

Abstract
Hydrogen sulfide is a cardioprotective signaling molecule but is toxic at elevated concentrations. Red blood cells can synthesize H2S but, lacking organelles, cannot dispose of H2S via the mitochondrial sulfide oxidation pathway. We have recently shown that at high sulfide concentrations, ferric hemoglobin oxidizes H2S to a mixture of thiosulfate and iron-bound polysulfides in which the latter species predominates. Here, we report the crystal structure of human hemoglobin containing low spin ferric sulfide, the first intermediate in heme-catalyzed sulfide oxidation. The structure provides molecular insights into why sulfide is susceptible to oxidation in human hemoglobin but is stabilized against it in HbI, a specialized sulfide-carrying hemoglobin from a mollusk adapted to life in a sulfide-rich environment. We have also captured a second sulfide bound at a postulated ligand entry/exit site in the α-subunit of hemoglobin, which, to the best of our knowledge, represents the first direct evidence for this site being used to access the heme iron. Hydrodisulfide, a postulated intermediate at the junction between thiosulfate and polysulfide formation, coordinates ferric hemoglobin and, in the presence of air, generated thiosulfate. At low sulfide/heme iron ratios, the product distribution between thiosulfate and iron-bound polysulfides was approximately equal. The iron-bound polysulfides were unstable at physiological glutathione concentrations and were reduced with concomitant formation of glutathione persulfide, glutathione disulfide, and H2S. Hence, although polysulfides are unlikely to be stable in the reducing intracellular milieu, glutathione persulfide could serve as a persulfide donor for protein persulfidation, a posttranslational modification by which H2S is postulated to signal.
Keywords: crystal structure, heme, hemoglobin, hydrogen sulfide, oxidation-reduction (redox)
Introduction
Hydrogen sulfide (H2S)2-dependent signaling occurs via persulfidation (Fig. 1A), a posttranslational modification of protein cysteine residues that leads to cysteine persulfide (Cys-SSH) formation (1). Persulfidation could occur via the reaction between H2S and an oxidized cysteine (e.g. cysteine sulfenic acid) (Fig. 1B). Alternatively, this modification could be catalyzed by thiol sulfurtransferases that stabilize Cys-SSH in active sites and transfer the persulfide group to a cysteine thiolate on a target protein (Fig. 1C) (2). Sulfur sources other than H2S that could potentially lead to protein persulfidation include (i) low molecular weight persulfides like Cys-SSH or glutathione persulfide (GSSH), (ii) sulfurtransferase substrates such as thiosulfate or mercaptopyruvate, and (iii) hydropolysulfides (hereafter referred to as polysulfides).
FIGURE 1.
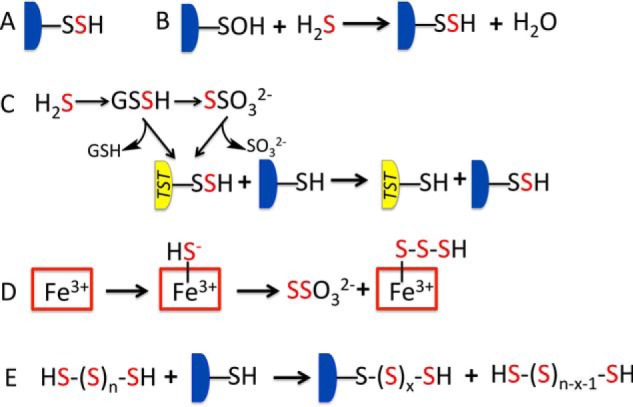
Schematic showing H2S-derived reactive sulfur species and persulfidation mechanisms. A, protein persulfidation is a posttranslational modification at a cysteine residue. B, persulfidation could occur via the reaction of H2S and an oxidized cysteine (e.g. cysteine sulfenic acid). C, oxidation of H2S via the mitochondrial sulfide oxidation pathway generates reactive sulfur species, such as GSSH and thiosulfate (S2O32−), which are substrates for thiol sulfurtransferases (TST) that stabilize active site persulfides and can transfer the outer sulfur to cysteines on target proteins. D, ferric heme-dependent oxidation of H2S by hemoglobin (or myoglobin) leads to thiosulfate and iron-bound polysulfide formation. E, the uncatalyzed reaction of cysteine thiols on target proteins with polysulfide could lead to the nonspecific transfer of one or more sulfur atoms, depending on which sulfur atom in the catenated chain is attacked.
Cystathionine β-synthase and γ-cystathionase can synthesize Cys-SSH (3, 4) in addition to H2S (5, 6). GSSH and thiosulfate are products of the sulfide oxidation pathway housed in the mitochondrion, which converts H2S ultimately to sulfate (7, 8) via reactive sulfur species (2). In bacteria, sulfide quinone oxidoreductase, the first enzyme in the sulfide oxidation pathway, generates polysulfides, which can serve as a periplasmic sulfur storage form (9, 10). The product of the mammalian sulfide quinone oxidoreductase is GSSH rather than polysulfides (7, 8). Hence, a biological source of polysulfides in mammals was not known until recently, when we demonstrated that hemeproteins such as hemoglobin and myoglobin can support the catalytic oxidation of H2S to thiosulfate and polysulfides (Fig. 1D) (11, 12). The ability of human hemoglobin to oxidize sulfide stands in intriguing contrast to other hemoglobins that function as sulfide carriers. Thus, organisms adapted to life in sulfide-rich environments use specialized hemoglobins (e.g. HbI in Lucina pectinata) to bind sulfide and deliver it to endosymbionts that utilize sulfide as an energy source (13).
Other systems have been reported to generate polysulfides as side products. For example, in the absence of a sulfur acceptor, mercaptopyruvate sulfur transferase generates polysulfides by catalyzing repeated sulfur transfers from the substrate, 3-mercaptopyruvate, to an active site cysteine (at pH 9.1). The bound polysulfide can eventually be released from the enzyme (14). Polysulfides can also be formed in solution (e.g. by the rapid reaction of hypochlorous acid, an oxidant produced by neutrophils, with sulfide to generate HSCl). The latter can be subsequently oxidized to a mixture of polysulfides, with HS4S− and HS3S− predicted to be the dominant species at physiological pH (15). In principle, polysulfide synthesis could occur via this route if H2S, which is typically present at very low concentrations, is transiently increased at sites of inflammation. Finally, H2S can reduce cytochrome c, the electron carrier between complexes III and IV. However, elemental sulfur, the expected product of this reaction, has not been characterized (16).
Polysulfides are considerably more reactive than H2S and, as expected, elicit cellular effects at lower concentrations. Hence, it is not surprising that polysulfides are increasingly invoked as substrates for protein persulfidation (17, 18). Depending on which sulfur atom in the catenated sulfur chain is attacked by the protein thiol, a string of posttranslational modifications would be generated (Fig. 1E). The drawback in using polysulfides is that nucleophilic attack at the terminal sulfur in a catenated sulfur chain must be precisely controlled. Strategies for how this control is achieved are not known and have not been addressed in studies invoking its relevance.
Oxidation of sulfide by ferric myoglobin and hemoglobin to generate iron-bound polysulfides and thiosulfate represent chemically challenging multistep reactions in which the reaction intermediates are poorly characterized (11, 12). The intracellular milieu is reducing, and it begs the question as to whether the polysulfides formed by hemoglobin evade reaction with low molecular weight thiols or succumb to reduction. Because red blood cells express mercaptopyruvate sulfurtransferase and synthesize H2S but lack mitochondria, understanding the mechanism of sulfide oxidation via the action of FeIII-Hb assumes even greater importance in this versus in other cell types.
In this study, we have captured the initially formed HS−-FeIII-Hb intermediate by X-ray crystallography in addition to a second sulfide at the entrance of a postulated ligand access channel in the α-subunit of hemoglobin. We have demonstrated that the postulated hydrodisulfide intermediate coordinates to ferric heme iron and is a substrate for further oxidation. We have found that the iron-bound polysulfides are susceptible to physiologically relevant reductants, revealing that they are unlikely to survive in the reducing conditions found in the cytoplasm. On the other hand, the predominant persulfide product of the reaction between polysulfides and reductants (i.e. GSSH) might play a role in signaling.
Results
Structure of Human Hemoglobin with Bound Sulfide
The crystal structure of human FeIII-Hb incubated with H2S was determined at 1.79 Å resolution (Fig. 2, Table 1). The globin fold structure of the HS−-FeIII-Hb complex is similar to the R-state structure of hemoglobin (19) with Cα root mean square deviations of <0.2 Å. To verify that the extra density observed at the distal side of the iron is a sulfur atom (Fig. 2A), sulfur anomalous dispersion signals were collected at a 1.77-Å wavelength, and diffraction was recorded to 2.8 Å resolution. Following molecular replacement, the sulfur anomalous difference map was calculated to locate sulfur atoms in hemoglobin. As shown in Fig. 2B, the sulfur anomalous signal overlaps with the electron density near the iron atom, confirming the presence of a sulfur ligand on the distal side of heme.
FIGURE 2.
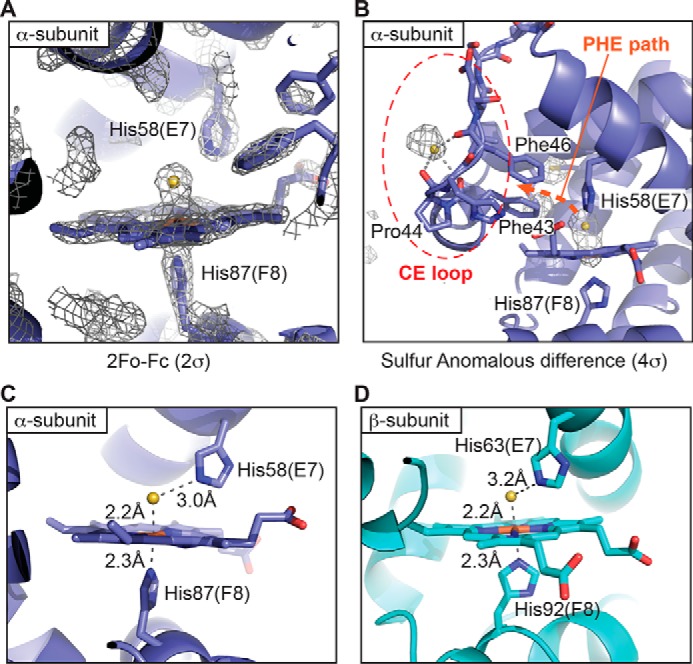
Crystal structure of the human ferric hemoglobin sulfide complex. A, the 2Fo − Fc electron density map (2σ contour level) near the heme group of the HS−-FeIII-Hb complex in the α-subunit. The density for the β-subunit was similar (not shown). B, the sulfur anomalous difference map (4σ contour level) of the HS−-FeIII-Hb complex identified two sulfur atoms at the distal side of the heme group and at a potential entry/exit point located near a path lined by Phe-43 and Phe-46 (PHE path, orange dashed arrow). The second sulfur atom at the surface was only seen in the α-subunit. The CE loop is marked as a red dashed circle. The additional observed electron densities belong to the sulfur atoms of methionine and cysteine residues. C and D, close-up showing the heme group and key residues in the α-subunit (C) and the β-subunit (D) of FeIII-Hb treated with sulfide. The α- and β-subunits of hemoglobin are colored dark blue and cyan, respectively. The sulfur ligand is shown as a yellow sphere.
TABLE 1.
Crystallographic data collection and refinement statistics
| Sulfur-bound hemoglobin | Sulfur-bound hemoglobin (sulfur anomalous) | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.1272 | 1.77 |
| Space group | P41212 | P41212 |
| Cell dimensions | ||
| a, b, c (Å) | 53,77, 53.77, 193.254 | 53.52, 53.52, 191.72 |
| α, β, γ (degrees) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50 to 1.8 (1.86 to 1.8)a | 48.08 to 2.81 (2.82 to 2.81) |
| Rsym or Rmerge (%) | 7.1 (146.3) | 6.1 (10.2) |
| Rrim or Rmeas (%) | 2.5 (65.4) | 6.3 (10.6) |
| I/σI | 28.97 (2.80) | 34.93 (19.29) |
| Completeness (%) | 99.9 (100) | 98.9 (93.4) |
| Redundancy | 11.9 | 13.7 |
| Refinement | ||
| Resolution (Å) | 37.30 to 1.80 | |
| No. of reflections | 27,405 | |
| Rwork/Rfree | 18.57/22.22 | |
| No. of atoms | ||
| Protein | 2458 | |
| Water | 169 | |
| B-factors | ||
| Protein | 41.15 | |
| Water | 46.16 | |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.004 | |
| Bond angles (degrees) | 0.688 | |
| Protein Data Bank code | 5UCU |
a Values in parentheses are for the highest resolution shell.
The distance between the proximal HisF8 (His-87 in the α-subunit and His-92 in the β-subunit) NE2 and the iron atoms is 2.3 Å in both the α- and β-subunits (Fig. 2, C and D). The bond length between the iron and the sulfur atoms with unrestrained distance refinement is 2.2 Å in both the α- and β-subunits. The HS−-FeIII-Hb intermediate forms a hydrogen bond with the distal HisE7 NE2 (His-58 in the α-subunit and His-63 in the β-subunit) with distances of 3.0 and 3.2 Å in the α- and β-subunits, respectively (Fig. 2, C and D).
Inspection of the sulfur anomalous difference map revealed the presence of another strong anomalous signal, which does not belong to the sulfur atoms in cysteine or methionine residues. Located at the surface of the α-subunit near the CE loop, this sulfur atom is involved in hydrogen bonding interactions with the backbone carbonyl groups of Phe-43, Pro-44, and Phe-46 (Fig. 2B). Interestingly, this sulfur atom is positioned at the mouth of the PHE path, one of proposed entry/exit sites for the hemoglobin ligands, CO and O2 (20, 21).
Reactivity of FeIII-Hb with Hydrodisulfide
We have postulated the presence of ferrous iron-bound hydrodisulfide (FeII-S-S−) as an intermediate in the globin-catalyzed sulfide oxidation reaction coordinate (11, 12). To test the possible formation of this species, we mixed FeIII-Hb with an excess of sodium hydrodisulfide (Na2S2). A shift in the Soret peak from 405 to 421 nm and the appearance of peaks at 543 and 575 nm were observed under anaerobic conditions (Fig. 3, A and B). Similar spectral changes were also observed under aerobic conditions (Fig. 3C). This spectrum is similar to that of FeIII-Hb treated with sulfide, which shows absorbance maxima at 423, 541, and 577 nm (11) and suggests direct coordination of the hydrodisulfide to ferric hemoglobin. In the absence of O2, slow reduction to FeII-Hb was observed as evidenced by a shift in the Soret peak to 429 nm and the appearance of a broad α/β band centered at 554 nm (Fig. 3B). A small increase in absorbance was also observed at 617 nm, indicating the formation of a small proportion of sulfhemoglobin. Aeration of the reaction mixture led to the formation of the oxy-FeII-Hb, with a Soret peak at 416 nm and α/β bands at 577 and 541 nm (Fig. 3D). Following exposure to air, thiosulfate formation was observed.
FIGURE 3.
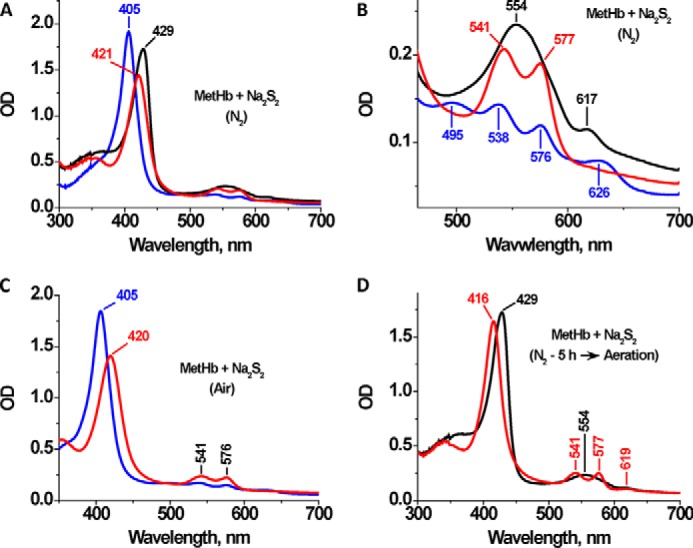
UV-visible spectral changes in FeIII-Hb by Na2S2. A, FeIII-Hb (10 μm heme) in 100 mm HEPES buffer, pH 7.4, was mixed with 100 μm Na2S2 at 25 °C under anaerobic conditions. The blue line represents the initial spectrum of FeIII-Hb, and the red and black spectra were recorded 1 min and 5 h, respectively, after the addition of Na2S2. The spectrum after 5 h is a mixture of predominantly FeII-Hb with low levels of sulfhemoglobin (as indicated by the 617-nm feature). B, close-up of the visible region of the spectra in A. C, FeIII-Hb (10 μm heme) in 100 mm HEPES buffer, pH 7.4, was mixed with 100 μm Na2S2 at 25 °C under aerobic conditions. The blue line represents the initial spectrum of FeIII-Hb, and the red spectrum was recorded 1 min after the addition of Na2S2. D, spectral shift induced upon exposure of the sample in A to air. The red spectrum corresponds to the anaerobic sample of FeII-Hb generated in A after a 5-h incubation of MetHb with Na2S2, and the black spectrum corresponds to O2-FeII-Hb formed upon exposure to air.
Characterization of Sulfide Oxidation Products at Low H2S/FeIII-Hb
We had previously characterized sulfide oxidation products at high H2S/FeIII-Hb ratios (11). However, under cellular conditions, the ratio of H2S to FeIII-Hb is expected to be low, and it is not known whether the product distribution between thiosulfate and polysulfides would be similar or different. To address this issue, we assessed the relative concentration of sulfide oxidation products that were formed when H2S/FeIII-Hb ratios were 1:1 or 2:1 (Fig. 4A). Under these conditions, low albeit detectable concentrations of polysulfides (3.0 ± 3.6 μm (1:1 ratio) and 20.9 ± 9.4 μm (2:1 ratio)) and thiosulfate (2.9 ± 0.3 μm (1:1 ratio) and 13.2 ± 0.8 μm (2:1 ratio)) were formed (Fig. 4A). These values correspond to the presence of 5.8 and 26.4 μm sulfur in the thiosulfate (S2O32−) product.
FIGURE 4.
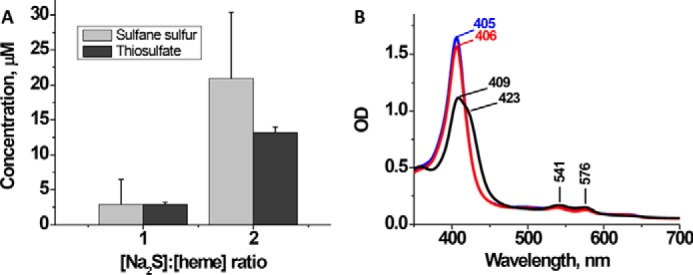
Interaction of FeIII-Hb with sulfide at low sulfide to heme ratios. A, concentrations of sulfane sulfur and thiosulfate formed after 30 min of incubation of FeIII-Hb (100 μm heme) in aerobic 100 mm HEPES buffer, pH 7.4, with 100 or 200 μm Na2S at 25 °C. The data represent the mean ± S.D. (error bars) of 3–4 independent experiments. B, representative spectral changes observed upon incubation of FeIII-Hb (10 μm heme) with Na2S (10 μm) under the same conditions as in A. The blue spectrum is of FeIII-Hb, and the black and red spectra were recorded at 20 min and 24 h, respectively, after the addition of Na2S.
The spectrum of FeIII-Hb treated with stoichiometric Na2S is indicative of the presence of a mixture of species, most likely FeIII-Hb and HS−-FeIII-Hb (Fig. 4B). The Soret peak diminishes in intensity as it shifts from 405 to 409 nm with a prominent shoulder at 423 nm. In the visible region of the spectrum, the peaks at 500 and 630 nm associated with FeIII-Hb decrease in intensity upon the addition of Na2S, and α/β bands at 576 and 541 nm appear (Fig. 4B, black trace). In contrast, in the presence of excess Na2S, the Soret peak shifts completely to 423 nm, indicating complete conversion to the HS−-FeIII-Hb species (11). The original FeIII-Hb spectrum was gradually restored after prolonged incubation of the sample under aerobic conditions (Fig. 4B, red trace).
Fate of Hemoglobin-bound Polysulfides in the Presence of Reductants
Unlike thiosulfate, the polysulfides formed during sulfide oxidation remain tightly bound to hemoglobin (11). Because the intracellular milieu is reducing, we assessed whether the polysulfide products are sequestered from reductants or are intercepted by them. First, we examined how a protein reductant, methionine synthase reductase (MSR), an NADPH-dependent diflavin oxidoreductase (22), affects hemoglobin-bound polysulfides. We have previously shown that MSR reduces HS−-FeIII-Hb to FeII-Hb, which is converted to O2-FeII-Hb in the presence of air (11). Polysulfides were allowed to accumulate for 30 min following treatment of FeIII-Hb with excess Na2S, after which NADPH and MSR were added to the reaction mixture. Following this treatment, the polysulfides remained associated with hemoglobin (Fig. 5A), although the heme spectrum reverted from the initial 423 nm to a 415-nm Soret peak, indicating formation of O2-FeII-Hb (Fig. 5B). This result indicates that although the polysulfides are no longer bound to the heme iron following reduction by MSR, they remain associated with hemoglobin.
FIGURE 5.
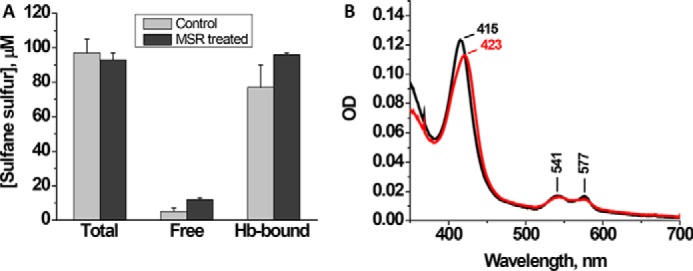
MSR reduces the heme- but not hemoglobin-bound polysulfides. A, FeIII-Hb (100 μm heme) was mixed with 0.5 mm Na2S in 100 mm HEPES buffer, pH 7.4, and incubated aerobically for 30 min at 25 °C before the addition of MSR and NADPH, after which incubation was continued for 3 h. Then the sulfane sulfur levels were measured in the protein-bound and -free fractions as described under “Experimental Procedures.” The data represent the mean ± S.D. (error bars) of three independent experiments. B, spectrum of FeIII-Hb after treatment with Na2S for 30 min (red) followed by a 1-h incubation with MSR and NADPH (black). Conditions are the same as in A except that the samples were diluted 1:10 with 100 mm HEPES, pH 7.4.
Next, we examined the effects of the artificial low molecular weight reductant tris(2-carboxyethyl)phosphine (TCEP) on the heme-bound polysulfide pool. TCEP resulted in release of the bulk (65–75%) of the hemoglobin-bound polysulfides (Fig. 6A). However, the polysulfides were not recovered quantitatively in the low molecular weight fraction, indicating that further chemical reaction of the polysulfides had occurred in the presence of TCEP. H2S is the expected product of polysulfide reduction, provided that the sulfur atoms are not oxygenated (e.g. as in Fe-S-SO32−). However, H2S was detected only at very low levels in the presence of TCEP (∼6%) (Fig. 6B).
FIGURE 6.
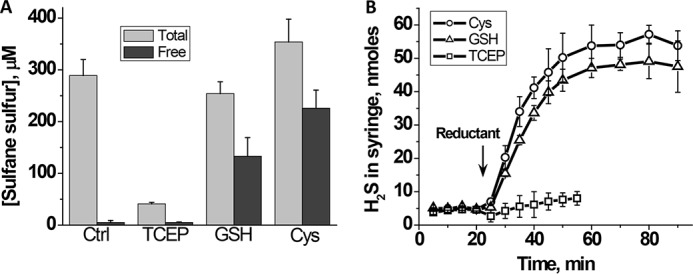
Effect of reductants on hemoglobin-bound sulfane sulfur and release of H2S. A, FeIII-Hb (100 μm heme) was incubated aerobically for 30 min with 1.0 mm Na2S in 100 mm HEPES buffer, pH 7.4, at 25 °C, followed by a 30-min incubation with or without the addition of a 2 mm concentration each of TCEP, GSH, or cysteine. The concentration of total and free sulfane sulfur was determined as described under “Experimental Procedures.” B, FeIII-Hb (100 μm heme) in 100 mm HEPES buffer, pH 7.4, was incubated aerobically for 30 min with 1.0 mm Na2S at 25 °C, following which H2S was determined by GC analysis as described under “Experimental Procedures.” The data represent the mean ± S.D. (error bars) of three or more independent experiments.
We then examined the effect of two naturally occurring low molecular weight reductants, cysteine and glutathione, on the fate of iron-bound polysulfide. A substantial fraction (52% with glutathione and 64% with cysteine) of the polysulfide pool was released from hemoglobin and recovered in solution. In addition, 32% (with glutathione) and 36% (with cysteine) of the sulfur was recovered as H2S following treatment with these reductants (Fig. 6B).
The low molecular weight products released from hemoglobin-bound polysulfides following treatment with glutathione or cysteine were characterized by LC/MS after alkylation with iodoacetamide giving the corresponding carbamidomethyl (CAM) derivative. GSSH (m/z = 397), glutathione (m/z = 365), glutathione disulfide (m/z = 613), cysteine (m/z = 179), and cysteine persulfide (m/z = 211) were identified in samples treated with the corresponding thiol (Fig. 7, A and B). Persulfides and disulfides were not observed in control samples (i.e. FeIII-Hb pretreated with Na2S alone or buffer incubated with the thiols for 30 min at 25 °C). The fragmentation patterns for Cys-S-CAM and Cys-SS-CAM are consistent with those seen previously (4) (Fig. 8A). The peak at m/z = 90 is assigned to the product of S–S bond heterolysis in the Cys-SS-CAM precursor peak (m/z = 211). The fragmentation pattern of GS-CAM and GSS-CAM confirmed the assignment of these species as described in the legend to Fig. 8B.
FIGURE 7.
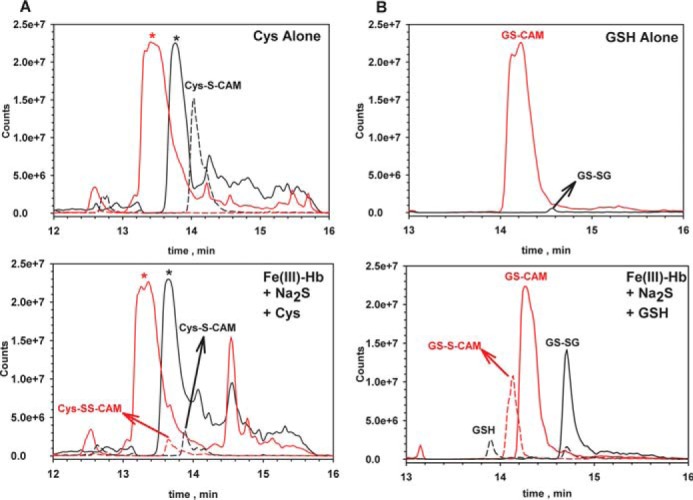
Mass spectrometric (LC-MS) analysis of the reaction of cysteine or glutathione with FeIII-Hb treated with Na2S. Hydrophilic interaction liquid ion chromatography was used to elute the components, as described under “Experimental Procedures.” A, traces are for the 0.5-atomic mass unit windows for Cys-S-CAM (dashed black line, m/z = 179.25) and Cys-SS-CAM (dotted red line, m/z = 211.25). B, traces are for the 0.5-atomic mass unit windows for GS-SG (solid black line, m/z = 613.25), GSH (dashed black line, m/z = 308.25), GS-CAM (solid red line, m/z = 365.25), and GSS-CAM (dotted red line, m/z = 397.25). In each panel, the top chromatogram represents samples containing reductant only (Cys or GSH), and the bottom one represents samples containing FeIII-Hb + Na2S that were treated with reductant. Scans were collected as described under “Experimental Procedures,” and masses for the selected ions were extracted using 0.5 Da windows. The peaks labeled with asterisks in A were present in all samples. They are likely to represent Tris (m/z = 122; red) and HEPES (m/z = 241; black), which were present in the samples.
FIGURE 8.

MS/MS spectra of the reactions of cysteine and glutathione with FeIII-Hb treated with sulfide. MS/MS spectra were extracted at 14.3 min for Cys-S-CAM (A; m/z = 179), at 14.5 min for GS-CAM (B; m/z = 365, top), at 13.8 min for Cys-SS-CAM (C; m/z = 211) and at 14.3 for GS-S-CAM (D; m/z = 397). The fragmentation of GS-S-CAM produced peaks corresponding to the loss of glycine (m/z = 322), pyroglutamic acid (m/z = 268), pyroglutamic acid + NH3 (m/z = 251), CAM persulfide + CO2 (m/z = 231), pyroglutamic acid + glycyl (m/z = 177), pyroglutamic acid + glycine + CO (m/z = 165), and loss of pyroglutamic acid + glycine + CO + NH3 (m/z = 148). We also observed the glyoxylic acid imine (m/z = 74) and S-S-CAM (m/z = 122) cation peaks. Fragmentation of GS-CAM shows the homologous fragments missing the additional sulfur atom (m/z = 290, 236, 219, 133, and 116). For the fragmentation of the protonated Cys-SS-CAM (m/z = 211), the main products are consistent with glyoxylic acid imine (m/z = 74), protonated 2-thiaglyoxamide (m/z = 90), 3-thia-alanine (m/z = 120), cysteine persulfide imine (m/z = 152), and protonated-3-thio-propenoic acid (m/z = 105) cation. The fragment peak at m/z = 120 is also consistent with the product of the loss of formic acid, CO, and NH3 from the persulfide precursor.
Discussion
Hemoglobins in organisms adapted to live in sulfide-rich habitats are specialized to carry sulfide to endosymbionts and protect against sulfide oxidation (23). The bivalve Lucina pectinata contains three hemoglobins (HbI, HbII, and HbIII) to transport O2 and H2S from the seawater to symbiotic bacteria (24, 25). The high sulfide affinity of monomeric HbI (KD = 3 nm) necessitates reduction of ferric to ferrous iron for sulfide release (24). In contrast, human ferric hemoglobin exhibits a relatively low affinity for sulfide (17 μm) and, in the presence of O2, catalyzes its conversion to a mixture of thiosulfate and polysulfides (11). Sulfide also undergoes spontaneous oxidation at neutral pH, and the product distribution is governed by the ratio of sulfide/O2. When the ratio is high, polysulfides predominate, and when it is low, thiosulfate and other oxyanions are formed (26). Our earlier study on catalytic sulfide oxidation by hemoglobin was performed at high sulfide concentrations, simulating a high sulfide/O2 ratio, and led to formation of higher polysulfide than thiosulfate product (11). In this study, we examined the distribution of products at lower sulfide/heme ratios (1:1 and 2:1) and found that polysulfides and thiosulfate were formed in approximately equal concentration (Fig. 4A).
Following H2S entry, the first step in the hemoglobin-catalyzed sulfide oxidation cycle is formation of a ferric sulfide intermediate (HS−-FeIII-Hb) (11). In the crystal structure of human hemoglobin, a sulfide ligand to the heme iron was observed in the α- and β-subunits (Fig. 2, C and D). In addition, a second sulfur atom was seen at the surface of the α-subunit of hemoglobin (Fig. 2B). The extra sulfur atom is located at the mouth of the postulated PHE path used by ligands to access the heme iron, which was identified using xenon to fill cavities in the crystal structure and by atomistic molecular dynamics simulations (20, 21). Simulations revealed that the PHE path is the major CO escape route from the α-subunit in the R-state of hemoglobin (20). We conclude that the extra sulfur atom in our crystal structure represents the entry/exit point for H2S. To the best of our knowledge, this is the first direct evidence for the use of the PHE path by a ligand for accessing the heme. The ligand entry/exit points are predicted to be different in the α- and β-subunits (20, 27, 28), in agreement with our observation that the extra sulfur anomalous signal was observed in the α-subunit but not in the β-subunit.
An Fe–S distance (unrestrained during refinement) of 2.2 Å was observed in the α- and β-subunits (Figs. 2 (C and D) and 9A). By comparison, the crystal structure of ferric sulfide HbI form L. pectinata revealed an Fe–S bond distance of 2.3 Å (Fig. 9B). Based on a computational analysis of sulfide-bound to ferric myoglobin, different Fe–S bond lengths were predicted, depending on whether the distal ligand is a sulfide anion (2.24 Å) or H2S (2.50 Å) (12). Our crystallographic results agree with the calculated bond lengths for FeIII-HS− (2.24 Å) and HisF8 NE2-FeIII (2.12 Å) in a low spin HS−-FeIII complex and indicate that we have captured the first postulated sulfide oxidation intermediate (11, 12).
FIGURE 9.
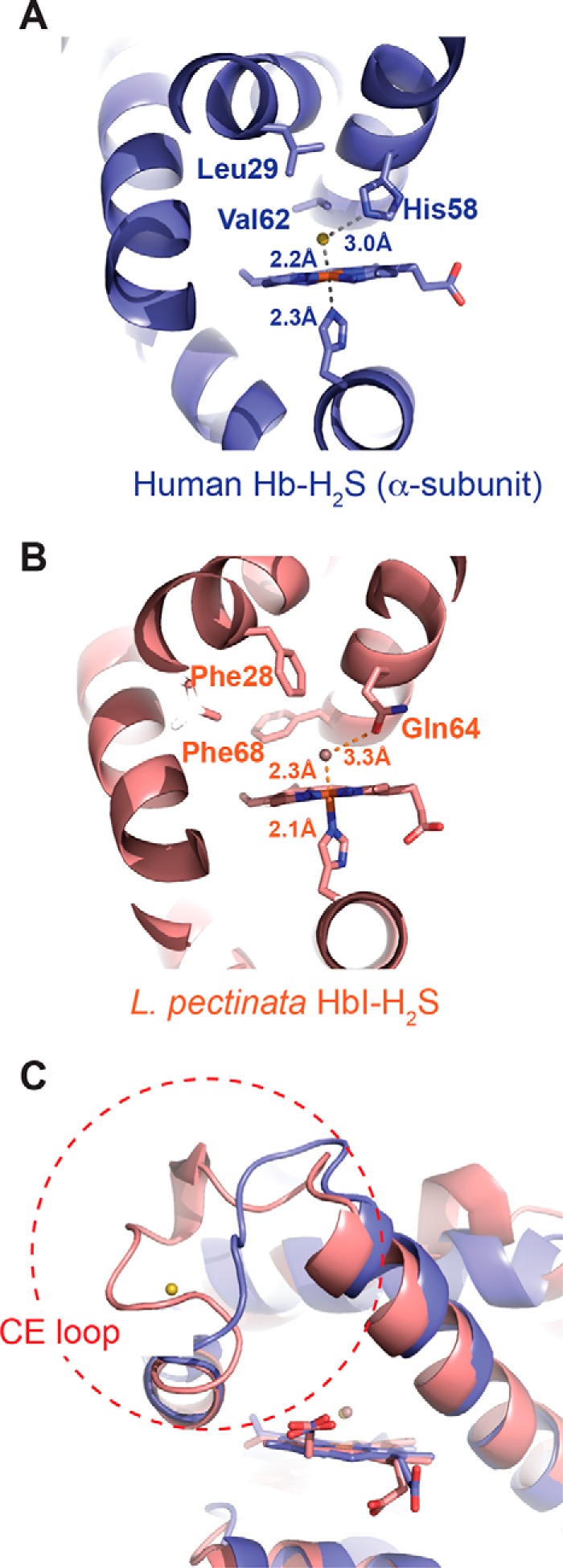
Comparison of the crystal structures of sulfide-bound human hemoglobin and HbI from L. pectinata. The distal heme pocket of the α-subunit human hemoglobin in complex with sulfide (A) and that of L. pectinata HbI in complex with sulfide (B). The distal heme site of L. pectinata HbI contains a “Phe cage” comprising Phe-28 and Phe-68, which are replaced by Leu-29 and Val-62 in human hemoglobin. The interaction between Gln-64 in L. pectinata HbI and sulfide is replaced by His-58 and sulfide in human hemoglobin. C, the conformation of the CE loop (enclosed by the dashed red circle) differs in the structures of HbI and human hemoglobin. The CE loop in human hemoglobin is postulated to be the entry/exit point for sulfide.
In human hemoglobin, the sulfide forms a hydrogen bond with the His-58 nitrogen (Fig. 9A). In the L. pectinata HbI, the corresponding interaction involves Gln-64, with a longer and more flexible side chain (Fig. 9B). Furthermore, in HbI, four phenylalanine residues form a hydrophobic cage around the sulfide ligand, whereas Gln-64 seals off access to the solvent. The tight aromatic pocket contributes to both the stabilization of and the high affinity for the sulfide ligand in HbI, properties suited for its role as a sulfide carrier. In human hemoglobin, smaller hydrophobic residues (valine and leucine) substitute for the phenylalanines in HbI (Fig. 9A). These less bulky amino acids with flexible side chains in the distal heme pocket of human hemoglobin accommodate binding of additional equivalents of sulfur and O2 and enable the observed oxidation chemistry. Another structural difference between sulfide-complexed HbI and human hemoglobin is in the conformation of the CE loop, which we have identified as the entry/exit site for H2S (Fig. 9C). It is not known whether the conformational difference has a bearing on the kinetics of H2S access and/or sulfide oxidation in human hemoglobin.
Of the two products of hemoglobin-catalyzed sulfide oxidation observed in vitro, thiosulfate is released into solution, whereas the polysulfides remain iron-bound. The fact that the intracellular environment is reducing begs the question of whether the polysulfides evade reduction by being sequestered in the distal pocket. We found that MSR, which can lead to heme iron reduction, does not lead to release of polysulfides into solution (Fig. 5). The artificial triphosphine reductant TCEP led to the loss of hemoglobin-bound polysulfides, albeit without the release of H2S, the expected product of polysulfide reduction. The lack of detectable H2S is explained by the reaction of TCEP (Ph3P) with polysulfides to form thiophosphines (Ph3P(S)), described previously (29). In contrast, polysulfides were released from hemoglobin with formation of H2S in the presence of the physiologically relevant low molecular weight reductants, cysteine and glutathione (Fig. 6, A and B). The other products of the reaction between glutathione or cysteine and polysulfides were the corresponding persulfides, GSSH and Cys-SSH. These persulfides, in turn, reacted with glutathione or cysteine, generating H2S and the oxidized products, glutathione disulfide and cystine, which were detected by mass spectrometry (Figs. 7 and 8). In contrast to cysteine (5 μm), the intracellular glutathione concentration in erythrocytes is high (3.2 mm) (30). Our results suggest that polysulfides, if formed in erythrocytes, would react with glutathione, forming GSSH. The latter, in turn, could serve as a persulfide donor for protein persulfidation, a posttranslational modification used for sulfide signaling (1). GSSH is a substrate for thiol sulfurtransferases, which could, in turn, catalyze protein persulfidation, as postulated (2).
In summary, we provide crystallographic evidence for the route used by sulfide to access the distal heme site in human hemoglobin and have captured the first intermediate in the sulfide oxidation reaction (i.e. HS−-FeIII-Hb). We have also demonstrated the ability of hydrodisulfide to bind to FeIII-Hb and be converted to products, consistent with its proposed relevance as an intermediate during sulfide oxidation. The susceptibility of iron-bound polysulfides to reduction by glutathione suggests that GSSH rather than polysulfides might be involved in sulfide signaling via protein persulfidation.
Experimental Procedures
Materials
Lyophilized human FeIII-Hb, glutathione, cysteine, NADPH, and sodium sulfide nonahydrate were purchased from Sigma; TCEP was from Gold Biotechnology (St. Louis, MO); and sodium disulfide (Na2S2) was from Dojindo Molecular Technologies (Rockville, MD). Recombinant human MSR was prepared as described previously (22).
Measurement of Polysulfides
Sulfane sulfur concentration was measured using the cold cyanolysis method as described previously (11). The concentration of sulfane sulfur was estimated using a calibration curve prepared using potassium thiocyanate samples of known concentration.
Treatment of Hemoglobin-Sulfide with MSR
FeIII-Hb (100 μm heme concentration in 100 mm HEPES-Na+ buffer, pH 7.4) was incubated aerobically for 30 min at 25 °C with Na2S (0.5 mm). Then, the reaction mixture was divided equally into two aliquots. To the first aliquot, MSR and NADPH were added to final concentrations of 8 μm and 1 mm, respectively, while the second aliquot was left untreated. Both aliquots were incubated aerobically at 25 °C for 3 h. Formation of O2-FeII-Hb in the reaction mixture was monitored spectrophotometrically. Free and protein-bound polysulfides were separated using centrifugal filters (Amicon Ultracell with a 10 kDa cut-off). Samples (400–450 μl) were placed on the filter and centrifuged at 10,000 × g and 4 °C for 10–15 min. The filtrate containing the low molecular weight sulfane sulfur fraction was collected separately from the protein fraction on the filter, and the concentration of sulfane sulfur in each fraction was determined.
Release of H2S from Iron-bound Polysulfides
FeIII-Hb (100 μm heme concentration) in 100 mm HEPES-Na+ buffer, pH 7.4, was incubated aerobically at 25 °C for 30 min with Na2S (1 mm) to generate iron-bound polysulfides as described (11). Then 0.5 ml of the reaction mixture was placed in the barrel of a 20-ml polypropylene syringe, and the headspace was flushed six times with N2 using a three-way stopcock and then filled with N2 to a total volume (gas plus liquid) of 20 ml. The syringe was kept at 25 °C, and 200-μl gas samples were aspirated at the desired times. Within 20 min, the H2S concentration in the gas phase stabilized at the level of ∼0.25–0.3 μm. Then the plunger was pushed to remove all but 2 ml of gas from the syringe, and 5 μl of 200 mm reductant (GSH, cysteine, or TCEP) in 100 mm HEPES-Na+ buffer, pH 7.4, was added to the reaction mixture to obtain a final concentration of 2 mm. The pH of the TCEP solution was adjusted to 7.0 with saturated potassium carbonate. The syringe was filled with N2 to a total volume of 20 ml and incubated at 25 °C. At the desired time intervals, 200-μl aliquots were removed from the gas phase. The quantity of H2S in the samples was measured using a gas chromatograph equipped with 355 sulfur chemiluminescence detector (GC-SCD) (Agilent) as described (31).
Mass Spectrometric Analysis
FeIII-Hb (100 μm in heme) in 100 mm HEPES-Na+ buffer, pH 7.4, was incubated with 1 mm Na2S for 30 min at 25 °C. Then the mixture was divided into three aliquots. The control sample received no further treatment; the second and third aliquots were treated with GSH or cysteine to a final concentration of 2 mm. All three aliquots were incubated for an additional 30 min at 25 °C. Then the reaction mixtures were filtered using Amicon filters (10 kDa molecular mass cut-off), and the filtrate was incubated with 10 mm iodoacetamide for 1 h at 25 °C in the dark, frozen, and stored at −80 °C. Control samples containing buffer with 2 mm GSH or cysteine were prepared and treated with iodoacetamide in parallel. The total and low molecular weight sulfane sulfur concentration was measured in all samples before and after incubation with reductants.
Aliquots (5 μl) of the reaction mixture were injected into a 4.6 × 100-mm amide XBridge column (Waters, Milford, MA) and eluted at a flow rate of 0.5 ml/min using Buffers A (20 mm ammonium acetate, 20 mm ammonium hydroxide, pH 9.0) and B (acetonitrile) and the following steps: isocratic for 2 min with 5% A, linear gradient from 5 to 95% A for 15 min, isocratic for 5 min with 5% B, and reequilibration for 10 min with 5% A. The effluent was coupled to a Sciex 4000 QTrap triple quadrupole mass spectrometer operating in either Q1 scan (MS) or MS2 (MS/MS) mode. Other instrument parameters were as follows: curtain gas = 20 liters/min, ion spray voltage = 5500 V, electrospray ionization temperature = 650 °C, GS1 = GS2 = 60 liters/min, declustering potential = 80 V, entrance potential = 10 V, exit potential = 15 V, collisional energy = 30 V (for MS/MS). Scans for MS were from m/z = 5 to 1005 in 1.0 s, and for MS/MS, they were from m/z = 5 to 650 in 0.25 s.
Reaction of FeIII-Hb with Hydrodisulfide
The experiments were performed inside an anaerobic chamber (Vacuum Atmospheres Co., Hawthorne, CA) with an atmosphere of N2 and containing <0.2–0.5 ppm O2. A stock solution of Na2S2 was prepared in anaerobic 100 mm Tris-HCl buffer, pH 8.0. Spectra were monitored following the addition of Na2S2 (100 μm final concentration) to FeIII-Hb (10 μm heme) in anaerobic 100 mm HEPES-Na+ buffer, pH 7.4, in a sealed cuvette.
X-ray Crystallography of FeIII-Hb in the Presence of H2S
Crystallization was carried as described previously (19) with the following modifications. Briefly, 50 mg of human FeIII-Hb was dissolved in 1 ml of 30 mm HEPES-Na+, pH 7.4, and dialyzed overnight against 1 liter of 1.6 m K2HPO4/NaH2PO4 buffer, pH 6.7, at 4 °C. The protein was concentrated to 60 mg/ml, and 200 μl of the protein solution was mixed with 40 μl of toluene and 300 μl of 2.8 m K2HPO4/NaH2PO4 buffer, pH 7.2, from which 5-μl drops were placed on coverslips. The latter were inverted on wells containing 2.3 m K2HPO4/NaH2PO4 reservoir buffer, pH 7.2, and sealed. Crystals were obtained at 20 °C in 1 week by the vapor diffusion method. The crystals were harvested by cryo-loops and soaked for 1 h at 20 °C in a cryoprotectant solution containing 50 mm Na2S, 360 mm K2HPO4/NaH2PO4 buffer, pH 7.2, and 14% (v/v) glycerol. The crystals were flash-frozen in liquid N2 and stored for data collection. The native (1.8 Å resolution) and sulfur anomalous (2.8 Å resolution) data sets of the crystals were collected at the LS-CAT beamline 21-ID-D (Advanced Photon Source, Argonne National Laboratory) at 1.13- and 1.77-Å wavelengths, respectively (Table 1).
Data sets were integrated and scaled using HKL2000. The space group and unit cell dimensions of the human HS−-FeIII-Hb complex were P41212 and a = b = 53.77 Å, c = 193.25 Å, α = β = γ = 90°. The phases of the HS−-FeIII-Hb complex were obtained by molecular replacement using Phaser (32) with the known structure of human hemoglobin as a search model (Protein Data Bank code 3D7O) (19). The model was built using COOT (33), and refinement calculations were carried out using the Phenix.Refine (34). The sulfur anomalous data set collected at 1.77-Å wavelength (2.8 Å resolution) was processed by XDS (35). After molecular replacement using Phaser (32), the sulfur anomalous difference map of the HS−-FeIII-Hb complex was calculated using the program phenix.maps (36). All molecular structure figures were prepared using PyMOL (Schrödinger, LLC, New York).
Author Contributions
V. V. designed and performed the experiments. P. K. Y., S. A., and U.-S. C. crystallized hemoglobin with sulfide and solved its structure. J. S. performed the mass spectrometric analysis. R. B. helped conceive the experiments, analyzed the data, and co-wrote the manuscript with V. V., U.-S. C., and J. S. All authors approved the final version of the manuscript.
This work was supported in part by National Institutes of Health Grants GM112455 (to R. B.) and DK111465 (to U.-S. C). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5UCU) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- H2S
- hydrogen sulfide
- Cys-SSH
- cysteine persulfide
- CAM
- carbamidomethyl
- FeIII-Hb
- methemoglobin or ferric hemoglobin
- MSR
- methionine synthase reductase
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., and Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mishanina T. V., Libiad M., and Banerjee R. (2015) Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 11, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., Suematsu M., Motohashi H., Fujii S., Matsunaga T., Yamamoto M., Ono K., Devarie-Baez N. O., Xian M., Fukuto J. M., and Akaike T. (2014) Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7606–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yadav P. K., Martinov M., Vitvitsky V., Seravalli J., Wedmann R., Filipovic M. R., and Banerjee R. (2016) Biosynthesis and reactivity of cysteine persulfides in signaling. J. Am. Chem. Soc. 138, 289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiku T., Padovani D., Zhu W., Singh S., Vitvitsky V., and Banerjee R. (2009) H2S biogenesis by cystathionine γ-lyase leads to the novel sulfur metabolites, lanthionine and homolanthionine, and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 284, 11601–11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh S., Padovani D., Leslie R. A., Chiku T., and Banerjee R. (2009) Relative contributions of cystathionine β-synthase and γ-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 284, 22457–22466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hildebrandt T. M., and Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 8. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., and Banerjee R. (2014) Organization of the human mitochondrial H2S oxidation pathway. J. Biol. Chem. 289, 30901–30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marcia M., Ermler U., Peng G., and Michel H. (2009) The structure of Aquifex aeolicus sulfide:quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proc. Natl. Acad. Sci. U.S.A. 106, 9625–9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brito J. A., Sousa F. L., Stelter M., Bandeiras T. M., Vonrhein C., Teixeira M., Pereira M. M., and Archer M. (2009) Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry 48, 5613–5622 [DOI] [PubMed] [Google Scholar]
- 11. Vitvitsky V., Yadav P. K., Kurthen A., and Banerjee R. (2015) Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J. Biol. Chem. 290, 8310–8320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bostelaar T., Vitvitsky V., Kumutima J., Lewis B. E., Yadav P. K., Brunold T. C., Filipovic M., Lehnert N., Stemmler T. L., and Banerjee R. (2016) Hydrogen sulfide oxidation by myoglobin. J. Am. Chem. Soc. 138, 8476–8488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pietri R., Román-Morales E., and López-Garriga J. (2011) Hydrogen sulfide and hemeproteins: knowledge and mysteries. Antioxid. Redox Signal. 15, 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hylin J. W., and Wood J. L. (1959) Enzymatic formation of polysulfides from mercaptopyruvate. J. Biol. Chem. 234, 2141–2144 [PubMed] [Google Scholar]
- 15. Nagy P., and Winterbourn C. C. (2010) Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem. Res. Toxicol. 23, 1541–1543 [DOI] [PubMed] [Google Scholar]
- 16. Collman J. P., Ghosh S., Dey A., and Decréau R. A. (2009) Using a functional enzyme model to understand the chemistry behind hydrogen sulfide induced hibernation. Proc. Natl. Acad. Sci. U.S.A. 106, 22090–22095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kimura H. (2015) Signaling of hydrogen sulfide and polysulfides. Antioxid. Redox Signal. 22, 347–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greiner R., Pálinkas Z., Bäsell K., Becher D., Antelmann H., Nagy P., and Dick T. P. (2013) Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox Signal. 19, 1749–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yi J., Safo M. K., and Richter-Addo G. B. (2008) The nitrite anion binds to human hemoglobin via the uncommon O-nitrito mode. Biochemistry 47, 8247–8249 [DOI] [PubMed] [Google Scholar]
- 20. Lucas M. F., and Guallar V. (2012) An atomistic view on human hemoglobin carbon monoxide migration processes. Biophys. J. 102, 887–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Savino C., Miele A. E., Draghi F., Johnson K. A., Sciara G., Brunori M., and Vallone B. (2009) Pattern of cavities in globins: the case of human hemoglobin. Biopolymers 91, 1097–1107 [DOI] [PubMed] [Google Scholar]
- 22. Olteanu H., and Banerjee R. (2001) Human methionine synthase reductase, a soluble P-450 reductase-like dual flavoprotein, is sufficient for NADPH-dependent methionine synthase activation. J. Biol. Chem. 276, 35558–35563 [DOI] [PubMed] [Google Scholar]
- 23. Bagarinao T., and Vetter R. D. (1992) Sulfide-hemoglobin interactions in the sulfide-tolerant salt marsh resident, the California killfish Fundulus parvipinnis. J. Comp. Physiol. B 162, 614–624 [Google Scholar]
- 24. Kraus D. W., and Wittenberg J. B. (1990) Hemoglobins of the Lucina pectinata/bacterial symbiosis. I. Molecular properties, kinetics, and equilibria of reactions with ligands. J. Biol. Chem. 265, 16043–16053 [PubMed] [Google Scholar]
- 25. Kraus D. W., Wittenberg J. B., Lu J. F., and Peisach J. (1990) Hemoglobins of the Lucina pectinata/bacteria symbiosis. II. An electron paramagnetic resonance and optical spectral study of the ferric proteins. J. Biol. Chem. 265, 16054–16059 [PubMed] [Google Scholar]
- 26. Chen K. Y., and Morris J. C. (1972) Kinetics of oxidation of aqueous sulfide by O2. Environ. Sci. Technol. 6 529–537 [Google Scholar]
- 27. Shadrina M. S., Peslherbe G. H., and English A. M. (2015) Quaternary-linked changes in structure and dynamics that modulate O2 migration within hemoglobin's gas diffusion tunnels. Biochemistry 54, 5268–5278 [DOI] [PubMed] [Google Scholar]
- 28. Shadrina M. S., Peslherbe G. H., and English A. M. (2015) O2 and water migration pathways between the solvent and heme pockets of hemoglobin with open and closed conformations of the distal HisE7. Biochemistry 54, 5279–5289 [DOI] [PubMed] [Google Scholar]
- 29. Cumnock K., Tully T., Cornell C., Hutchinson M., Gorrell J., Skidmore K., Chen Y., and Jacobson F. (2013) Trisulfide modification impacts the reduction step in antibody-drug conjugation process. Bioconjug. Chem. 24, 1154–1160 [DOI] [PubMed] [Google Scholar]
- 30. Raftos J. E., Whillier S., and Kuchel P. W. (2010) Glutathione synthesis and turnover in the human erythrocyte: alignment of a model based on detailed enzyme kinetics with experimental data. J. Biol. Chem. 285, 23557–23567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vitvitsky V., and Banerjee R. (2015) H2S analysis in biological samples using gas chromatography with sulfur chemiluminescence detection. Methods Enzymol. 554, 111–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kabsch W. (2010) XDS. Acta Crystallogr. D 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pražnikar J., Afonine P. V., Guncar G., Adams P. D., and Turk D. (2009) Averaged kick maps: less noise, more signal, and probably less bias. Acta Crystallogr. D Biol. Crystallogr. 65, 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]