

Summary
The Drosophila hnRNP F/H homolog, Glorund (Glo), regulates nanos mRNA translation by interacting with a structured UA-rich motif in the nanos 3′ untranslated region. Glo regulates additional RNAs however, and mammalian homologs bind G-tract sequences to regulate alternative splicing, suggesting that Glo also recognizes G-tract RNA. To gain insight into how Glo recognizes both structured UA-rich and G-tract RNAs, we used mutational analysis guided by crystal structures of Glo’s RNA-binding domains and identified two discrete RNA-binding surfaces that allow Glo to recognize both RNA motifs. By engineering Glo variants that favor a single RNA-binding mode, we show that a subset of Glo’s functions in vivo is mediated solely by the G-tract binding mode, whereas regulation of nanos requires both recognition modes. Our findings suggest a molecular mechanism for the evolution of dual RNA motif recognition in Glo that may be applied to understanding the functional diversity of other RNA-binding proteins.
Keywords: Glorund, nanos, Drosophila, hnRNP, hnRNP F, hnRNP H, RNA-binding protein, translational control, translational repressor, post-transcriptional regulation, development
Graphical abstract
INTRODUCTION
Post-transcriptional gene regulation relies on the association of RNA with RNA-binding proteins (RBPs) that direct RNA processing, turnover, localization, and translation. Many RBPs have evolved to function in multiple aspects of RNA regulation and recognize numerous different target RNAs (Ascano et al., 2013; Gerstberger et al., 2014). These multifunctional RBPs must therefore specifically select and distinguish among their diverse target RNAs. Such multifunctionality is common among RBPs belonging to the heterogeneous nuclear ribonucleoprotein (hnRNP) family (Han et al., 2010). Although hnRNPs were first defined as a complex of proteins involved in packaging and processing nascent pre-mRNA transcripts (Dreyfuss et al., 1993), the identification of homologous proteins expanded the hnRNP family (Han et al., 2010). An increasing number of hnRNPs with general roles in pre-mRNA metabolism have now been shown to play additional, specialized roles in the regulation of specific transcripts (Piccolo et al., 2014). To understand how hnRNPs and other RBPs carry out their varied functions in vivo, it is essential to elucidate their RNA recognition properties as well as how they use these properties to regulate diverse RNA networks.
The hnRNP F/H family of proteins comprises five mammalian homologs (hnRNP F, hnRNP H, hnRNP H′, hnRNP 2H9 and G-rich sequence factor 1 (GRSF-1)) with roles in alternative splicing, polyadenylation, and translational activation (Buratti et al., 2004; Jablonski et al., 2008; Martinez-Contreras et al., 2006; Veraldi et al., 2001). HnRNP F/H proteins recognize a stretch of guanine nucleotides called a G-tract using two or three quasi-RNA-recognition motifs (qRRMs) (Caputi and Zahler, 2001; Dominguez and Allain, 2006; Dominguez et al., 2010). Solution structures of hnRNP F qRRMs bound to G-tract RNA showed that the qRRMs are structurally homologous to classical RRMs, with a four-stranded β-sheet packed against two α-helices. However, whereas RRMs typically interact with RNA using conserved aromatic residues on the surface of the β-sheet (Lunde et al., 2007; Maris et al., 2005), these residues are poorly conserved in the hnRNP F/H qRRMs. Recognition of the G-tract is instead mediated by residues located in three loops (Dominguez and Allain, 2006; Dominguez et al., 2010; Honore et al., 1995). The loop residues encage the G-tract, sequestering it in a single-stranded conformation (Dominguez et al., 2010; Samatanga et al., 2013).
In Drosophila, the hnRNP F/H family is represented by a single protein, Glorund (Glo). Glo was first identified as a repressor of nanos (nos) mRNA translation through its interaction with a translational control element (TCE) in the nos 3′ untranslated region (3′UTR). The TCE comprises two stem-loops, designated TCEII and TCEIII, joined to a base stem (TCEI) (Gavis et al., 1996; Kalifa et al., 2006). Previous mutational analyses of the TCE indicated that both the sequence and structure of the UA-rich stem of the TCEIII stem-loop are required for TCE function in vivo and Glo binding in vitro (Kalifa et al., 2006). Specifically, mutations in the double-stranded UA-rich motif that disrupt Glo binding or mutation of Glo itself abrogate TCE-mediated repression of nos during oogenesis (Forrest et al., 2004; Kalifa et al., 2006). Like its mammalian counterparts, however, Glo has multiple functions suggesting additional mRNA targets. Glo is required for viability to adulthood (Kalifa et al. 2006), and in addition to nos regulation, Glo has recently been implicated in translational repression of nuclear-encoded mitochondrial respiratory chain complex transcripts (Gehrke et al., 2015). Glo also plays roles in ovarian nurse cell chromosome organization and dorsal-ventral patterning, most likely as a splicing regulator (Kalifa et al., 2009; Kalifa et al., 2006). How Glo distinguishes among its targets to exert the necessary regulatory activity is not known.
To further probe the mechanistic bases for Glo’s multiple functions and, in particular, the role of RNA recognition, we took a structure-based functional approach. We determined the crystal structures of Glo’s three qRRMs and, based on these structures, identified two distinct modes of RNA recognition. We show that, like hnRNP F qRRMs, Glo qRRMs bind to single-stranded G-tracts and that mutation of loop residues equivalent to those in hnRNP F disrupt G-tract binding. We also identify a second, noncanonical RNA-binding interface present on the surface of Glo qRRM β-sheets and show that mutation of this interface disrupts recognition of the double-stranded UA-rich motif in the TCE. By engineering mutations in Glo that disable each RNA recognition mode in vitro and evaluating the effect of these mutations on Glo function in vivo, we demonstrate that regulation of a subset of Glo targets is mediated solely using the G-tract binding mode, whereas regulation of nos unexpectedly requires both modes of recognition. Consistent with this finding, we identified a G-tract sequence within the TCE and show that higher affinity binding of Glo in vitro and TCE function in vivo require both the G-tract and the UA-rich stem. The use of two distinct RNA-binding modes to recognize different sets of target RNAs provides a molecular mechanism to explain the diverse functions of Glo and possibly other hnRNP F/H proteins. Moreover, it illustrates a general principle that RBPs may use different combinations of RNA recognition interfaces to regulate subsets of target RNAs that correlate with particular biological functions.
RESULTS
Crystal Structures of Glo qRRMs Predict that Glo Recognizes G-tract RNA
As a first step toward understanding how Glo interacts with in vivo RNA targets, we determined the crystal structures of each of its three qRRMs: qRRM1, qRRM2 and qRRM3 (Figure 1; Table S1). Despite limited overall sequence similarity between Glo and its human homologs (24% amino acid identity/33% similarity), the residues in hnRNP F that mediate interaction with G-tract RNA are conserved in Glo (Dominguez et al., 2010; Kalifa et al., 2006). Comparison of the three crystal structures with the solution structures of hnRNP F qRRMs in complex with single-stranded G-tract RNA indicated that the G-tract RNA interaction features are conserved in Glo (Figure 1B, C). Thus, Glo qRRMs likely retain the G-tract RNA-binding ability of the hnRNP F/H family.
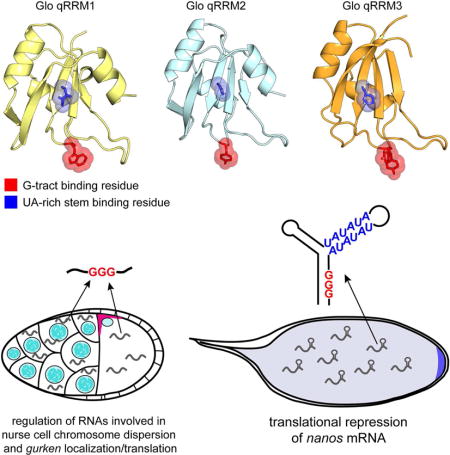
Figure 1. Crystal structures of Glo qRRM1, qRRM2 and qRRM3 domains identify RNA-binding motifs.
(A) Schematic representation of the domain structure of Glo. Amino acid residues at domain boundaries are numbered, and the qRRM domains are color coded: qRRM1 (yellow), qRRM2 (cyan) and qRRM3 (orange). (B) Ribbon diagrams of the solution structure of hnRNP F qRRM1 bound to single-stranded G-tract RNA (PDB code 2KFY; Dominguez et al., 2010) and crystal structures of Glo qRRMs. The ribbon diagram of hnRNP F qRRM1 is shown in grey with a cartoon representation of bound G-tract RNA in yellow. Three critical G-tract binding residues in hnRNP F qRRM1 are displayed as stick models superimposed with transparent red space-filling spheres (R16, W20, and Y82); the structurally equivalent residues in Glo qRRM1 (yellow), qRRM2 (cyan), and qRRM3 (orange) are displayed similarly. N- and C-termini of proteins, 5′ and 3′ ends of G-tract RNA, and loops 1, 3, and 5, which contain residues that may interact with G-tract RNA, are indicated. (C) Conservation of G-tract RNA-binding residues in Glo. Comparison of the consensus G-tract binding motif sequence of hnRNP F qRRMs with corresponding residues in Glo qRRMs and hnRNP F qRRM1. Consensus residues important for stacking interaction with G-tract RNA (shown in B) are highlighted in red. Additional residues involved in G-tract RNA binding are colored magenta in the consensus sequence. (D) Ribbon diagrams of the crystal structures of classical U1A RRM bound to a fragment of U1 small nuclear RNA (PDB code 1URN;(Oubridge et al., 1994) and Glo qRRMs. The ribbon diagram of U1A RRM is shown in grey with a cartoon representation of bound U1 small nuclear RNA sequence in yellow. Critical residues within the RNP1 and RNP2 motifs of U1A are displayed as stick models superimposed with transparent green and blue space-filling spheres, respectively (Y13 and F56); the structurally equivalent residues in Glo qRRM1, qRRM2, and qRRM3 are displayed similarly. αC helices in qRRM1 and qRRM2 that occlude the RNP1 motif are colored red. N- and C-termini of proteins and 5′ and 3′ ends of the fragment of U1 small nuclear RNA are indicated; the 5′ end of the RNA is truncated in this rendering. (E) Conservation of classical RRM RNA-binding residues in Glo. Comparison of the consensus RNP motif sequences of classical RRMs with corresponding residues in Glo qRRMs and U1A RRM. Consensus RNP1 and RNP2 residues important for RNA binding by classical RRMs are colored green and blue, respectively. See also Table S1.
To evaluate whether Glo can bind to G-tract sequences, we performed electrophoretic mobility shift assays (EMSAs) and determined the dissociation constants (Kd) of each domain with single-stranded G-tract RNA (5′-AGGGA). Each qRRM domain bound independently to G-tract RNA (Figure 2), and qRRM1 and qRRM3 exhibited several-fold higher affinity than qRRM2 (Kd=0.18 μM and 0.17 μM, respectively, vs 0.62 μM). This is similar to the measurement of binding affinities for the qRRMs of hnRNP F, where qRRM2 bound to G-tract RNA with lower affinity than qRRMs 1 and 3 (kd=4.6 μM vs kd=0.4 μM; Dominguez et al. 2010). We were unable to quantitatively evaluate binding to G-tract RNA by full-length Glo protein due to poor expression. However, a truncated protein containing the two N-terminal qRRMs (Glo qRRM1,2) bound two 5′-AGGGA RNAs with an affinity intermediate to that of the individual domains (Kd = 0.28 μM; Figure 2 and data not shown; see Supplemental Methods). This finding is consistent with independent binding of each qRRM to a distinct RNA. We conclude that, like hnRNP F qRRMs, Glo qRRMs are each capable of interacting with G-tract RNA.
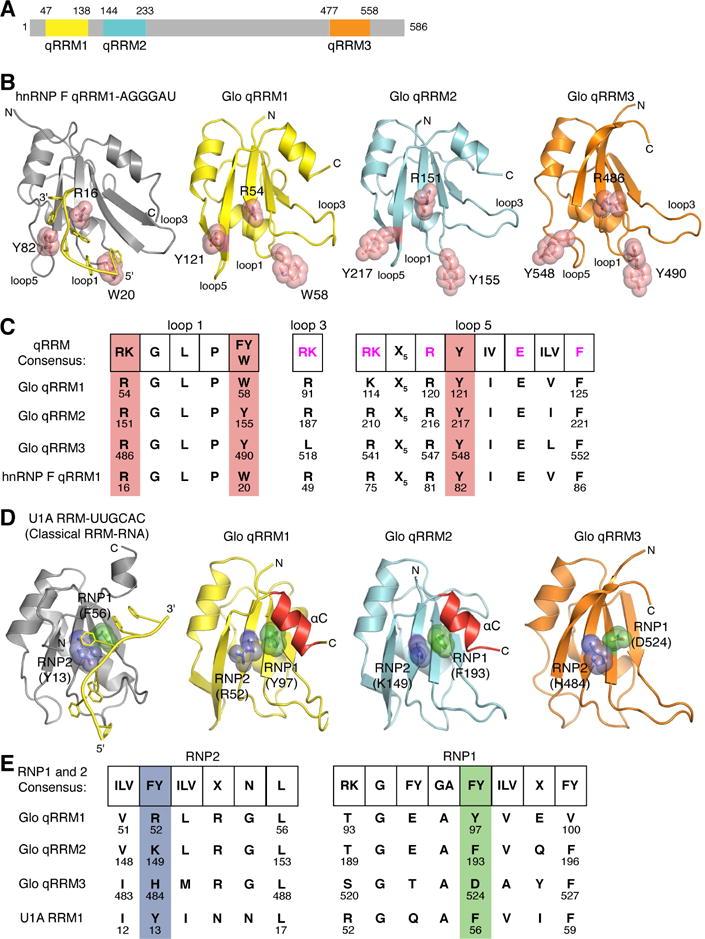
Figure 2. Glo qRRMs bind to a single-stranded G-tract sequence.
Binding of purified recombinant Glo qRRM1, qRRM2, qRRM3, and qRRM1,2 proteins to radiolabeled G-tract (5′-AGGGA) RNA. Representative EMSA gels are shown, with data plotted below. The highest protein concentration (right-most lanes) was 3.1 μM and 2-fold serial dilutions were assayed. No protein was added to the reactions in the lanes marked ‘-’. The positions of unbound RNA (asterisk) and Glo qRRM:RNA complexes (arrowhead) are indicated. Three technical replicates of EMSAs were performed, and apparent Kd values shown are mean ± SEM.
Crystal Structures of Glo qRRMs Suggest a Second, Distinct RNA Interaction Mode
Given the conservation of the G-tract RNA interaction, it is surprising that Glo was identified by its interaction with the double-stranded UA-rich motif in the nos TCEIII stem-loop (Kalifa et al., 2006). Therefore, we inspected the Glo qRRM crystal structures for residues that might mediate interaction with TCEIII. The three qRRMs adopt a core classical RRM fold (β1α1β2β3α2β4) with a four-stranded antiparallel β-sheet on one surface and two α-helices on the opposite surface (Figure 1D). The classical RRM recognizes two RNA bases using hydrophobic residues within two conserved motifs, designated RNP1 and RNP2, that are located on the face of its β-sheet surface (Figure 1D, E). We therefore examined whether Glo could also use these motifs to interact with nos TCE RNA.
Conserved aromatic groups are present within the RNP1 motifs of Glo qRRM1 and qRRM2 (Y97 or F193, respectively) (Figure 1E). However, unlike classical RRMs, the aromatic RNP1 residues in Glo qRRM1 and qRRM2 are not available for RNA recognition. Glo qRRM1 and qRRM2 each possess a third C-terminal α-helix (αC; Figure 1D) that forms hydrophobic interactions with the respective aromatic RNP1 residues. Mutation of either Y97 or F193 to alanine resulted in aggregated proteins, suggesting that these hydrophobic interactions are important for structural stability of the protein. In contrast, Glo qRRM3 lacks both an αC helix and a conserved aromatic residue in its RNP1 motif (Figures 1D, E). Together, the crystal structures suggest that the RNP1 motifs of Glo may not interact with RNA.
Basic residues in all three Glo qRRMs (R52 in qRRM1, K149 in qRRM2, and H484 in qRRM3) replace the aromatic residues that are conserved in RNP2 motifs of classical RRMs (Figure 1E). These basic residues are exposed on the protein surface (Figure 1D), suggesting that they are available for RNA binding. We propose that these Glo RNP2 residues, which are conserved in hnRNP F, interact with RNA, but in a manner that is distinct from that of classical RRMs. Together, our structural analysis predicts that each of the Glo qRRMs has two RNA-binding interfaces that differ from classical RRMs: the G-tract binding loops identified in hnRNP F and a basic β-strand interface within the RNP2 motif. Moreover, the exposed basic residues in the RNP2 motifs suggest a possible mechanism for recognition of the UA-rich TCEIII stem-loop by Glo.
Glo Recognizes Two Different TCE Features
In order to assess whether Glo qRRMs use either of their RNA-binding interfaces to interact with the nos TCE, we first tested the ability of individual Glo qRRMs or the tandem domain Glo qRRM1,2 protein to bind TCE RNA by EMSA. In contrast to their ability to bind G-tract RNA, single qRRM domains did not bind stably to a TCE RNA probe (Figure 3A; Kd>100 μM for qRRM1 and qRRM3; Kd>20 μM for qRRM2; data not shown). However, Glo qRRM1,2 did bind stably to the TCE, with two plateaus apparent in the binding curve (Figure 3B, C). These data fit best to a curve assuming two binding sites with higher (Kd=0.26 μM) and lower (Kd=13.5 μM) affinity components, suggesting that the TCE contains two Glo recognition sites with distinct properties. We therefore re-examined the sequences that comprise the complete TCE for a second Glo binding site in addition to the UA-rich TCEIII stem-loop. This analysis identified a G-tract embedded in the TCEI base stem (Figure 3A) that could potentially mediate Glo interaction. Notably, the higher affinity binding of Glo qRRM1,2 to the TCE (Kd=0.26 μM) is similar to the affinity of Glo qRRM1,2 for single-stranded G-tract RNA (Kd=0.28 μM).
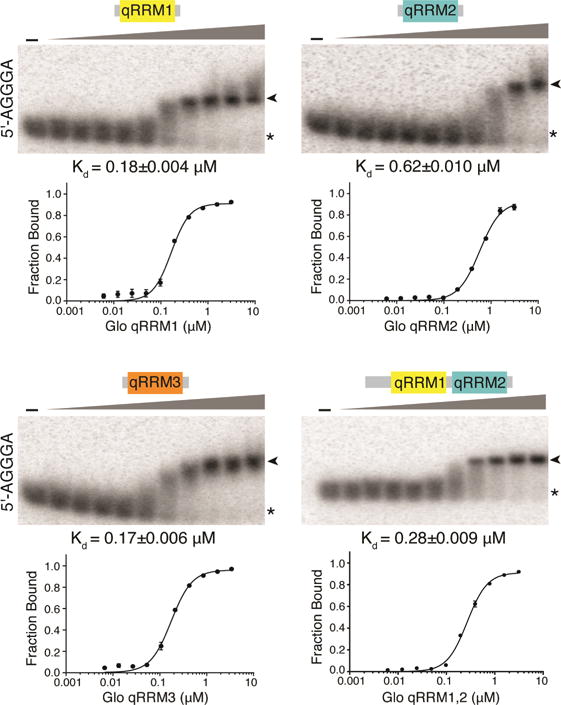
Figure 3. Glo recognizes two elements in the nos TCE that contribute higher and lower affinity components.
(A) Predicted secondary structures of TCE and TCEI RNAs. RNA secondary structure prediction was calculated using Mfold (Zuker, 2003). TCEI, II, and III elements are indicated. The complete sequences of the transcribed RNAs are shown. The G-tract in TCEI is boxed and TCEGAG/UUC mutations are indicated in red. (B) Representative EMSA gels of purified recombinant Glo qRRM1,2 binding to TCE (top), TCEGAG/UUC (middle), or TCEI (bottom) RNAs. The highest Glo qRRM1,2 protein concentration (right-most lanes) was 100 μM for the TCE and TCEGAG/UUC binding assays and 144 μM for the TCEI binding assays with 2-fold serial dilutions. No protein was added to the reactions in the lanes marked ‘-’. The positions of unbound RNA (asterisk) and Glo qRRM1,2:RNA complex (arrowhead) RNA are indicated. (C) Superposition of plots for binding of Glo qRRM1,2 to TCE (red) or TCEGAG/UUC (black) RNA. Higher affinity binding to the TCE is selectively disrupted by mutation of the G-tract sequence in the TCEI stem. Three technical replicates of EMSAs were performed, and apparent Kd values shown are mean ± SEM. (D) Superposition of plots for binding of Glo qRRM1,2 to TCE (red) or TCEI (dotted black) RNA. The TCEI stem alone retains the higher affinity binding component of the Glo-TCE interaction, but the lower affinity component is diminished. See also Figure S1.
To determine whether the TCEI GGG sequence contributes to TCE recognition by Glo qRRM1,2, we mutated it to GAG, while maintaining base pairing with a compensatory change on the 5′ strand (TCEGAG/UUC; Figure 3A). Glo qRRM1,2 bound to TCEGAG/UUC RNA with a Kd of 8.9 μM, similar to the lower affinity binding to TCE RNA (Kd=13.5 μM), and curve fitting now assumed a single binding site (Figure 3B, C). Strikingly, higher affinity binding was absent, suggesting that this component could be assigned to recognition of the TCEI G-tract sequence and the lower affinity binding that remained was due to recognition of the UA-rich TCEIII stem-loop. The GAG mutation alone eliminated higher affinity binding (Kd=18.6 μM) (Figure S1A), and the compensatory mutation alone displayed both higher and lower affinity binding (Figure S1B). In addition, two mutants with an intact G-tract but predicted to disrupt TCEI stem base pairing, TCEUAC and TCEGAC, retained both higher and lower affinity binding components (Figure S1C, D), indicating that the G-tract sequence is responsible for the higher affinity binding. These data also indicate that Glo can interact with the G-tract regardless of whether it is base-paired and suggest that, like hnRNP F (Dominguez et al. 2010; Samatanga et al., 2013), Glo binding may promote a single-stranded configuration of G-tracts.
To further assign higher and lower affinity binding to the TCE recognition sites, we tested binding of Glo qRRM1,2 to RNAs comprising TCEI and TCEIII (TCEI_III) or only TCEI. Glo qRRM1,2 bound to TCEI_III RNA with higher (Kd=0.24 μM) and lower (Kd=27.4 μM) affinity components, similar to binding to the intact TCE (Figure S1E). As expected, TCEII, which is recognized by the embryonic repressor Smaug (Smg; see below), is not required for recognition by Glo. When the RNA was further truncated to the TCEI stem alone (Figure 3A), Glo qRRM1,2 bound with a Kd of 0.17 μM, similar to the Kd for the higher affinity site in TCE RNA (Figure 3B, D) whereas the lower affinity binding component was considerably reduced (Kd>87 μM). We therefore conclude that binding of Glo qRRM1,2 to the TCE includes two components: higher affinity binding (Kd≈0.3 μM) to the TCEI G-tract sequence and lower affinity binding (Kd≈13 μM) to the UA-rich TCEIII stem-loop.
Identification of Glo Residues that Mediate G-tract or TCE RNA Binding
Our crystallographic analysis suggested that Glo has two distinct RNA-binding interfaces within each of its qRRMs, and we hypothesized that each of the two binding interfaces confers one component of Glo’s dual RNA-binding specificity. If correct, the two RNA-binding activities should be separable by mutation of the respective interfaces. We therefore generated mutations in residues within the loop and RNP2 binding surfaces and tested whether they differentially alter the ability of Glo to bind single-stranded G-tract (5′-AGGGA) and TCE RNAs in vitro.
We first mutated to alanine Glo W58 (qRRM1) and Y155 (qRRM2), which are equivalent to hnRNP F G-tract binding residues W20 (qRRM1) and Y120 (qRRM2), respectively (Figure 1B, C, Figure S2). Binding to G-tract RNA by Glo qRRM1,2W58A,Y155A protein with mutations in both qRRMs was >200-fold weaker than binding by the wild-type Glo qRRM1,2 protein, suggesting that Glo uses these equivalent residues for G-tract interaction (Figures 4A, S2A, and Table S2). Consistent with the behavior of individual qRRM domains, a single qRRM was sufficient to bind to the single-stranded G-tract RNA, as the individual W58A and Y155A mutations individually had little to no effect on G-tract binding by Glo qRRM1,2, and the Glo qRRM1,2 mutant with an intact qRRM1 (qRRM1,2Y155A) bound with slightly higher affinity than the mutant with an intact qRRM2 (qRRM1,2W58A) (Figures 4A, S2A, and Table S2).
Figure 4. Identification of Glo residues critical for G-tract or TCEIII recognition.
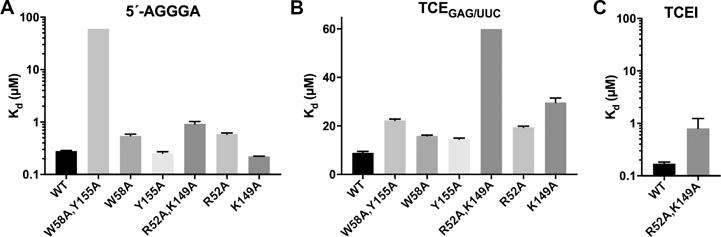
Bar graphs of mean Kd values for binding of wild-type (WT) and mutant qRRM1,2 proteins to either TCEGAG/UUC, 5′-AGGGA, or TCEI RNAs determined from three technical replicates. Error bars indicate SEM. P-values were calculated for mutant binding to the TCEGAG/UUC or 5′-AGGGA RNA relative to the WT protein using a one-way ANOVA, excluding the double mutants whose binding was too weak to determine precisely and are reported as a lower limit of >60 μM. The p-values for qRRMR52A,K149A mutant binding to TCEGAG/UUC or TCEI RNA relative to the WT were calculated using an unpaired, two-tailed t-test with Welch’s correction for unequal variances, and the p-value for qRRMW58A,Y155A binding to 5′-AGGGA RNA relative to the WT was calculated using an unpaired, one-tailed t-test with Welch’s correction. P-values were <0.01 with the following exceptions: binding to 5′-AGGGA RNA by qRRMW58A,Y155A (p=0.024); qRRMY155 (p=0.996); and qRRMK149A (p=0.846) mutants; and binding to TCEI RNA by the qRRMR52A,K149A mutant (p=0.28). Representative EMSA gels and plots are presented in Figures S2 and S3. See also Table S2.
We next tested whether mutation of these residues affects the binding of Glo to TCE RNA. For these experiments, we used primarily a TCE RNA lacking the embedded G-tract sequence, TCEGAG/UUC, to probe the lower affinity interaction with the UA-rich TCEIII stem-loop. Mutation of either one or both of the G-tract binding residues had only a small effect (<2.5 fold) on binding of Glo qRRM1,2 to TCEGAG/UUC RNA, indicating that these residues are not critical for recognition of the TCEIII UA-rich motif (Figures 4B, S2B, and Table S2). We also tested binding of Glo qRRM1,2W58A,Y155A to wild-type TCE RNA. Whereas wild-type Glo qRRM1,2 protein exhibited both lower and higher-affinity binding to the TCE, Glo qRRM1,2W58A,Y155A protein showed only the lower affinity component, indicating that the mutations disrupted the higher-affinity binding component attributed to recognition of the embedded TCEI G-tract sequence (Figure S2C). This behavior is similar to that observed for Glo qRRM1,2 binding to TCEGAG RNAs (Figures 3C, S1A) and consistent with the idea that the Glo qRRM loop residues recognize both single-stranded G-tract RNA and the G-tract present in the TCEI base stem.
We next focused on the basic residues in Glo’s divergent RNP2 motif as potential candidate residues for recognition of the UA-rich TCEIII stem-loop. Mutation of both RNP2 residues (R52A, K149A) in Glo qRRM1,2 (Figures 1D, 1E, and S3) greatly diminished binding to TCEGAG/UUC RNA by more than 7-fold (a lower limit due to the protein solubility at high concentration) as compared to the wild-type protein (Figures 4B, S3A, and Table S2). Moreover, these mutations preferentially affected recognition of the UA-rich TCEIII stem-loop, since strong binding to single-stranded G-tract RNA and the TCEI base stem was retained (Figures 4C, S3B, and Table S2). Together, these results demonstrate that Glo has two different RNA recognition modalities and uses these to recognize two distinct features of the TCE. Residues within the qRRM loops are critical for G-tract recognition in the TCEI stem, and basic residues in the divergent RNP2 motifs are important for recognition of the UA-rich TCEIII stem-loop.
Translational Repression of nos Requires Both Modes of RNA Recognition
The ability of Glo to recognize both the G-tract and the double-stranded UA-rich motif in the TCE suggests that Glo employs both modes of RNA recognition to regulate nos in vivo. We therefore sought to determine whether mutations that preferentially disrupt TCEIII stem-loop or TCEI G-tract RNA binding by Glo in vitro affect translational repression of nos in vivo (Figure 5A–C). Nos protein is required in the posterior of the early embryo for abdominal development but must be excluded from the anterior where it inhibits head development (Gavis and Lehmann, 1992; Wang and Lehmann, 1991). This distribution is achieved through the selective translation of nos mRNA localized at the posterior of the embryo and the repression of nos mRNA remaining within the bulk cytoplasm (Gavis and Lehmann, 1992, 1994). Localization of nos occurs during late stages of oogenesis, and Glo acts at this time to establish the repressed state of nos (Kalifa et al., 2006). Because translation of nos is restricted to the posteriorly localized transcripts, only a small amount of Nos protein accumulates in late-stage oocytes (Figure 5D, E; Forrest et al., 2004; Forrest and Gavis, 2003). By contrast, Nos protein levels are dramatically increased when repression of unlocalized nos is abolished by mutation of glo (glo−; Figure 5D, E).
Figure 5. Glo uses both modes of RNA recognition for translational repression of nos.
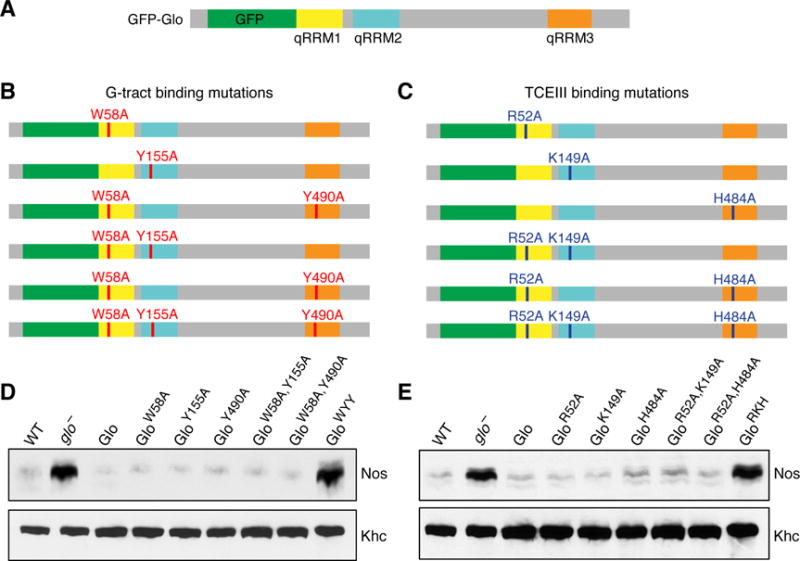
(A) Schematic representation of the wild-type GFP-Glo protein, with the N-terminal GFP tag (green) and qRRM1 (yellow), qRRM2 (cyan), and qRRM3 (orange) indicated. (B, C) GFP-Glo proteins with alanine substitutions that were assayed for function in vivo. G-tract binding mutations are shown in red (B), TCEIII binding mutations are shown in dark blue (C). All gfp-glo transgenes were inserted into the same chromosomal location. Two independent lines of each transgene were tested for their ability to rescue the lethality of the glo null mutant and in all cases, the independent lines behaved similarly. (D, E) Immunoblot analysis of Nos protein in extracts of late ovaries from glo mutant females expressing GFP-Glo proteins with mutations in either (D) G-tract binding residues (corresponding to panel C) or (E) TCEIII binding mutations (corresponding to panel D). Kinesin heavy chain (Khc) was used as a loading control.
We expressed wild-type GFP-Glo or mutant GFP-Glo proteins in glo mutants and tested their ability to repress nos translation by assaying Nos protein levels in ovaries enriched for late-stage oocytes, referred to as late ovaries (Andrews et al., 2011). GFP-Glo proteins with a single alanine substitution in either a residue that affects G-tract binding (qRRM1: W58A; qRRM2: Y155A; qRRM3: Y490A) or a residue that affects TCEIII binding (qRRM1: R52A; qRRM2: K149A; qRRM3: H484A), behaved identically to wild-type GFP-Glo in their ability to confer translational repression of nos (Figure 5D, E). Similarly, GFP-Glo proteins with alanine substitutions in both qRRM1 and qRRM2 (G-tract binding: GloW58A,Y155A; TCEIII binding: GloR52A,K149A) or qRRM1 and qRRM3 (G-tract binding: GloW58A,Y490A; TCEIII binding: GloR52A,H484A) retained the ability to repress nos (Figure 5D, E). In contrast, Nos protein levels were dramatically increased in late ovaries from glo mutant females expressing either the G-tract binding triple mutant (GFP-GloW58A,Y155A,Y490A, abbreviated GloWYY) or the TCEIII binding triple mutant (GFP-GloR52A,K149A,H484A, abbreviated GloRKH) protein (Figure 5D, E). Immunoblot analysis showed that both GFP-GloWYY and GFP-GloRKH were expressed at levels comparable to proteins that do confer nos repression (Figure S4), indicating that their failure to repress nos translation did not result from reduced protein expression.
Together, these results indicate that, consistent with the in vitro binding studies, both modes of RNA recognition are required for translational repression of nos during oogenesis. Because Glo binds with only modest affinity to TCEIII in vitro and the qRRM1,2R52A,K149A mutation that abolishes this binding has an additional, albeit minor, effect on TCEI binding, we considered an alternative model in which another protein, instead of Glo, binds to the TCEIII UA-rich motif to regulate nos. This model predicts that: 1) TCEI G-tract mutations would eliminate binding of Glo to the TCE; 2) binding of Glo to the G-tract would be the same as binding to the full TCE; and 3) the GloRKH mutation, which selectively abrogates binding to the TCEIII UA-rich stem, would have only a minor effect on Glo function in vivo compared to the GloWYY mutation, which selectively abrogates binding to the TCEI G-tract. None of these predictions is borne out. Furthermore, the binding specificity might well be higher for the full-length proteins in vivo. Thus, although we cannot entirely rule out the possibility that only G-tract binding is relevant to Glo regulation of nos translation in vivo, our data best support a dual recognition model for Glo interaction with the TCE.
TCE Function in vivo Requires Both Glo Binding Motifs
To determine whether the requirements for TCE function are the same as or different from the requirements for recognition of the TCE by Glo, we generated a series of mutations that alter either the TCEI G-tract, the TCEIII UA-rich motif, or both in an otherwise wild-type nos transgene (Figure 6A). These TCE mutants were then assayed for their ability to repress nos translation by analyzing Nos protein levels in late ovaries from transgenic females. Mutation of either the G-tract (GAG) or TCE UA-rich stem (IIIA) resulted in a modest increase in Nos protein as compared to the wild-type nos transgene, indicating that both motifs contribute to translational repression of nos (Figure 6B). Consistent with this, the TCEGAG.IIIA double mutation had an additive effect, resulting in a dramatic increase in Nos protein levels (Figure 6B). We therefore conclude that nos translational regulation depends upon both the UA-rich and G-tract motifs in the nos TCE and their interaction with Glo qRRMs.
Figure 6. TCE function in vivo requires both Glo recognition sites.
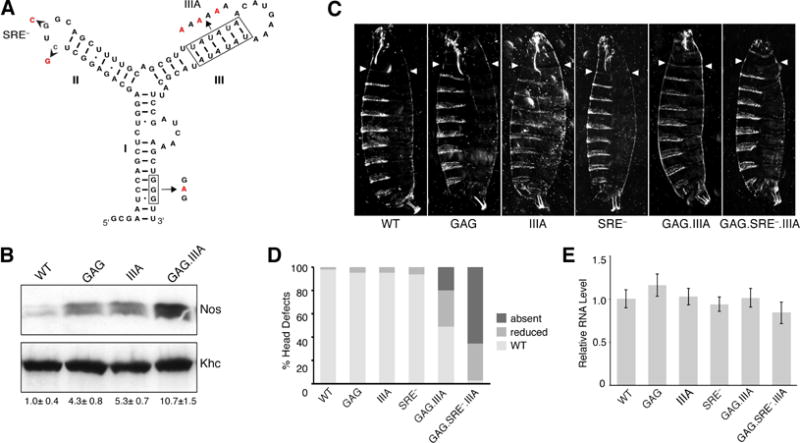
(A) Predicted TCE secondary structure, with the TCEI G-tract and TCEIII UA-rich motifs indicated by boxes. Nucleotide changes creating the GAG, IIIA, and SRE− mutations are indicated. (B) Immunoblot analysis of Nos protein in extracts of late ovaries from females expressing either a wild-type nos transgene (WT) or nos transgenes with the GAG, IIIA, or GAG.IIIA mutation as indicated. Nos protein levels were normalized to the Khc loading control. The mean value ± SEM for the relative abundance of Nos shown below each lane was determined from three independent experiments. A one-way ANOVA showed no significant difference. (C) Representative cuticular phenotypes of embryos from females expressing either the WT or the indicated TCE mutant transgenes. Arrowheads indicate the extent of the head skeleton formed, which reaches the third thoracic segment in WT. Head structures in the TCEGAG.IIIA cuticle are foreshortened and malformed (reduced classification in D); no head structures are visible in the TCEGAG.III.SRE− cuticle (absent classification in D). (D) Quantification of the fraction of embryos exhibiting defects in the development of head structures for the TCE mutants shown in (C). Values represent the average percentages obtained from analysis of two independent lines (n>200 embryos/line) (E) Quantitative RT-PCR of transgenic nos RNA. Values are the percent mean number of transcripts ± SEM of three technical replicates from each of three biological replicates.
The ultimate physiological consequence of the failure to repress nos is the suppression of head development (Gavis and Lehmann, 1992, 1994). This defect can be easily visualized in the larval cuticle, providing a sensitive, cumulative readout of nos regulation in the oocyte and early embryo (Dahanukar and Wharton, 1996; Gavis et al., 1996; Smibert et al., 1996). The repression of unlocalized nos that is initiated by Glo during oogenesis is maintained in the early embryo by the SAM-domain protein Smg, which binds to a motif in TCE stem-loop II called the Smg Recognition Element (SRE; Dahanukar et al., 1999; Smibert et al., 1999; Smibert et al., 1996). To evaluate the relative contributions of the TCEI G-tract, TCEIII UA-rich motif, and SRE to nos regulation, we assayed the effects of mutations in these sequences, individually and in combination, on embryonic head development.
Mutation of either the TCE G-tract (TCEGAG), TCE UA-rich stem (TCEIIIA), or SRE (TCESRE−) individually did not significantly affect head development (Figure 6C, D), indicating that the elevated Nos protein levels observed by immunoblot analysis are not sufficient to suppress the formation of anterior structures. Strikingly, mutation of both Glo recognition motifs (TCEGAG.IIIA) was sufficient to disrupt head development, even though Smg binding was not disrupted (Figure 6C, D). Consistent with the requirement for Glo and Smg, however, the most severe phenotype was produced by combining the GAG, IIIA, and SRE− mutations (TCEGAG.IIIA.SRE−), with over 95% of embryos exhibiting loss of head structures (Figure 6D). Similar effects on head development were observed when these transgenes were introduced into nos mutants, indicating that the increase in nos activity is not due simply to increased gene dosage (data not shown). Furthermore, all transgenes examined produce similar levels of RNA, as indicated by quantitative RT-PCR analysis (Figure 6E). Therefore, differences in the behavior of the transgenes are unlikely to result from differences in the levels of expression or stabilities of the RNAs. Finally, all transgenes behaved similarly in oskar (osk) mutants, where nos localization is abolished, confirming that the assay monitors de-repression of unlocalized nos RNA (data not shown). Taken together, these results confirm the physiological relevance of the in vitro binding data and establish a critical role for both the TCEI G-tract and TCEIII UA-rich motif in TCE function during oogenesis. Moreover these results further demonstrate that translational repression of unlocalized nos during oogenesis is critical to achieve the spatially restricted synthesis of Nos needed for embryonic patterning.
Glo Functions During Mid-oogenesis Are Mediated Solely by the G-tract Binding Mode
Like many hnRNPs, Glo has diverse biological functions. Indeed, the pleiotropy of the glo mutant phenotype, which includes dorsal-ventral and nurse cell nuclear morphology defects, suggests that Glo acts at different stages of oogenesis to regulate RNAs in addition to nos. Although the specific target RNAs whose regulation is important for these processes are not known, the identification of mutations that discriminate the two Glo binding modes in vivo allowed us to investigate how Glo recognizes the RNAs relevant for these functions.
In wild-type Drosophila ovaries, the nurse cell chromosomes initially exhibit a polytene morphology, but subsequently become dispersed throughout the nucleus (Figure S5) (Dej and Spradling, 1999). This chromosomal dispersion fails to occur in glo mutant ovaries (Kalifa et al., 2009; see also Figure 7). In addition, Glo indirectly regulates the localization and translation of gurken (grk) mRNA, which is required for dorsal fate specification. As a result, the dorsally located respiratory appendages of the egg shell are broad and often laterally expanded in glo mutants as compared to wild-type (Kalifa et al., 2009). To better understand how Glo interacts with the RNAs required for these processes, we assayed the functionality of mutant GFP-Glo proteins during mid-oogenesis (Figure 7). GFP-Glo proteins carrying a single mutation in residues that affect either G-tract or TCEIII binding behaved like wild-type GFP-Glo and completely rescued the glo mutant chromosome organization, grk localization, and dorsal appendage defects (Figures 7B, 7C, 7F, and S5). Similarly, expression of GFP-Glo proteins with mutations in both qRRM1 and qRRM2 (G-tract binding: GloW58A,Y155A ; TCEIII binding: GloR52A,K149A) or qRRM1 and qRRM3 (G-tract binding: GloW58A,Y490A ; TCEIII binding: GloR52A,H484A) also rescued these defects (Figure S5). Strikingly, while expression of the GFP-GloRKH protein fully rescued the chromosome organization and grk regulation defects, expression of the GFP-GloWYY protein failed to rescue either of these glo mutant phenotypes (Figure 7D–F). Similarly to glo mutants, the nurse cell chromosomes of ovaries expressing GFP-GloWYY remained polytene throughout oogenesis (Figure 7D). In addition, grk was mislocalized along the anterior cortex (Figure 7D) and the resulting eggs exhibited abnormal dorsal appendages (Figure 7F). We therefore conclude that regulation of Glo target RNAs required for chromosome organization and grk regulation specifically require the G-tract RNA binding mode whereas the TCEIII binding mode is dispensable for these Glo functions. Furthermore, the behavior of the qRRM1,2 (GloW58A,Y155A) and qRRM1,3 (GloW58A,Y490A) mutant proteins indicate that RNA binding by a single qRRM is sufficient for regulating these targets.
Figure 7. Regulation of chromosome dispersion and dorsal-ventral patterning are mediated solely by the G-tract binding mode of Glo.
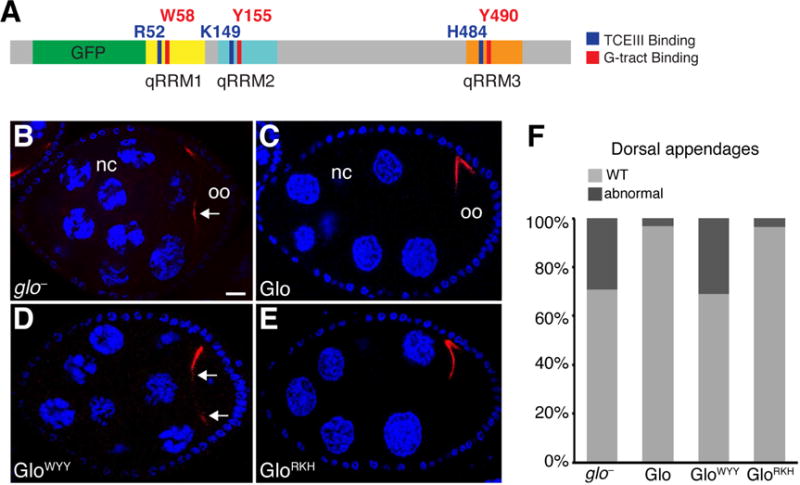
(A) Schematic representation of the GFP-Glo protein with a summary map of the alanine substitutions shown in Figure 5B, C and analyzed in (B–F). (B–E) Confocal sections of a glo mutant egg chamber (B) and glo mutant egg chambers expressing GFP-Glo (B), GFP-GloWYY(C), or GFP-GloRKH (D). Nurse cells (nc) and oocyte (oo) are indicated. grk mRNA (red) is detected by smFISH, nurse cell and follicle cell nuclei (blue) are stained with DAPI. Egg chambers are oriented with anterior to the left, dorsal up. Arrows indicate mislocalized grk mRNA. Scale bar=10 μM. (F) Quantification of the fraction of eggs laid by glo mutant females expressing GFP-Glo, GFP-GloWYY, or GFP-GloRKH that show dorsal appendage defects, including shortened, laterally expanded, fused, and/or missing appendages. See also Figures S5 and S6.
DISCUSSION
RNA-binding proteins are often modular, with multiple RNA-binding domains that in combination can increase both specificity and affinity (Lunde et al., 2007). Our structure/function analysis of Glo uncovers another layer of complexity by showing that a single RNA-binding domain, the Glo qRRM, encompasses two discrete RNA-binding interfaces. These binding surfaces allow Glo to specifically recognize two very different RNA substrates, single-stranded G-tract RNA and a structured UA-rich motif. By assaying the effect of mutations that preferentially disrupt each recognition mode in vitro on Glo function in vivo, we show that regulation of a subset of Glo’s targets in vivo is mediated solely using the G-tract binding mode whereas regulation of nos requires both modes of recognition. The discovery of distinct RNA-binding modes within a single RNA-binding domain suggests a mechanism for the diversification of target RNA recognition that expands the functional repertoire of individual RNA-binding proteins.
Recognition of G-tracts by qRRMs differs from core RNA recognition by classical RRMs in utilization of loop residues rather than β-sheet residues. Moreover, β-sheet basic residues in Glo qRRMs have diverged from the canonical RNP1/RNP2 interface of the classical RRMs. Analysis of both protein and RNA mutations confirmed that these residues endow Glo with a second RNA-binding mode that interacts with the double-stranded UA-rich motif in nos, thereby diversifying the repertoire of Glo target RNAs and its biological functions. Interestingly, these basic RNP2 residues are conserved in mammalian hnRNP F/H proteins, and a recent study using crosslinking and immunoprecipitation coupled with high-throughput sequencing demonstrated that hnRNP F often bound G-tract motifs near UA-rich regions (Huelga et al., 2012). It is therefore possible that dual modes of recognition also impart functional versatility on the human proteins.
Recognition of the nos TCE by Glo requires at least two qRRMs, and both Glo binding modes are required for regulation of nos in vivo. In contrast, recognition of targets involved in nurse cell chromosome dispersion and dorsal-ventral patterning requires binding of a single qRRM to G-tract motifs. Thus, it is the combination of the two binding modes within the three qRRMs that confers multifunctionality at the level of target RNA selection and, potentially at the level of regulatory function. Glo qRRM1,2 protein binds with >50-fold higher affinity to the TCEI G-tract than to the TCEIII UA-rich motif in vitro, suggesting that G-tract binding dominates stable TCE interaction whereas TCEIII recognition dominates target specificity. Moreover, the finding that G-tract or TCEIII binding residues must be mutated in all three qRRMs to abolish nos repression suggests that, unlike other proteins containing RRMs, the G-tract or TCEIII motif interaction may not map to individual domains.
In addition to discriminating among RNA targets, the use of different binding modes may influence how Glo regulates those targets. For example, the engagement of Glo with both TCE motifs may be required for the recruitment of a larger repressor complex and/or for interactions with components of the translation machinery. HnRNP F/H proteins are best known for their roles in the regulation of alternative splicing by binding to G-tracts, and G-tract recognition by single hnRNP F qRRMs is sufficient for alternative splicing of Bcl-x (Dominguez et al., 2010). We showed previously that Glo forms a complex with two Drosophila splicing factors, Half-pint (Hfp)/PUF68 and Hrp48 (Kalifa et al., 2009). Mutations in glo, hfp, or hrp48 cause similar defects in dorsal-ventral patterning and nurse cell chromosome dispersion, suggesting that Glo might collaborate with Hfp and Hrp48 to regulate the splicing of transcripts needed for these processes. Our finding that the G-tract binding mode alone is required for Glo function in dorsal-ventral patterning and chromosome dispersion supports this idea. Interaction of Glo with Hfp and Hrp48 is RNA-independent and although GFP-GloWYY cannot bind to G-tracts, it retains the ability to bind Hfp and Hrp48 as assayed by coimmunoprecipitation (Figure S6). Thus, it is possible that Glo can only bind to G-tracts and not to TCEIII-like motifs when complexed with Hfp and Hrp48, thereby targeting the relevant Glo regulatory function to the proper targets.
Glo is required for viability to adulthood and although the relevant target RNAs are not known, we found that, as for nos repression, both the Glo G-tract and TCEIII binding modes are essential. This suggests that Glo similarly recognizes other RNAs with bipartite motifs in addition to nos. As other RNA targets of Glo are identified, computational analyses should search for both G-tract and TCEIII-like motifs. Analysis of how the new targets are regulated and what accessory factors are engaged will deepen our understanding of how recognition of G-tract or TCEIII-like elements influences RNA metabolism.
EXPERIMENTAL PROCEDURES
Methods are described in detail in the Supplemental Experimental Procedures.
Protein Expression and Purification
The cDNA sequences encoding the qRRM domains of Glo were subcloned into pET15b (Novagen), which encodes an N-terminal His6-tag. Individual qRRM domains were expressed in E. coli strain BL21-CodonPlus (DE3)-RIL (Agilent Technologies) at 20 °C overnight after induction with 0.5 mM IPTG. His6-tagged proteins were purified using Ni-NTA agarose (Thermo Scientific). For crystallization, the His6 tag was cleaved overnight with 5 U of thrombin (Novagen). Glo qRRM proteins were further purified using a HiLoad 16/60 Superdex 75 column (GE Healthcare) followed by a HiTrap Heparin column (GE Healthcare). Purified protein was dialyzed against a buffer containing 20 mM Tris-HCl, pH 8.0, 100 mM NaCl and then concentrated to 10 mg/ml. For Glo qRRM2 purification, purified protein was dialyzed against a buffer containing 20 mM Tris-HCl, pH 8.0, 500 mM NaCl and then concentrated to 9 mg/ml. For Glo qRRM3 purification, purified protein was also applied to HiTrap Q (GE Healthcare) and HiTrap SP columns (GE Healthcare) sequentially to remove contaminating proteins that bound to these columns. Final purified Glo qRRM3 protein was dialyzed against a buffer containing 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, then concentrated to 10 mg/ml.
Individual Glo qRRM domains for in vitro binding assays were purified by Ni-NTA agarose chromatography as described above, followed by purification on a HiLoad 16/60 Superdex 75 column (GE Healthcare). Wild-type Glo qRRM1,2 protein and qRRM1,2 mutants generated by site-directed mutagenesis (Agilent Technologies) were purified by the same procedure as individual Glo qRRM proteins for in vitro binding assays. Purified Glo qRRM1,2WT, qRRM1,2W58A,Y155A, and qRRM1,2R52A,K149A proteins were analyzed by circular dichroism to assess folding, and no differences were observed between wild-type and mutant proteins (Figure S2D, Figure S3C).
Crystallization, Data Collection, Structure Determination and Refinement
Crystals of Glo qRRM1 were prepared by the sitting-drop vapor diffusion method at 4 °C. Crystals of Glo qRRM2 or qRRM3 were prepared by the hanging-drop vapor diffusion method at 20 °C. X-ray diffraction data for crystals of qRRM1 and qRRM3 were collected using a conventional X-ray source and for crystals of qRRM2 were collected at the SER-CAT Beamline 22-ID at the Advanced Photon Source, Argonne National Laboratories. Diffraction data were processed using the program package HKL2000 (HKL Research Inc. (Otwinowski and Minor, 1997). The structures were determined by molecular replacement using the program Molrep (Vagin and Teplyakov, 2000). Model building was carried out with the program Coot (Emsley and Cowtan, 2004). The programs Refmac5 (Murshudov et al., 1997) and Phenix.refine (Adams et al., 2010) were used for refinement. The structures displayed good geometry when analyzed by MolProbity (Chen et al., 2010).
In vitro Transcription
Templates for in vitro transcription of TCE and TCE mutant RNAs were engineered with a GAG at the transcript start to stimulate in vitro transcription by T7. Thus, all transcripts begin with GAG (see Figure 3A). TCE and TCE mutant RNAs were produced by in vitro transcription and purified by electrophoresis on a 10% polyacrylamide-urea gel (Invitrogen) and a HiTrap Q column (GE Healthcare). Peak fractions containing target RNAs were pooled, ethanol precipitated, and resuspended in DEPC-treated water.
Electrophoretic Mobility Shift Assay
TCE and TCE mutant RNAs were produced by in vitro transcription as described above, and a 5′-AGGGA RNA oligonucleotide was generated by RNA synthesis (GE Dharmacon). RNAs were radiolabeled at the 5′ end using [γ-32P] ATP and T4 polynucleotide kinase, then purified using an Illustra MicroSpin G-25 column (GE Healthcare). TCE and TCE mutant RNAs were prepared by heating to 90 °C for 5 min, and then slowly cooling to room temperature. The 5′-AGGGA RNA oligonucleotide was incubated at 90 °C for 5 min in 50 mM Tris, pH 8.0 and then cooled on ice to produce single-stranded RNA that bound to qRRMs. RNA-binding reactions included 0.9 nM radiolabeled RNA and increasing concentrations (2-fold) of protein. Binding reactions were incubated for 1 hr at room temperature and separated by electrophoresis on 10% polyacrylamide gels (Invitrogen). Gels were dried and exposed to storage phosphor screens for 6–20 hr, scanned with a Typhoon 8600 Imager, and the band intensities were quantified with ImageQuant 5.2. The data were analyzed and Kd values were calculated via non-linear regression analysis for one- or two-site binding with GraphPad Prism 6.
Construction of gfp-glo and nos Transgenes and Transgenic Lines
A functional gfp-glo transgene was generated by insertion of the egfp coding sequences at the start codon of a 5.6 kb genomic glo rescue fragment (g-glo; Kalifa et al., 2009; Kalifa et al., 2006) and cloned into the pattB vector (http://www.flyc31.org/sequences_and_vectors.php). Point mutations were introduced into the glo sequences by PCR-based site-directed mutagenesis. All transgenes were inserted into the attp40 landing site by the phiC31 integrase method to eliminate position effects on gene expression. Phenotypic analysis was performed by crossing transgenes into the null glo162x background (Kalifa et al., 2006). Mutations in the nos 3′UTR were engineered by PCR into a 4.3 kb genomic nos rescue fragment (Gavis and Lehmann, 1992) and cloned into the pattB vector (Bischof et al., 2013). All nos transgenes were inserted into the attP40 landing site.
Immunoblotting
For analysis of GFP-Glo levels during oogenesis, ovaries were dissected from well-fed females in PBS and frozen in liquid nitrogen. For analysis of Nos levels during late-stages of oogenesis, ovaries enriched for late-stage oocytes were generated as described previously (Andrews et al., 2011), dissected in PBS, and frozen in liquid nitrogen. Ovary extract preparation and immunoblotting followed the procedures of Forrest et al. (2004), using nitrocellulose membrane. The following primary antibodies were used: 1:1000 rabbit anti-Nos (gift of A. Nakamura); 1:1000 anti-Glo (5B7; (Kalifa et al., 2006); 1:10,000 rabbit anti-kinesin heavy chain (Cytoskeleton). Proteins were visualized by chemiluminescence (Roche) and quantified using ImageJ.
qPCR
Total RNA was extracted and reverse transcribed from dissected ovaries using a standard random-primed cDNA protocol (Qiagen). qPCR was conducted using gene-specific primers and three technical replicates from each of three biological replicates were performed per genotype (TaqMan, ThermoFisher). Data are expressed as the amount of target cDNA relative to the amount of rpl7 cDNA and normalized to the wild-type transgene.
smFISH
smFISH was performed on ovaries dissected from well-fed females as described previously (Abbaszadeh and Gavis, 2016) using probes for the coding region of grk conjugated to ATTO-565 (ATTO-Tec). DNA was visualized with DAPI (1:1000; Molecular Probes). Oocytes were imaged using a Nikon A1R confocal microscope with a 60x/1.4 NA oil objective.
Analysis of Eggshell and Embryonic Phenotypes
Embryos were collected on yeasted apple juice plates (Wieschaus and Nüsslein-Volhard, 1986) at RT and immediately analyzed for eggshell phenotypes. For the analysis of embryonic phenotypes, the embryos were aged for >24 hours at RT, after which larval cuticle preparations were made (Wieschaus and Nüsslein-Volhard, 1986).
Statistical methods
The Kd values are reported as mean +/− standard error of the mean (SEM), which indicates the discrepancy among the technical replicates. We also examined the error of the Kd values for each technical replicate and calculated mean errors for the sets of three technical replicates. This analysis indicated that the SEM is a reasonable representation of the accuracy of the Kd values. P-values for Glo qRRM1,2 mutant binding to the TCEGAG/UUC or AGGGA RNA relative to wild-type Glo qRRM1,2 protein were calculated using a one-way ANOVA, excluding the double mutants (qRRM1,2W58A,Y155A and qRRM1,2R52A,K149A) whose binding was too weak to determine precisely and are reported as a lower limit of >60 μM. The p-value for qRRM1,2W58A,Y155A binding to 5′-AGGGA RNA relative to wild-type qRRM1,2 was calculated using an unpaired, one-tailed t-test with Welch’s correction and the p-values for qRRM1,2R52A,K149A binding to TCEGAG/UUC and TCEI RNAs relative to wild-type qRRM1,2 were calculated using an unpaired, two-tailed t-test with Welch’s correction for unequal variances. Differences in Nos protein levels in late ovaries expressing nos transgenes were analyzed by one-way ANOVA.
Supplementary Material
Acknowledgments
The authors would like to thank D. Snowflack and Y. Kalifa for initial studies, G. Laevsky for assistance with confocal microscopy, M. Niepielko for help with qPCR analysis, R. Petrovich of the NIEHS Protein Expression Core Facility for assistance with circular dichroism analysis, and L. Pedersen and the staff of the Southeast Regional Collaborative Access Team beamlines for assistance with X-ray data collection. We are grateful to G. Kissling for advice on statistical analyses, F. Hughson for helpful discussions, and E. Abbaszadeh and F. Hughson for critical comments on the manuscript. This research was supported in part by a National Science Foundation Graduate Research Fellowship awarded to J.V.T. (DGE 1148900), a Japan Society for the Promotion of Science fellowship awarded to T.T., grants from the National Institutes of Health to E.R.G. (R01 GM061107) and the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences (T.M.T.H.). The Advanced Photon Source used for this study is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31-109-Eng-38.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
J.V.T., T.T., T.M.T.H. and E.R.G. designed the experiments. J.V.T., T.T., and S.C. conducted the experiments. J.V.T., T.T., T.M.T.H. and E.R.G. analyzed the data. J.V.T., T.T., T.M.T.H. and E.R.G participated in writing and editing the manuscript.
Accession Numbers
Coordinates and structure factors for qRRMs 1–3 have been deposited in the RCSB Protein Data Bank (www.rcsb.org) with the accession codes: 5UZG, 5UZM, 5UZN.
References
- Abbaszadeh EK, Gavis ER. Fixed and live visualization of RNAs in Drosophila oocytes and embryos. Methods. 2016;98:34–41. doi: 10.1016/j.ymeth.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S, Snowflack DR, Clark IE, Gavis ER. Multiple mechanisms collaborate to repress nanos translation in the Drosophila ovary and embryo. RNA. 2011;17:967–977. doi: 10.1261/rna.2478611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Gerstberger S, Tuschl T. Multi-disciplinary methods to define RNA-protein interactions and regulatory networks. Curr Opin Genet Dev. 2013;23:20–28. doi: 10.1016/j.gde.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Bjorklund M, Furger E, Schertel C, Taipale J, Basler K. A versatile platform for creating a comprehensive UAS-ORFeome library in Drosophila. Development. 2013;140:2434–2442. doi: 10.1242/dev.088757. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle M, De Conti L, Baralle D, Romano M, Ayala YM, Baralle FE. hnRNP H binding at the 5′ splice site correlates with the pathological effect of two intronic mutations in the NF-1 and TSHbeta genes. Nucleic Acids Res. 2004;32:4224–4236. doi: 10.1093/nar/gkh752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputi M, Zahler AM. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H′/F/2H9 family. J Biol Chem. 2001;276:43850–43859. doi: 10.1074/jbc.M102861200. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahanukar A, Walker JA, Wharton RP. Smaug, a novel RNA-binding protein that operates a translational switch in Drosophila. Mol Cell. 1999;4:209–218. doi: 10.1016/s1097-2765(00)80368-8. [DOI] [PubMed] [Google Scholar]
- Dahanukar A, Wharton RP. The Nanos gradient in Drosophila embryos is generated by translational regulation. Genes Dev. 1996;10:2610–2620. doi: 10.1101/gad.10.20.2610. [DOI] [PubMed] [Google Scholar]
- Dej KJ, Spradling AC. The endocycle controls nurse cell polytene chromosome structure during Drosophila oogenesis. Development. 1999;126:293–303. doi: 10.1242/dev.126.2.293. [DOI] [PubMed] [Google Scholar]
- Dominguez C, Allain FH. NMR structure of the three quasi RNA recognition motifs (qRRMs) of human hnRNP F and interaction studies with Bcl-x G-tract RNA: a novel mode of RNA recognition. Nucleic Acids Res. 2006;34:3634–3645. doi: 10.1093/nar/gkl488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C, Fisette JF, Chabot B, Allain FH. Structural basis of G-tract recognition and encaging by hnRNP F quasi-RRMs. Nat Struct Mol Biol. 2010;17:853–861. doi: 10.1038/nsmb.1814. [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Forrest KM, Clark IE, Jain RA, Gavis ER. Temporal complexity within a translational control element in the nanos mRNA. Development. 2004;131:5849–5857. doi: 10.1242/dev.01460. [DOI] [PubMed] [Google Scholar]
- Forrest KM, Gavis ER. Live imaging of endogenous RNA reveals a diffusion and entrapment mechanism for nanos mRNA localization in Drosophila. Curr Biol. 2003;13:1159–1168. doi: 10.1016/s0960-9822(03)00451-2. [DOI] [PubMed] [Google Scholar]
- Gavis ER, Lehmann R. Localization of nanos RNA controls embryonic polarity. Cell. 1992;71:301–313. doi: 10.1016/0092-8674(92)90358-j. [DOI] [PubMed] [Google Scholar]
- Gavis ER, Lehmann R. Translational regulation of nanos by RNA localization. Nature. 1994;369:315–318. doi: 10.1038/369315a0. [DOI] [PubMed] [Google Scholar]
- Gavis ER, Lunsford L, Bergsten SE, Lehmann R. A conserved 90 nucleotide element mediates translational repression of nanos RNA. Development. 1996;122:2791–2800. doi: 10.1242/dev.122.9.2791. [DOI] [PubMed] [Google Scholar]
- Gehrke S, Wu Z, Klinkenberg M, Sun Y, Auburger G, Guo S, Lu B. INK1 and Parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell Metab. 2015;21:95–108. doi: 10.1016/j.cmet.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SP, Tang YH, Smith R. Functional diversity of the hnRNPs: past, present and perspectives. Biochem J. 2010;430:379–392. doi: 10.1042/BJ20100396. [DOI] [PubMed] [Google Scholar]
- Honore B, Rasmussen HH, Vorum H, Dejgaard K, Liu X, Gromov P, Madsen P, Gesser B, Tommerup N, Celis JE. Heterogeneous nuclear ribonucleoproteins H, H′, and F are members of a ubiquitously expressed subfamily of related but distinct proteins encoded by genes mapping to different chromosomes. J Biol Chem. 1995;270:28780–28789. doi: 10.1074/jbc.270.48.28780. [DOI] [PubMed] [Google Scholar]
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012;1:167–178. doi: 10.1016/j.celrep.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski JA, Buratti E, Stuani C, Caputi M. The secondary structure of the human immunodeficiency virus type 1 transcript modulates viral splicing and infectivity. J Virol. 2008;82:8038–8050. doi: 10.1128/JVI.00721-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalifa Y, Armenti ST, Gavis ER. Glorund interactions in the regulation of gurken and oskar mRNAs. Dev Biol. 2009;326:68–74. doi: 10.1016/j.ydbio.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalifa Y, Huang T, Rosen LN, Chatterjee S, Gavis ER. Glorund, a Drosophila hnRNP F/H homolog, is an ovarian repressor of nanos translation. Dev Cell. 2006;10:291–301. doi: 10.1016/j.devcel.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- Martinez-Contreras R, Fisette JF, Nasim FU, Madden R, Cordeau M, Chabot B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006;4:e21. doi: 10.1371/journal.pbio.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- Piccolo LL, Corona D, Onorati MC. Emerging roles for hnRNPs in post-transcriptional regulation: what can we learn from flies? Chromosoma. 2014;123:515–527. doi: 10.1007/s00412-014-0470-0. [DOI] [PubMed] [Google Scholar]
- Samatanga B, Dominguez C, Jelesarov I, Allain FH. The high kinetic stability of a G-quadruplex limits hnRNP F qRRM3 binding to G-tract RNA. Nucleic Acids Res. 2013;41:2505–2516. doi: 10.1093/nar/gks1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smibert CA, Lie YS, Shillinglaw W, Henzel WJ, Macdonald PM. Smaug, a novel and conserved protein, contributes to repression of nanos mRNA translation in vitro. RNA. 1999;5:1535–1547. doi: 10.1017/s1355838299991392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smibert CA, Wilson JE, Kerr K, Macdonald PM. smaug protein represses translation of unlocalized nanos mRNA in the Drosophila embryo. Genes Dev. 1996;10:2600–2609. doi: 10.1101/gad.10.20.2600. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- Veraldi KL, Arhin GK, Martincic K, Chung-Ganster LH, Wilusz J, Milcarek C. hnRNP F influences binding of a 64-kilodalton subunit of cleavage stimulation factor to mRNA precursors in mouse B cells. Mol Cell Biol. 2001;21:1228–1238. doi: 10.1128/MCB.21.4.1228-1238.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Lehmann R. Nanos is the localized posterior determinant in Drosophila. Cell. 1991;66:637–647. doi: 10.1016/0092-8674(91)90110-k. [DOI] [PubMed] [Google Scholar]
- Wieschaus EF, Nüsslein-Volhard C. Looking at embryos. In: Roberts DB, editor. Drosophila: A Practical Approach. Oxford: IRL Press; 1986. pp. 199–227. [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.