

Abstract
Many family 4 cytochrome P450s play key roles in fatty acid hydroxylation at the terminal, or ω, carbon, but the mechanistic basis for this energetically disfavored regiostereochemistry has been less clear. A co-crystal structure of the rabbit family 4 enzyme CYP4B1 with its substrate octane reveals that the propensity for ω-hydroxylation is orchestrated by active-site sterics, partially mediated by an unusual heme-polypeptide ester bond.
Introduction
Cytochrome P450 enzymes perform a variety of chemically arduous tasks in biosynthesis, metabolism, and detoxification, mediated by a catalytic intermediate formed by the heme cofactor and refined by the unique characteristics of each polypeptide. Many cytochrome P450 enzymes in family 4 hydroxylate fatty acids specifically at the terminal, or ω, carbon. Such hydroxylation involves hydrogen abstraction, which is energetically much more favorable to do at the penultimate, or ω-1, position. Thus, biochemists have long wondered how these enzymes block the chemically more tractable ω-1 position and favor the ω position. Although biochemical evidence has provided some clues, the field has eagerly awaited structures that could help elucidate how this is accomplished. One member of this family, CYP4B1, hydroxylates C7-C10 fatty acids and alkanes and has yielded a new X-ray structure with octane (1) that now provides these answers.
One significant challenge in getting this structure, as with many membrane-bound cytochromes P450, is that the human CYP4B1 enzyme has proved recalcitrant to recombinant expression (2). To bypass this difficulty, the new report by Hsu et al. (1) used the rabbit homolog of CYP4B1, which had already been obtained in a functional form via partial truncation of the single, N-terminal transmembrane helix followed by liberation from the membrane with detergents (3). Hsu et al. (1) first confirmed that the truncated rabbit CYP4B1 binds octane with spectral changes and an affinity similar to that of the full-length enzyme. They then used conventional cryocrystallography and molecular replacement to solve the CYP4B1/octane structure.
The overall features of CYP4B1 mirror those of other cytochromes P450, but the 2.7 Å complex further revealed that the octane substrate approaches the heme iron—the site of the catalytic oxygen intermediate during turnover—end on (Fig. 1). The linear hydrocarbon is constrained to an extended conformation within a “slot-like” active site cavity that narrows near the heme so that the (ω-1)- hydrogens are more shielded from the heme than those ω-carbons, thus promoting the observed regioselectivity of the hydroxylation reaction. A key portion of the protein constraining this orientation is an ester bond between one of the heme methyls and the side chain of Glu-310. Formed autocatalytically, this type of interaction is unusual for cytochrome P450 enzymes. It is restricted to those in family 4 and associated with the capacity for ω-hydroxylation. This stabilizes the Glu-310 conformation such that side chain atoms help restrict substrate access to the heme. Consistent with this idea, an E310A mutation yields a 4-fold decrease in the regioselectivity of octane hydroxylation.
FIGURE 1.
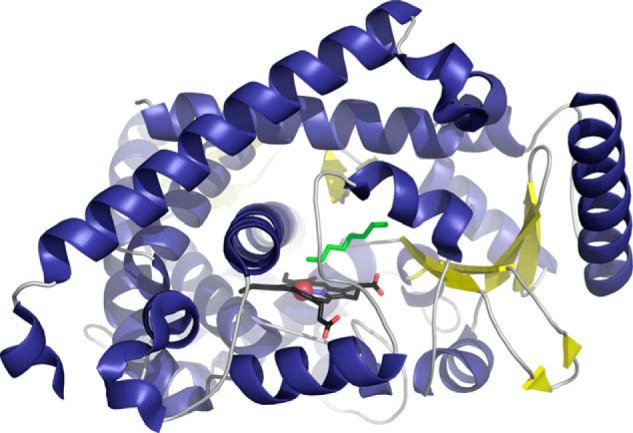
Cytochrome P450 4B1 (purple helices and yellow strands) orients octane (green sticks) such that the terminal atom is over the active site heme iron (black sticks and red sphere) to favor hydroxylation at this energetically less favored ω position.
While extrapolation from the rabbit enzyme to the human CYP4B1 (2) and to other CYP4 enzymes can be uncertain, the new structure provides opportunities to form testable hypotheses about the structure/function relationships in these enzymes. Many of the residues surrounding octane, including Glu-310, are highly conserved in human and other mammalian CYP4B1 enzymes. The authors point out, however, that Glu-310 is not conserved throughout human family 4—those CYP4 enzymes that are not ω-hydroxylases have either a Gly or Ala at this position, generally consistent with altered activity observed in tests of rabbit CYP4B1 mutants (1). In addition to identifying amino acids of interest in the active site, the authors used this new structure as a template for homology modeling of human CYP4A11, which performs ω-hydroxylation on fatty acids as long as C16. The authors' model is consistent with lauric acid binding in an orientation that would promote such regioselectivity and provides some indication as to how the longer chain might be accommodated.
Although the physiological necessity of CYP4B1 is uncertain—knock-out mice are phenotypically normal (4)—there are obvious translational motivations to understand the mechanism of this enzyme. First, in many species CYP4B1 first came to attention because it often selectively bioactivates protoxins to active toxins in the extrahepatic tissues in which it is expressed (2). In humans, CYP4B1 has been investigated for possible associations with bladder cancer (5). In fact, this toxin-activating activity has even been evaluated as a potential gene therapy, in which a CYP4B1 transgene is delivered to tumors to activate a systemic protoxin in a site-specific manner (6). However, a number of human hepatic P450 enzymes also have some propensity to activate this same protoxin, likely explaining the liver toxicity observed in cancer patients (7). The new structural information for CYP4B1 versus hepatic, drug-metabolizing P450 enzymes could support more selective prodrug development. Second, the CYP4B1 1-octanol product has desirable fuel characteristics similar to diesel and has been explored as a biofuel (8). Although mammalian membrane-bound P450 enzymes are typically unsuitable for biotechnology applications due to their poor stability and low turnover, the current structural information has the potential to broadly assist in advanced engineering of soluble P450 enzyme systems being studied for this purpose (9). Overall, however, the most satisfying aspect of the new CYP4B1/octane structure is likely the visualization of two of the most intriguing aspects of CYP4 enzymes—the propensity for energetically unfavorable ω-hydroxylation and an unusual heme-polypeptide ester bond.
Footnotes
The author declares that she has no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
References
- 1. Hsu M.-H., Baer B. R., Rettie A. E., and Johnson E. F. (2017) The crystal structure of cytochrome P450 4B1 (CYP4B1) monooxygenase complexed with octane discloses several structural adaptations for ω-hydroxylation. J. Biol. Chem. 292, 5610–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baer B. R., and Rettie A. E. (2006) CYP4B1: An enigmatic P450 at the interface between xenobiotic and endobiotic metabolism. Drug Metab. Rev. 38, 451–476 [DOI] [PubMed] [Google Scholar]
- 3. Cheesman M. J., Baer B. R., Zheng Y. M., Gillam E. M., and Rettie A. E. (2003) Rabbit CYP4B1 engineered for high-level expression in Escherichia coli: Ligand stabilization and processing of the N-terminus and heme prosthetic group. Arch. Biochem. Biophys. 416, 17–24 [DOI] [PubMed] [Google Scholar]
- 4. Parkinson O. T., Liggitt H. D., Rettie A. E., and Kelly E. J. (2013) Generation and characterization of a Cyp4b1 null mouse and the role of CYP4B1 in the activation and toxicity of ipomeanol. Toxicol. Sci. 134, 243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Imaoka S., Yoneda Y., Sugimoto T., Hiroi T., Yamamoto K., Nakatani T., and Funae Y. (2000) CYP4B1 is a possible risk factor for bladder cancer in humans. Biochem. Biophys. Res. Commun. 277, 776–780 [DOI] [PubMed] [Google Scholar]
- 6. Rainov N. G., Dobberstein K. U., Sena-Esteves M., Herrlinger U., Kramm C. M., Philpot R. M., Hilton J., Chiocca E. A., and Breakefield X. O. (1998) New prodrug activation gene therapy for cancer using cytochrome P450 4B1 and 2-aminoanthracene/4-ipomeanol. Hum. Gene Ther. 9, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 7. Rowinsky E. K., Noe D. A., Ettinger D. S., Christian M. C., Lubejko B. G., Fishman E. K., Sartorius S. E., Boyd M. R., and Donehower R. C. (1993) Phase I and pharmacological study of the pulmonary cytotoxin 4-ipomeanol on a single dose schedule in lung cancer patients: Hepatotoxicity is dose limiting in humans. Cancer Res. 53, 1794–1801 [PubMed] [Google Scholar]
- 8. Akhtar M. K., Dandapani H., Thiel K., and Jones P. R. (2015) Microbial production of 1- octanol: A naturally excreted biofuel with diesel-like properties. Metab. Eng. Commun. 2, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lonsdale T. H., Lauterbach L., Honda Malca S., Nestl B. M., Hauer B., and Lenz O. (2015) H2-driven biotransformation of n-octane to 1-octanol by a recombinant Pseudomonas putida strain co-synthesizing an O2-tolerant hydrogenase and a P450 monooxygenase. Chem. Commun. (Camb.) 51, 16173–16175 [DOI] [PubMed] [Google Scholar]