

Abstract
Phenazines are a class of redox-active molecules produced by diverse bacteria and archaea. Many of the biological functions of phenazines, such as mediating signaling, iron acquisition, and redox homeostasis, derive from their redox activity. Although prior studies have focused on extracellular phenazine oxidation by oxygen and iron, here we report a search for reductants and catalysts of intracellular phenazine reduction in Pseudomonas aeruginosa. Enzymatic assays in cell-free lysate, together with crude fractionation and chemical inhibition, indicate that P. aeruginosa contains multiple enzymes that catalyze the reduction of the endogenous phenazines pyocyanin and phenazine-1-carboxylic acid in both cytosolic and membrane fractions. We used chemical inhibitors to target general enzyme classes and found that an inhibitor of flavoproteins and heme-containing proteins, diphenyleneiodonium, effectively inhibited phenazine reduction in vitro, suggesting that most phenazine reduction derives from these enzymes. Using natively purified proteins, we demonstrate that the pyruvate and α-ketoglutarate dehydrogenase complexes directly catalyze phenazine reduction with pyruvate or α-ketoglutarate as electron donors. Both complexes transfer electrons to phenazines through the common subunit dihydrolipoamide dehydrogenase, a flavoprotein encoded by the gene lpdG. Although we were unable to co-crystallize LpdG with an endogenous phenazine, we report its X-ray crystal structure in the apo-form (refined to 1.35 Å), bound to NAD+ (1.45 Å), and bound to NADH (1.79 Å). In contrast to the notion that phenazines support intracellular redox homeostasis by oxidizing NADH, our work suggests that phenazines may substitute for NAD+ in LpdG and other enzymes, achieving the same end by a different mechanism.
Keywords: enzyme kinetics, metabolism, Pseudomonas aeruginosa (P. aeruginosa), pyruvate dehydrogenase complex (PDC), X-ray crystallography, dihydrolipoamide dehydrogenase, phenazine
Introduction
Over a century ago, Pseudomonas aeruginosa was identified as the source of a blue pigment observed in infected wounds (1). This pigment, a phenazine derivative known as pyocyanin (Fig. 1), was the first natural organic molecule shown to undergo one-electron reduction (2–4). This distinctive property inspired early studies into bacterial respiration. As early as 1931, Friedheim and Michaelis (5, 6) demonstrated that pyocyanin stimulates oxygen consumption and carbon dioxide production in both P. aeruginosa (5) and human tissue (6), and so initially pyocyanin was considered an accessory respiratory pigment analogous to quinones (5). This interpretation predated an understanding of the electron transport chain, and today pyocyanin is understood to alter redox homeostasis more generally (7, 8), although methanophenazine can substitute for quinones in the electron transport chains of certain methanogenic archaea (9). Later researchers recognized that reduced pyocyanin and other phenazines react with molecular oxygen to form toxic superoxide radicals (10). Phenazines also poison cells by oxidizing iron-sulfur clusters (11), short-circuiting electron transfer (12), covalently modifying proteins (13), and intercalating into DNA (14). This broad spectrum of toxicity became the focus of phenazine research for several decades, and so today phenazines are best known as antibiotics and virulence factors.
FIGURE 1.

Chemical structures and redox chemistry of the redox-active substrates employed in this study.
Our interest in phenazines stems from the discovery that the P. aeruginosa phenazines can reduce extracellular minerals (15, 16). Rather than merely inhibiting the growth of competing organisms, we hypothesized that phenazine “antibiotics” could directly benefit P. aeruginosa by promoting iron acquisition and redox homeostasis (17, 18). This hypothesis has motivated several experiments that illustrate physiological functions for the endogenous phenazines of P. aeruginosa. We previously showed that phenazine redox cycling promotes anaerobic survival on glucose (19, 20). Although P. aeruginosa does not ferment glucose, phenazines serve as an electron acceptor that enables glucose oxidation to acetate with concomitant ATP synthesis (20). We also showed that phenazines, by acting as electron shuttles, expand the habitable zone of colony biofilms into the anoxic portion (8). These findings paint a nuanced view where phenazines are multifunctional molecules, in some contexts possessing antibiotic activity and in others supporting redox homeostasis or iron acquisition.
In addition to pyocyanin, P. aeruginosa synthesizes at least six other phenazine derivatives. The precursor molecule, phenazine-1-carboxylic acid (PCA)4 (Fig. 1), is synthesized from chorismic acid by enzymes encoded in the homologous phzABCDEFG1 and phzABCDEFG2 operons (21, 22). Additional enzymes catalyze structural modifications of PCA to pyocyanin, phenazine-1-hydroxide, and phenazine-1-carboxamide (23). Other endogenous P. aeruginosa phenazines have been observed with amination (24) and sulfonation (25) modifications, although the pathways for these modifications are unknown. Beyond P. aeruginosa, over 180 naturally occurring phenazines have been described in the literature from a variety of bacteria and archaea (Dictionary of Natural Products, Chemnetbase). This diverse array of structurally similar molecules suggests particular phenazines might be tuned for certain functions or properties. In support of this interpretation, the endogenous P. aeruginosa phenazines have different pH-dependent solubilities, toxicities, and reactivities toward oxygen and iron (16, 27).
Despite their structural diversity, the key biological functions of phenazines, be it promoting anaerobic energy generation or facilitating iron acquisition, derive from their redox activity. The environmentally relevant extracellular oxidants, molecular oxygen and ferric iron, are well characterized (16). In stark contrast, there is a dearth of information about biologically catalyzed phenazine reduction. Although pyocyanin and PCA are known to oxidize the electron donors NAD(P)H, dihydrolipoamide, and glutathione in vitro (15, 28), it is unknown whether this is relevant in vivo or whether it is enzymatically catalyzed. To address this unknown, previous work in our laboratory attempted to identify phenazine reductases by screening transposon mutants (29, 30). Mutants in glycerol-3-phosphate dehydrogenase and the cytochrome bc1 complex were found to be deficient in pyocyanin reduction (29), but these mutants are generally unhealthy and carry a number of pleiotropic effects (29), rendering it impossible to ascribe pyocyanin reduction to these proteins from the physiological data alone.
As an alternative to our prior genetic screens, in this study we used a biochemical approach to identify P. aeruginosa enzymes with endogenous phenazine reductase activity. Our results suggest that P. aeruginosa might not express an enzyme evolved to specifically reduce phenazines; rather, our findings implicate flavoproteins and/or heme-containing proteins as important nonspecific catalysts for phenazine reduction. Illustrating this principle, we identified and characterized the pyruvate and α-ketoglutarate dehydrogenase complexes as models for flavoprotein-mediated phenazine reduction. Our data indicate that under energy-limited anoxic conditions, phenazine substitution for NAD+ in these enzymes might contribute to the maintenance of intracellular redox homeostasis.
Results
Phenazine Reduction Is Catalyzed by Cell Lysate
We first asked whether P. aeruginosa produces enzymes that catalyze phenazine reduction. We focused on PCA (Fig. 1) because it is the precursor for all biological phenazines in P. aeruginosa (23), and it promotes anaerobic energy generation by redox cycling (19, 20). It conveniently undergoes a color change to bright yellow after reduction (31), enabling a colorimetric detection of PCA reduction. To avoid re-oxidation by atmospheric oxygen, we monitored PCA reduction using a plate reader housed within an anaerobic chamber. Among the many possible intracellular electron carriers, we tested NADH, NADPH, and glutathione as electron donors that are important determinants of the intracellular redox state (7, 32). Compared with pyocyanin, PCA reacts relatively slowly with these donors, minimizing the risk of misinterpreting non-enzymatic reduction for enzyme activity. Because phenazines alter the metabolic flux of P. aeruginosa (7, 20), we reasoned that intermediates of central metabolism might also serve as electron donors, and so we also tested pyruvate, citrate, isocitrate, α-ketoglutarate, succinate, and malate. We prepared cell lysate from late-exponential phase cultures grown on succinate, a preferred carbon source of P. aeruginosa (33). Because phenazines induce a transcriptional response in P. aeruginosa (34), and so may regulate the activity of their endogenous reductases, we harvested the cultures several hours after the onset of phenazine production as indicated by the blue color of pyocyanin.
In the absence of cell lysate, PCA did not react with the organic acid metabolic intermediates, and it reacted only slowly with NADH, NADPH, and glutathione (Fig. 2). Similarly, in the absence of an exogenous electron donor, cell lysate slowly reduced PCA over time (Fig. 2), likely deriving some reducing equivalents from metabolites already present within the lysate. Relative to lysate or electron donor alone, lysate rapidly reduced PCA when supplemented with NADH, NADPH, citrate, isocitrate, pyruvate, or α-ketoglutarate (Fig. 2). Glutathione slightly increased the rate of PCA reduction, whereas malate and succinate had no stimulatory effect when added to lysate (Fig. 2). Boiling the cell lysate beforehand effectively eliminated PCA reduction even with the addition of exogenous electron donors (Fig. 2), demonstrating that the lysate catalyzed reduction rather than indirectly accelerating reduction through a change in ionic strength or pH. Additionally, we found that adding boiled lysate to un-boiled lysate could further accelerate reduction with pyruvate and α-ketoglutarate (Fig. 2), suggesting the presence of additional cofactors or substrates in cell lysate that are required for PCA reduction. Together, these results demonstrate that P. aeruginosa produces one or more enzymes with PCA reduction activity.
FIGURE 2.
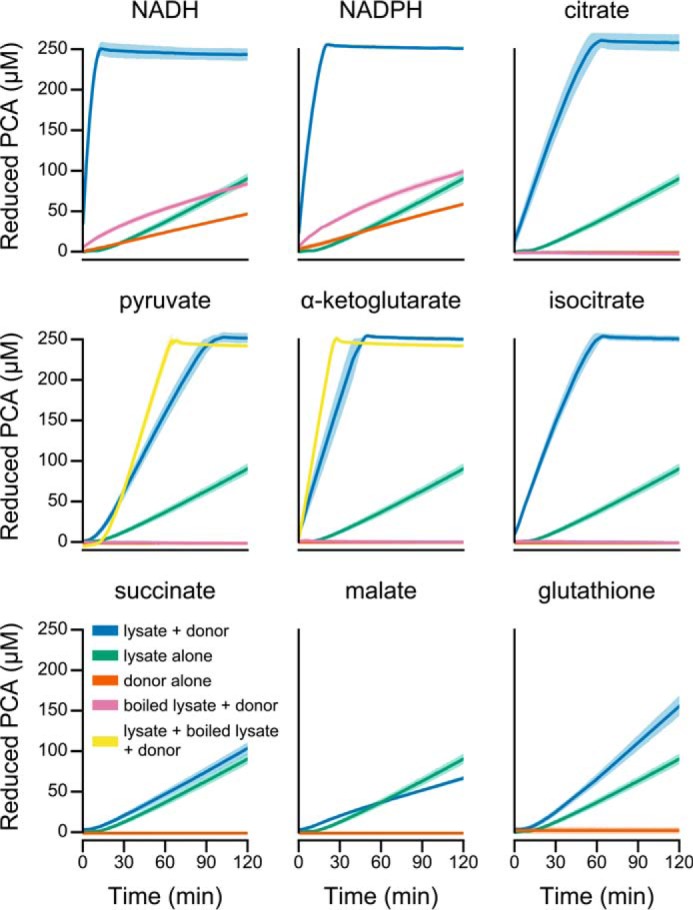
Catalytic reduction of PCA by P. aeruginosa cell-free lysate. Each panel illustrates a time course of PCA reduction using the indicated electron donor. The line and shaded region represent the mean and standard deviation, respectively, from three independent assays using lysate prepared from independent cultures. The final concentrations in the assay were 1 mm for NADH and NADPH, 5 mm for racemic dl-isocitrate (to give 2.5 mm d-isocitrate), and 2.5 mm for all other donors.
We attempted to purify an NADH:PCA or NADPH:PCA oxidoreductase from P. aeruginosa cell lysate using anion-exchange, cation-exchange, and hydrophobic interaction chromatography. Unfortunately, our efforts were hampered by the abiotic reaction between NAD(P)H and PCA, which is greatly accelerated at higher salt concentrations involved in protein chromatography. We struggled to locate chromatographic peaks of activity from the high background. To learn more about the types of proteins that might catalyze phenazine reduction, we opted to characterize the phenazine reduction activity in terms of cellular localization and sensitivity toward chemical inhibitors.
Multiple Enzymes Can Catalyze Phenazine Reduction
We wondered whether the electron transport chain catalyzes phenazine reduction, because pyocyanin is reported to stimulate respiration (5, 6) and phenazines induce morphological changes in colony biofilms that are consistent with increased respiration (8). To test whether PCA reduction activity derives mostly from the membrane-bound electron transport chain, we separated membrane-associated components from the lysate by ultracentrifugation (note that some soluble components, such as ribosomes, β-oxidation machinery, and 2-oxoacid dehydrogenases, can also be pelleted by ultracentrifugation). We saved the supernatant containing soluble proteins and resuspended the membrane pellet with a gentle non-ionic detergent. We then tested the three fractions (crude lysate, soluble proteins, and membrane-associated proteins) for PCA reduction in terms of absolute rate and specific activity.
Both the soluble and membrane fractions contained approximately equal activity, but specific activity (activity normalized by protein concentration) was 2-fold greater in the soluble fraction relative to the membrane fraction (Fig. 3). In the membrane samples, the calculated final concentration of reduced PCA (300–350 μm) was greater than the amount initially added to the spectrophotometric assay (250 μm) (Fig. 3). This artifact may be due to redox-active cytochromes absorbing at the same wavelength as PCA or residual endogenous phenazines associated with the membrane, and so our data might overestimate the rate of PCA reduction in membrane fractions. Citrate and isocitrate stimulated PCA reduction primarily in the soluble fraction (Fig. 3). Notably, for the 2-oxoacids pyruvate and α-ketoglutarate, ultracentrifugation significantly lowered PCA reduction activity, but we did not recover this activity in the membrane fraction (Fig. 3). Because many enzymes in central metabolism use NAD(P)H as an electron carrier, from these data we infer that the cell lysate contains at least two NAD(P)H:PCA oxidoreductases that are distributed between the soluble and membrane fractions, with more activity residing in the soluble fraction. This implies that the electron transport chain does not exclusively reduce PCA in vitro. Additional enzymes might also use citrate, isocitrate, pyruvate, or α-ketoglutarate as electron donors.
FIGURE 3.

Fractionation of PCA reductase activity in cell-free lysate. Cell-free lysate was fractionated into soluble- and membrane-associated components by ultracentrifugation. The fractions were assayed for their ability to catalyze PCA reduction. Left, each panel illustrates a time course of PCA reduction using the indicated electron donor. The line and shaded region represent the mean and standard deviation, respectively, from four independent assays using lysate prepared from independent cultures. The solid portion of each line indicates the linear region used for quantification. Right, specific activity for each fraction was determined by normalizing the rate of PCA reduction, illustrated with a solid line in the left panels, by the protein concentration of each fraction. Error bars represent the standard deviation from four independent biological replicates.
We further probed the lysate using three inhibitors as follows: (a) cyanide to inhibit metalloproteins and heme-containing proteins (35); (b) diphenyleneiodonium to inhibit flavoproteins and some heme-containing proteins (36); and (c) arsenite to inhibit thiol-dependent proteins (37). Cyanide only slightly inhibited activity, whereas diphenyleneiodonium greatly inhibited activity with all of the electron donors tested (Fig. 4). Arsenite specifically inhibited activity only for pyruvate and α-ketoglutarate (Fig. 4). Together, the differential localization and inhibitor activities suggest that P. aeruginosa lysate contains multiple enzymes that catalyze PCA reduction. The strong inhibition by diphenyleneiodonium (Fig. 4) further suggests that flavoproteins and/or heme-containing proteins may catalyze PCA reduction.
FIGURE 4.

Inhibition of PCA reduction activity in cell-free lysate. Unfractionated cell-free lysate was assayed for its ability to catalyze PCA reduction in the presence of the indicated inhibitors. Left, each panel illustrates a time course of PCA reduction using the indicated electron donor. The line and shaded region represent the mean and standard deviation, respectively, from four independent assays using lysate prepared from independent cultures. The solid portion of each line indicates the linear region used for quantification. Right, specific activity for each fraction was determined by normalizing the rate of PCA reduction, illustrated with a solid line in the left panels, by the protein concentration of each sample. Error bars represent the standard deviation of four independent biological replicates.
Pyruvate and α-Ketoglutarate Dehydrogenase Catalyze Phenazine Reduction
Our experiments with cell lysate suggested a role for the pyruvate and α-ketoglutarate dehydrogenases in PCA reduction. Not only did pyruvate and α-ketoglutarate stimulate PCA reduction (Fig. 3), but the pattern of localization and inhibition was consistent with the conserved mechanism of the 2-oxoacid dehydrogenase complexes (38, 39). In the 2-oxoacid dehydrogenase complexes, the E1 subunit, the substrate dehydrogenase, conjugates the substrate to pyrophosphate (TPP) with concomitant decarboxylation. The TPP cofactor is regenerated by transferring the substrate onto a lipoamide cofactor covalently attached to the E2 subunit. The E2 subunit, dihydrolipoyl transacetylase, then regenerates lipoamide by transferring the substrate to CoA, producing dihydrolipoamide. The dihydrolipoamide is finally re-oxidized by the E3 subunit, dihydrolipoamide dehydrogenase (DLDH), by transferring electrons to NAD+ through a flavin in the form of a tightly bound FAD.
We recognized that the megadalton-sized 2-oxoacid dehydrogenases can be pelleted by ultracentrifugation, which would explain why 2-oxoacid-dependent activity was depleted by ultracentrifugation (Fig. 3), and the cofactor requirement could explain why activity was not recovered in the membrane fraction (Fig. 3) because cofactors would remain in the soluble fraction. The obligate reduction and oxidation of the lipoamide thiols is consistent with inhibition by arsenite (Fig. 4). Moreover, oxidation of the lipoamide cofactor requires a flavoprotein subunit, consistent with inhibition by diphenyleneiodonium (Fig. 4). We therefore asked whether the pyruvate and α-ketoglutarate dehydrogenase complexes were sufficient to catalyze phenazine reduction and, if so, what electron transfer pathway was employed.
To answer this question, we isolated the native pyruvate and α-ketoglutarate dehydrogenase complexes from P. aeruginosa (detailed under “Experimental Procedures”). As judged by Coomassie staining of an SDS-polyacrylamide gel, the complexes were ∼90% pure (Fig. 5A). The α-ketoglutarate dehydrogenase preparation contained no detectable pyruvate dehydrogenase (<0.1%), whereas the pyruvate dehydrogenase preparation contained a small amount of contaminating α-ketoglutarate dehydrogenase (∼4%) as measured by NAD+ reduction activity.
FIGURE 5.
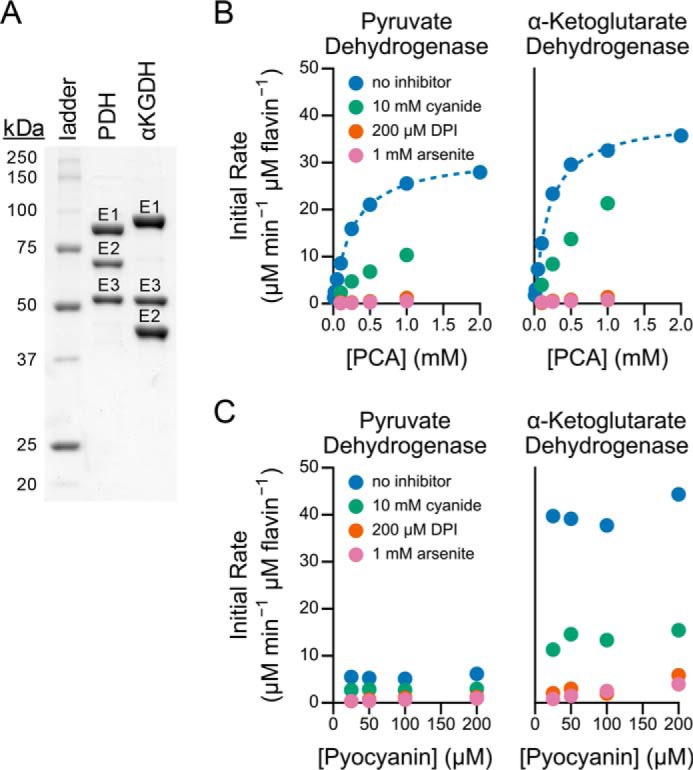
Preparation and characterization of the pyruvate and α-ketoglutarate dehydrogenase complexes. A, Coomassie Blue-stained SDS-polyacrylamide gel of the purified pyruvate dehydrogenase (PDH) and α-ketoglutarate (αKGDH) complexes. The three prominent bands represent the three subunits of the complex as labeled. PDH, E1 (pyruvate dehydrogenase; AceE, E2 (dihydrolipoamide acetyltransferase, AceF); E3 (dihydrolipoamide dehydrogenase, LpdG); αKGDH, E1 (α-ketoglutarate dehydrogenase, SucA), E2 (dihydrolipoamide succinyltransferase, SucB); E3 (dihydrolipoamide dehydrogenase, LpdG). B and C, PCA (B) and pyocyanin (C) reduction by the purified complexes in the absence or presence of the indicated inhibitors. The dashed line represents the fitted Michaelis-Menten binding curve.
The purified complexes catalyzed PCA (Fig. 5B) and pyocyanin (Fig. 5C) reduction using their respective electron donors. We normalized the reaction rate to the protein flavin content (as a measure of dihydrolipoamide dehydrogenase) rather than defining the number of active sites in a large, potentially heterogeneous complex with multiple reaction steps. A Michaelis-Menten kinetic model fit to the initial rate of PCA reduction (Fig. 5B) yielded a Km of 260 μm and Vmax of 32 min−1 for pyruvate dehydrogenase and a Km of 190 μm and Vmax of 40 min−1 for α-ketoglutarate dehydrogenase. The initial rates of pyocyanin reduction did not depend on the pyocyanin concentration (Fig. 5C), implying a Km of less than 25 μm for pyocyanin. Interestingly, although the pyruvate and α-ketoglutarate dehydrogenase complexes reduced PCA at similar rates (Fig. 5B), pyruvate dehydrogenase reduced pyocyanin considerably slower than did α-ketoglutarate dehydrogenase (Fig. 5C).
Cyanide, diphenyleneiodonium, and arsenite inhibited PCA and pyocyanin reduction by the purified complexes (Fig. 5, B and C). The relative inhibition of the purified complexes mirrored that of whole cell lysate (Fig. 4), with cyanide partially inhibiting PCA reduction, and both arsenite and diphenyleneiodonium more fully inhibiting PCA reduction (Fig. 5, B and C), indicating that the 2-oxoacid dehydrogenase complexes can account for the activity in lysate.
In control experiments, PCA and pyocyanin reduction by the purified complexes required all known substrates and cofactors. We detected no activity when the electron donor (pyruvate or α-ketoglutarate), coenzyme A, or magnesium were omitted from the reaction mixture. We observed residual activity (<1%) in the absence of TPP, possibly due to co-purification of trace amounts of this cofactor with the protein. Although we cannot rule out catalysis by a contaminant in our enzyme preparation, the inhibition by arsenite and diphenyleneiodonium and the requirement for the known cofactors of pyruvate and α-ketoglutarate dehydrogenase indicate that the 2-oxoacid dehydrogenase complexes directly catalyze PCA and pyocyanin reduction.
Dihydrolipoamide Dehydrogenase Catalyzes Phenazine Reduction
We next asked which 2-oxoacid dehydrogenase subunit catalyzes phenazine reduction. In the absence of CoA, the E1 subunit catalyzes ferricyanide and dichloroindophenol reduction (40, 41). However, we observed no PCA or pyocyanin reduction in the absence of CoA, suggesting these phenazines are not reduced by the E1 subunit. As the E3 subunit has long been recognized to reduce methylene blue (41), a molecule structurally similar to phenazines (Fig. 1), and the porcine and malarial E3 subunits have been shown to catalyze pyocyanin reduction (28), we hypothesized that the P. aeruginosa E3 subunit (DLDH) catalyzes phenazine reduction.
Strain PA14 encodes the following three DLDH homologs in its genome: LpdG (PA14_43970); Lpd3 (PA14_63850); and LpdV (PA14_35490) (42, 43). We purified His-tagged versions of each protein. All three enzymes catalyzed PCA and pyocyanin reduction using both NADH and dihydrolipoamide as electron donors (Fig. 6A). Phenazine reduction by both dihydrolipoamide and NADH, the substrate and product of the physiological reaction, respectively, is consistent with the FAD cofactor acting as a bidirectional electron-transfer intermediate, suggesting that phenazines interact with the flavin during the catalytic cycle of DLDH. In the context of the full 2-oxoacid dehydrogenase complexes, we propose that phenazines can substitute for NAD+ as the final electron acceptor of pyruvate and α-ketoglutarate oxidation. This contrasts with our previous assumption that phenazines serve as an oxidant for NADH (20).
FIGURE 6.
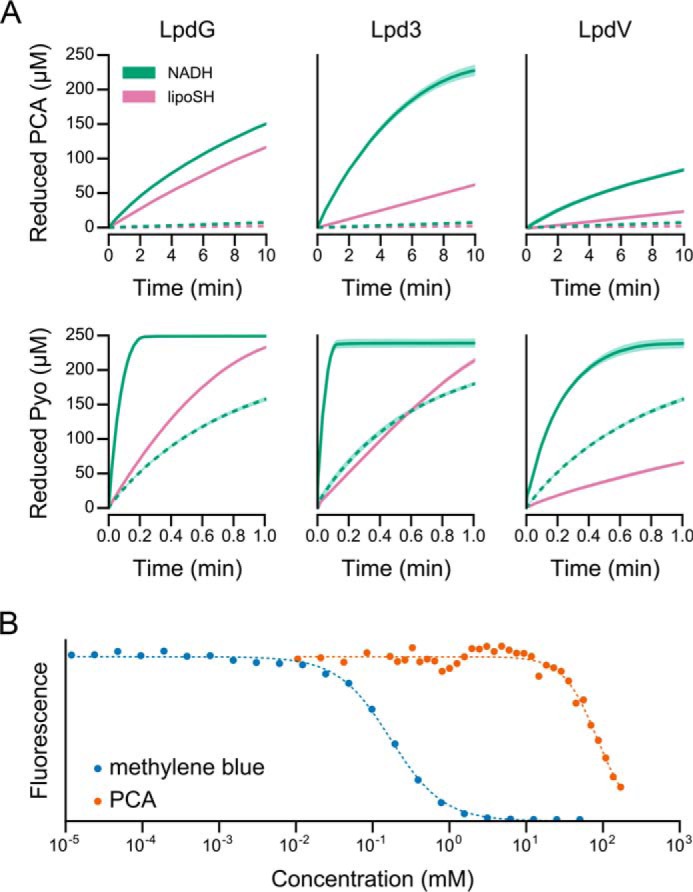
Phenazine reduction and binding by DLDH homologs of P. aeruginosa. A, time courses of PCA and pyocyanin reduction by DLDH using both NADH and dihydrolipoamide (lipoSH) as electron donors. Solid lines indicate the presence of 2 μm enzyme. Dashed lines indicate the absence of enzyme. Shaded regions indicate the standard deviation of three technical replicates. B, affinity of LpdG for PCA and methylene blue as measured by fluorescence quenching. The dashed line indicates the binding curve fitted to the data. The data points are normalized to the maximum and minimum values of the fitted curve.
Before probing the interaction between phenazines and DLDH, we determined the enzymatically relevant DLDH homolog for the 2-oxoacid dehydrogenase complexes. Although the pyruvate dehydrogenase complex of P. aeruginosa has been purified before (44), the precise identity of its constituent DLDH was not determined, and the operon encoding pyruvate dehydrogenase does not contain an associated DLDH. In contrast, the operon encoding α-ketoglutarate dehydrogenase encodes all three subunits (E1, E2, and E3 (LpdG)). Phenotypic evidence in Pseudomonas putida suggests the E3 component of pyruvate dehydrogenase is LpdG, but Lpd3 may substitute in the absence of LpdG (45). To address this ambiguity, we excised the band corresponding to DLDH from an SDS-polyacrylamide gel (Fig. 5A), performed an in-gel trypsin digest, and analyzed the resulting peptide fragments using LC-MS/MS. As expected from its operon structure, the DLDH peptides from α-ketoglutarate dehydrogenase mapped to LpdG (Table 1). The DLDH peptides from pyruvate dehydrogenase also mapped to LpdG (Table 1), with only 0.03% of peptide intensity mapping to Lpd3, indicating that LpdG is the primary DLDH of the P. aeruginosa PA14 pyruvate dehydrogenase. We detected no peptides corresponding to LpdV in either complex (Table 1). We also confirmed the identity of dihydrolipoamide acetyltransferase in our pyruvate dehydrogenase preparation (Table 1), as this band migrated slower than expected for its predicted size of 56.7 kDa (Fig. 5A).
TABLE 1.
LC-MS/MS confirmation of constituent proteins in the purified 2-oxoacid dehydrogenase complexes
ND, not detected; iBAQ, intensity-base absolute quantification.
| Banda | Unique peptidesb | Intensityc | iBAQd |
|---|---|---|---|
| Pyruvate dehydrogenase E3 | |||
| LpdG | 45 | 85.1% | 83.9% |
| Lpd3 | 9 | 0.030% | 0.035% |
| LpdV | 0 | ND | ND |
| α-Ketoglutarate dehydrogenase E2 | |||
| LpdG | 39 | 85.9% | 82.0% |
| Lpd3 | 0 | ND | ND |
| LpdV | 0 | ND | ND |
| Pyruvate dehydrogenase E2 | |||
| AceE | 46 | 80.4% | 81.6% |
a Band labels correspond to the gel shown in Fig. 5A. Each sub-row shows information about proteins of interest for a given gel band.
b Number of unique peptides observed for the indicated protein within the indicated band is shown.
c Percent peptide abundance was based on the raw signal intensity of identified peptides.
d Percent peptide abundance using the iBAQ method (26) is shown.
Having established LpdG as the enzymatically relevant DLDH for both pyruvate and α-ketoglutarate dehydrogenase, we quantified the interaction between LpdG and phenazines in more detail. We measured the binding affinity, Kd, by exploiting the intrinsic fluorescence of the flavin moiety in the FAD cofactor of LpdG. We anticipated that binding of an aromatic group near the flavin would quench fluorescence, thereby providing a direct measure of substrate binding. This prediction was borne out experimentally. High concentrations of PCA were sufficient to quench fluorescence (Fig. 6B), consistent with binding near the flavin. The extrapolated Kd is 84 mm with a Hill coefficient of 1.95 (Fig. 6B). In support of a cooperative model of binding, a non-cooperative model (Hill coefficient = 1) required a nonsensical large negative minimum fluorescence and an unrealistically large Kd (>1000 mm). In contrast to PCA, the positively charged methylene blue quenched fluorescence with a Kd of 170 μm and a Hill coefficient of 1.27 (Fig. 6B). We were unable to apply this method to pyocyanin, which is water-soluble at pH 7.5 only to ∼1 mm or to phenazine-1-hydroxide and phenazine-1-carboxamide, which are even less soluble. Hill coefficients of less than two are consistent with the dimeric structure of DLDH. Cooperative substrate binding in DLDH has been observed for NADH (46) and may originate from the architecture of the active site, which contains residues from both chains of the DLDH homodimer (47, 48), potentially allowing for substrate binding to induce long range conformational changes within the protein.
Structural Insights into the Interaction between LpdG and Phenazines
Although the affinity of the P. aeruginosa LpdG for PCA appears to be low, we were encouraged by the comparatively high affinity for methylene blue, and so we attempted to solve the X-ray crystal structure of LpdG bound to a phenazine or methylene blue. LpdG crystallized in multiple screens using polyethylene glycol as a precipitant, similar to previously discovered conditions for DLDH. During optimization, we discovered a new crystal form in which over 90% of crystals diffracted to 1.8 Å or better in the space group P21. One crystal diffracted to 1.43 Å (Fig. 7A) with a signal-to-noise cutoff of 2.0, which we refined to 1.35 Å using paired refinement (49), representing the highest published resolution of any DLDH to date.
FIGURE 7.

Structural analysis of LpdG. Where visible, the embedded FAD cofactor is shown in red. Protein side chain carbons are shown in purple. A, overview of the LpdG dimer. The two polypeptides of the biological assembly are shown in shades of blue. B, close-up of DMSO bound in the lipoamide binding pocket. The orange mesh represents the anomalous difference map contoured at 4σ, which highlights sulfur atoms and the phosphorus atoms in FAD. The DMSO carbon atoms are shown in green. C and D, close-up of NADH (C) and NAD+ (D) bound to LpdG. Electron density was not visible for the oxidized nicotinamide group in NAD+, and so this group has not been modeled. The blue mesh represents the 2mFo − DFc map of electron density for FAD and NAD(H) contoured at 1.5σ. The NAD(H) carbon atoms are shown in yellow. The dashed line illustrates a hydrogen bond interaction. E, example of π-stacking interactions at a surface residue in a crystal soaked with 225 mm PCA. The green mesh represents positive density in the 2mFo − DFc difference map contoured at 3σ. F, hypothetical illustration of a phenazine (orange) substituting for NADH, demonstrating a steric clash with Ile-192. The phenazine was placed manually in Coot and does not represent a real dataset.
Apart from minor rotamer corrections and the orientations of some surface residues, our model of LpdG at 1.35 Å is essentially identical to the previously reported structure at 2.8 Å (the root mean square deviation is less than 0.4 over 470 α-carbons), and so we defer to the original publication by Mattevi et al. (47) for a full structural description. A noteworthy feature unique to our model is the association of LpdG with the cryoprotectant DMSO. We observed two molecules of DMSO inside the active site where dihydrolipoamide is postulated to bind (Fig. 7B). This orientation appears to be stabilized by a sulfur-π interaction between the central sulfur atom of DMSO and a histidine side chain. This configuration might mimic the binding of the dihydrolipoamide sulfhydryl groups in the active site of LpdG. Crystals soaked for 1 h with 10 mm NADH or NAD+ yielded electron densities corresponding to the soaked substrate (Fig. 7, C and D). As observed previously with human DLDH (50), we could not resolve electron density for the nicotinamide group of NAD+ (Fig. 7D), suggesting the oxidized cofactor is disordered or present in multiple conformations. Statistics for the apo-, NAD+-, and NADH-bound structures are shown in Table 2.
TABLE 2.
Crystallography data collection and refinement statistics
Values in parentheses are for the highest resolution shell.
| LpdG | LpdG with NAD+ | LpdG with NADH | |
|---|---|---|---|
| Data collection | |||
| Space group | P21 | P21 | P21 |
| Unit cell | |||
| a, b, c (Å) | 65.32, 116.28, 136.59 | 65.61, 116.69, 136.86 | 65.51, 117.83, 139.73 |
| α, β, γ (°) | 90, 94.735, 90 | 90, 94.871, 90 | 90, 94.302, 90 |
| Resolution range (Å) | 37.52–1.35 (1.398–1.35) | 38.90–1.45 (1.502–1.45) | 34.93–1.79 (1.854–1.79) |
| Unique reflections | 443,431 (44,051) | 361,916 (36,058) | 198,611 (19,792) |
| Rpim (%) | 1.72 (48.7) | 1.91 (48.0) | 2.39 (39.11) |
| 〈I/σ(I)〉 | 19.81 (1.40) | 21.06 (1.53) | 18.36 (2.65) |
| Completeness (%) | 99.88 (99.46) | 99.98 (99.98) | 99.98 (99.98) |
| Multiplicity | 13.6 (13.4) | 13.6 (13.0) | 13.5 (13.8) |
| Wilson B-factor (Å2) | 17.93 | 19.42 | 28.19 |
| Refinement | |||
| Resolution range (Å) | 37.52–1.35 | 38.9–1.45 | 34.93–1.79 |
| Rwork/Rfreea | 0.1325/0.1565 | 0.1521/0.1736 | 0.1667/0.1785 |
| No. of non-H atoms | |||
| Protein | 13,905 | 13,863 | 13,661 |
| Water | 2,383 | 2105 | 1228 |
| DMSO | 156 | 128 | 80 |
| Root mean square deviation | |||
| Bond lengths (Å) | 0.012 | 0.004 | 0.004 |
| Bond angles (°) | 1.21 | 0.768 | 0.745 |
| Average B-factors (Å2) | |||
| Protein | 24.37 | 24.88 | 40.01 |
| Water | 40.72 | 37.43 | 43.78 |
| Ligandb | 27.31 | 28.39 | 43.58 |
| Ramachandran (%) | |||
| Favored | 97.51 | 97.14 | 96.55 |
| Allowed | 2.49 | 2.86 | 3.45 |
| Outliers | 0 | 0 | 0 |
| PDB accession code | 5U8U | 5U8V | 5U8W |
a A total of 1.18% of reflections were excluded from refinement for cross-validation.
b Data includes DMSO.
Our initial screens yielded no crystals when 2 mm methylene blue or 250 mm PCA was included in the protein solution, so for these substrates we focused on soaking into preformed crystals. Unfortunately, we observed no electron density assignable to a phenazine after soaking for up to 1 week with 10 mm pyocyanin, 10 mm phenazine-1-carboxamide, 10 mm phenazine-1-hydroxide, 5 mm methylene blue, or up to 175 mm PCA. We were similarly unsuccessful when the soaks included up to 100 mm dithionite or 100 mm dihydrolipoamide to generate the reduced forms of LpdG and substrate. Above 200 mm PCA, we observed an electron density characteristic of a π-stacking interaction between PCA and aromatic residues on the surface of the protein (Fig. 7E), but this interaction is unlikely to be physiologically relevant because of the high PCA concentration it requires.
The crystal structure reveals a carefully structured nicotinamide-binding site that sterically excludes larger molecules such as phenazines (Fig. 7, C and D), likely explaining why we were unable to obtain crystals bound to a phenazine. In particular, an isoleucine side chain (Ile-192) restricts access to the flavin. The smaller nicotinamide moiety of NADH forms a π-stack with the flavin that is further stabilized by hydrogen bonding of the amide group with Glu-195 and the peptide backbone (Fig. 7C). Assuming no other conformational changes, extending nicotinamide to contain three rings yields a steric clash with Ile-192 and disrupts the stabilizing hydrogen bonds (Fig. 7F). Based on this structural interpretation, we hypothesized that mutating Ile-192 to a smaller residue might increase accessibility of the flavin to phenazines, thereby accelerating DLDH-catalyzed phenazine reduction. We were also interested in Val-191, which sits above the flavin-nicotinamide π-stack. In other homologs, and the closely related glutathione reductase, this residue is a tyrosine that may regulate NADH binding or reactivity through additional π-stacking and compression (51). We therefore generated two site-directed mutants of LpdG, I192G and V191Y. Contrary to our expectations, the I192G mutant catalyzed PCA reduction more slowly than wild type LpdG and also appeared to be substrate-inhibited (Fig. 8A). The V191Y mutation also lowered enzyme activity and conferred substrate inhibition (Fig. 8A). The mutations did not affect the PCA concentration required to quench LpdG fluorescence (Fig. 8B), demonstrating that the binding affinity remained unchanged in the mutants. Together, these results indicate that residues Val-191 and Ile-192 do not sterically hinder PCA from binding to LpdG.
FIGURE 8.
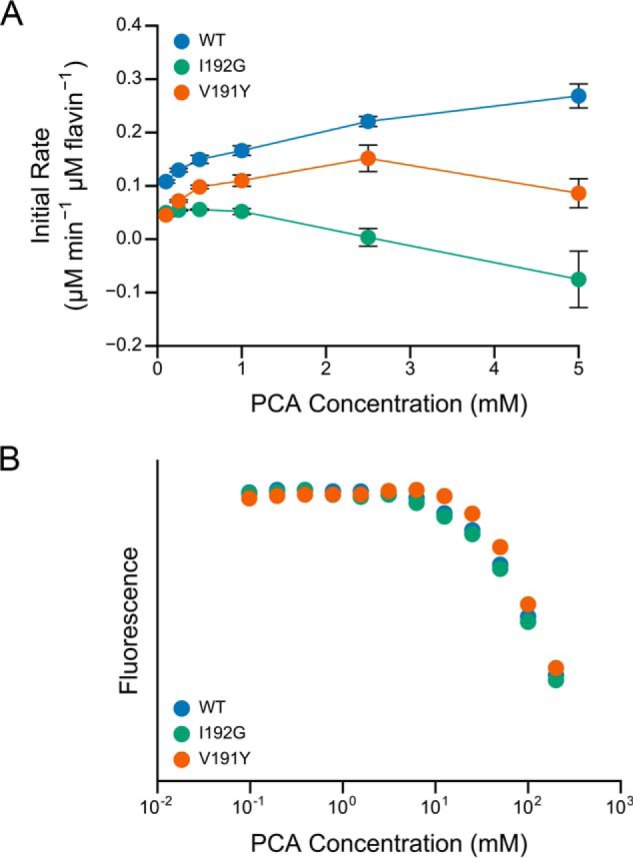
Site-directed mutagenesis of LpdG. A, PCA reduction by site-directed mutants of LpdG. The initial rate of reduction was normalized by the concentration of protein (2 μm) and subtracted by the abiotic rate of reduction as measured without protein. Error bars represent the standard deviation of four technical replicates. B, affinity of site-directed mutants of LpdG for PCA as measured by fluorescence quenching. The fluorescence values are normalized to the maximum fluorescence for each mutant.
Discussion
The search for phenazine reductases was motivated by our interest in phenazine-mediated anaerobic survival and energy generation (19, 20). Our work here suggests that, rather than possessing an enzyme with specific phenazine reductase activity, P. aeruginosa makes a variety of redox-active enzymes that adventitiously reduce phenazines under anoxic conditions. By subsequently reacting with molecular oxygen or ferric iron (16), phenazines would enable the oxidation of metabolites in a manner independent of the electron transport chain. The electron donor for this process could be the metabolite itself, for example pyruvate or α-ketoglutarate, without involvement of another cofactor such as NAD(P)H. Through extracellular electron transport in a microbial community (8), this mechanism would allow multiple metabolic pathways, such as glycolysis or pyruvate oxidation (20), to support energy generation for cells locally starved of an electron acceptor (i.e. oxygen or nitrate).
We identified the pyruvate and α-ketoglutarate dehydrogenase complexes as phenazine reductases (Fig. 5) based on activity assays in whole cell lysate (Fig. 2) and the pattern of fractionation and inhibition (Figs. 3 and 4). We focused on this specific subset of enzymes because we previously found that phenazines enable ATP synthesis through the conversion of glucose to acetate by serving as an electron acceptor (20). Notably, this pathway requires pyruvate dehydrogenase. In our prior work, we hypothesized that phenazines stimulate pyruvate dehydrogenase by converting NADH to NAD+ (20), a limiting substrate in the absence of a terminal electron acceptor. Our results here suggest that phenazines may instead directly substitute for NAD+ in the pyruvate dehydrogenase complex during anaerobic survival. Under NAD+-limiting conditions (e.g. hypoxia), phenazines could therefore increase metabolic flux through pyruvate dehydrogenase. The phenazine reduction activity of pyruvate dehydrogenase is admittedly low (Vmax = 32 min−1 for PCA) (Fig. 5), but it may be compatible with the low metabolic activities observed in anoxic planktonic models of survival (20, 52).
Importantly, the affinity we measured of pyruvate dehydrogenase for phenazines (Km of 260 μm for PCA and <25 μm for pyocyanin) is within the physiological range produced by P. aeruginosa, which can reach up to 200 μm within infection contexts (53). We observed a large mismatch between the ligand binding affinity of the catalytic subunit LpdG (i.e. Kd, Fig. 6B) and the concentration required to saturate the reaction rate in the full complex (i.e. Km, Fig. 5, B and C). LpdG is natively associated with the larger complex, and so the rate saturation of the full complex likely represents the more physiologically meaningful result. The large disparity in magnitude suggests that LpdG is not rate-limiting within the full complex and/or that binding to the complex induces substantial changes in the affinity of LpdG for its substrates. Given the high Kd value for PCA, we would not expect PCA to outcompete NAD+ unless the concentrations of available NAD+ were very low, but we note that this is precisely the condition where phenazines manifest a physiological benefit for P. aeruginosa (7, 8, 19, 20).
The phenazine reductase activity of pyruvate dehydrogenase is particularly intriguing in the context of biofilm maintenance and development. The inner core of established biofilms is anoxic (8, 54) and enters a metabolically repressed state with decreased respiration and increased substrate-level phosphorylation (54). In the absence of oxygen, P. aeruginosa can survive by mixed-acid fermentation of pyruvate to acetate and lactate (52), a process that is enhanced by phenazine redox cycling (20). This suggests a model where cells at the anoxic core of a biofilm may use pyruvate dehydrogenase, with phenazines as an electron acceptor and coupled to substrate-level phosphorylation, to maintain a basal level of energy generation; reduced phenazines could then be re-oxidized at the oxygen-rich surface of the biofilm. In addition to maintaining energy generation in anoxic regions of biofilms (8), phenazines can also stimulate biofilm formation through iron acquisition (55), extracellular DNA release (56), and other unknown mechanisms (55–57). In the context of our work here, it is striking that early biofilm development in P. aeruginosa requires pyruvate fermentation and redox homeostasis (58). It is tempting to speculate that the biofilm-promoting behavior of phenazines and pyruvate fermentation could be mechanistically linked through the activity of pyruvate dehydrogenase.
We do not mean to suggest that pyruvate and α-ketoglutarate dehydrogenase are the only relevant phenazine reductases in vivo. Indeed, NADH and NADPH are more effective than the 2-oxoacids at stimulating PCA reduction in cell lysate (Figs. 2–4). The 2-oxoacid:PCA oxidoreductase activity in lysate could be explained by the 2-oxoacid dehydrogenases reducing NAD+ to NADH, enabling the activity of an NADH:PCA oxidoreductase. Similarly, the stimulation by citrate and isocitrate (Figs. 2–4) could result from the downstream production of NAD(P)H. Therefore, our results with cell lysate are best interpreted as an upper limit to the overall PCA reductase activity of pyruvate and α-ketoglutarate dehydrogenase. With that caveat, we also found that diphenyleneiodonium is a strong inhibitor of PCA reduction (Fig. 4). Notably, diphenyleneiodonium is a known flavoprotein inhibitor that conjugates to reduced flavins via a radical-mediated mechanism, although it can also inhibit some heme proteins (36). This is consistent with our finding that the flavoenzyme LpdG, the E3 subunit of the 2-oxoacid dehydrogenases studied here (Table 1), directly catalyzes PCA and pyocyanin reduction (Fig. 6). Together, these findings suggest that flavoproteins, and possibly some heme-containing proteins, account for the majority of phenazine reductase activity in P. aeruginosa.
Ultimately, pyruvate and α-ketoglutarate dehydrogenase serve to demonstrate that many electron donors, through the activity of their catabolic enzymes, could support phenazine reduction. By accepting electrons from flavoproteins, a ubiquitous class of proteins important for metabolism, phenazines could potentially alter metabolic flux through a number of pathways. A quantitative prediction of this activity de novo is presently impossible. Even for the adventitious reduction of oxygen, the most studied side-reaction of flavoproteins, the parameters that govern reaction rates (which differ by at least 6 orders of magnitude) are still unclear (59). Mirroring this uncertainty, our prediction that sterics are the rate-limiting factor for phenazine reduction by LpdG did not hold up to site-directed mutagenesis (Fig. 8), suggesting that even for the restricted active site of LpdG, the enzyme-specific particulars of electronic structure and transition state are important. Beyond enzyme kinetics, phenazine reduction varies by growth phase (7), and so we expect the overall effects of phenazines on metabolism to be heavily context-dependent. For example, within a ok;4heterogeneous and stratified biofilm community, different enzymes likely catalyze phenazine reduction under (hyp)oxic and anoxic conditions. The results of this study, indicating that oxidized PCA and pyocyanin can substitute for NAD+, allow us to postulate that under strictly anaerobic conditions, where NAD+ may be limited (7, 20), substitution of phenazines for NAD+ in enzymes like LpdG would facilitate redox homeostasis. This is in a manner distinct from what we have previously assumed (the coupling of NADH oxidation to phenazine reduction) but results in the same net effect, maintenance of a higher NAD+/NADH ratio. Such substitution would allow for flux through pathways that would otherwise not proceed, including, but not limited to, energy-generation pathways such as pyruvate oxidation (20). A comprehensive understanding of phenazine metabolism in vivo will require a synthesis of biochemical and genetic approaches, yet this study helps focus our attention on potentially important enzymatic catalysts and pathways.
Experimental Procedures
Materials
The reagents used for growth media and protein purification were from Sigma or Acros Organics and were of ACS grade or better. Pyocyanin was from Cayman Chemicals. Phenazine-1-carboxylic acid and phenazine-1-carboxamide were from Princeton Biosciences. Phenazine-1-hydroxide was from TCI America. NAD+, NADH, thiamine pyrophosphate, coenzyme A, MgCl2, lipoamide, potassium cyanide, diphenyleneiodonium chloride, and sodium (meta)arsenite were from Sigma. Dihydrolipoamide was synthesized from lipoamide by reduction with sodium borohydride (60) and stored desiccated at −20 °C. For crystallography, PEG-3350 (50% solution) was from Hampton Research, and DMSO, HEPES, KOH, and KCl were from Sigma. Nickel-Sepharose and Sephacryl S-400 were from GE Healthcare. CHT hydroxyapatite was from Bio-Rad. Thrombin was from EMD Millipore. Other chemicals were from Sigma unless stated otherwise. All phosphate buffers were prepared with KH2PO4 and adjusted to the appropriate pH with KOH at room temperature. Anaerobic buffers were prepared by autoclaving and transferring the still-warm solution into an anaerobic chamber; other anaerobic solutions were prepared by dissolving solid compounds with anaerobic buffers within the anaerobic chamber.
Growth Conditions
For routine culturing, P. aeruginosa and Escherichia coli were grown in lysogeny broth containing 10 g/liter tryptone, 5 g/liter yeast extract, and 10 g/liter NaCl. Solid agar plates contained 15 g/liter agar. The succinate minimal medium for P. aeruginosa contained 14.15 mm KH2PO4, 35.85 mm K2HPO4, 42.8 mm NaCl, 9.3 mm NH4Cl, 40 mm disodium succinate, 1 mm MgSO4, 7.5 μm FeCl2·4H2O, 0.5 μm ZnCl2, 0.5 μm MnCl2·4H2O, 0.1 μm H3BO3, 0.8 μm CoCl2·6H2O, 0.01 μm CuCl2·2H2O, 0.1 μm NiCl2·6H2O, and 0.15 μm Na2MoO4. The minimal medium was sterilized by autoclaving; to avoid precipitation, the metals were prepared as a separate solution (1000×) and added immediately prior to inoculation. All liquid cultures were incubated in a New Brunswick Innova 44R incubator at 37 °C shaking at 250 rpm (2.54-cm stroke length).
PCA Reduction by Cell Lysate
Wild type P. aeruginosa PA14 was incubated in 250-ml Erlenmeyer flasks containing 50 ml of succinate minimal medium and grown to an A500 of 2.4–2.8 (∼11 h from a starting A500 of 0.01). The cells were harvested by centrifugation for 15 min at 6800 × g, washed twice with 1 ml of 50 mm KH2PO4, pH 7.5, and resuspended to a final volume of 1 ml with 50 mm KH2PO4, pH 7.5, and 1 mm phenylmethylsulfonyl fluoride (PMSF). The cells were lysed by sonication on ice using a Thermo Fisher Scientific Sonic Dismembrator 550 set to power level 3 with a pulse setting of 1 s on, 4 s off for 3 min total sonication time. Large debris and unlysed cells were removed by centrifugation for 15 min at 8000 × g. Where applicable, membrane-associated proteins were separated by ultracentrifugation using a Beckman Optima ultracentrifuge with a TLA-100.3 fixed-angle rotor. The lysate was centrifuged for 1 h at 70,000 rpm (208,000 × g average), and the supernatant was saved. The pellet was resuspended by gently pipetting an equivalent volume of 50 mm KH2PO4, pH 7.5, with 1% n-dodecyl β-d-maltoside and 1 mm PMSF. Undissolved material was removed by centrifugation for 15 min at 8000 × g. Protein concentration was measured using the bicinchoninic acid assay (Pierce) (61) calibrated with BSA standards (Pierce). Boiled lysate was prepared by placing an aliquot into a boiling water bath for 5 min; the precipitated material was removed by centrifugation at 17,000 × g for 5 min.
PCA reduction assays were performed inside an anaerobic glove box (Coy) with an atmosphere of 3–4% H2 and 97–96% N2. Reactions were performed at 28 °C in a 96-well plate with each well containing 50 μl of lysate, 50 μl of electron donor (dissolved in 50 mm KH2PO4, pH 7.5), and 50 μl of buffer (50 mm KH2PO4, pH 7.5) or boiled lysate. Where applicable, inhibitors were added using 2 μl of a 100× stock solution (KCN and NaAsO2 were dissolved in water; diphenyleneiodonium chloride was dissolved in DMSO). The reaction was initiated by adding 50 μl of PCA (1 mm stock in 50 mm KH2PO4, pH 7.5; 250 μm final concentration). The reaction was monitored by measuring absorbance at 440 nm using a BioTek Synergy 4 plate reader housed within the anaerobic glove box. The reaction progress was normalized against four averaged control wells containing 250 μm PCA and 10 mm sodium dithionite as a measure of 100% PCA reduction.
Pyruvate and α-Ketoglutarate Dehydrogenase Purification
The pyruvate and α-ketoglutarate dehydrogenase complexes were prepared natively from P. aeruginosa PA14 Δphz1/2 (34). This phenazine-null strain was used to minimize the generation of reactive oxygen species during the initial stages of purification. Cells were grown in 1 liter of the succinate minimal medium (with 80 mm disodium succinate to achieve higher cell density) in a 2.8-liter baffled Fernbach flask to an A500 of 4–6. In a typical preparation, 6 liters of cells were harvested by centrifugation for 15 min at 8000 × g. The cells were resuspended in 200 ml of 50 mm KH2PO4, adjusted to pH 7.5 with KOH, with 1 mm EDTA, and a cOmplete ULTRA protease inhibitor tablet (Roche Applied Science). The cells were lysed using 3 or 4 passes through an Avestin Emulsiflex-C3 operating at 20,000 p.s.i. The lysate was centrifuged for 30 min at 30,000 × g, and the pellet was discarded. In subsequent steps, the activity of the complexes was followed using a reaction mix containing 50 mm KH2PO4, pH 7.5, 1 mm MgCl2, 100 μm TPP, 100 μm CoA, 500 μm NAD+, and 2.5 mm pyruvate or α-ketoglutarate. Activity was indicated by an absorbance increase over time at 340 nm after adding 1–5 μl of lysate or protein to 199–195 μl of the reaction mix.
After adjusting the lysate dropwise with 5% acetic acid to pH 6.0 on ice, 0.17 volumes of 2% protamine sulfate were added, and the mixture was gently stirred on ice for 30 min. The mixture was centrifuged for 30 min at 30,000 × g, and the pellet was discarded. The lysate was further clarified by adding 0.2 volumes of ice-cold acetone, stirring the mixture on ice for 15 min, centrifuging for 15 min at 30,000 × g, and discarding the pellet. The dehydrogenase complexes were then precipitated by slowly adding an additional 0.4 volumes of ice-cold acetone. The mixture was gently stirred for 30 min on ice and centrifuged for 30 min at 30,000 × g. The supernatant was discarded.
The precipitated proteins were dissolved in 50 ml of ice-cold 100 mm KH2PO4, pH 7.5, with 0.5 mm PMSF. Undissolved material was removed by centrifugation for 15 min at 15,000 × g. The supernatant was applied to a CHT ceramic hydroxyapatite (type I, 40 μm particle size) column (dimensions 2.2 × 16.5 cm) at a flow rate of 2.0 ml/min using an Äkta Express system at room temperature. The column was washed with 2 column volumes of 100 mm KH2PO4, pH 7.5. The complexes were eluted at a flow rate of 5 ml/min using a linear gradient over 2 column volumes to 500 mm KH2PO4, pH 7.5, with 2-ml fraction collection. Fractions were assayed for pyruvate and α-ketoglutarate dehydrogenase activity and pooled accordingly. The complexes were concentrated using an Amicon ultra-centrifugal filter (30-kDa cutoff) to a volume of less than 1 ml. The complexes were further purified using gel filtration over a Sephacryl S-400 column (dimensions 1.6 × 38.0 cm) run isocratically with 100 mm KH2PO4, pH 7.5, and 1 mm EDTA at 0.5 ml/min with an Äkta Purifier system at 4 °C. Fractions containing pyruvate or α-ketoglutarate dehydrogenase activity were again pooled and concentrated using an Amicon ultra-centrifugal filter. Purity was assessed using an SDS-polyacrylamide gel stained with Coomassie Blue.
PCA and Pyocyanin Reduction by Purified Proteins
Phenazine reduction by purified proteins was measured using a Thermo Fisher Scientific Evolution 260 Bio-spectrophotometer housed within an anaerobic chamber. PCA reduction was followed at 440 nm at 5 Hz with an integration time of 0.2 s. Pyocyanin reduction was followed at 690 nm at 5 or 20 Hz with an integration time of 0.2 or 0.05 s, respectively. The extinction coefficients used were ϵ440 = 2206 m−1 cm−1 for reduced PCA, ϵ440 = 35.4 m−1 cm−1 for oxidized PCA, ϵ690 = 4306 m−1 cm−1 for oxidized pyocyanin, and ϵ690 = 0 m−1 cm−1 for reduced pyocyanin. PCA was dissolved to a stock concentration of 20 mm in 300 mm KH2PO4, pH 7.5. Pyocyanin was dissolved to a stock concentration of 10 mm in 20 mm HCl. All reactions were performed at 30 °C. The limit of detection was ∼25 μm, corresponding to a change in absorbance of ∼0.01 before the rate became non-linear.
The pyruvate and α-ketoglutarate dehydrogenase complexes were assayed using 0.5–0.9 μm LpdG (as measured by flavin content) in 50 mm KH2PO4, pH 7.5, 1 mm EDTA, 3 mm MgCl2, 200 μm TPP, 200 μm CoA, and 2.5 mm pyruvate or α-ketoglutarate. Where applicable, inhibitors were added from a 100×stock solution. The reaction was initiated by adding an equal volume of a 2× solution of phenazine.
DLDH was assayed using 2 μm protein (as measured by flavin content) in phosphate buffer, pH 7.5, with 1 mm EDTA and 1 mm NADH or 1 mm dihydrolipoamide (dissolved in 50% isopropyl alcohol as a 100 mm stock solution). Because the enzymes required different ionic strengths for stability, LpdG and Lpd3 were assayed using 300 mm KH2PO4, and LpdV was assayed using 50 mm KH2PO4. For PCA, the reaction was initiated by adding an equal volume of 2× PCA; for pyocyanin, the reaction was initiated by adding an equal volume of 2× NADH or dihydrolipoamide.
Fluorescence Quenching
The fluorescence of LpdG was measured in a black half-area microtiter plate using a BioTek Synergy 4 plate reader. Each well contained 100 μl of 2 μm protein in 300 mm KH2PO4, pH 7.5, with the appropriate concentration of ligand. Fluorescence was measured using a tungsten lamp with a 485 ± 10-nm excitation filter and a 528 ± 10-nm emission filter. The background fluorescence of a control well, without protein but with the same ligand concentration, was subtracted from the protein fluorescence.
Curve Fitting
Non-linear curve fitting was performed using the curve_fit function from SciPy's optimization package. Fluorescence quenching data were fit with the formula F = ([S]N/([S]N + KdN)) × (Fs − Fo) + Fo, where F is the observed fluorescence; [S] is the substrate concentration; N is the measure of cooperativity (Hill coefficient); Kd is the dissociation constant; Fs is the fluorescence at a saturating substrate concentration; and Fo is the fluorescence in the absence of substrate. Kinetic data were fit with the equation R = Vmax·[S]/(Km + [S]), where R is the rate of reaction; Vmax is the maximum reaction velocity; [S] is the substrate concentration; and Km is the substrate concentration at half of Vmax.
Cloning
Heterologous expression vectors for LpdG, Lpd3, and LpdV were prepared similarly as follows. The full ORFs were amplified with PCR from P. aeruginosa PA14 genomic DNA. The forward and reverse primers contained an additional stop codon and restriction sites for cloning into the Novagen pET-15b expression vector (Millipore, Sigma). For LpdG, the forward primer was AGAGAGcatatgAGCCAGAAATTCGACGTG, and the reverse primer was ACACACggatccTTATTATCAGCGCTTCTTGCGGTT. For Lpd3, the forward primer was AGAGAGcatatgATGGAAAGCTATGACGTGATCGTG, and the reverse primer was ACACACggatccTTATTATCAGTTCTGCATGGCCCG. For LpdV, the forward primer was AGAGAGcatatgAGCCAGATCCTGAAGACTTC, and the reverse primer was ACACACctcgagTTATTATCAGATGTGCAGGGCGTG. The products for LpdG and Lpd3, and the pET15b vector, were digested with NdeI and BamHI. The product for LpdV and the pET15b vector were digested with NdeI and XhoI. The digests were purified using the Qiagen PCR purification kit, and the resulting ORF and plasmid products were ligated together with T4 DNA ligase. The ligated product was transformed into E. coli Top10 (Invitrogen) and selected for on LB agar plates with 50 μg/ml carbenicillin. Individual colonies were culture, and the constructed plasmid was isolated using a Qiagen miniprep kit and sequenced (Retrogen) to verify that no mutations were introduced. The plasmid was then transformed into E. coli BL21(DE3) (62) for protein expression.
Site-directed mutants of LpdG were created by PCR amplification of the LpdG-pET15b vector. The wild type vector was amplified using primers that contained appropriate mismatches. The resulting PCR product was phosphorylated using T4 polynucleotide kinase, blunt-end ligated using T4 DNA ligase, and transformed into E. coli Top10 and BL21 as described above. For LpdG I192G, the primers were GGCCTGGAGCTGGGTTC and GCCGACGCCGGCGCCGAT. For LpdG V191Y, the primers were ATTGGCCTGGAGCTGGGT and ATAGCCGGCGCCGATCACAC.
Dihydrolipoamide Dehydrogenase Purification
His6-tagged DLDH homologs and mutants were purified using the same procedure. E. coli BL21 harboring the expression plasmid (see under “Cloning”) was cultured at 37 °C to an A600 of ∼0.8 in terrific broth containing 12 g/liter tryptone, 24 g/liter yeast extract, 4 ml/liter glycerol, 17 mm KH2PO4, 72 mm K2HPO4, 8 mm MgSO4, and 100 μg/ml ampicillin. A typical purification used 1 liter of cells grown in a 2.8-liter baffled Fernbach flask. All steps after culturing were performed at 4 °C. The cells were harvested by centrifugation for 15 min at 5000 × g. The pellet was resuspended in 2.5 volumes of 50 mm KH2PO4, pH 7.5, 200 mm KCl, 50 mm imidazole, and 1 mm PMSF. The cells were lysed using 3–4 passes through an Avestin Emulsiflex-C3 operating at 15,000 p.s.i., and the lysate was clarified by centrifugation for 30 min at 30,000 × g. The supernatant was applied to a 5-ml HisTrap HP column (GE Healthcare) with an Äkta Purifier system running at 3 ml/min. The column was washed with 5 column volumes of 50 mm KH2PO4, pH 7.5, 200 mm KCl, and 50 mm imidazole, and proteins were eluted using a linear gradient over 5 column volumes to 50 mm KH2PO4, pH 7.5, 200 mm KCl, and 500 mm imidazole. The yellow fractions were pooled, concentrated using an Amicon ultra-centrifugal filter (30 kDa) to a volume of less than 3 ml, and desalted using a HiPrep 26/10 desalting column (GE Healthcare) into a buffer containing 300 mm KH2PO4, pH 7.5. The yellow fractions were again pooled and stored at 4 °C. All of the preparations were at least 95% pure as judged by Coomassie Blue staining of an SDS-polyacrylamide gel.
For crystallography, LpdG was purified as described above except that protein was purified from 6 liters of cell culture using a 20-ml HisPrep FF 16/10 column (GE Healthcare). Prior to desalting, the protein was incubated overnight at 4 °C with 50 units of thrombin (EMD Millipore). The protein was concentrated and desalted into 50 mm KH2PO4, pH 7.5, 500 mm KCl, and 20 mm imidazole. Uncleaved protein, and other contaminants that bind to the HisPrep column, were removed by passing the protein back through the HisPrep column and collecting the flow-through. The flow-through was applied to a CHT ceramic hydroxyapatite (type I, 40 μm particle size) column (dimensions 2.2 × 16.5 cm) at a flow rate of 5.0 ml/min. The column was washed with 2 column volumes of 100 mm KH2PO4, pH 7.5, and highly pure LpdG was eluted using a linear gradient over 2 column volumes to 500 mm KH2PO4, pH 7.5. The yellow fractions were pooled, concentrated, and desalted into 25 mm HEPES, pH 7.5, with 600 mm KCl, and stored at 4 °C.
Flavoprotein concentrations were determined spectrophotometically. An aliquot of protein was first boiled in the dark for 5 min. The precipitated protein was removed by centrifugation, and the concentration of liberated flavin was determined using the FAD extinction coefficient of ϵ450 = 11,300 m−1 cm−1.
Proteomics
In-gel digests were performed as described previously (63), and LC-MS/MS analysis was performed at the Caltech Proteome Exploration Laboratory (64).
Crystallography
Initial crystal screens were performed using vapor diffusion in sitting drop plates. The five screens used were Crystal Screen HT (Hampton), Index HT (Hampton), PEGRx HT (Hampton), JCSG-plus HT-96 (Molecular Dimensions), and Wizard Classic 1 and 2 (Rigaku). An Art Robins Gryphon nano-liquid-handling robot was used to mix 0.2 μl of screen solution with 0.2 μl of protein solution (30 mg/ml in 25 mm HEPES, pH 7.5, with 600 mm KCl). The plates were sealed with transparent film and incubated at 20 °C. The plates were monitored every several days for crystal growth using a stereoscopic microscope. For each well that formed crystals, a secondary screen was performed using similar conditions with a wider concentration of salt, pH, and PEG.
The optimized crystals were grown in a hanging-drop format using Hampton VDX plates with sealant and siliconized glass cover slides. Drops were created by mixing 1.5 μl of mother liquor with 2 μl of protein solution and 0.5 μl of seed solution. The mother liquor contained 500 μl of 22% PEG-3350 with 530 mm HEPES, pH 6.1. The protein solution contained 6 mg/ml LpdG (as measured by the BCA assay) in 400 mm HEPES, pH 6.1, with 100 mm KCl. The seed solution contained several crystals, which were crushed by vortexing with a glass bead and diluted ∼1000-fold, in 22% PEG-3350 with 10 mm HEPES, pH 6.5. The pH 6.1 HEPES buffers were prepared using a stock of 1 m HEPES with 50 mm MES, and so the final drop solution contained 11% PEG-3350, 400 mm HEPES, 50 mm KCl, and 23 mm MES. The initial seed crystal formed under similar conditions at pH 7.5 and was propagated to the lower pH through several rounds of crystallization and seeding. Crystals formed as yellow cubes or bricks and were apparent after 5 h at pH 6.5–7.0 or overnight at pH 6.1. The best diffracting crystals were grown for 24–48 h and were 50–100 μm in each dimension.
Ligand soaking and cryoprotection were performed delicately because the crystals were highly susceptible to cracking from osmotic strength changes. Crystals were first transferred to 8 μl of mother liquor on a coverslip. Serial dilutions (2-fold) were created of the soaking or cryo solution with the mother liquor, and the crystals were slowly acclimated by mixing in 0.5–1 μl of new solution with a pipette tip. The same volume was removed from the drop and iteratively replaced until Schlieren lines were no longer visible, and the process was repeated using a higher concentration of ligand or cryoprotectant. Typically, batches of 10 crystals were acclimated at a time over 30–45 min. The final cryoprotection solution contained 50 mm HEPES, pH 7.0, 20% PEG-3350, 20% DMSO, and the final desired ligand concentration. Crystals were flash-frozen by plunging into liquid nitrogen.
Data collection was performed at the Stanford Synchrotron Radiation Lightsource, beamline 12-2. Diffraction images were collected every 0.15° for a full 360° around a single axis (2400 images total) using X-rays at 12,658 eV. Data completion was frequently low (<90%) because of the low symmetry in P21 and the dead spaces between detector tiles of the Pilatus 6 M, and so a second dataset was collected for each crystal utilizing a detector offset of 10 mm in the X and Y directions. For well diffracting crystals with minimal radiation damage, this process was repeated with X-rays at 6900 eV to capture the anomalous signal from sulfur. The datasets were integrated with XDS (65), scaled together with XSCALE, and then processed with POINTLESS (66), merged with AIMLESS (67), and converted to structure factors with CTRUNCATE in the CCP4 suite (68). The structure was solved by molecular replacement with Phaser (69) using a search model consisting of chain A from the structure 1LPF in the Protein Data Bank. The structure was refined using phenix.refine (70), and ligands (FAD and NADH) were placed using phenix.ligandfit (71). For the structure containing NAD+, a fragment of the NAD+ ligand was placed manually in Coot (72) because electron density was not apparent for the full ligand. Side chains were trimmed in Coot where there was undefined electron density. Further refinement was performed with phenix.refine and iterative model building in Coot to the resolution that provided an arbitrary signal-to-noise cutoff of 2.0. Paired refinement (49) was then used in 0.05–0.08 Å increments to establish the resolution limit of useful data. Riding hydrogens considerably improved Rfree and overall geometry and so they were included in all models. Compared with a simpler TLS model of anisotropy, a fully anisotropic model improved Rfree and overall geometry for the apo- and NAD+ structures, and so these models were refined using anisotropic B-factors. TLS parameters for the lower resolution NADH structure were determined and refined with PHENIX. A large number of DMSO molecules, originating from the cryoprotectant, were placed manually in Coot with guidance from the low-energy anomalous difference map. The high resolution apo-structure suggested deviations from planarity in the flavin ring of FAD, and so the final rounds of refinement in the apo- and NAD+ structures were performed using relaxed planarity restraints for FAD.
Author Contributions
N. R. G. and D. K. N. designed the experiments and wrote the manuscript. N. R.G. collected the data and refined the protein structures. B. X. W. created the protein expression vectors, assisted with protein purification, and performed preliminary enzyme kinetics. B. X. W. and J. A. H. performed the initial crystallography screens and optimized crystal growth conditions. J. A. H. discovered the high resolution crystal form of LpdG and provided crystallography training. N. R. G. further optimized crystallization and cryoprotection. D. K. N. coordinated the project. All authors reviewed the manuscript.
Acknowledgments
We thank Dr. Doug Rees and the Caltech Molecular Observatory for providing essential guidance, training, and materials for X-ray crystallography; the Caltech Proteome Exploration Laboratory for its services; and the SSRL staff. Members of the Newman laboratory (past and present) provided constructive feedback on the manuscript.
This work was supported in part by Howard Hughes Medical Institute, National Institutes of Health Grant 5R01HL117328-03, National Science Foundation Grant 1144469, and the Amgen Scholars Program. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5U8U, 5U8V, and 5U8W) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PCA
- phenazine-1-carboxylic acid
- TPP
- thiamine pyrophosphate
- DLDH
- dihydrolipoamide dehydrogenase.
References
- 1. Gessard C. (1882) De la Pyocyanine et de Son Microbe. Colorations Qui en Dépendant de les Liquides Organiques (pus et Sérosités, Suer, Liquides des Culture). Doctoral thesis, Faculté de Médicine de Paris, Paris, France [Google Scholar]
- 2. Elema B. (1931) Studies on the oxidation-reduction of pyocyanine: Part II. Redox potentials of pyocyanine. Recueil des Travaux Chimiques des Pays-Bas 50, 807–826 [Google Scholar]
- 3. Friedheim E., and Michaelis L. (1931) Potentiometric study of pyocyanine. J. Biol. Chem. 91, 355–368 [Google Scholar]
- 4. Michaelis L. (1931) The formation of semiquinones as intermediary reduction products from pyocyanine and some other dyestuffs. J. Biol. Chem. 92, 211–232 [Google Scholar]
- 5. Friedheim E. (1931) Pyocyanine, an accessory respiratory enzyme. J. Exp. Med. 54, 207–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedheim E. (1934) The effect of pyocyanine on the respiration of some normal tissues and tumours. Biochem. J. 28, 173–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Price-Whelan A., Dietrich L. E., and Newman D. K. (2007) Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J. Bacteriol. 189, 6372–6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dietrich L. E., Okegbe C., Price-Whelan A., Sakhtah H., Hunter R. C., and Newman D. K. (2013) Bacterial community morphogenesis is intimately linked to the intracellular redox state. J. Bacteriol. 195, 1371–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abken H.-J., Tietze M., Brodersen J., Bäumer S., Beifuss U., and Deppenmeier U. (1998) Isolation and characterization of methanophenazine and function of phenazines in membrane-bound electron transport of Methanosarcina mazei Gö1. J. Bacteriol. 180, 2027–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hassan H. M., and Fridovich I. (1979) Intracellular production of superoxide radical and of hydrogen peroxide by redox active compounds. Arch. Biochem. Biophys. 196, 385–395 [DOI] [PubMed] [Google Scholar]
- 11. Gu M., and Imlay J. (2011) The SoxRS response of Escherichia coli is directly activated by redox-cycling drugs rather than by superoxide. Mol. Microbiol. 79, 1136–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Armstrong A. V., and Stewart-Tull D. E. (1971) The site of the activity of extracellular products of Pseudomonas aeruginosa in the electron-transport chain in mammalian cell respiration. J. Med. Microbiol. 4, 263–270 [DOI] [PubMed] [Google Scholar]
- 13. Morales D. K., Jacobs N. J., Rajamani S., Krishnamurthy M., Cubillos-Ruiz J. R., and Hogan D. A. (2010) Antifungal mechanisms by which a novel Pseudomonas aeruginosa phenazine toxin kills Candida albicans in biofilms. Mol. Microbiol. 78, 1379–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hollstein U., and Van Gemert R. J. Jr. (1971) Interaction of phenazines with polydeoxyribonucleotides. Biochemistry 10, 497–504 [DOI] [PubMed] [Google Scholar]
- 15. Cox C. D. (1986) Role of pyocyanin in the acquisition of iron from transferrin. Infect. Immun. 52, 263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y., and Newman D. K. (2008) Redox reactions of phenazine antibiotics with ferric (hydr)oxides and molecular oxygen. Environ. Sci. Technol. 42, 2380–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hernandez M. E., and Newman D. K. (2001) Extracellular electron transfer. Cell. Mol. Life Sci. 58, 1562–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Price-Whelan A., Dietrich L. E., and Newman D. K. (2006) Rethinking 'secondary' metabolism: physiological roles for phenazine antibiotics. Nat. Chem. Biol. 2, 71–78 [DOI] [PubMed] [Google Scholar]
- 19. Wang Y., Kern S. E., and Newman D. K. (2010) Endogenous phenazine antibiotics promote anaerobic survival of Pseudomonas aeruginosa via extracellular electron transfer. J. Bacteriol. 192, 365–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glasser N. R., Kern S. E., and Newman D. K. (2014) Phenazine redox cycling enhances anaerobic survival in Pseudomonas aeruginosa by facilitating generation of ATP and a proton-motive force. Mol. Microbiol. 92, 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mavrodi D. V., Ksenzenko V. N., Bonsall R. F., Cook R. J., Boronin A. M., and Thomashow L. S. (1998) A seven-gene locus for synthesis of phenazine-1-carboxylic acid by Pseudomonas fluorescens 2-79. J. Bacteriol. 180, 2541–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McDonald M., Mavrodi D. V., Thomashow L. S., and Floss H. G. (2001) Phenazine biosynthesis in Pseudomonas fluorescens: branchpoint from the primary shikimate biosynthetic pathway and role of phenazine-1,6-dicarboxylic acid. J. Am. Chem. Soc. 123, 9459–9460 [DOI] [PubMed] [Google Scholar]
- 23. Mavrodi D. V., Bonsall R. F., Delaney S. M., Soule M. J., Phillips G., and Thomashow L. S. (2001) Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J. Bacteriol. 183, 6454–6465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holliman F. G. (1969) Pigments of Pseudomonas species: Part I. Structure and synthesis of aeruginosin A. J. Chem. Soc. Perkin 1 18, 2514–2516 [DOI] [PubMed] [Google Scholar]
- 25. Herbert R. B., and Holliman F. G. (1969) Pigments of Pseudomonas species: Part II. Structure of aeruginosin B. J. Chem. Soc. Perkin 1 18, 2517–2520 [DOI] [PubMed] [Google Scholar]
- 26. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 27. Cezairliyan B., Vinayavekhin N., Grenfell-Lee D., Yuen G. J., Saghatelian A., and Ausubel F. M. (2013) Identification of Pseudomonas aeruginosa phenazines that kill Caenorhabditis elegans. PLoS Pathog. 9, e1003101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kasozi D. M., Gromer S., Adler H., Zocher K., Rahlfs S., Wittlin S., Fritz-Wolf K., Schirmer R. H., and Becker K. (2011) The bacterial redox signaller pyocyanin as an antiplasmodial agent: comparisons with its thioanalog methylene blue. Redox Rep. 16, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Price-Whelan A. M. (2009) Physiology and Mechanisms of Pyocyanin Reduction in Pseudomonas aeruginosa. Ph.D. thesis, California Institute of Technology, Pasadena [Google Scholar]
- 30. Kern S. E. (2013) Consequences of Redox-active Phenazines on the Physiology of the Opportunistic Pathogen Pseudomonas aeruginosa. Ph.D. thesis, Massachusetts Institute of Technology [Google Scholar]
- 31. Sullivan N. L., Tzeranis D. S., Wang Y., So P. T., and Newman D. (2011) Quantifying the dynamics of bacterial secondary metabolites by spectral multiphoton microscopy. ACS Chem. Biol. 6, 893–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Masip L., Veeravalli K., and Georgiou G. (2006) The many faces of glutathione in bacteria. Antioxid. Redox Signal. 8, 753–762 [DOI] [PubMed] [Google Scholar]
- 33. Huang J., Sonnleitner E., Ren B., Xu Y., and Haas D. (2012) Catabolite repression control of pyocyanin biosynthesis at an intersection of primary and secondary metabolism in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 78, 5016–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dietrich L. E., Price-Whelan A., Petersen A., Whiteley M., and Newman D. K. (2006) The phenazine pyocyanin is a terminal signalling factor in the quorum sensing network of Pseudomonas aeruginosa. Mol. Microbiol. 61, 1308–1321 [DOI] [PubMed] [Google Scholar]
- 35. Williams H. D., Zlosnik J. E., and Ryall B. (2007) Oxygen, cyanide and energy generation in the cystic fibrosis pathogen Pseudomonas aeruginosa. Adv. Microb. Physiol. 52, 1–71 [DOI] [PubMed] [Google Scholar]
- 36. O'Donnell B. V., Tew D. G., Jones O. T., and England P. J. (1993) Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem. J. 290, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carter D. E., Aposhian H. V., and Gandolfi A. J. (2003) The metabolism of inorganic arsenic oxides, gallium arsenide, and arsine: a toxicochemical review. Toxicol. Appl. Pharmacol. 193, 309–334 [DOI] [PubMed] [Google Scholar]
- 38. Reed L. J. (1974) Multienzyme complexes. Acc. Chem. Res. 7, 40–46 [Google Scholar]
- 39. de Kok A., Hengeveld A. F., Martin A., and Westphal A. H. (1998) The pyruvate dehydrogenase multi-enzyme complex from Gram-negative bacteria. Biochim. Biophys. Acta 1385, 353–366 [DOI] [PubMed] [Google Scholar]
- 40. Gruys K. J., Datta A., and Frey P. A. (1989) 2-Acetylthiamin pyrophosphate (acetyl-TPP) pH-rate profile for hydrolysis of acetyl-TPP and isolation of acetyl-TPP as a transient species in pyruvate dehydrogenase catalyzed reactions. Biochemistry 28, 9071–9080 [DOI] [PubMed] [Google Scholar]
- 41. Corran H. S., Green D. E., and Straub F. B. (1939) On the catalytic function of heart flavoprotein. Biochem. J. 33, 793–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee D. G., Urbach J. M., Wu G., Liberati N. T., Feinbaum R. L., Miyata S., Diggins L. T., He J., Saucier M., Déziel E., Friedman L., Li L., Grills G., Montgomery K., Kucherlapati R., Rahme L. G., and Ausubel F. M. (2006) Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 7, R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Winsor G. L., Griffiths E. J., Lo R., Dhillon B. K., Shay J. A., and Brinkman F. S. (2016) Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 44, D646–D653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jeyaseelan K., Guest J. R., and Visser J. (1980) The pyruvate dehydrogenase complex of Pseudomonas aeruginosa PAO. Purification, properties and characterization of mutants. J. Gen. Microbiol. 120, 393–402 [DOI] [PubMed] [Google Scholar]
- 45. Burns G., Sykes P. J., Hatter K., and Sokatch J. R. (1989) Isolation of a third lipoamide dehydrogenase from Pseudomonas putida. J. Bacteriol. 171, 665–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sahlman L., and Williams C. H. Jr. (1989) Lipoamide dehydrogenase from Escherichia coli. Steady-state kinetics of the physiological reaction. J. Biol. Chem. 264, 8039–8045 [PubMed] [Google Scholar]
- 47. Mattevi A., Obmolova G., Kalk K. H., van Berkel W. J., and Hol W. G. (1993) Three-dimensional structure of lipoamide dehydrogenase from Pseudomonas fluorescens at 2.8 Å resolution: Analysis of redox and thermostability properties. J. Mol. Biol. 230, 1200–1215 [DOI] [PubMed] [Google Scholar]
- 48. Mattevi A., Obmolova G., Sokatch J. R., Betzel C., and Hol W. G. (1992) The refined crystal structure of Pseudomonas putida lipoamide dehydrogenase complexed with NAD+ at 2.45 Å resolution. Proteins 13, 336–351 [DOI] [PubMed] [Google Scholar]
- 49. Karplus P. A., and Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brautigam C. A., Chuang J. L., Tomchick D. R., Machius M., and Chuang D. T. (2005) Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J. Mol. Biol. 350, 543–552 [DOI] [PubMed] [Google Scholar]
- 51. Berkholz D. S., Faber H. R., Savvides S. N., and Karplus P. A. (2008) Catalytic cycle of human glutathione reductase near 1 Å resolution. J. Mol. Biol. 382, 371–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Eschbach M., Schreiber K., Trunk K., Buer J., Jahn D., and Schobert M. (2004) Long-term anaerobic survival of the opportunistic pathogen Pseudomonas aeruginosa via pyruvate fermentation. J. Bacteriol. 186, 4596–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hunter R. C., Asfour F., Dingemans J., Osuna B. L., Samad T., Malfroot A., Cornelis P., and Newman D. K. (2013) Ferrous iron is a significant component of bioavailable iron in cystic fibrosis airways. MBio 4, e00557–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Williamson K. S., Richards L. A., Perez-Osorio A. C., Pitts B., McInnerney K., Stewart P. S., and Franklin M. J. (2012) Heterogeneity in Pseudomonas aeruginosa biofilms includes expression of ribosome hibernation factors in the antibiotic-tolerant subpopulation and hypoxia-induced stress response in the metabolically active population. J. Bacteriol. 194, 2062–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Y., Wilks J. C., Danhorn T., Ramos I., Croal L., and Newman D. K. (2011) Phenazine-1-carboxylic acid promotes bacterial biofilm development via ferrous iron acquisition. J. Bacteriol. 193, 3606–3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Das T., Ibugo A. I., Klare W., and Manefield M. (2016) in Microbial Biofilms–Importance and Applications (Dhanasekaran D., and Thajuddin N., eds) pp. 23–42, InTech [Google Scholar]
- 57. Ramos I., Dietrich L. E., Price-Whelan A., and Newman D. K. (2010) Phenazines affect biofilm formation by Pseudomonas aeruginosa in similar ways at various scales. Res. Microbiol. 161, 187–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Petrova O. E., Schurr J. R., Schurr M. J., and Sauer K. (2012) Microcolony formation by the opportunistic pathogen Pseudomonas aeruginosa requires pyruvate and pyruvate fermentation. Mol. Microbiol. 86, 819–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Massey V. (1994) Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 269, 22459–22462 [PubMed] [Google Scholar]
- 60. Patel M. S., Vettakkorumakankav N. N., and Liu T.-C. (1995) Dihydrolipoamide dehydrogenase: activity assays. Methods Enzymol. 252, 186–195 [DOI] [PubMed] [Google Scholar]
- 61. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., and Klenk D. C. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 62. Studier F. W., and Moffatt B. A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130 [DOI] [PubMed] [Google Scholar]
- 63. Babin B. M., Bergkessel M., Sweredoski M. J., Moradian A., Hess S., Newman D. K., and Tirrell D. A. (2016) SutA is a bacterial transcription factor expressed during slow growth in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 113, E597–E605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kalli A., and Hess S. (2012) Effect of mass spectrometric parameters on peptide and protein identification rates for shotgun proteomic experiments on an LTQ-orbitrap mass analyzer. Proteomics 12, 21–31 [DOI] [PubMed] [Google Scholar]
- 65. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 67. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Terwilliger T. C., Klei H., Adams P. D., Moriarty N. W., and Cohn J. D. (2006) Automated ligand fitting by core-fragment fitting and extension into density. Acta Crystallogr. D Biol. Crystallogr. 62, 915–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]