferritin iron, with modest levels of mitochondrial iron. This makes sense because the newborn does not use its brain extensively. Then, during the first few weeks of life, the animal opens its eyes, and begins to sense and interact with its environment, increasing its need for energy to drive brain function. To accommodate these needs, ferritin iron in the brain is transformed into mitochondrial iron. In fact, the overall iron concentration in the brain declines during the first few weeks of life, as the combined rate of mitochondriogenesis and brain volume growth outpaces the import rate of new iron (39). Gradually, as the animal ages, the brain, like the heart, accumulates iron in the form of ferritin.
The changes in the iron content of the liver differ from those of brain and heart. The newborn liver contains very high concentrations of ferritin iron, much of which is exported from the liver during the first week of life (34). This burst of exported iron is most likely used to help other organs develop (including the heart but perhaps not the brain). In healthy mice raised under iron-sufficient conditions, the liver accumulates a modest amount of iron as it ages, but not an excessive amount (this might depend on the iron concentration in the diet).
Another result that highlights an important difference between heart, brain, and liver is the absolute concentration of mitochondrial iron in these organs. The concentrations of mitochondrial iron in the developing brain and liver maximize at about 110 and 180 μm, respectively, whereas it maximizes at about 280 μm in the heart. These differences may again reflect the high and sustained level of chemical energy required by the heart relative to these other organs.
Another difference between mouse hearts, brains, livers, and human Jurkat cells is the concentration of nonheme high-spin (NHHS) FeII in these organs/cells. Jurkat cells are used for this comparison because they have been characterized by Mössbauer spectroscopy (41, 44) and so the concentration of NHHS FeII can be quantified. The concentration of such ions in the brain is 10 μm of 170 μm total iron (38). Their concentration in the liver is 20–40 μm of 300–650 μm total iron (34). In Jurkat cells, the concentration of NHHS FeII is 40 μm of 400 μm total iron (41). By contrast, we had difficulty detecting the NHHS FeII doublet in most heart spectra. The heart contains high levels of mt-ferritin, which is thought to sequester FeII. It is interesting to consider that the concentration of NHHS FeII ions is so low in heart tissue because mt-ferritin sequesters these ions.
It is also interesting to compare the total iron concentrations in heart, brain, liver, and Jurkat cells (given above). Heart, liver, and Jurkat cells contain about 400 μm iron, whereas the brain contains less than half of that concentration. Perhaps the danger associated with Fenton chemistry has minimized the use of this metal in the most complicated and delicate of all organs.
Relative to age-matched controls, HFE−/− hearts contained slightly more total iron, including more ferritin and less mitochondrial iron; these differences were typical of somewhat older hearts, perhaps reflecting an increased burden on the heart due to hemochromatosis. The HFE−/− liver was iron-overloaded, as expected, but it also contained lower than normal concentrations of mitochondrial iron, which was not expected. Given the literature suggesting that hemosiderin ought to be present in diseased livers, we were surprised not to detect it in our samples.
The lack of IRP2 in IRP2−/− mutant mice should allow ferritin transcripts to be translated and TfR1 transcripts to be degraded quickly under all nutrient iron conditions. Under iron-deficient conditions, the absence of IRP2 should lead to insufficient cellular iron, whereas under iron-excess conditions, the same absence should lead to excess cellular iron. In our study, mice were fed iron-sufficient chow (but just barely so) and so cellular iron would be expected to be near normal levels. Consistent with this, mutant hearts contained about 85% of the iron found in WT hearts, with an iron content similar to that of WT hearts. Similarly, spectra of the brains of IRP2−/− mice were virtually indistinguishable from controls (34, 38). By contrast, IRP2−/− livers were iron-overloaded with ferritin (34). Perhaps the IRP1 system is sufficient to retain iron homeostasis in the mutant heart and brain, whereas the liver relies more on the IRP2 system for regulation, and so dysregulation was more severe. Interestingly, the level of mitochondrial iron in IRP2−/− livers was substantially higher than in the HFE−/− livers, even though both livers were overloaded with iron.
We also examined the effect of iron deficiency on a pregnant mouse and her offspring. As expected, the iron-deficient mother contained subnormal levels of ferritin iron, but normal levels of mitochondrial iron. A similar phenomenon was observed in spectra of iron-deficient brains (38), in which the CD was actually more intense than it was in spectra of iron-sufficient brains. The hearts of the 3-week-old pups from the iron-deficient mother contained normal levels of mitochondrial iron. This suggests that the iron-deficient mother does not limit the transfer of iron to her offspring during pregnancy or lactation. Interestingly, the iron-deficient mother also had a higher concentration of mitochondrial iron in the heart than did the iron-sufficient control.
Our study provides “control” spectra for the published MB spectrum of a frataxin KO mouse heart (23). That spectrum contained only a doublet species due to FeIII oxyhydroxide nanoparticles (no ferritin or CD was observed). It should also be compared with the MB spectrum of human Jurkat cells in which frataxin expression was knocked-down by RNAi (44). That spectrum also consisted of a nanoparticle doublet with parameters (δ = 0.48 mm/s, ΔEQ = 0.57 mm/s) very near to those reported for the frataxin KO heart doublet. We conclude that the type of nanoparticles in both mouse hearts and human Jurkat cells must be similar.
Finally, we have pondered as to why previously published MB spectra of human hearts lacked the mitochondrial doublet and the blood doublet that we have observed with such strong intensities. Published spectra only exhibit features due to ferritin and hemosiderin. Our samples certainly contained ferritin, but none appeared to contain hemosiderin. Perhaps the iron associated with mitochondria and blood in published samples of human organs was converted into ferritin and/or hemosiderin iron during sample preparation. Few details of sample preparation were provided in those studies, but we suspect that the period between death and freezing samples for MB analysis was longer for human (days?) than mouse tissues (30 min). Also, embalming solutions and/or exposure to O2 might alter the iron content of human tissues. In contrast, our mouse tissues were dissected immediately in an anaerobic and refrigerated glove box. They were flushed with an isotonic aqueous buffer, and were frozen within 30 min of death. Clearly there are advantages to using non-human mammals in MB studies besides the ability to enrich them with 57Fe. These advantages, along with the availability of many iron-related transgenic strains of mice, make these small mammals ideal for future MB investigations of iron-related diseases.
Experimental Procedures
All procedures involving mice were approved by the Animal Care and Use committee at Texas A&M University. The original C57BL/6 mice used in the study were a gift from Louise Abbott (Texas A&M University). IRP2−/− mice were a gift from Tracey Rouault (National Institutes of Health). C57BL/6 and IRP2−/− mice were bred as described (38). They were fed iron-deficient chow (Harlan Teklad, Madison, WI; number 80396) to which 50 mg of 57FeIII (Isoflex USA, San Francisco, CA) citrate per kg of chow was added. An iron-deficient pregnant mouse was raised similarly except that she was fed iron-deficient chow unenriched in 57Fe for 1 month, starting a week before giving birth. Water was distilled and deionized. Three 8-week-old male HFE−/− (B6.129S6-Hfe<tm2Nca>/J; 017784) mice and matching C57BL/6 controls were purchased from The Jackson Laboratory. These mice were fed iron-deficient chow to which 200 mg of 57Fe/kg of chow and 1.2 g of ascorbic acid/kg of chow were added. They were also given triple-distilled water to which 100 μm 57FeIII citrate and 1 mm ascorbic acid had been added. These 6 mice were euthanized 1 month after arrival, at 12 weeks of age. Mice were raised in iron-deficient cages, and euthanized with ketamine and xylazine, as described (38). Immediately after death, carcasses were imported into a refrigerated (8 °C) nitrogen-atmosphere glove box (Mbraun Labmaster) containing <10 ppm O2. Preparing samples anaerobically prevented any oxidation or degradation of iron centers in these samples. Animals older than 1 week were flushed by puncturing the heart with a needle, cutting the caudal vena cava, and flowing Ringer's buffer into the heart at 700 μl/min for 5 min per 10 g of animal mass. Organs were removed, weighed, transferred to MB cups, frozen, and removed from the box. EPR samples were prepared by thawing frozen samples anaerobically, adding ∼5 ml of buffer, homogenizing using a tissue grinder, transferring to an EPR tube, capping the tube, removing it from the glove box, and spinning the tubes to pack the material. MB spectroscopy was performed as described (38). Continuous-wave X-band EPR spectra were obtained using a Bruker Elexsys E500A spectrometer with a cryogen-free cooling system. After spectra were collected, samples were thawed and transferred quantitatively to 15-ml plastic tubes with screw-tops. An equal volume of concentrated trace metal-grade nitric acid was added to each sample, and sealed samples were heated to 90 °C overnight. Samples were diluted with 6.0 ml of distilled deionized water and analyzed by ICP-MS (Agilent 7700x).
Author Contributions
M. C. led in raising the mice, dissections, sample preparation, and early spectroscopy characterization. J. D. W. collected the remaining MB spectra, the EPR spectra, performed the metal analysis, prepared figures, and helped write the paper. P. A. L. designed the study, helped with all aspects of it, and wrote most of the paper. All authors approved the final version of the manuscript.
Acknowledgments
We thank current and former members of the Lindahl lab who helped raise and dissect the mice used in this study, including Allison L. Cockrell, Nathaniel M. Dziuba, Gregory P. Holmes-Hampton, Lora S. Lindahl, Cody Lupardus, Sean P. McCormick, Michael J. Moore, and Jinkyu Park.
This work was supported, in whole or in part, by National Institutes of Health Grant GM084266 (for the non-vertebrate animal portion) and Robert A. Welch Foundation Grant A1170. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ISC
- iron-sulfur cluster
- BD
- blood doublet
- ΔEQ
- quadrupole splitting
- FA
- Friedreich ataxia
- HFE
- high iron gene
- HFE−/−
- genotype in which both alleles of the HFE gene have been mutated
- HH
- hereditary hemochromatosis
- ICP-MS
- inductively coupled plasma mass spectrometry
- IRE
- iron responsive element
- IRP1/2
- iron responsive element proteins 1 and 2
- IRP2−/−
- genotype in which both alleles of the IRP2 gene have been mutated
- MB
- Mössbauer
- NHHS
- nonheme high-spin
- ROS
- reactive oxygen species
- TfR1
- transferrin receptor 1
- mt-ferritin
- mitochondrial ferritin.
References
- 1. Sanchis-Gomar F., Perez-Quilis C., Leischik R., and Lucia A. (2016) Epidemiology of coronary heart disease and acute coronary syndrome. Ann. Transl. Med. 4, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Theil E. C., Tosha T., and Behera R. K. (2016) Solving biology's iron chemistry problem with ferritin nanocages. Acc. Chem. Res. 49, 784–791 [DOI] [PubMed] [Google Scholar]
- 3. Ferreira C., Bucchini D., Martin M. E., Levi S., Arosio P., Grandchamp B., and Beaumont C. (2000) Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 275, 3021–3024 [DOI] [PubMed] [Google Scholar]
- 4. Ahmad S., Moriconi F., Naz N., Sultan S., Sheikh N., Ramadori G., and Malik I. A. (2013) Ferritin L and ferritin H are differentially located within hepatic and extra hepatic organs under physiological and acute phase conditions. Int. J. Clin. Exp. Pathol. 6, 622–629 [PMC free article] [PubMed] [Google Scholar]
- 5. Kistler M., Even A., Wagner S., Becker C., Darshan D., Vanoaica L., Kühn L. C., and Schümann K. (2014) Fe-59-distribution in conditional ferritin-H-deleted mice. Exp. Hematol. 42, 59–69 [DOI] [PubMed] [Google Scholar]
- 6. Omiya S., Hikoso S., Imanishi Y., Saito A., Yamaguchi O., Takeda T., Mizote I., Oka T., Taneike M., Nakano Y., Matsumura Y., Nishida K., Sawa Y., Hori M., and Otsu K. (2009) Downregulation of ferritin heavy chain increases labile iron pool, oxidative stress and cell death in cardiomyocytes. J. Mol. Cell. Cardiol. 46, 59–66 [DOI] [PubMed] [Google Scholar]
- 7. Santambrogio P., Biasiotto G., Sanvito F., Olivieri S., Arosio P., and Levi S. (2007) Mitochondrial ferritin expression in adult mouse tissues. J. Histochem. Cytochem. 55, 1129–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campanella A., Rovelli E., Santambrogio P., Cozzi A., Taroni F., and Levi S. (2009) Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 18, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kohgo Y., Ikuta K., Ohtake T., Torimoto Y., and Kato J. (2008) Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 88, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nam H., Wang C. Y., Zhang L., Zhang W., Hojyo S., Fukada T., and Knutson M. D. (2013) ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: implications for tissue iron uptake in iron-related disorders. Haematologica 98, 1049–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chua-anusorn W., Tran K. C., Webb J., Macey D. J., and St. Pierre T. G. (2000) Chemical speciation of iron deposits in thalassemic heart tissue. Inorg. Chim. Acta 300, 932–936 [Google Scholar]
- 12. Neves J. V., Olsson I. A., Porto G., and Rodrigues P. N. (2010) Hemochromatosis and pregnancy: iron stores in the HFE−/− mouse are not reduced by multiple pregnancies. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G525-G529 [DOI] [PubMed] [Google Scholar]
- 13. Gutiérrez L., Quintana C., Patiño C., Bueno J., Coppin H., Roth M. P., and Lázaro F. J. (2009) Iron speciation study in HFE knockout mice tissues: magnetic and ultrastructural characterization. Biochim. Biophys. Acta 1792, 541–547 [DOI] [PubMed] [Google Scholar]
- 14. Subramaniam V. N., McDonald C. J., Ostini L., Lusby P. E., Wockner L. F., Ramm G. A., and Wallace D. F. (2012) Hepatic iron deposition does not predict iron loading in mouse models of hereditary hemochromatosis. Am. J. Pathol. 181, 1173–1179 [DOI] [PubMed] [Google Scholar]
- 15. St. Pierre T. G., Tran K. C., Webb J., Macey D. J., Pootrakul P., and Dickson D. P. (1992) Core structures of haemosiderins deposited in various organs in β-thalassemia hemoglobin E disease. Hyperfine Interact. 71, 1279–1282 [Google Scholar]
- 16. Bell S. H., Weir M. P., Dickson D. P., Gibson J. F., Sharp G. A., and Peters T. J. (1984) Mössbauer spectroscopic studies of human haemosiderin and ferritin. Biochim. Biophys. Acta 787, 227–236 [DOI] [PubMed] [Google Scholar]
- 17. Webb J., Macey D. J., Chua-anusorn W., St. Pierre T. G., Brooker L. R., Rahman I., and Noller B. (1999) Iron biominerals in medicine and the environment. Coord. Chem. Rev. 190–192, 1199–1215 [Google Scholar]
- 18. St Pierre T. G., Tran K. C., Webb J., Macey D. J., Heywood B. R., Sparks N. H., Wade V. J., Mann S., and Pootrakul P. (1991) Organ-specific crystalline structures of ferritin cores in β-thalassemia hemoglobin E. Biol. Met. 4, 162–165 [DOI] [PubMed] [Google Scholar]
- 19. Kaufman K. S., Papaefthymiou G. C., Frankel R. B., and Rosenthal A. (1980) Nature of iron deposits on the cardiac walls in β-thalassemia by Mössbauer spectroscopy. Biochim. Biophys. Acta 629, 522–529 [DOI] [PubMed] [Google Scholar]
- 20. Perdomini M., Belbellaa B., Monassier L., Reutenauer L., Messaddeq N., Cartier N., Crystal R. G., Aubourg P., and Puccio H. (2014) Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich's ataxia. Nat. Med. 20, 542–547 [DOI] [PubMed] [Google Scholar]
- 21. Michael S., Petrocine S. V., Qian J., Lamarche J. B., Knutson M. D., Garrick M. D., and Koeppen A. H. (2006) Iron and iron-responsive proteins in the cardiomyopathy of Friedreich's ataxia. Cerebellum 5, 257–267 [DOI] [PubMed] [Google Scholar]
- 22. Payne R. M. (2011) The heart in Friedreich's ataxia: basic findings and clinical implications. Prog Pediatr. Cardiol. 31, 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whitnall M., Suryo Rahmanto Y., Huang M. L., Saletta F., Lok H. C., Gutiérrez L., Lázaro F. J., Fleming A. J., St Pierre T. G., Mikhael M. R., Ponka P., and Richardson D. R. (2012) Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc. Natl. Acad. Sci. U.S.A., 109, 20590–20595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pousset F., Legrand L., Monin M. L., Ewenczyk C., Charles P., Komajda M., Brice A., Pandolfo M., Isnard R., Tezenas du Montcel S., and Durr A. (2015) A 22-year follow-up study of long-term cardiac outcome and predictors of survival in Friedreich ataxia. JAMA Neurol. 72, 1334–1341 [DOI] [PubMed] [Google Scholar]
- 25. Whitnall M., Suryo Rahmanto Y., Sutak R., Xu X., Becker E. M., Mikhael M. R., Ponka P., and Richardson D. R. (2008) The MCK mouse heart model of Friedreich's ataxia: Alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc. Natl. Acad. Sci. U.S.A. 105, 9757–9762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Puccio H., Simon D., Cossée M., Criqui-Filipe P., Tiziano F., Melki J., Hindelang C., Matyas R., Rustin P., and Koenig M. (2001) Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 27, 181–186 [DOI] [PubMed] [Google Scholar]
- 27. Sutak R., Xu X., Whitnall M., Kashem M. A., Vyoral D., and Richardson D. R. (2008) Proteomic analysis of hearts from frataxin knockout mice: marked rearrangement of energy metabolism, a response to cellular stress and altered expression of proteins involved in cell structure, motility and metabolism. Proteomics 8, 1731–1741 [DOI] [PubMed] [Google Scholar]
- 28. Zhao N., Sun Z., Mao Y., Hang P., Jiang X., Sun L., Zhao J., and Du Z. (2010) Myocardial iron metabolism in the regulation of cardiovascular diseases in rats. Cell. Physiol. Biochem. 25, 587–594 [DOI] [PubMed] [Google Scholar]
- 29. Huang M. L., Becker E. M., Whitnall M., Suryo Rahmanto Y., Ponka P., and Richardson D. R. (2009) Elucidation of the mechanism of mitochondrial iron loading in Friedreich's ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. U.S.A. 106, 16381–16386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schulz T. J., Westermann D., Isken F., Voigt A., Laube B., Thierbach R., Kuhlow D., Zarse K., Schomburg L., Pfeiffer A. F., Tschöpe C., and Ristow M. (2010) Activation of mitochondrial energy metabolism protects against cardiac failure. Aging 2, 843–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Elas M., Bielanska J., Pustelny K., Plonka P. M., Drelicharz L., Skorka T., Tyrankiewicz U., Wozniak M., Heinze-Paluchowska S., Walski M., Wojnar L., Fortin D., Ventura-Clapier R., and Chlopicki S. (2008) Detection of mitochondrial dysfunction by EPR technique in mouse model of dilated cardiomyopathy. Free Rad. Biol. Med. 45, 321–328 [DOI] [PubMed] [Google Scholar]
- 32. Lesnefsky E. J., Gudz T. I., Migita C. T., Ikeda-Saito M., Hassan M. O., Turkaly P. J., and Hoppel C. L. (2001) Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch. Biochem. Biophys. 385, 117–128 [DOI] [PubMed] [Google Scholar]
- 33. Zhang D. L., Ghosh M. C., and Rouault T. A. (2014) The physiological functions of iron regulatory proteins in iron homeostasis: an update. Front. Pharmacol. 5, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chakrabarti M., Cockrell A. L., Park J., McCormick S. P., Lindahl L. S., and Lindahl P. A. (2015) Speciation of iron in mouse liver during development, iron deficiency, IRP2 deletion, and inflammatory hepatitis. Metallomics 7, 93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Galy B., Ferring D., Minana B., Bell O., Janser H. G., Muckenthaler M., Schümann K., and Hentze M. W. (2005) Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 106, 2580–2589 [DOI] [PubMed] [Google Scholar]
- 36. Zumbrennen-Bullough K. B., Becker L., Garrett L., Hölter S. M., Calzada-Wack J., Mossbrugger I., Quintanilla-Fend L., Racz I., Rathkolb B., Klopstock T., Wurst W., Zimmer A., Wolf E., Fuchs H., Gailus-Durner V., et al. (2014) Abnormal brain iron metabolism in IRP2 deficient mice is associated with mild neurological and behavioral impairments. PLOS ONE 9, e98072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chakrabarti M., and Lindahl P. A. (2014) The Utility of Mössbauer Spectroscopy in Eukaryotic Cell Biology and Animal Physiology, in Iron-sulfur Clusters (Rouault T. A., ed) pp. 49–75, Walter de Gruyter Publisher, Berlin Germany [Google Scholar]
- 38. Holmes-Hampton G. P., Chakrabarti M., Cockrell A. L., McCormick S. P., Abbott L. C., Lindahl L. S., and Lindahl P. A. (2012) Changing iron content of the mouse brain during development. Metallomics 4, 761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chakrabarti M., Barlas M. N., McCormick S. P., Lindahl L. S., and Lindahl P. A. (2015) Kinetic of iron import into developing mouse organs determined by a pup-swapping method. J. Biol. Chem. 290, 520–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chua-anusorn W, St. Pierre T. G., Webb J., Macey D. J., Yansukon P., and Pootrakul P. (1993) Mössbauer spectroscopic study of the forms of iron in normal liver and spleen tissue. Hyperfine Interact. 91, 905–910 [Google Scholar]
- 41. Jhurry N. D., Chakrabarti M., McCormick S. P., Holmes-Hampton G. P., and Lindahl P. A. (2012) Biophysical investigation of the ironome of human Jurkat cells and mitochondria. Biochemistry 51, 5276–5284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ramos E., Ruchala P., Goodnough J. B., Kautz L., Preza G. C., Nemeth E., and Ganz T. (2012) Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 120, 3829–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Truksa J., Gelbart T., Peng H., Beutler E., Beutler B., and Lee P. (2009) Suppression of the hepcidin-encoding gene Hamp permits iron overload in mice lacking both hemojuvelin and matriptase-2/TMPRSS6. Br. J. Haematol. 147, 571–581 [DOI] [PubMed] [Google Scholar]
- 44. Jhurry N. D., Chakrabarti M., McCormick S. P., Gohil V. M., and Lindahl P. A. (2013) Mössbauer study and modeling of iron import and trafficking in human Jurkat cells. Biochemistry 52, 7926–7942 [DOI] [PMC free article] [PubMed] [Google Scholar]