

Abstract
The 4E10 antibody displays an extreme breadth of HIV-1 neutralization and therefore constitutes a suitable model system for structure-guided vaccine design and immunotherapeutics against AIDS. In this regard, the relevance of autoreactivity with membrane lipids for the biological function of this antibody is still a subject of controversy. To address this dispute, herein we have compared the membrane partitioning ability of the 4E10 antibody and several of its variants, which were mutated at the region of the paratope surface in contact with the membrane interface. We first employed a physical separation approach (vesicle flotation) and subsequently carried out quantitative fluorescence measurements in an intact system (spectroscopic titration), using 4E10 Fab labeled with a polarity-sensitive fluorescent probe. Moreover, recognition of epitope peptide in membrane was demonstrated by photo-cross-linking assays using a Fab that incorporated the genetically encoded unnatural amino acid p-benzoylphenylalanine. The experimental data ruled out that the proposed stereospecific recognition of viral lipids was necessary for the function of the antibody. In contrast, our data suggest that nonspecific electrostatic interactions between basic residues of 4E10 and acidic phospholipids in the membranes contribute to the observed biological function. Moreover, the energetics of membrane partitioning indicated that 4E10 behaves as a peripheral membrane protein, tightening the binding to the ligand epitope inserted in the viral membrane. The implications of these findings for the natural production and biological function of this antibody are discussed.
Keywords: antibody, human immunodeficiency virus (HIV), membrane protein, protein-lipid interaction, vaccine development, 4E10 antibody, MPER vaccine, broadly neutralizing antibody, lipid polyreactivity, protein-membrane interaction
Introduction
Engagement by the 4E10 antibody of a conserved helical epitope on the membrane-proximal external region (MPER)5 of the Env gp41 subunit results in one of the broadest neutralization levels of HIV-1 reported to date (98% of viruses blocked in standard infectivity tests (1–3)). Prevalence of anti-MPER antibodies has been linked to neutralization breadth and potency of certain sera from chronically infected individuals (3, 4). Thus, solving the molecular mechanism that leads to viral blocking after MPER engagement by broadly neutralizing antibody (bNAb) 4E10 is critical in the fields of vaccine design and immunotherapy (3, 5–7). Because the viral membrane takes part in the stabilization of the MPER helix, it is generally accepted that antibodies targeting the MPER membrane site are polyspecific, showing a capacity to bind lipid moieties (8–12). In that respect, anti-MPER antibodies have been proposed to resemble natural antibodies that bind to phospholipids (13). Furthermore, it has been argued that HIV-1 may have evolved to escape anti-MPER responses by structural mimicry of the phospholipid ligands bound to autoantibodies (14). Following this line of evidence, some researchers consider anti-MPER antibodies as generally autoreactive and postulate that their natural production is limited by B-cell tolerance mechanisms (14–16).
Some experimental evidence suggesting that bNAb 4E10 binds to phospholipids, particularly anionic species such as phosphatidylserine (PS), cardiolipin (CL), or phosphatidylinositol (PI), has been obtained in several laboratories during the last decade (9, 10, 14, 17), which supported the notion of structural mimicry of phospholipid-binding sites by the antibody (13, 14). In agreement with that idea, the existence of a binding pocket accommodating the polar headgroup of PS or CL within the paratope of 4E10 was proposed (10). Strikingly, the structure of the paratope with lipid bound would differ from that of the paratope with peptide epitope bound (10). Nonetheless, some of these concepts have been challenged by the recent resolution of crystal structures of the unbound Fab form (18, 19) and Fab-lipid complexes (12, 20).
In particular, Irimia et al. (12) determined the crystal structure of the complex between 4E10 Fab and lipids such as phosphatidic acid (PA), phosphatidylglycerol (PG), and the lipid moiety glycerol phosphate, which revealed two binding sites on the paratope surface in contact with the membrane interface. Lipid recognition occurred primarily at the heavy chain complementarity determining region 1 (CDRH1) between the backbone atoms of the protein and the glycerol and phosphate moieties, which are the common components of phosphoglycerides. Thus, from these results it appears that the 4E10 Fab does not behave as a target-specific phospholipid-binding domain that performs selective-stereospecific recognition of ligand molecules (21). The crystallographic data were also consistent with the simultaneous accommodation of the phospholipid headgroup moieties and the bound peptide epitope within the 4E10 paratope and further underscored the role of the hydrophobic CDRH3 apex in establishing interactions with the lipid tails (12).
In an attempt to discern the role played by lipid interactions in the 4E10 neutralization mechanism, we have herein determined the specificity and intensity of its interactions with phospholipids in the context of biologically relevant bilayer systems. To that end, we have employed liposome flotation assays (a physical separation method) that were subsequently complemented with spectroscopic titration assays using Fabs labeled with the polarity-sensitive probe NBD. Moreover, a Fab 4E10 variant incorporating the UV-sensitive unnatural amino acid pBPA was used to monitor the specific recognition of the epitope peptide in a bilayer milieu. Our data indicated that direct partitioning of the 4E10 antibody from the aqueous phase into membranes is driven by favorable electrostatic interactions between the surfaces of the membrane and the paratope. Although not strictly required for its biological function, electrostatic forces were also beneficial for binding to the membrane-anchored epitope peptide, and these favorable interactions correlated with the neutralizing potency of the 4E10 antibody. Collectively, our observations support the view that favorable, albeit unspecific, interactions between 4E10 and lipids play a central role in the neutralization mechanism by increasing potency of the antibody, while keeping its broad coverage.
Results
Partitioning of 4E10 into Membranes Depends on Anionic Phospholipids
We first determined the partitioning of the 4E10 antibody into membranes using a vesicle flotation assay (Fig. 1A). We note that in these experiments the amount of antibody incubated with vesicles was kept the same under all conditions tested, even though the intensity of the bands among the different samples slightly fluctuated. The results displayed in Fig. 1B first reflected the different capacities of 10E8 and 4E10 antibodies for partitioning into virus-like (VL) membranes mimicking the viral envelope composition (22). The 10E8 antibody does not show lipid polyreactivity (3, 23) or insertion into membranes devoid of peptide epitope (24) and, consequently, was used as a negative control for membrane binding in our assays. Thus, whereas most of the 10E8 antibody was recovered from the high density fractions (Fig. 1B, first row), a major fraction of the input 4E10 antibody was recovered from the upper low density fractions, i.e. cofloating with the VL vesicles (Fig. 1B, second row). In search for a possible dependence of 4E10 binding to membranes on specific lipids, we subsequently established the contribution of each lipid present in the VL mixture to the process. Subtraction from the VL mixture of individual zwitterionic phospholipids (sphingomyelin or phosphatidylethanolamine (PE)), or cholesterol one at a time, did not abolish membrane binding by 4E10 (Fig. 1B, third, fourth, and fifth rows; lipid compositions displayed in Table 1). Control experiments indicated that removal of zwitterionic and ubiquitous lipid phosphatidylcholine (PC) did not alter this antibody-vesicle binding pattern (data not shown). In contrast, removal of the anionic PS from the composition of the vesicles prevented 4E10 to bind to the liposomes (Fig. 1C, first row). To determine whether binding was specific for PS or resulted from unspecific electrostatic effects, we replaced PS with other anionic phospholipid species (i.e. PG, PI, PA, or CL in Fig. 1C). The results convincingly showed that binding of 4E10 to liposomes was restored for all anionic phospholipids tested, indicating that membrane association did not require stereospecific recognition of a particular headgroup (21).
FIGURE 1.

Partitioning of the anti-MPER 4E10 Fab into membranes. A, 4E10 membrane partitioning detected in a sucrose gradient. Lipid vesicles incubated with the Fab were subjected to centrifugation. The sample was divided into four different fractions based on their different densities. An additional fraction employing SDS was collected, representing the material attached to the surface of the tube. The locations of the liposomes in the third and fourth fractions (i.e. floating fractions) were verified from the Rho-PE emission (bottom panels). The presence of Fab was probed by Western blotting. B, effect of the constituent lipids of the VL mixture on the process (lipid compositions are given in Table 1). 10E8 antibody was used as negative control (first row). C, effect of removing PS from the composition (first row) and recovery of membrane binding by its replacement with other anionic phospholipids: PG, PI, PA, or CL. The band corresponding to the Fabs comigrated in the gels with the 25-kDa molecular mass marker (indicated by the arrowheads, only in the first rows in B and C). SM, sphingomyelin; Chol, cholesterol.
TABLE 1.
Composition of VL lipid mixtures used in this work
The values are mole percentages of lipid. Chol, cholesterol.
| PC | Chol | SM | PE | PS | |
|---|---|---|---|---|---|
| VL | 14 | 46 | 17 | 16 | 7 |
| Without SMa | 31 | 46 | 16 | 7 | |
| Without PEa | 30 | 46 | 17 | 7 | |
| Without Cholb | 27 | 30 | 29 | 14 | |
| Without PSa | 21 | 46 | 17 | 16 |
a Contribution of phospholipids added to PC.
b Contribution of Chol distributed proportionally among the other lipids.
Basic Residues of the Paratope Promote Partitioning of 4E10 into the Membrane
The previous flotation experiments suggested that electrostatic interactions of 4E10 with anionic phospholipids promote its partitioning into membranes. Inspection of the surface of the paratope in contact with the membrane interface as inferred from the structure of Fab in complex with lipids (12) revealed a positively charged region (Fig. 2A). We sought to elucidate the relevance of this charged patch for the electrostatic interaction of 4E10 with the membrane by separately mutating the basic residues Arg-73HC or Lys-100eHC by a Glu residue (R73E and K100eE mutants, respectively; Fig. 2A). We assumed that such substitutions, reversing the charge of the affected residues, would reduce the net positive charge at the base of the paratope, making the antibody less prone to interact electrostatically with the negatively charged membranes. The analysis included a negative control designated as BS, in which residues Ser-28 and Ser-30 of the CDRH1 were mutated to Ala. These substitutions involved uncharged residues and therefore did not presumably alter the surface charge of the paratope even if involving residues located in the lipid-binding sites (12).
FIGURE 2.

Design of 4E10 mutants with exposed basic residues of the paratope. A, effect of R73E and K100eE substitutions on the surface charge of the paratope. B, membrane partitioning as measured by flotation assays using vesicles containing 50 mol % of anionic lipid (PS) and PC. BS stands for a negative control with a double Ser to Ala substitution at positions 28 and 30 of the heavy chain.
Fig. 2B illustrates the phenotypic traits resulting from the mutations with regard to membrane binding. To obtain a more robust comparison, these assays employed membranes containing high levels of PS (PC:PS 1:1, mole ratio). As expected from a model dominated by electrostatic interactions, most of the input WT antibody associated with the vesicles. In contrast, the binding of R73E to the vesicles was reduced by ∼50%. The K100eE mutant was not present in the floating fractions containing the vesicles, indicating an even weaker tendency to partition into membranes. The negative BS control reproduced rather well the behavior of the WT antibody. We arrived at similar conclusions using VL vesicles (see below).
Charge-reversing Mutations Interfere with the Biological Function of 4E10
We next examined the role of electrostatic forces in the biological function of the 4E10 antibody using the same collection of antibodies described immediately above (Figs. 3 and 4 and Table 2). First, ITC experiments were conducted to evaluate the effect of the mutations on the affinity for the epitope peptide MPER(664–690) (Fig. 3A and Table 2). We have shown in previous ITC experiments that this peptide mimics better the C-terminal MPER epitopes than peptides truncated at position 683 (24). The values of KD determined from the binding isotherms were 4.6, 12.2, and 12.8 nm for the WT, BS, and R73E antibodies, respectively (Table 1), i.e. within the same range of affinity previously determined for the 10E8 antibody (KD, 9.6 nm (24)). A more significant reduction of affinity (≈20-fold) was observed in the case of the K100eE mutant (KD = 91 nm).
FIGURE 3.

Functional characterization of 4E10 mutants: binding to epitope peptide. A, binding isotherms of the MPER(664–690) epitope peptide to Fab 4E10 examined by ITC. The upper panel indicates the heat released upon consecutive injections of 10 μl of peptide solution (40 μm) into Fab (3 μm) in the calorimeter cell, and the lower panel indicates the integrated heats (symbols) and non-linear least squares fit (solid line) to the data using a one site binding model with the program ORIGIN 7.0. The thermodynamic parameters of binding are displayed in Table 1. Each titration was carried out once. B, flotation experiments in the presence of VL LUVs (left panel) or VL LUVs containing MPER(671–693) peptide (right panel). C, recognition of VL LUVs (left panel) or VL LUVs containing MPER(671–693) peptide (right panel) as determined by dot blot analysis. 2-fold serial dilutions of 500 μm LUVs and 10 μm peptide was spotted on the filters. BS, negative control with a double Ser to Ala substitution at positions 28 and 30 of the heavy chain.
FIGURE 4.

Functional characterization of 4E10 mutants: biological activity. A, PsV neutralization potency of the different mutants. The means of six measurements ± S.D. are represented in the plots. The experimental points were fitted to saturation curves. B, JRCSF PsV recognition as determined by dot blot analysis. Decreasing amounts of PsV (from left to right) were spotted onto nitrocellulose membranes and probed with 4E10 Fab WT and its mutants.
TABLE 2.
Thermodynamic parameters of binding of Fab 4E10 mutants to the MPER(664–690) peptide and their neutralization potencies
The temperature was 298 K.
| Antibody (4E10) | KDa | ΔG°a | ΔH°a | −TΔS° | nb | HXB2- IC50c | JRCSF- IC50c |
|---|---|---|---|---|---|---|---|
| nm | kcal mol−1 | kcal mol−1 | kcal mol−1 | μg/ml | μg/ml | ||
| 4E10_WT | 4.6 ± 1.5 | −11.4 ± 0.2 | −15.1 ± 0.2 | 3.7 | 1.0 ± 0.1 | 0.21 ± 0.02 | 1.26 ± 0.09 |
| BS | 12.2 ± 2.5 | −10.8 ± 0.1 | −11.7 ± 0.1 | 0.9 | 1.3 ± 0.1 | 0.38 ± 0.03 | 1.59 ± 0.27 |
| R73E | 12.8 ± 3.5 | −10.8 ± 0.1 | −14.7 ± 0.2 | 3.9 | 1.1 ± 0.1 | 5.3 ± 0.4 | 13.2 ± 3.5 |
| K100eE | 91 ± 14 | −9.6 ± 0.2 | −10.4 ± 0.2 | 0.7 | 1.4 ± 0.1 | 3.3 ± 0.2 | 12.7 ± 4.0 |
aThe errors were calculated from the fitting procedure with ORIGIN 7.0.
bn refers to the molar ratio peptide/protein.
cThe values determined from the fitting curves in Fig. 4A.
To assess the effect of the mutations on epitope binding in a membrane context, flotation experiments were next carried out comparing the location of the Fab in the presence of bare VL vesicles or VL vesicles decorated with the peptide MPER(671–693) (Fig. 3B). This peptide has been shown to expose optimally the epitope region on the surface of lipid vesicles (24, 25). In the absence of peptide (Fig. 3B, left panel), the most conspicuous effect was observed for the K100eE mutant, which again did not appreciably associate with membranes in the VL system. As compared with WT or BS, a lower level of binding to LUVs was also evident for the R73E mutant. Incubation with VL-peptide complexes (Fig. 3B, right panel) resulted in the complete association of the WT and BS antibodies with vesicles. However, although a significant increase in binding to vesicles with respect to the bare vesicles was evident, association of R73E and K100eE variants with vesicles containing peptide was not complete.
The previous flotation results were obtained under conditions that promoted membrane partitioning (i.e. high lipid concentration (26)). To establish further differences in membrane affinities, binding to the vesicles as a function of the lipid concentration was monitored in dot blot assays (Fig. 3C). In this approach, measuring conditions were initially set up to reproduce the relative binding signals observed in flotation assays (i.e. to match signals of the Fabs recovered in the floating fractions). Titration of Fab-s with VL or VL-peptide complexes (Fig. 3C, left and right panels, respectively) confirmed the poor performance of the charge-reversing R73E and K100eE mutants for partitioning into membranes, as well as their lower affinity for vesicles containing the epitope peptide.
To establish a correlation between biological function, binding to membrane-embedded epitope, and membrane partitioning, the potency of the antibody and mutants was evaluated in neutralization assays (Fig. 4A). The IC50 values determined for the mutants R73E and K100eE increased by approximately 1 order of magnitude with respect to WT and BS (Fig. 4A and Table 1). Interestingly, the R73E and BS mutants bound to peptide by ITC with almost equal affinity, but their neutralization potencies were significantly different (Table 1). These data demonstrated that mutations reducing the electrostatic charge in the vicinity of the paratope have a negative impact not only in the binding to the epitope inserted in vesicles, but also in the biological function of the antibody. In contrast, the BS mutant reproduced for the most part the behavior of the WT Fab. Dot blot assays indicated that the reduced levels of activity of the mutants correlated well with lower binding of the mutant antibodies to pseudovirus immobilized on a solid substrate (Fig. 4B). Overall, these results revealed a correlation between direct binding to immobilized pseudovirus (Fig. 4B) and direct binding to immobilized vesicles containing the epitope peptide (Fig. 3C, right panel).
Electrostatic Interactions Contribute to the Efficient Partitioning of 4E10 into Membranes
The left panel of Fig. 5A illustrates the difference in the conformation of the 4E10 CDRH3 loop in solution (PDB entry code 2FX7, gray) or embedded in a vesicle-like micellar structure made of dihexanoyl phosphatidic acid (PDB entry code 4XBG, white). Based on this observation, a movement of the CDRH3 loop was proposed to occur upon interaction of the antibody with the membrane (12). According to this model, the Trp-100bHC at the tip of the CDRH3 would relocate within the membrane interface to a position roughly equidistant from the headgroups and first methylene groups of the acyl chains of the lipids. Moreover, in a recent work we have shown that the polarity-sensitive NBD probe replacing Trp at the position 100b of the CDRH3 can be employed to monitor the insertion of the Fab 10E8 into the membrane (24). Here we have followed a similar strategy to characterize the association of 4E10 with membranes.
FIGURE 5.

Binding of 4E10 to VL LUVs monitored by changes in NBD fluorescence. A, left panel, repositioning of CDRH3 loop Trp-100bHC residue upon 4E10 interaction with membranes. The loops were modeled according to crystal structures with PDB entries 2FX7 and 4XBG (gray and white, respectively). A second phospholipid molecule was aligned with that bound in 4XBG crystal. Right panel, changes in fluorescence that occur upon incubation of the Fab 4E10 labeled with NBD at position 100bHC with lipid vesicles. Right top panel, the solid lines correspond to NBD-4E10 incubated in solution (black) or in presence of 100 or 200 μm VL liposomes (red and blue, respectively). The dotted lines correspond to incubations with VL liposomes containing 0.25 mol % of Rho-PE. Right bottom panel, NBD-Fab was incubated with VL LUVs or VL LUVs containing 10 mol % 16:0–5 doxyl-PC (red solid and red dotted lines, respectively). B, titration of NBD-labeled Fab with increasing concentrations of VL liposomes as indicated in the panels. C, plot of the fraction of Fab bound as a function of the concentration of lipid accessible (half the total lipid concentration). The molar fraction partition coefficient, Kx, was calculated from the best fit of Equation 1 to the data (curve). Each symbol on the plot represents an average of three independent experiments (±S.D. if larger than symbol). 150 nm of NBD-Fab was used in these experiments.
The right panels of Fig. 5A display the changes in fluorescence that occur upon incubation of the NBD-Fab 4E10 with lipid vesicles. In the presence of VL vesicles, the fluorescence emission of the NBD attached to 4E10 shifted toward shorter wavelengths, and its intensity increased. These two properties indicated that the dye (and therefore the apex of the CDRH3) its being transferred from a solvent-exposed environment to a more hydrophobic environment (27). The right top panel of Fig. 5A additionally illustrates the shift of emission to longer wavelengths when vesicles contained Rho-PE. This phenomenon occurred as a consequence of the resonance energy transfer between the NBD(donor)-Rhodamine(acceptor) pair (27). As displayed in the right bottom panel of Fig. 5A, NBD fluorescence emission was also quenched by inclusion of 16:0–5 doxyl-PC in the lipid composition. The effects induced by these probes residing within the lipid bilayer demonstrated that NBD-Fab fluorescence changes were caused by the antibody-membrane interaction. Such effects were further consistent with the interfacial location of the Trp-100bHC predicted by the model displayed in the left panel of Fig. 5A.
We next analyzed the partitioning process quantitatively by monitoring the changes in NBD-Fab fluorescence that occur upon incubation of the Fab with increasing concentrations of VL large unilamellar vesicles (LUVs) (Fig. 5B, left panel). Consistent with the restrained interaction deduced from the flotation assays (Fig. 1C), the fluorescence of the NBD-Fab did not significantly change upon incubation with vesicles devoid of PS (Fig. 5B, right). The titration values adjusted to Equation 1 (Fig. 5C) from which a mole fraction partition coefficient (Kx) of 0.6 × 106 was determined, a value that falls in the range of that observed for peripheral membrane proteins (28–30).
To evaluate the relative contribution of electrostatic interactions to the 4E10 partitioning process, titration experiments were conducted in a PC:PS model system (30, 31). Thus, Kx values were calculated for vesicles containing different mol percentages of monovalent acidic PS (Fig. 6). Association with vesicles increased with the PS content (Fig. 6, A and B), following the pattern predicted from a favorable contribution of electrostatic interactions (30). The plot of the observed partitioning free energy (ΔGobs) versus the surface potential (Ψ0) calculated using Equations 2 and 3, respectively (32, 33), yielded through extrapolation a ΔGobs value of approximately −5 kcal mol−1 for membranes lacking net charge (Fig. 6C). This value indicates that, in the absence of anionic PS, a lipid concentration of ∼15 mm would be required for 50% of the Fab initially in solution be bound to membrane. In contrast, the value measured for pure PS vesicles was much more favorable (ΔGobs = −8.8 kcal mol−1). A simple calculation using Equation 2 reveals that lipid diluted 3 orders of magnitude (i.e. 17.5 μm) would be sufficient to attain a similar extent of Fab-membrane binding at room temperature.
FIGURE 6.
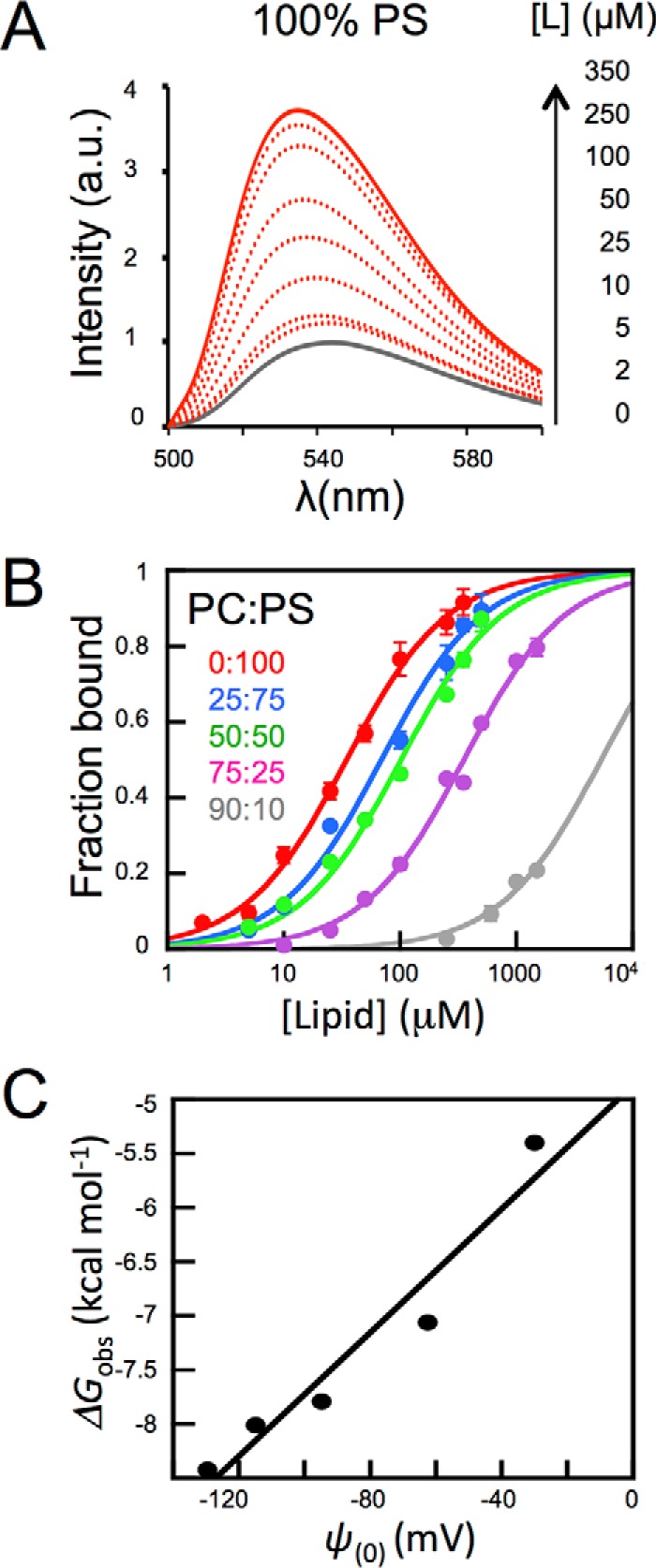
Effect of anionic phospholipid density on 4E10 binding to LUVs. A, changes of NBD-4E10 fluorescence in the presence of increasing concentrations PS LUVs as indicated in the panel. B, titration of NBD-labeled Fab with vesicles containing different mol percentages of PC:PS as follows: 90:10 (gray), 75:25 (magenta), 50:50 (green), 25:75 (blue), and 0:100 (red). The molar fraction partition coefficients, Kx, were calculated from the best fit of Equation 1 to the data (curves). Each symbol on the plot represents the average (±S.D.) of three independent experiments as the one displayed in the previous panel. C, plot of the free energy of partitioning versus the membrane surface potential in the previous lipid vesicles, estimated according to Equations 2 and 3, respectively. Conditions otherwise as in Fig. 5.
Electrostatic Interactions Enhance Epitope Peptide Binding by the 4E10 Antibody
To infer the contribution of membrane partitioning to epitope recognition, we conducted experiments with PC:PS vesicles decorated with peptide epitope (Fig. 7). Consistent with previous flotation results (Fig. 4), the NBD emission spectra obtained upon incubation with vesicles containing 25 or 50% of PS revealed a favorable contribution of both anionic phospholipids and peptide epitope to Fab partitioning (Fig. 7A). Overall, the presence of peptide resulted in an increment of the partition constant, the effect being more pronounced with the highest proportion of PS (Fig. 7B).
FIGURE 7.
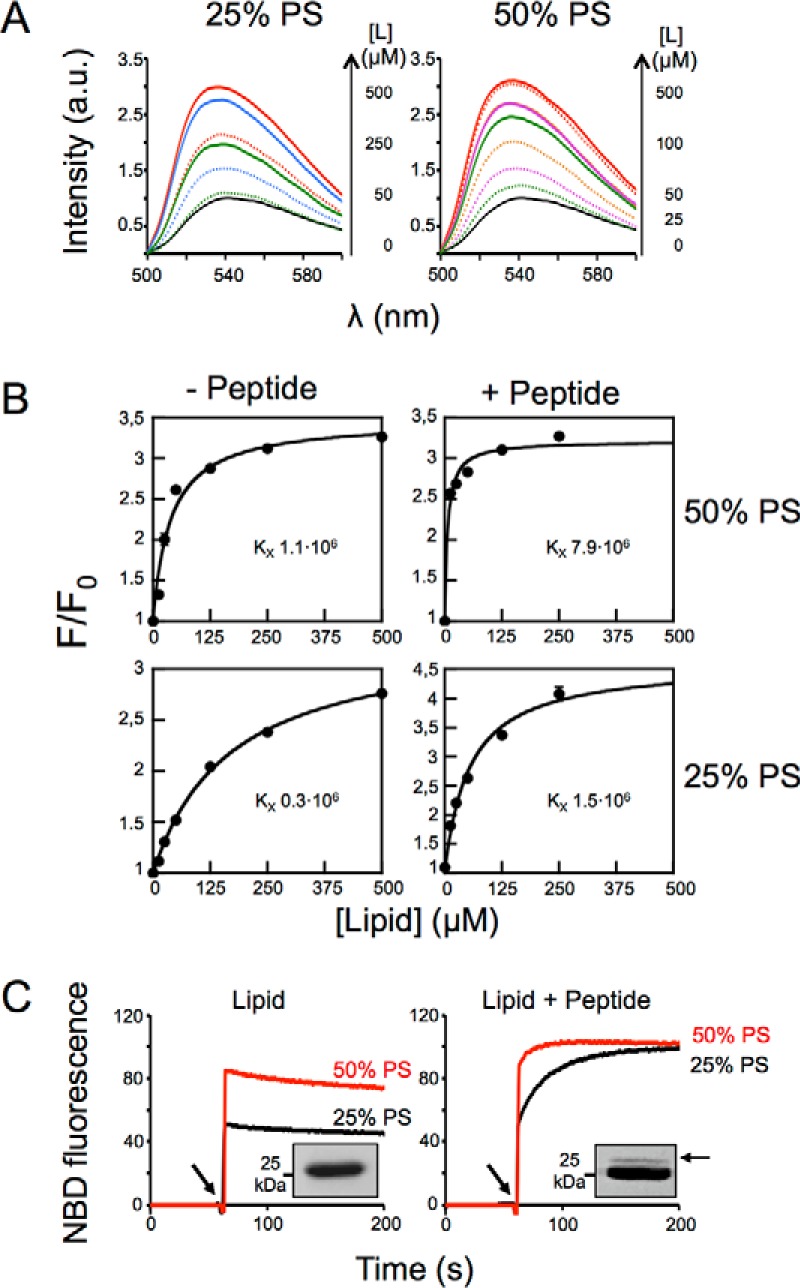
Effect of electrostatic interactions on epitope recognition at the membrane surface. A, changes of NBD-4E10 fluorescence emission spectra in the presence of increasing concentrations of vesicles as indicated in the panel were measured in the absence (dotted lines) or in the presence of 1.7 μm of MPER(671–693) peptide inserted in the membrane (solid lines). B, effect of PS on Fab 4E10 partitioning into vesicles with and without MPER(671–693)peptide inserted in the membrane. NBD-Fab was titrated with PC:PS LUVs as indicated. Each data point corresponds to the average of three titrations (± S.D.) as the ones displayed in the previous panel. C, kinetics of incorporation of NBD-Fab into bare vesicles containing 25 or 50% of PS (left panel) or into the same vesicles decorated with 1.7 μm MPER(671–693) peptide (right panel). The arrow indicates NBD-Fab addition. Fab-peptide interaction was assayed by photo-cross-linking using Fab-pBPA and 25% PS vesicles. The presence of an adduct band confirmed Fab-peptide interaction in peptide-containing vesicles (insets). Lipid concentration was 100 μm. The results representative of two replicas are presented.
To study in more detail the combined effect of electrostatic interactions with the membrane surface and peptide recognition, we next performed time course binding assays. Fig. 7C displays the kinetic traces of the changes of fluorescence intensity that occurred upon injection of NBD-Fab (arrow) in a solution containing LUVs (left panel), or a solution containing LUV-peptide complexes (right panel). The addition of antibody to vesicles containing PS (25 or 50%) resulted in both cases in a sudden increase of fluorescence intensity followed by a signal plateau, consistent with a fast association to the membrane.
In contrast, upon addition of vesicles of the same composition containing the peptide, the sudden increase was followed by a second phase, in which the fluorescence signal increased gradually, leveling off at later times (Fig. 7C, right panel). Thus, whereas the fast phase of the NBD change can be explained by the spontaneous insertion into the membrane, the slower phase appears to be driven by specific peptide recognition. Interestingly, the peptide-dependent slower phase observed for the 50% mol PS LUVs (Fig. 7C, right panel, red line) was faster than that observed with 25% mol PS. Thus, the PS appeared to have two effects. On the one hand, and as previously described, PS promoted higher levels of spontaneous incorporation. On the other hand, PS promoted faster recognition of the epitope peptide by the fraction of the Fab that had remained in solution.
Finally, to demonstrate peptide engagement under these conditions, we employed photo-cross-linking assays. To that end, the 4E10 Fab was modified with the UV-sensitive unnatural amino acid pBPA (34, 35), which was genetically encoded at position Trp-100bHC of the antibody as previously described for the Fab 10E8 (24). Formation of covalent adducts were detected only when the lipid vesicles harbored the MPER(671–693) peptide but not with vesicles devoid of peptide (Fig. 7C, insets).
4E10 Antibody Pre-equilibrated on the Membrane Is Competent for Epitope Engagement
Attachment of 4E10 molecules to virions without engagement to the epitope has been postulated as a mechanism favoring viral neutralization (36, 37). This two-step model assumes that the fraction of 4E10 antibody prebound to the membrane engages with gp41 when viral fusion is activated. Fig. 8 illustrates experiments to evaluate the capacity of the Fab for encountering and effectively engage the membrane-inserted epitope. To ensure that all Fab actually bound to membranes, we increased the lipid concentration in the assay, as well as the PS content in the vesicles. As explained in the diagram displayed in Fig. 8A, under these conditions the subsequent process of peptide engagement can be scored by photo-cross-linking assays.
FIGURE 8.

Epitope recognition by 4E10 antibody bound to membrane. A, schematic representation of the assay showing partitioning of the Fab into the membrane (top panel) and its movement through the surface until the encounter and efficient engagement with epitope peptide (bottom panel). This latter event can be scored by photo-cross-linking. B, kinetics of NBD-Fab incorporation into pure PS vesicles in the presence (red line) or absence of MPER(671–693) peptide (black solid line). Lipid and peptide concentrations were 250 and 1.7 μm, respectively. The dotted line corresponds to the signal of the NBD-Fab in solution. The arrow indicates the NBD-Fab addition time. C, photo-cross-linking experiments. Lane 1, pBPA-Fab4E10 was added to PS vesicles decorated with MPER(671–693) (red kinetic trace in previous panel); lane 2, pBPA-Fab4E10 and MPER(671–693) peptide were incubated in the absence of PS vesicles; lane 3, pBPA-Fab4E10 was incubated with vesicles that contained the MPER(671–693)/W672A-F673A peptide; lane 4, pBPA-Fab4E10 was first incorporated into vesicles (see black kinetic trace in previous panel) and after 1 min MPER(671–693) was added. Protein bands were detected by Coomassie staining or Western blotting (top and bottom panels, respectively). Lipid and peptide concentrations were as in the previous panel. Results representative of two replicas are presented in B and C.
As expected, NBD-Fab addition to vesicles composed of 100% PS resulted in an abrupt and comparable increase of fluorescence in the presence and in the absence of epitope peptides (Fig. 8B). The Fab-peptide complexes that could form under these conditions were subjected to UV light, and the formation of adducts was analyzed by SDS-PAGE and Western blotting (Fig. 8C). An additional band corresponding to cross-linked peptide (MPER(671–693)) and Fab HC was observed in samples of pBPA-Fab incubated with liposome-peptide complexes (lane 1). A similar outcome was observed when pBPA-Fab and peptide were incubated in the absence of vesicles (lane 2). In contrast, the adduct band was not detected upon pBPA-Fab incubation with liposomes that contained a peptide with crucial residues for antibody binding Trp-672 and Phe-673 mutated to Ala (lane 3). This negative control was included to assure that adducts formed within vesicles came from specific peptide binding and not from spontaneous cross-linking potentially promoted by the high densities of Fab and peptide attained at the membrane surface. Finally, a band corresponding to the Fab HC-peptide adduct was also observed when Fab was first pre-equilibrated on the membrane and peptide subsequently added to resulting liposome-Fab complexes (lane 4). These data clearly indicate that Fab prebound to membrane is competent in engagement with epitope.
Discussion
The Env complex embodies the entry machinery of the HIV-1 and constitutes its main antigen (38, 39). The gp41 MPER is highly conserved across the different HIV-1 strains and isolates and is functionally involved in assisting the membrane fusion process that culminates with cell infection (40–42). Because of its high degree of sequence conservation and the key role played by this element in the viral cycle, the bNAb 4E10 raised against the MPER exhibits an unusual neutralization breadth (5, 43, 44). It has been hypothesized that interactions with viral membrane lipids are required for the fulfillment of 4E10 biological function (9–12, 17, 36, 45–49). However, a systematic and quantitative assessment of 4E10 partitioning into membranes was lacking. A detailed examination of this process is important for two reasons: (i) to establish the lipid specificity range, which helps to evaluate the assumption that lipid autoreactivity can prevent the production of 4E10-like antibodies through vaccination, and (ii) to establish the contribution of membrane-assisted MPER engagement to the biological function of 4E10.
Relevance of Spontaneous Partitioning for the Biological Function of 4E10
Our data in Figs. 1–6 demonstrate that 4E10 partitioning into membranes is promoted by unspecific electrostatic interactions between the negatively charged membrane surface and the basic residues exposed on the paratope and not by the stereospecific recognition of a particular phospholipid headgroup (21). In this regard, 4E10 can be considered a peripheral membrane protein employing concerted electrostatic and hydrophobic forces to insert into membranes and engage its epitope (for reviews, see Refs. 29, 50, and 51). Herein we have examined the factors intervening in the electrostatically driven partitioning of 4E10 in the membrane by employing two complementary approaches: (i) by altering the surface charge of the Fab paratope via mutagenesis (Figs. 2–4) and (ii) by modifying the density of monovalent anionic phospholipid in the membrane, particularly that of PS (Figs. 6–8).
As expected from the unspecific nature of electrostatic interactions, the mutation of basic residues as in R73E or K100eE clearly diminished the partitioning of the antibody into membranes. However, albeit less potent, these mutants were still active in neutralization assays, an effect that correlated with weaker binding to immobilized PsVs or VL vesicles decorated with peptide. Thus, although comprising a modulatory factor for the amount of membrane-bound antibody and hence potency, partitioning driven by electrostatic interactions was not an absolute requirement for the neutralizing activity of the antibody. This key observation strongly suggests that the ability of the antibody to interact electrostatically with viral membranes needs an adaptation during the maturation pathway.
Our data employing vesicles with different surface charge densities also support the spontaneous interaction of 4E10 with negatively charged membranes (Fig. 6) and that such interaction can contribute to the more efficient recognition of the membrane-bound epitope (Figs. 7 and 8). This is in contrast to the 10E8 interaction with membranes, which appears to depend on binding to peptide ligand anchored to the bilayer (24). However, the bNAb 10E8 displays higher potency than the antibody 4E10, and this phenomenon occurs without exhibiting binding to anionic phospholipid (Fig. 1B and Refs. 3 and 32) or higher affinity for the epitope peptide MPER(664–690) by ITC (24). This important difference suggests that 4E10 and 10E8 follow different adaptive mechanisms to efficiently engage with the MPER helix at membrane interfaces, where the antibody exerts its viral neutralization activity.
Implications for the Mechanism of Neutralization by 4E10
Collectively, our data support the idea that direct interaction of 4E10 with the membrane is of high relevance for its neutralizing activity. Fig. 9 illustrates three non-exclusive mechanisms that have been previously proposed for the role of membrane interactions in the mechanism of 4E10 neutralization. We discuss below the possible ways by which the peripheral membrane interactions described in this work may support these mechanisms.
FIGURE 9.
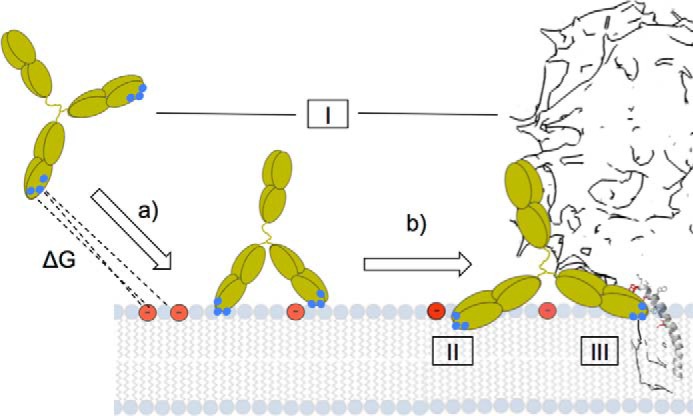
Summary of mechanisms to explain the involvement of electrostatic interactions in MPER helix engagement and ensuing neutralization. See “Discussion” for details.
Accumulation in the Viral Membrane (Fig. 9, Left Structure)
Several authors have proposed a two-step mechanism for neutralization, in which 4E10 first attaches to the viral membrane (36, 37). This reversible step would be required to ensure subsequent binding to target MPER epitope, which is transiently exposed after fusion activation. Preattachment implies first sufficient free energy for driving spontaneous partitioning (Fig. 9, arrow a) and second competence of the Fab prebound to membranes for subsequent specific recognition of the epitope ligand (Fig. 9, arrow b). Our quantitative measurements (Figs. 5–7) are consistent with partitioning constants in the range of those measured for peripheral membrane proteins (28–30). Moreover, by combining fluorescence spectroscopy and photo-cross-linking assays, we were able to demonstrate the competence of antibody prebound to membrane for specific epitope binding (Fig. 8). Thus, in principle, our in vitro data employing model systems are consistent with this mechanism.
Avidity Effect (Fig. 9, Center Structure)
The surface of one virion particle only contains approximately 10 Env complexes, and consequently the non-immunogenic lipid component of the envelope constitutes the main structural-functional element of the HIV particle that is accessible from the external milieu (52). The reduced accessibility to the epitope precludes bifunctional binding and has been thus regarded to as an evasion mechanism (6). Lipid polyreactivity may increase the avidity of IgG by endowing 4E10 with the capacity to directly interact with membranes (6, 37). Our data support this notion, although we note that the association of 4E10 with the membrane is driven by unspecific electrostatic interactions.
Structural Adaptation (Fig. 9, Right Structure)
The insertion of helical MPER epitope into the membrane interface imposes structural adaptations to the mechanism of recognition within the two-dimensional membrane milieu (17, 49). It has been reported that PS becomes accessible at the external leaflet of membranes in mature virions (for a discussion on this issue, see Ref. 22). Our data suggest that 4E10 takes the advantage of favorable electrostatic interactions with PS to enhance affinity for the membrane-bound epitope ligand (Figs. 3 and 7), most likely by providing an optimal orientation to the epitope-binding pocket relative to the membrane plane and sticking out MPER-helix.
In summary, our data support that electrostatic interactions with the negatively charged viral membrane surface contributes to enhance the affinity of 4E10 for the MPER helix and, hence, its potency. Assuming a constant lipid composition of virions across the different strains and isolates of HIV-1, we speculate that this adaptation could have evolved through maturation without compromising the neutralization breadth of the antibody.
Experimental Procedures
Materials
The peptides MPER(664–690), KKKK-DKWASLWNWFDITNWLWYIKLFIMIVG-KKKKK; MPER(671–693), KKK-NWFDITNWLWYIKLFIMIVGGLV-KK; and MPER(671–693)/W672A-F673A, KKK-NAADITNWLWYIKLFIMIVGGLV-KK were synthesized in C-terminal carboxamide form by solid phase methods using Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry, purified by reverse phase HPLC, and characterized by MALDI-TOF mass spectrometry (purity > 95%). Peptides were routinely dissolved in DMSO (spectroscopy grade), and their concentrations were determined by the bicinchoninic acid microassay (Pierce). The experimental procedures described in Refs. 4 and 53 were followed for the production and purification of Fabs. Vector pEVOL, encoding a Tyr-tRNA synthetase suitable for the incorporation of photoreactive amino acid p-benzoylphenylalanine (pBPA), was a gift from Prof. P. G. Schultz (Scripps Research Institute). pBPA was purchased from Bachem (Bubendorf, Switzerland), and NBD was from Thermo Fisher. All lipids including Rho-PE and doxyl-PC (16:0–5) were purchased from Avanti Polar Lipids (Alabaster, AL).
Liposome Flotation Assays
LUVs were prepared following the extrusion method of Hope et al. (54). Phospholipids and cholesterol were mixed in chloroform and dried under a N2 stream. Traces of organic solvent were removed by overnight vacuum pumping. Subsequently, the dried lipid films were dispersed in buffer and subjected to 10 freeze-thaw cycles prior to extrusion 10 times through 2 stacked polycarbonate membranes with a nominal pore size of 100 nm (Nuclepore, Inc., Pleasanton, CA). Phospholipid concentration of liposome suspensions was determined by phosphate analysis. Cholesterol content in vesicles was determined after extrusion by the cholesterol oxidase/peroxidase method (BioSystems, Barcelona, Spain) and found to be within the experimental error. Vesicle flotation experiments in sucrose gradients were subsequently performed following the method described by Yethon et al. (55). In brief, 100 μl of a sample containing rhodamine-labeled liposomes and Fab (1.5 mm lipid and 1.5 μm Fab) was adjusted to a sucrose concentration of 1.4 m in a final volume of 300 μl and subsequently overlaid with 400- and 300-μl layers of 0.8 and 0.5 m sucrose, respectively. The gradient was centrifuged at 436,000 × g for 3 h in a TLA 120.2 rotor (Beckman Coulter, Brea, CA). After centrifugation, four 250-μl fractions were collected as depicted in Fig. 1A. The material adhered to the tubes was collected into a fifth fraction by washing with 250 μl of hot (100 °C) 1% (w/v) SDS. The different fractions were run on SDS-PAGE, and the presence of Fab was probed by Western blotting using a sandwich comprising a goat (anti-human Fab) antibody (Sigma) and a mouse (anti-goat) antibody-HRP conjugate (Santa Cruz). The results displayed in the figures are representative of at least two replicates.
Isothermal Titration Calorimetry
Titration experiments were performed with a VP-ITC microcalorimeter (MicroCal, Northampton, MA) at 25 °C. Prior to the experiment, the proteins were dialyzed overnight at 4 °C against 10 mm sodium phosphate (pH 7.5), 150 mm NaCl, and 10% glycerol. Samples containing protein and peptide solubilized in dialysis buffer were supplemented with 5 mm DPC and degassed immediately before each measurement. Fab 4E10 and mutant versions (3 μm) were titrated with peptide (40 μm). The volume of each injection was 10 μl. Peptide dilution heat was subtracted for data analysis. The binding isotherms were fitted to a one-site binding model using the program ORIGIN 7.0. The fitting procedure yields the stoichiometry (n), the binding constant (KD), and the enthalpy (ΔH°) of the binding reaction. Each experiment was carried out once, where errors were obtained from the fitting procedure.
Cell Entry Inhibition
The cell entry inhibition assays were carried out as previously described (25, 53, 56). To run the experiments, HIV-1 pseudoviruses were first produced by transfection of human kidney HEK293T cells with the full-length env clones HXB2 or JRCSF (kindly provided by Jamie K. Scott and Naveed Gulzar, Simon Fraser University, Burnaby, Canada) using calcium phosphate. The cells were cotransfected with vectors pWXLP-GFP and pCMV8.91, encoding a green fluorescent protein and an env-deficient HIV-1 genome, respectively (generously provided by Patricia Villace, Consejo Superior de Investigaciones Científicas, Madrid, Spain). After 24 h, the medium was replaced with Optimem-GlutaMAX II (Invitrogen) without serum. Three days after transfection, the pseudovirus particles were harvested, passed through 0.45-μm pore sterile filters (Millex® HV; Millipore NV, Brussels, Belgium), and finally concentrated by ultracentrifugation in a sucrose gradient. Neutralization was determined using TZM-bl target cells (AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, contributed by J. Kappes). Samples were set up in duplicate in 96-well plates and incubated for 1.5 h at 37 °C with a 10–15% tissue culture infectious dose of pseudovirus. After antibody-pseudovirus coincubation, 11,000 target cells were added in the presence of 30 μg/ml DEAE-dextran (Sigma-Aldrich). Neutralization levels after 72 h were inferred from the reduction in the number of GFP-positive cells as determined by flow cytometry using a BD FACSCalibur flow cytometer (Becton Dickinson).
Fab Labeling with the NBD Fluorescent Probe and Spectroscopic Titration
Labeling with the polarity-sensitive NBD probe was performed as described (57, 58). In brief, a cysteine-substituted Fab derivative (W100bHCC) was first generated by site-directed mutagenesis and modified with a sulfhydryl-specific iodoacetamide derivative of NBD. Fluorescence emission spectra were recorded with the excitation wavelength fixed at 470 nm. An emission spectrum of a sample lacking the fluorophore was subtracted from the spectrum of the equivalent sample containing the fluorophore. Partitioning curves were computed from the fractional changes in emitted NBD fluorescence when titrated with increasing lipid concentrations. The mole fraction partition coefficients, Kx, were determined by fitting the experimental values to a hyperbolic function (26),
| (Eq. 1) |
where [L] is the concentration of accessible lipid, and K is the lipid concentration at which the bound peptide fraction is 0.5. Therefore, Kx = [W]/K, where [W] is the molar concentration of water. The observed free energy of water membrane partitioning was subsequently calculated according to the following expression.
| (Eq. 2) |
For the estimation of the electrical potential at the membrane surface as a function of the PS content, the following equation was used,
| (Eq. 3) |
where c is the number of ions per volume, and σ is the surface charge density (33). To calculate the latter parameter, a surface area/phospholipid of 69.5 Å2 was considered, and net charges of 0 and −1 were assigned to PC and PS, respectively (59).
Fab Labeling with Photo-activable Amino Acid
For the photo-cross-linking experiments, an amber codon specific for an engineered tRNA that translates the unnatural amino acid pBPA was encoded in the DNA sequence of the heavy chain of the 4E10 Fab. Procedures to express a 4E10 Fab mutant bearing pBPA instead of Trp at position 100bHC were adapted from previous reports (34, 35). Synthesis of Fab and the engineered tRNA was induced with 0.4 mm isopropyl-d-thiogalactopyranoside and 4% (w/v) arabinose, respectively, in LB medium supplemented with 0.2 mg/liter pBPA. For the photo-cross-linking experiment, samples containing Fab bearing the Trp-100bHC × pBPA substitution at 1.5 μm and peptides at 10 μm were irradiated with UV light at 365 nm for 20 min at 4 °C using a UVP B-100AP lamp.
Author Contributions
E. R., J. M. M. C., K. T., and J. L. N. conceived and designed the experiments; E. R. produced Fabs and performed biophysical and biochemical characterizations with assistance of S. I. and M. G.-M.; E. R., J. M. M. C., and J. L. N. wrote the paper with input from K. T., and all of the authors reviewed it.
Acknowledgment
We thank Dr. Apellaniz for helpful discussions.
This work was supported by the National Institutes of Health and Basque Government Grants AI097051 and IT838-13 (to J. L. N.). Work in the Tsumoto laboratory was supported by Japanese Society for the Promotion of Science (JSPS) KAKENHI-A Grants 25249115 and 16H02420 (to K. T.), by the Platform for Drug Discovery, Informatics and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by JSPS KAKENHI-C Grant 15K06962 (to J. M. M. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MPER
- membrane-proximal external region
- bNAb
- broadly neutralizing antibody
- CDR
- complementarity determining region
- NBD
- 4-chloro-7-nitrobenz-2-oxa-1,3-diazole
- pBPA
- p-benzoylphenylalanine
- PS
- phosphatidylserine
- VL
- virus-like
- CL
- cardiolipin
- PI
- phosphatidylinositol
- PA
- phosphatidic acid
- PG
- phosphatidylglycerol
- PE
- phosphatidylethanolamine
- PC
- phosphatidylcholine
- ITC
- isothermal titration calorimetry
- PDB
- Protein Data Bank
- LUV
- large unilamellar vesicle
- Rho-PE
- 1,2-dioleoyl-sn-glycerol-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl)
- doxyl-PC
- 1-palmitoyl-2-stearoyl-(5-doxyl)-sn-glycero-3-phosphocholine (16:0–5)
- PsV
- pseudovirus.
References
- 1. Zwick M. B., Labrijn A. F., Wang M., Spenlehauer C., Saphire E. O., Binley J. M., Moore J. P., Stiegler G., Katinger H., Burton D. R., and Parren P. W. (2001) Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol. 75, 10892–10905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Binley J. M., Wrin T., Korber B., Zwick M. B., Wang M., Chappey C., Stiegler G., Kunert R., Zolla-Pazner S., Katinger H., Petropoulos C. J., and Burton D. R. (2004) Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 78, 13232–13252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang J., Ofek G., Laub L., Louder M. K., Doria-Rose N. A., Longo N. S., Imamichi H., Bailer R. T., Chakrabarti B., Sharma S. K., Alam S. M., Wang T., Yang Y., Zhang B., Migueles S. A., et al. (2012) Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491, 406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jacob R. A., Moyo T., Schomaker M., Abrahams F., Grau Pujol B., and Dorfman J. R. (2015) Anti-V3/glycan and anti-MPER neutralizing antibodies, but not anti-V2/glycan site antibodies, are strongly associated with greater anti-HIV-1 neutralization breadth and potency. J. Virol. 89, 5264–5275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Montero M., van Houten N. E., Wang X., and Scott J. K. (2008) The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol. Mol. Biol. Rev. 72, 54–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klein F., Mouquet H., Dosenovic P., Scheid J. F., Scharf L., and Nussenzweig M. C. (2013) Antibodies in HIV-1 vaccine development and therapy. Science 341, 1199–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim A. S., Leaman D. P., and Zwick M. B. (2014) Antibody to gp41 MPER alters functional properties of HIV-1 Env without complete neutralization. PLoS Pathog. 10, e1004271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sánchez-Martínez S., Lorizate M., Hermann K., Kunert R., Basañez G., and Nieva J. L. (2006) Specific phospholipid recognition by human immunodeficiency virus type-1 neutralizing anti-gp41 2F5 antibody. FEBS Lett. 580, 2395–2399 [DOI] [PubMed] [Google Scholar]
- 9. Matyas G. R., Beck Z., Karasavvas N., and Alving C. R. (2009) Lipid binding properties of 4E10, 2F5, and WR304 monoclonal antibodies that neutralize HIV-1. Biochim. Biophys. Acta 1788, 660–665 [DOI] [PubMed] [Google Scholar]
- 10. Finton K. A., Larimore K., Larman H. B., Friend D., Correnti C., Rupert P. B., Elledge S. J., Greenberg P. D., and Strong R. K. (2013) Autoreactivity and exceptional CDR plasticity (but not unusual polyspecificity) hinder elicitation of the anti-HIV antibody 4E10. PLoS Pathog. 9, e1003639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen J., Frey G., Peng H., Rits-Volloch S., Garrity J., Seaman M. S., and Chen B. (2014) Mechanism of HIV-1 neutralization by antibodies targeting a membrane-proximal region of gp41. J. Virol. 88, 1249–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Irimia A., Sarkar A., Stanfield R. L., and Wilson I. A. (2016) Crystallographic identification of lipid as an integral component of the epitope of HIV broadly neutralizing antibody 4E10. Immunity 44, 21–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alving C. R. (2008) 4E10 and 2F5 monoclonal antibodies: binding specificities to phospholipids, tolerance, and clinical safety issues. AIDS 22, 649–651 [DOI] [PubMed] [Google Scholar]
- 14. Haynes B. F., Fleming J., St Clair E. W., Katinger H., Stiegler G., Kunert R., Robinson J., Scearce R. M., Plonk K., Staats H. F., Ortel T. L., Liao H. X., and Alam S. M. (2005) Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308, 1906–1908 [DOI] [PubMed] [Google Scholar]
- 15. Doyle-Cooper C., Hudson K. E., Cooper A. B., Ota T., Skog P., Dawson P. E., Zwick M. B., Schief W. R., Burton D. R., and Nemazee D. (2013) Immune tolerance negatively regulates B cells in knock-in mice expressing broadly neutralizing HIV antibody 4E10. J. Immunol. 191, 3186–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Verkoczy L., Kelsoe G., and Haynes B. F. (2014) HIV-1 envelope gp41 broadly neutralizing antibodies: hurdles for vaccine development. PLoS Pathog. 10, e1004073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sánchez-Martínez S., Lorizate M., Katinger H., Kunert R., and Nieva J. L. (2006) Membrane association and epitope recognition by HIV-1 neutralizing anti-gp41 2F5 and 4E10 antibodies. AIDS Res. Hum. Retroviruses 22, 998–1006 [DOI] [PubMed] [Google Scholar]
- 18. Rujas E., Gulzar N., Morante K., Tsumoto K., Scott J. K., Nieva J. L., and Caaveiro J. M. (2015) Structural and thermodynamic basis of epitope binding by neutralizing and nonneutralizing forms of the anti-HIV-1 antibody 4E10. J. Virol. 89, 11975–11989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rujas E., Gulzar N., Morante K., Tsumoto K., Scott J. K., Nieva J. L., and Caaveiro J. M. (2016) Reply to “The broadly neutralizing, anti-HIV antibody 4E10: an open and shut case?”. J. Virol. 90, 3276–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bird G. H., Irimia A., Ofek G., Kwong P. D., Wilson I. A., and Walensky L. D. (2014) Stapled HIV-1 peptides recapitulate antigenic structures and engage broadly neutralizing antibodies. Nat. Struct. Mol. Biol. 21, 1058–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lemmon M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 [DOI] [PubMed] [Google Scholar]
- 22. Huarte N., Carravilla P., Cruz A., Lorizate M., Nieto-Garai J. A., Kräusslich H. G., Pérez-Gil J., Requejo-Isidro J., and Nieva J. L. (2016) Functional organization of the HIV lipid envelope. Sci. Rep. 6, 34190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwon Y. D., Georgiev I. S., Ofek G., Zhang B., Asokan M., Bailer R. T., Bao A., Caruso W., Chen X., Choe M., Druz A., Ko S. Y., Louder M. K., McKee K., O'Dell S., et al. (2016) Optimization of the solubility of HIV-1-neutralizing antibody 10E8 through somatic variation and structure-based design. J. Virol. 90, 5899–5914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rujas E., Caaveiro J. M., Partida-Hanon A., Gulzar N., Morante K., Apellániz B., García-Porras M., Bruix M., Tsumoto K., Scott J. K., Jiménez M. A., and Nieva J. L. (2016) Structural basis for broad neutralization of HIV-1 through the molecular recognition of 10E8 helical epitope at the membrane interface. Sci. Rep. 6, 38177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Apellániz B., Rujas E., Serrano S., Morante K., Tsumoto K., Caaveiro J. M., Jiménez M. A., and Nieva J. L. (2015) The atomic structure of the HIV-1 gp41 transmembrane domain and its connection to the immunogenic membrane-proximal external region. J. Biol. Chem. 290, 12999–13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. White S. H., Wimley W. C., Ladokhin A. S., and Hristova K. (1998) Protein folding in membranes: determining energetics of peptide-bilayer interactions. Methods Enzymol. 295, 62–87 [DOI] [PubMed] [Google Scholar]
- 27. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., pp. 205–235, Springer-Verlag, New York Inc., New York [Google Scholar]
- 28. Nomikos M., Mulgrew-Nesbitt A., Pallavi P., Mihalyne G., Zaitseva I., Swann K., Lai F. A., Murray D., and McLaughlin S. (2007) Binding of phosphoinositide-specific phospholipase C-ζ (PLC-ζ) to phospholipid membranes: potential role of an unstructured cluster of basic residues. J. Biol. Chem. 282, 16644–16653 [DOI] [PubMed] [Google Scholar]
- 29. Mulgrew-Nesbitt A., Diraviyam K., Wang J., Singh S., Murray P., Li Z., Rogers L., Mirkovic N., and Murray D. (2006) The role of electrostatics in protein-membrane interactions. Biochim. Biophys. Acta 1761, 812–826 [DOI] [PubMed] [Google Scholar]
- 30. Arbuzova A., Wang J., Murray D., Jacob J., Cafiso D. S., and McLaughlin S. (1997) Kinetics of interaction of the myristoylated alanine-rich C kinase substrate, membranes, and calmodulin. J. Biol. Chem. 272, 27167–27177 [DOI] [PubMed] [Google Scholar]
- 31. Arbuzova A., Wang L., Wang J., Hangyás-Mihályné G., Murray D., Honig B., and McLaughlin S. (2000) Membrane binding of peptides containing both basic and aromatic residues. Experimental studies with peptides corresponding to the scaffolding region of caveolin and the effector region of MARCKS. Biochemistry 39, 10330–10339 [DOI] [PubMed] [Google Scholar]
- 32. White S. H., and Wimley W. C. (1999) Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28, 319–365 [DOI] [PubMed] [Google Scholar]
- 33. McLaughlin S. (1989) The electrostatic properties of membranes. Annu. Rev. Biophys. Biophys. Chem. 18, 113–136 [DOI] [PubMed] [Google Scholar]
- 34. Young T. S., Ahmad I., Yin J. A., and Schultz P. G. (2010) An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374 [DOI] [PubMed] [Google Scholar]
- 35. Abe R., Caaveiro J. M., Kozuka-Hata H., Oyama M., and Tsumoto K. (2012) Mapping ultra-weak protein-protein interactions between heme transporters of Staphylococcus aureus. J. Biol. Chem. 287, 16477–16487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alam S. M., Morelli M., Dennison S. M., Liao H. X., Zhang R., Xia S. M., Rits-Volloch S., Sun L., Harrison S. C., Haynes B. F., and Chen B. (2009) Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proc. Natl. Acad. Sci. U.S.A. 106, 20234–20239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haynes B. F., Nicely N. I., and Alam S. M. (2010) HIV-1 autoreactive antibodies: are they good or bad for HIV-1 prevention? Nat. Struct. Mol. Biol. 17, 543–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wyatt R., and Sodroski J. (1998) The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280, 1884–1888 [DOI] [PubMed] [Google Scholar]
- 39. Karlsson Hedestam G. B., Fouchier R. A., Phogat S., Burton D. R., Sodroski J., and Wyatt R. T. (2008) The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nat. Rev. Microbiol. 6, 143–155 [DOI] [PubMed] [Google Scholar]
- 40. Salzwedel K., West J. T., and Hunter E. (1999) A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol. 73, 2469–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sáez-Cirión A., Nir S., Lorizate M., Agirre A., Cruz A., Pérez-Gil J., and Nieva J. L. (2002) Sphingomyelin and cholesterol promote HIV-1 gp41 pretransmembrane sequence surface aggregation and membrane restructuring. J. Biol. Chem. 277, 21776–21785 [DOI] [PubMed] [Google Scholar]
- 42. Apellániz B., Rujas E., Carravilla P., Requejo-Isidro J., Huarte N., Domene C., and Nieva J. L. (2014) Cholesterol-dependent membrane fusion induced by the gp41 membrane-proximal external region-transmembrane domain connection suggests a mechanism for broad HIV-1 neutralization. J. Virol. 88, 13367–13377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zwick M. B. (2005) The membrane-proximal external region of HIV-1 gp41: a vaccine target worth exploring. AIDS 19, 1725–1737 [DOI] [PubMed] [Google Scholar]
- 44. Kwong P. D., and Mascola J. R. (2012) Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity 37, 412–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cardoso R. M., Zwick M. B., Stanfield R. L., Kunert R., Binley J. M., Katinger H., Burton D. R., and Wilson I. A. (2005) Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22, 163–173 [DOI] [PubMed] [Google Scholar]
- 46. Huarte N., Lorizate M., Maeso R., Kunert R., Arranz R., Valpuesta J. M., and Nieva J. L. (2008) The broadly neutralizing anti-human immunodeficiency virus type 1 4E10 monoclonal antibody is better adapted to membrane-bound epitope recognition and blocking than 2F5. J. Virol. 82, 8986–8996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun Z. Y., Oh K. J., Kim M., Yu J., Brusic V., Song L., Qiao Z., Wang J. H., Wagner G., and Reinherz E. L. (2008) HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity 28, 52–63 [DOI] [PubMed] [Google Scholar]
- 48. Dennison S. M., Stewart S. M., Stempel K. C., Liao H. X., Haynes B. F., and Alam S. M. (2009) Stable docking of neutralizing human immunodeficiency virus type 1 gp41 membrane-proximal external region monoclonal antibodies 2F5 and 4E10 is dependent on the membrane immersion depth of their epitope regions. J. Virol. 83, 10211–10223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scherer E. M., Leaman D. P., Zwick M. B., McMichael A. J., and Burton D. R. (2010) Aromatic residues at the edge of the antibody combining site facilitate viral glycoprotein recognition through membrane interactions. Proc. Natl. Acad. Sci. U.S.A. 107, 1529–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McLaughlin S., and Aderem A. (1995) The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 20, 272–276 [DOI] [PubMed] [Google Scholar]
- 51. Gelb M. H., Cho W., and Wilton D. C. (1999) Interfacial binding of secreted phospholipases A2: more than electrostatics and a major role for tryptophan. Curr. Opin. Struct. Biol. 9, 428–432 [DOI] [PubMed] [Google Scholar]
- 52. Zhu P., Liu J., Bess J. Jr, Chertova E., Lifson J. D., Grisé H., Ofek G. A., Taylor K. A., and Roux K. H. (2006) Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441, 847–852 [DOI] [PubMed] [Google Scholar]
- 53. Julien J. P., Huarte N., Maeso R., Taneva S. G., Cunningham A., Nieva J. L., and Pai E. F. (2010) Ablation of the complementarity-determining region H3 apex of the anti-HIV-1 broadly neutralizing antibody 2F5 abrogates neutralizing capacity without affecting core epitope binding. J. Virol. 84, 4136–4147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hope M. J., Bally M. B., Webb G., and Cullis P. R. (1985) Production of large unilamellar vesicles by a rapid extrusion procedure: characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta 812, 55–65 [DOI] [PubMed] [Google Scholar]
- 55. Yethon J. A., Epand R. F., Leber B., Epand R. M., and Andrews D. W. (2003) Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J. Biol. Chem. 278, 48935–48941 [DOI] [PubMed] [Google Scholar]
- 56. Serrano S., Araujo A., Apellániz B., Bryson S., Carravilla P., de la Arada I., Huarte N., Rujas E., Pai E. F., Arrondo J. L., Domene C., Jiménez M. A., and Nieva J. L. (2014) Structure and immunogenicity of a peptide vaccine, including the complete HIV-1 gp41 2F5 epitope: implications for antibody recognition mechanism and immunogen design. J. Biol. Chem. 289, 6565–6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shepard L. A., Heuck A. P., Hamman B. D., Rossjohn J., Parker M. W., Ryan K. R., Johnson A. E., and Tweten R. K. (1998) Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry 37, 14563–14574 [DOI] [PubMed] [Google Scholar]
- 58. Heuck A. P., Hotze E. M., Tweten R. K., and Johnson A. E. (2000) Mechanism of membrane insertion of a multimeric beta-barrel protein: perfringolysin O creates a pore using ordered and coupled conformational changes. Mol. Cell 6, 1233–1242 [DOI] [PubMed] [Google Scholar]
- 59. Rand R. P., and Parsegian V. A. (1989) Hydration forces between phospholipid bilayers. Biochim. Biophys. Acta 988, 351–376 [Google Scholar]