

Abstract
Protein kinase C in Saccharomyces cerevisiae, i.e., Pkc1, is an enzyme that plays an important role in signal transduction and the regulation of lipid metabolic enzymes. Pkc1 is structurally similar to its counterparts in higher eukaryotes, but its requirement of phosphatidylserine (PS) and diacylglycerol (DAG) for catalytic activity has been unclear. In this work, we examined the role of these lipids in Pkc1 activity with protein and peptide substrates. In agreement with previous findings, yeast Pkc1 did not require PS and DAG for its activity on the peptide substrates derived from lipid metabolic proteins such as Pah1 [phosphatidate (PA) phosphatase], Nem1 (PA phosphatase phosphatase), and Spo7 (protein phosphatase regulatory subunit). However, the lipids were required for Pkc1 activity on the protein substrates Pah1, Nem1, and Spo7. Compared with DAG, PS had a greater effect on Pkc1 activity, and its dose-dependent interaction with the protein kinase was shown by the liposome binding assay. The Pkc1-mediated degradation of Pah1 was attenuated in the cho1Δ mutant, which is deficient in PS synthase, supporting the notion that the phospholipid regulates Pkc1 activity in vivo.
Keywords: Pah1 phosphatidate phosphatase, Nem1-Spo7 protein phosphatase, Cki1 choline kinase, Ura7 CTP synthetase, yeast
The model eukaryote yeast, Saccharomyces cerevisiae, synthesizes membrane phospholipids by pathways that are generally common to those of higher eukaryotes (1, 2). Its phospholipid synthesis is a complex process that is regulated by genetic and biochemical mechanisms, and is interrelated with the synthesis of other major lipid classes that include fatty acids, triacylglycerol, sphingolipids, and sterols (1, 2). On a biochemical level, the posttranslational modification of phosphorylation has emerged as a key mechanism by which lipid metabolic enzymes or transcription factors are regulated (1, 2). Our laboratory has shown the phosphorylation-mediated regulation of lipid metabolic proteins (1, 2) by conserved protein kinases such as protein kinase C (PKC) (3–10).
S. cerevisiae PKC (also known as Pkc1) is required for progression through the cell cycle (11) and plays a role in regulating lipid synthesis (5–10) and in maintaining cell wall integrity (11–13). Pkc1 (11, 14) is considered to be a prototypic form of PKC (15) by containing all of the conserved domains (e.g., kinase domain, pseudosubstrate domain, calcium-dependent phospholipid binding domain, diacylglycerol (DAG) binding domain, Rho-binding domain, and regulatory region) found in the conventional (e.g., α, βI, βII, γ), novel (e.g., δ, ε, θ, η), or atypical (e.g., ξ, ι, λ) PKC enzymes of higher eukaryotic organisms (16–19). In spite of its domain structure similarities to PKC enzymes of higher eukaryotes, the yeast enzyme is not supposed to be dependent on lipids [e.g., phosphatidylserine (PS) and DAG] for activity (20, 21) unless it is bound to Rho1 GTPase (22).
In previous studies (3, 5, 6, 10, 23), we have rationalized the use of rat brain PKC (mixture of α, β, γ isoforms) (24, 25) to study the in vitro phosphorylation of yeast lipid metabolic proteins; rat liver PKC has catalytic properties common to Pkc1 (7, 21, 26, 27). Studies using the rat brain enzyme, which is commercially available, have identified the Pkc1 target sites in Pah1 phosphatidate (PA) phosphatase (3), Cki1 choline kinase (5), Ura7 CTP synthetase (8, 9), and Opi1 transcriptional repressor (10). Moreover, the in vivo analyses of the proteins lacking phosphorylation by Pkc1 have revealed the importance of the protein kinase in the regulation of lipid metabolism in S. cerevisiae (3, 5, 8–10).
Recently, we have been studying the Pkc1-mediated phosphorylation of additional enzymes in lipid metabolism of S. cerevisiae. During the course of this work, we examined the notion that Pkc1, unlike mammalian PKCs, does not require the lipids PS and DAG for catalytic activity. Using the protein and peptide substrates, we found that Pkc1 has a different requirement of PS and DAG for catalytic activity: the lipids were required for Pkc1 activity on Pah1, but not for its peptide. This novel observation was recapitulated with two newly identified targets of Pkc1, Nem1 and Spo7, which form a protein phosphatase complex (28) catalyzing the dephosphorylation of Pah1 (29–31). Of the two lipids, PS had a greater stimulatory effect on Pkc1 activity, and it was shown to interact with the protein kinase in the liposome-binding assay. Moreover, we demonstrated that the Pkc1-mediated degradation of Pah1 in vivo (3) is attenuated in the cho1Δ mutant, which lacks the synthesis of PS, supporting the regulatory role of PS on Pkc1 activity.
MATERIALS AND METHODS
Materials
All chemicals were reagent grade or better. Growth medium supplies were purchased from Difco. Protein assay reagents, electrophoresis reagents, DNA and protein size standards, and Coomassie Blue R-250 were from Bio-Rad. IgG-Sepharose, polyvinylidene difluoride membrane, and the enhanced chemifluorescence Western blotting reagent were purchased from GE Healthcare. New England Biolabs was the supplier of restriction endonucleases, modifying enzymes, Phusion high fidelity DNA polymerase, and T4 polynucleotide kinase. DNA gel extraction and plasmid DNA purification kits and nickel-nitrilotriacetic acid-agarose resin were from Qiagen. Sigma-Aldrich was the source of ampicillin, kanamycin, carbenicillin, PCR primers, nucleotides, protease inhibitors (phenylmethylsulfonyl fluoride, benzamidine, aprotinin, leupeptin, and pepstatin), 2-mercaptoethanol, Ponceau S stain, BSA, phosphoamino acid standards, isopropyl-β-D-thiogalactoside, L-1-tosylamido-2-phenylethyl chloromethyl ketone-trypsin, and rabbit anti-protein A antibody (product P3775, lot 025K4777). Peptides were prepared by EZBiolab. P81 phosphocellulose paper was from Whatman. PerkinElmer Life Science and National Diagnostics were the sources of radiochemicals and scintillation counting supplies, respectively. Avanti Polar Lipids was the source of lipids. Cellulose TLC plates were from EMD Millipore. SYPRO Ruby fluorescent stain and alkaline phosphatase-conjugated goat anti-rabbit IgG antibodies (product 31340, lot NJ178812) were from Thermo Fisher Scientific.
Plasmids and DNA manipulations
Plasmids pWM122 and pWM123 direct the isopropyl β-D-thiogalactoside-induced expression of the transmembrane-deletion mutants of Nem1 (Nem1-ΔTM) and His6-tagged Spo7 (Spo7-ΔTM), respectively. pWM122 was constructed by insertion of the NEM1 DNA lacking codons 87-126, which was generated from YCplac111-NEM1-PtA (29) by overlap extension PCR into pSBETA (32) at the NdeI/BamHI sites. pWM123 was constructed by insertion of the SPO7 DNA lacking codons 73-127, which was generated from pRS314-SPO7-Myc3 (28) by overlap extension PCR into pET-15b (Novagen) at the NdeI/BamHI sites. Standard methods were used for the isolation of DNA and its manipulation with restriction enzymes, DNA ligase, and modifying enzymes (33). PCR reactions were optimized as described by Innis and Gelfand (34). All plasmid constructions were confirmed by DNA sequencing, which was performed by GENEWIZ, Inc. Escherichia coli transformation was performed as described previously (33).
Strains and growth conditions
E. coli DH5α (F−ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rk−mk+) phoA supE44λ−thi-1gyrA96 relA1) (33) was used for the propagation of plasmids. E. coli strain BL21(DE3) [F− ompT hsdSB (rB−mB−) gal dcm (DE3)] (Invitrogen) was transformed with pWM122 and pWM123 for the expression of Nem1-ΔTM and His6-Spo7-ΔTM. The E. coli transformant was grown at 30°C in LB medium containing 100 μg/ml carbenicillin and 30 μg/ml kanamycin. When the culture reached A600 nm of 0.6, the expressions of Nem1-ΔTM and His6-Spo7-ΔTM were induced for 2 h by the addition of 1 mM isopropyl β-D-thiogalactoside. The cho1Δ::URA3 deletion mutant, derivative of strain W303-1A (MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1) (35) was constructed (36) and used to examine the role of PS in the degradation of Pah1. Yeast cells were grown at 30°C in standard synthetic complete medium (37) containing 2% glucose and 1 mM choline (38).
Preparation of yeast cell lysates
Yeast cells were grown to the stationary phase and, at the indicated time of growth, cells were harvested by centrifugation. Cell lysates were prepared by boiling 3 × 107 cells suspended in 30 μl of Laemmli buffer (39) supplemented with protease (5 mM EDTA, 5 μg/ml leupeptin, 5 μg/ml pepstatin A, 5 μg/ml aprotinin, 1 mM benzamidine, and 0.5 mM phenylmethylsulfonyl fluoride) and phosphatase (1 mM sodium vanadate and 1 mM sodium fluoride) inhibitors (40).
Purification of enzymes
ZZ-tagged (two repeats of the 60 amino acid IgG-binding domain of Staphylococcus aureus protein A) Pkc1 was expressed in yeast and purified by chromatography with DE53 and IgG-Sepharose as described (20) with modifications (7). The purified enzyme was dialyzed against 50 mM Tris-HCl (pH 7.5) buffer containing 15% glycerol. His6-tagged Pah1 PA phosphatase was expressed in E. coli and purified by affinity chromatography with nickel-nitrilotriacetic acid-agarose (41). Nem1-ΔTM and His6-tagged Spo7-ΔTM were coexpressed in E. coli and purified as a complex [because Nem1 and Spo7 form a tight association (28)] by affinity chromatography with nickel-nitrilotriacetic acid-agarose (41), followed by ion-exchange chromatography with Q-Sepharose (31). Cki1 choline kinase was expressed in Sf9 insect cells and purified from the cytosolic fraction by chromatography with Con A, Affi-Gel Blue, and Mono Q (4). Ura7 CTP synthetase was purified from the cytosolic fraction of yeast by ammonium sulfate fractionation, followed by chromatography with Sephacryl 300 HR, Q-Sepharose, Affi-Gel Blue, and Superose 6 (42).
PKC assays
PKC assays were performed in triplicate for 20 min at 30°C in a total volume of 20 μl. The standard reaction mixture contained 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 10 mM 2-mercaptoethanol, 1.7 mM CaCl2, 500 μM PS, 150 μM DAG, 50 μM [γ-32P]ATP (3,000 cpm/pmol), and the indicated amounts of substrate (protein or peptide) and enzyme (Pkc1 or rat brain PKC). At the end of the kinase reaction with a protein substrate, the reaction mixture was treated with Laemmli buffer (39), resolved by SDS-PAGE, and transferred to a polyvinylidene difluoride membrane for phosphorimaging analysis. Alternatively, the SDS-polyacrylamide gel was dried and subjected to the analysis, and the radioactive phosphorylation was quantified with ImageQuant software. In the kinase assay with a peptide substrate, the enzyme reaction was terminated by spotting the reaction mixture onto a P81 phosphocellulose paper. The paper was washed three times with 75 mM phosphoric acid and then subjected to scintillation counting.
SDS-PAGE and Western blotting
Proteins were separated by SDS-PAGE (39) using 8, 10, or 12% slab gels. SDS-polyacrylamide gels were stained with Coomassie Brilliant Blue R-250 or SYPRO Ruby stain. The samples for immunoblotting were normalized to total protein loading as determined by the Coomassie Blue-based assay (43). Immunoblotting with polyvinylidene difluoride membrane was performed as described previously (44–46). Ponceau S staining was used to monitor the protein transfer from the polyacrylamide gels to the polyvinylidene difluoride membrane. The membrane blots were probed with rabbit anti-protein A antibody (dilution 1:3,000), rabbit anti-Pah1 antibody (47) (2 μg/ml), or rabbit anti-Cho1 antibody (36) (2 μg/ml) followed by goat anti-rabbit IgG antibody conjugated with alkaline phosphatase (dilution of 1:5,000). Immune complexes were detected using the enhanced chemifluorescence immunoblotting substrate. Fluoroimaging was used to acquire fluorescence signals from immunoblots, and the intensities of the images were analyzed by ImageQuant software. A standard curve was used to ensure that the immunoblot signals were in the linear range of detection.
Analysis of phosphoamino acids and phosphopeptides
32P-labeled Nem1-ΔTM or Spo7-ΔTM was resolved by SDS-PAGE, transferred to the polyvinylidene difluoride membrane, and hydrolyzed with 6 N HCl at 110°C (for phosphoamino acid analysis) or proteolytically digested with L-1-tosylamido-2-phenylethyl chloromethyl ketone-trypsin (for phosphopeptide mapping analysis) (6, 48, 49). The acid hydrolysates were mixed with standard phosphoamino acids, and were separated by two-dimensional electrophoresis on cellulose TLC plates, whereas the tryptic digests were separated on the cellulose plates first by electrophoresis and then by TLC (6, 48, 49). Radioactive phosphoamino acids and peptides were visualized by phosphorimaging analysis. Nonradioactive phosphoamino acid standards were visualized by ninhydrin staining.
Liposome binding assay
Liposomes (unilamellar phospholipid vesicles) consisting of phosphatidylcholine (PC) and the indicated amounts of PS were prepared by the lipid extrusion method (50). The solution of phospholipids in chloroform was evaporated under nitrogen to form a thin film. The dried phospholipids were then resuspended in aqueous solution consisting of 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1.7 mM CaCl2, 10 mM 2-mercaptoethanol, and 150 mM NaCl. After five cycles of freezing and thawing, the phospholipid suspensions were extruded 11 times through a polycarbonate filter with a 100 nm diameter. The final concentration of phospholipids in the PC-PS liposomes was 1 mM. The amount of PS in the liposomes was expressed as a surface concentration in mole percent.
Pkc1 (560 ng) was incubated for 10 min with PC-PS liposomes in a total volume of 0.3 ml. Following the incubation, the liposomes were collected by centrifugation at 100,000 g for 1 h. The liposomal pellet was subjected to SDS-PAGE, followed by staining with SYPRO Ruby. Fluoroimaging was used to acquire the images of the stained proteins and their intensities were analyzed by ImageQuant software.
Data analyses
SigmaPlot software was used for the statistical analysis of data; P < 0.05 was taken as a significant difference.
RESULTS
Phosphorylation of Pah1 and its peptide by rat brain PKC requires PS and DAG
The S. cerevisiae Pah1 PA phosphatase has been shown to be phosphorylated in vitro and in vivo by PKC (3, 23). This enzyme catalyzes the dephosphorylation of PA to generate DAG (41, 51), which is used for the synthesis of the neutral lipid, triacylglycerol, or the phospholipids, PC and phosphatidylethanolamine (1, 2). The in vitro kinase assay has utilized recombinant Pah1 and rat brain PKC (3, 23). Because conventional PKC activity requires PS and DAG (24, 25), these lipids have been included in the phosphorylation reaction of Pah1 (3, 23). Here, we examined the effects of PS and DAG on rat brain PKC activity using Pah1 and its peptide as substrates. When Pah1 was used as a substrate, the omission of PS or DAG from the standard PKC reaction resulted in a 70% reduction or a 10% reduction, respectively, of the enzyme phosphorylation (Fig. 1A). The lack of both lipids in the kinase reaction showed a 93% reduction in the enzyme phosphorylation (Fig. 1A). In Pah1, Ser-769 is a major target site of phosphorylation by PKC (3). When a Pah1 peptide (763-NYNRTKSRRA-772) containing the serine residue was used as a substrate, the dependence of its phosphorylation on PS and DAG mirrored that shown by phosphorylation of Pah1 (Fig. 1B).
Fig. 1.

Effects of PS and DAG on the phosphorylation of Pah1 and its peptide by rat brain PKC. A: Recombinant Pah1 (50 μg/ml) was incubated with rat brain PKC (0.25 ng) for 20 min in the presence of 50 μM [γ-32P]ATP, 10 mM MgCl2, and 1.7 mM CaCl2 by the inclusion of 0.5 mM PS, 0.15 mM DAG, or both. The reaction mixtures were resolved by SDS-PAGE (10% gel) and the gel was dried and subjected to phosphorimaging. The levels of Pah1 phosphorylation were normalized to its maximum phosphorylation in the presence of PS and DAG. B: The peptide of Pah1 (763-NYNRTKSRRA-772, 100 μM) was incubated with rat brain PKC in the same conditions as in (A). The reaction mixtures were spotted onto the P81 phosphocellulose paper, which was washed with 75 mM phosphoric acid and subjected to scintillation counting. The levels of the peptide phosphorylation were normalized to its maximum phosphorylation (2.5 μmol/min/mg) in the presence of PS and DAG. The data shown are the averages of three experiments ± SD (error bars).
Pkc1 requires PS and DAG for its activity on Pah1, but not on its peptide
We also examined the phosphorylation of Pah1 and its peptide by Pkc1, which was purified as the ZZ-tagged enzyme (20). The ZZ-tag facilitates the purification of Pkc1, but does not alter the catalytic properties of the enzyme (20, 21). The Pkc1 preparation used in our study was shown to be highly pure by the SDS-PAGE analysis, and its identity was confirmed by Western blot analysis with anti-protein A antibody (Fig. 2A). We examined the phosphorylation of Pah1 by Pkc1 under the same conditions used for its phosphorylation by rat brain PKC. Pkc1 catalyzed the incorporation of the γ-phosphate of [γ-32P]ATP into Pah1 (Fig. 2B). When Pah1 was incubated with Pkc1 in the absence of PS and DAG, its phosphorylation was 87% lower when compared with that in the presence of PS and DAG (Fig. 3A). This finding was unexpected, given that yeast Pkc1 is not supposed to require the lipids for catalytic activity (20, 21). Indeed, the addition of PS or DAG to the kinase assay resulted in a dose-dependent increase in the phosphorylation of Pah1 by 6.4- or 2.8-fold, respectively (Fig. 4). PS and DAG together stimulated the phosphorylation of Pah1 by 7.4-fold when compared with its phosphorylation in the absence of these lipids (Fig. 3A). The omission of calcium from the standard assay did not have a significant effect on the Pkc1-mediated phosphorylation of Pah1 (Fig. 3A). The pH optimum for the phosphorylation of Pah1 by Pkc1 was 7.5 (Fig. 5A), and the maximum phosphorylation was obtained with 10 mM magnesium (Fig. 5C).
Fig. 2.
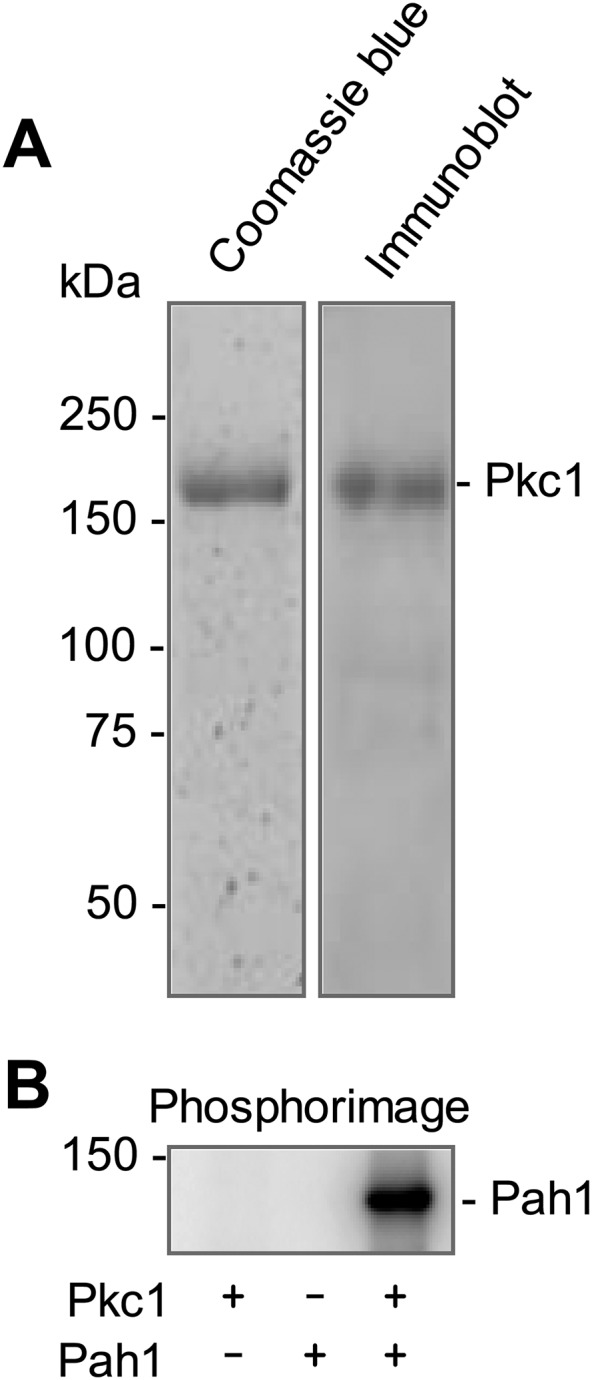
Pkc1 phosphorylates Pah1. A: SDS-PAGE and immunoblot analyses of the ZZ-tagged Pkc1 purified from yeast. The purified enzyme resolved in the SDS-polyacrylamide gel (8%) was stained with Coomassie Brilliant Blue R-250 (left, 0.75 μg Pkc1) or subjected to immunoblot analysis with anti-protein A antibody (right, 15 ng Pkc1). The positions of Pkc1 and the molecular mass standards are indicated. B: The purified Pkc1 (70 ng) was incubated with Pah1 (50 μg/ml) for 20 min in the presence of 50 μM [γ-32P]ATP, 10 mM MgCl2, 0.5 mM PS, 0.15 mM DAG, and 1.7 mM CaCl2. Pkc1 or Pah1 was omitted from the reaction where indicated. The reaction mixtures were resolved by SDS-PAGE and the gel was dried and subjected to phosphorimaging analysis. The positions of the 150 kDa molecular mass standard and phosphorylated Pah1 are indicated. The data are representative of three experiments.
Fig. 3.

Effects of PS, DAG, and calcium on the Pkc1 phosphorylation of Pah1 and its peptide. A: Pah1 (50 μg/ml) was incubated for 20 min with 70 ng Pkc1, 50 μM [γ-32P]ATP, and 10 mM MgCl2 in the presence of 0.5 mM PS, 0.15 mM DAG, or 1.7 mM CaCl2. The reaction mixtures were resolved by SDS-PAGE (10% gel) and the gel was dried and subjected to phosphorimaging. The levels of Pah1 phosphorylation were normalized to its maximum phosphorylation in the presence of PS, DAG, and calcium. B: The peptide of Pah1 (763-NYNRTKSRRA-772, 100 μM) was incubated with Pkc1 in the same conditions as in (A). The reaction mixtures were spotted onto the P81 phosphocellulose paper, which was washed with 75 mM phosphoric acid and subjected to scintillation counting. The levels of the peptide phosphorylation were normalized to its maximum phosphorylation (70 nmol/min/mg) in the presence of PS and DAG. The data shown are the averages of three experiments ± SD (error bars).
Fig. 4.

Effects of PS and DAG on the Pkc1 phosphorylation of Pah1. Pah1 (50 μg/ml) was incubated with 70 ng Pkc1 for 20 min in the presence of 50 μM [γ-32P]ATP, 10 mM MgCl2, 1.7 mM CaCl2, and the indicated concentration of PS (A) or DAG (B). The reaction mixtures were resolved by SDS-PAGE (10% gel) and the gel was dried and subjected to phosphorimaging analysis. The levels of Pah1 phosphorylation were normalized to that (shown in Fig. 3A) in the presence of 0.5 mM PS and 0.15 mM DAG. The data shown are the averages of three experiments ± SD (error bars).
Fig. 5.

Effects of pH and MgCl2 on the Pkc1 phosphorylation of Pah1 and its peptide. Pah1 (50 μg/ml) (A, C) or its peptide (763-NYNRTKSRRA-772, 100 μM) (B, D) was incubated for 20 min with 70 ng Pkc1 and 50 μM [γ-32P]ATP. The reaction of Pah1 phosphorylation also contained 0.5 mM PS, 0.15 mM DAG, and 1.7 mM CaCl2, and was carried out at the indicated pH of 50 mM Tris-maleate-glycine buffer with 10 mM MgCl2 (A) or at pH 7.5 of the composite buffer with the indicated concentrations of MgCl2 (C). The reaction mixtures were resolved by SDS-PAGE (10% gel) and the gel was dried and subjected to phosphorimaging analysis. The levels of Pah1 phosphorylation were normalized to the maximum phosphorylation at pH 7.5 in the presence of PS, DAG, CaCl2, and 10 mM MgCl2. The phosphorylation reaction of the Pah1 peptide did not contain PS, DAG, and CaCl2, and was carried out at the indicated pH of 50 mM Tris-maleate-glycine buffer with 10 mM MgCl2 (B) or at pH 7.5 of the composite buffer with the indicated concentrations of MgCl2 (D). The reaction mixtures were spotted onto the P81 phosphocellulose paper, which was washed with 75 mM phosphoric acid and subjected to scintillation counting. The levels of the peptide phosphorylation were normalized to the maximum phosphorylation (75 nmol/min/mg) at pH 7.5 in the presence of 10 mM MgCl2. The data shown are the averages of three experiments ± SD (error bars).
The phosphorylation of the Pah1 peptide by Pkc1 was examined in the presence and absence of PS or DAG. In contrast to rat brain PKC, Pkc1 did not require PS or DAG for its activity on the Pah1 peptide (Fig. 3B). Calcium had no stimulatory effect on the Pkc1-mediated phosphorylation by the Pah1 peptide. In fact, it inhibited the peptide phosphorylation by ∼60% (Fig. 3B). Like Pah1, the Pah1 peptide showed its maximum phosphorylation by Pkc1 at pH 7.5 (Fig. 5B) in the presence of 10 mM magnesium (Fig. 5D).
Yeast Pkc1 requires PS and DAG for its activity on Nem1-ΔTM and Spo7-ΔTM, but not on their peptides
The above data indicated that Pkc1 activity requires PS and DAG on the protein substrate, but does not require the lipids on the peptide substrate. We sought evidence that this novel property is common to other Pkc1 substrates. To address this question, we examined the Pkc1 phosphorylation of Nem1 and Spo7, proteins that are intimately related to the function of Pah1. Nem1 (catalytic subunit) and Spo7 (regulatory subunit) in the endoplasmic reticulum form a protein phosphatase complex (28) that catalyzes the dephosphorylation of Pah1 and thereby regulates its location, abundance, and PA phosphatase activity (29–31, 47, 52). Phosphoproteomic studies (53, 54) indicate that Nem1 (e.g., Ser-201) and Spo7 (Ser-22) are phosphoproteins, and bioinformatics indicate that both proteins contain putative target sites for Pkc1. Nem1-ΔTM and Spo7-ΔTM, which lack transmembrane domains, were expressed in E. coli and then purified as a soluble protein complex (Fig. 6A). The purified complex of Nem1-ΔTM and Spo7-ΔTM was examined for its phosphorylation by Pkc1 in the standard reaction mixture containing PS and DAG. Both subunits of the phosphatase complex were phosphorylated by Pkc1 (Fig. 6B). Phosphoamino acid analysis showed that they are phosphorylated on a serine residue (Fig. 6C), and phosphopeptide mapping indicated that the phosphorylation site is located in one major peptide of Nem1-ΔTM and on multiple peptides of Spo7-ΔTM (Fig. 6D). Moreover, similar to that of Pah1, the phosphorylations of Nem1-ΔTM (Fig. 7A) and Spo7-ΔTM (Fig. 7B) by Pkc1 required PS and DAG for their maximum phosphorylations, and showed that PS has a stronger regulatory effect than DAG.
Fig. 6.

Nem1 and Spo7 are phosphorylated by Pkc1 on serine residues. A: SDS-PAGE (12% gel) of Nem1-ΔTM and His6-tagged Spo7-ΔTM expressed in E. coli (lane 1), purified by affinity chromatography with nickel-nitrilotriacetic acid-agarose (lane 2), and further purified by ion-exchange chromatography with Q-Sepharose (lane 3). The stoichiometry of Nem1-ΔTM to Spo7-ΔTM in the preparation was 1.5:1 (mol/mol). B: The complex of Nem1-ΔTM (80 μg/ml) and His6-tagged Spo7-ΔTM (30 μg/ml) was incubated for 20 min with 70 ng Pkc1 in the presence of 50 μM [γ-32P]ATP, 10 mM MgCl2, 0.5 mM PS, 0.15 mM DAG, and 1.7 mM CaCl2. Pkc1 or the complex of Nem1-ΔTM and Spo7-ΔTM was omitted from the reaction where indicated. Following the incubation, the reaction mixtures were resolved by SDS-PAGE (12% gel) and the gel was dried and subjected to phosphorimaging analysis. The positions of phosphorylated Nem1-ΔTM and Spo7-ΔTM are indicated. C, D: 32P-labeled Nem1-ΔTM and Spo7-ΔTM in the polyacrylamide gel were transferred to a polyvinylidene difluoride membrane and then treated with HCl or L-1-tosylamido-2-phenylethyl chloromethyl ketone-treated trypsin. Phosphoamino acids in the acid hydrolysate were separated on cellulose TLC plates by two-dimensional electrophoresis (C), whereas phosphopeptides in the tryptic digest were separated by electrophoresis (from left to right) in the first dimension and by chromatography (from bottom to top) in the second dimension (D). Phosphoamino acids and phosphopeptides resolved on the TLC plates were subjected to phosphorimaging analysis. The positions of the standard phosphoamino acids, phosphoserine (p-Ser), phosphothreonine (p-Thr), and phosphotyrosine (p-Tyr) are indicated by dashed line circles. The data shown are representative of three experiments.
Fig. 7.

Effects of PS and DAG on the Pkc1 phosphorylations of Nem1-ΔTM, Spo7-ΔTM, and the Nem1 and Spo7 peptides. Nem1-ΔTM (80 μg/ml) (A), Spo7-ΔTM (30 μg/ml) (B), Nem1 peptide (197-LRAQSVKSRPR-207, 100 μM) (C), or Spo7 peptide (17-SASIVSGPRRR-27, 100 μM) (D) was incubated for 20 min with 70 ng Pkc1, 50 μM [γ-32P]ATP, and 10 mM MgCl2 in the presence of 0.5 mM PS, 0.15 mM DAG, or both. A, B: CaCl2 (1.7 mM) was also included in the phosphorylation of Nem1-ΔTM and Spo7-ΔTM. The reaction mixtures were resolved by SDS-PAGE (12% gel) and the gel was dried and subjected to phosphorimaging analysis. The levels of Nem1-ΔTM and Spo7-ΔTM phosphorylations were normalized to their maximum phosphorylations in the presence of DAG and PS. C, D: CaCl2 was not included in the phosphorylation of the Nem1 and Spo7 peptides. The reaction mixtures were spotted onto the P81 phosphocellulose paper, which was washed with 75 mM phosphoric acid and subjected to scintillation counting. The levels of the peptide phosphorylation were normalized to their maximum phosphorylations (Nem1 peptide, 82 nmol/min/mg; Spo7 peptide, 22 nmol/min/mg) in the presence of DAG and PS. The data shown are the averages of three experiments ± SD (error bars).
Pkc1 was also examined for its activity on the peptides of Nem1 and Spo7: a Nem1 peptide (197-LRAQSVKSRPR-207) containing Ser-201 and a Spo7 peptide (17-SASIVSGPRRR-27) containing Ser-22. It phosphorylated the Nem1 and Spo7 peptides and their phosphorylations (Fig. 7C, D), like that of the Pah1 peptide, did not require PS and/or DAG for their maximum phosphorylations.
Pkc1 requires PS and DAG for its activity on Cki1 choline kinase and Ura7 CTP synthetase
To further confirm the requirement of PS and DAG for Pkc1 activity on lipid metabolic enzymes, we examined the phosphorylations of Cki1 choline kinase and Ura7 CTP synthetase. Cki1 choline kinase catalyzes the phosphorylation of choline to produce phosphocholine (4, 55), the committed step in the synthesis of PC via the CDP-choline branch of the Kennedy pathway (1, 2). Ura7 CTP synthetase catalyzes the ATP-dependent transfer of the amide nitrogen from glutamine to the C-4 position of UTP to form CTP (56, 57). The CTP generated by the reaction may be used to synthesize the CDP-choline needed to synthesize PC via the Kennedy pathway (1, 2). These enzymes are phosphorylated by rat brain PKC (5, 6). The maximum phosphorylation of Cki1 (Fig. 8A) or Ura7 (Fig. 8B) (7) by Pkc1 was observed when both PS and DAG were present in the reaction. When the kinase reactions were performed in the absence of the lipids, the extents of Cki1 and Ura7 phosphorylations were reduced by 96 and 70%, respectively (Fig. 8). Cki1 (5) and Ura7 (8, 9) peptide substrates have been used to examine phosphorylation by rat brain PKC. Unfortunately, these peptides are no longer available and we did not resynthesize them for analysis with Pkc1. Nonetheless, the current study demonstrates that PS and DAG are required for the maximum phosphorylations of the Cki1 and Ura7 proteins by Pkc1.
Fig. 8.

Effects of PS and DAG on the Pkc1 phosphorylation of Cki1 choline kinase and Ura7 CTP synthetase. Cki1 (50 μg/ml) (A) or Ura7 (45 μg/ml) (B) was incubated with 70 ng Pkc1 for 20 min in the presence of 50 μM [γ-32P]ATP, 10 mM MgCl2, and 1.7 mM CaCl2 by the inclusion of 0.5 mM PS, 0.15 mM DAG, or both. The reaction mixtures were resolved by SDS-PAGE and the gel was dried and subjected to phosphorimaging analysis. The levels of Cki1 and Ura7 phosphorylations were normalized to their maximum phosphorylations in the presence of PS and DAG. The data shown are the averages of three experiments ± SD (error bars).
Pkc1 interacts with PS
Because PS had a great stimulatory effect on the Pkc1 phosphorylation of lipid metabolic proteins, we examined the physical interaction of the protein kinase with PS in the liposome-binding assay. PC liposomes containing various amounts of PS were incubated with Pkc1, and then were separated by centrifugation from the unbound form of the protein kinase. Pkc1 bound to the liposomes was resolved by SDS-PAGE, and was subjected to fluoroimaging after its staining with SYPRO Ruby. In this analysis, the association of Pkc1 with the liposomes increased in a PS-dependent manner (Fig. 9), indicating that Pkc1 binds to PS. At the highest surface concentration of PS (20 mol%) used in the assay, about 15% of the Pkc1 was shown to bind to the liposomes with the Kd value of 3.3 mol%.
Fig. 9.

Pkc1 binds to the PS-containing liposomes. Pkc1 (560 ng) was incubated for 10 min with PC liposomes containing the indicated amounts of PS. Following the incubation, the reaction mixtures were centrifuged to collect the liposomes. The liposomal pellets were resolved by SDS-PAGE and the gel was stained with SYPRO Ruby and subjected to fluoroimaging for quantification. The data shown are the averages of three experiments ± SD (error bars). The dissociation constant (Kd) of the Pkc1-PS interaction is shown in the inset.
Pah1 abundance is stabilized in yeast cells lacking PS
In S. cerevisiae, Pah1 PA phosphatase is regulated for its abundance by the proteasome-mediated degradation as the cells progress from the exponential to the stationary phase of growth (58). In vitro, the phosphorylation of Pah1 by rat brain PKC stimulates its degradation by the 20S proteasome (59). The phosphorylation-mediated degradation of Pah1 in vivo is supported by the observation that the enzyme abundance is stabilized by alanine mutations of the phosphorylation sites (3). We reasoned that if the stimulation of Pkc1 activity by PS enhances the degradation of Pah1, its abundance in the cell would be stabilized by a mutation that blocks PS synthesis. To address this hypothesis, we examined the abundance of Pah1 in the cho1Δ mutant, which lacks the enzyme catalyzing the synthesis of PS (38, 60, 61). As described previously (58), by the late exponential to stationary phase, the Pah1 protein in wild-type cells was hardly detected by Western blot analysis (Fig. 10). In the cho1Δ mutant, however, the Pah1 protein was stabilized as the mutant progressed from the exponential to stationary phases of growth (Fig. 10).
Fig. 10.

Effect of the cho1Δ mutation on the level of Pah1 during cell growth. Wild-type and the cho1Δ cells were grown in synthetic complete medium supplemented with 1 mM choline. The cells at the indicated time of growth were lysed and subjected to immunoblot analysis with anti-Pah1 antibody. The amounts of Pah1 on the immunoblot were determined by fluoroimaging. The amount of Pah1 from the wild-type or cho1Δ mutant was normalized to the amount of that found at the 16 h time point. The lack of the Cho1 protein in the cho1Δ mutant was confirmed by immunoblot analysis with anti-Cho1 antibody. The yeast cells, grown for 16–20 h, were in the exponential phase, whereas those grown for 36 h were in the stationary phase. The data shown are the averages of three experiments ± SD (error bars).
DISCUSSION
In contrast to current belief (20, 21), the work presented here showed that S. cerevisiae PKC, i.e., Pkc1, requires PS and DAG for its maximum activity. The lipid requirement of the protein kinase, however, was governed by the nature of its substrate. Pkc1 required PS and DAG for the phosphorylation of lipid metabolic proteins (e.g., Pah1, Nem1, Spo7, Cki1, and Ura7), but not for the phosphorylation of peptides derived from the protein substrates (e.g., Pah1, Nem1, and Spo7). The lack of lipid requirement for Pkc1 activity on the peptide substrate is consistent with the prior observation that the protein kinase does not require PS or DAG when the enzyme activity is assayed with its pseudosubstrate peptide containing Thr-401 and the Bck1 peptide containing Ser-939 (20, 21). The binding of Rho1 GTPase to Pkc1 renders the protein kinase dependent on PS when the Bck1 peptide is utilized as substrate (22), and data indicate that the phospholipid facilitates the interaction of Rho1 with Pkc1 (62). It is yet unclear whether Pkc1 activity on the Bck1 protein requires PS in the absence or presence of Rho1 GTPase.
Mammalian PKC has been extensively studied for its structure, function, and regulatory properties (16–19). With respect to catalytic function, the interaction of PKC with lipids is necessary to dissociate the pseudosubstrate domain from the substrate-binding cavity of the enzyme; the lipid requirement persists regardless of whether PKC acts on protein or peptide substrates (17–19). Our data from the analysis of Pah1 and its peptide phosphorylation by rat brain PKC was consistent with this mechanism. That Pkc1 does not require lipids for its activity on the peptide substrates indicates that the yeast and mammalian PKC enzymes differ with respect to catalytic mechanism. In any event, when a lipid metabolic protein was used as substrate, its maximum phosphorylation by Pkc1 was dependent on PS and DAG and, in particular, on the phospholipid. In a protein-lipid overlay assay, the GST-C1 (lipid binding domain of Pkc1) has been shown to interact with PS, but not with DAG (62). Here, we demonstrated that Pkc1 binds to liposomes in a PS-dependent manner, and the protein-lipid interaction occurs in the absence of Rho1 GTPase. In fact, Rho1 GTPase was not included in any of the assays performed in this study. While Rho1 GTPase is clearly an important component of the cell wall integrity (MAP kinase) pathway, the cellular process to which the most attention has been paid with respect to Pkc1 (63, 64), its role in the regulation of lipid metabolism, is yet unclear.
The current focus of our laboratory is to understand how the Pah1/Nem1-Spo7 phosphatase cascade is controlled by phosphorylation. Pah1 PA phosphatase plays an important role in the regulation of lipid metabolism; the enzyme controls the cellular levels of its substrate PA, which is used for the synthesis of membrane phospholipids (e.g., PS and phosphatidylinositol) via CDP-DAG, and its product DAG, which is used for the synthesis of triacylglycerol and phospholipids via the Kennedy pathway (1, 2, 65, 66). The phosphorylation of Pah1 by the cyclin-dependent protein kinases, Pho85 (52) and Cdc28 (29, 47) as well as by protein kinase A (67), inactivates the enzyme; these phosphorylations cause the retention of Pah1 in the cytosol apart from its substrate PA that is localized at the nuclear/endoplasmic reticulum membrane. Pah1 associates with the nuclear/endoplasmic reticulum membrane through its dephosphorylation catalyzed by the Nem1-Spo7 protein phosphatase complex (i.e., PA phosphatase phosphatase) (28, 29, 31). The phosphorylation of Pah1 by Pho85 (52) and protein kinase A (67) also causes the inhibition of its PA phosphatase activity; whereas, the dephosphorylation of the enzyme by the Nem1-Spo7 phosphatase complex (30, 31) stimulates its activity. The phosphorylation of Pah1 by Pkc1 does not affect its location or PA phosphatase activity, but instead affects its stability (3). The Pkc1 phosphorylation of Pah1, which is favored when the enzyme is not phosphorylated on the target sites of Pho85 and Cdc28, promotes its degradation by the 20S proteasome (3, 59). Here we showed that the abundance of Pah1 is stabilized in cho1Δ mutant cells lacking the ability to synthesize PS, which indirectly supports the notion that PS regulates Pkc1 activity in vivo. We acknowledge that this finding alone does not generalize the PS dependency of Pkc1 activity on the protein targets in vivo. Yet, it is known that the C1 lipid-binding domain of Pkc1 is needed for the stress-mediated activation of the MAP kinase pathway (68), and that a role for PS is supported by the attenuation of the pathway in cho1Δ mutant cells (62).
Another new finding reported here is that the Nem1-Spo7 phosphatase complex is a target of Pkc1. Current studies are directed at defining how this regulation affects the function of Pah1 PA phosphatase in lipid metabolism. Finally, there is a plethora of potential protein targets for Pkc1 in S. cerevisiae that are involved in a variety of physiological processes [e.g., carbohydrate metabolism, mating, cell aging, cytoskeleton biogenesis, and spore wall synthesis (15)] that have yet to be explored experimentally. An important take-home message from the current work is that the conditions for the phosphorylation of new Pkc1 targets should consider the lipid requirement(s) of the enzyme phosphorylation activity.
Acknowledgments
The authors acknowledge David Levin and Alexandra Newton for helpful discussions during the course of this work.
Footnotes
Abbreviations:
- DAG
- diacylglycerol
- PA
- phosphatidate
- PC
- phosphatidylcholine
- PKC
- protein kinase C
- Pkc1
- yeast protein kinase C
- PS
- phosphatidylserine
This work was supported in whole or in part by National Institutes of Health Grants GM028140 and GM050679 from the United States Public Health Service. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Carman G. M., and Han G-S.. 2011. Regulation of phospholipid synthesis in the yeast Saccharomyces cerevisiae. Annu. Rev. Biochem. 80: 859–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henry S. A., Kohlwein S., and Carman G. M.. 2012. Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics. 190: 317–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Su W-M., Han G-S., and Carman G. M.. 2014. Cross-talk phosphorylations by protein kinase C and Pho85p-Pho80p protein kinase regulate Pah1p phosphatidate phosphatase abundance in Saccharomyces cerevisiae. J. Biol. Chem. 289: 18818–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim K-H., Voelker D. R., Flocco M. T., and Carman G. M.. 1998. Expression, purification, and characterization of choline kinase, product of the CKI gene from Saccharomyces cerevisiae. J. Biol. Chem. 273: 6844–6852. [DOI] [PubMed] [Google Scholar]
- 5.Choi M-G., Kurnov V., Kersting M. C., Sreenivas A., and Carman G. M.. 2005. Phosphorylation of the yeast choline kinase by protein kinase C. Identification of Ser25 and Ser30 as major sites of phosphorylation. J. Biol. Chem. 280: 26105–26112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang W-L., and Carman G. M.. 1995. Phosphorylation of CTP synthetase from Saccharomyces cerevisiae by protein kinase C. J. Biol. Chem. 270: 14983–14988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang W-L., Bruno M. E. C., and Carman G. M.. 1996. Regulation of yeast CTP synthetase activity by protein kinase C. J. Biol. Chem. 271: 11113–11119. [DOI] [PubMed] [Google Scholar]
- 8.Park T-S., O’Brien D. J., and Carman G. M.. 2003. Phosphorylation of CTP synthetase on Ser36, Ser330, Ser354, and Ser454 regulates the levels of CTP and phosphatidylcholine synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 278: 20785–20794. [DOI] [PubMed] [Google Scholar]
- 9.Choi M-G., Park T. S., and Carman G. M.. 2003. Phosphorylation of Saccharomyces cerevisiae CTP synthetase at Ser424 by protein kinases A and C regulates phosphatidylcholine synthesis by the CDP-choline pathway. J. Biol. Chem. 278: 23610–23616. [DOI] [PubMed] [Google Scholar]
- 10.Sreenivas A., Villa-Garcia M. J., Henry S. A., and Carman G. M.. 2001. Phosphorylation of the yeast phospholipid synthesis regulatory protein Opi1p by protein kinase C. J. Biol. Chem. 276: 29915–29923. [DOI] [PubMed] [Google Scholar]
- 11.Levin D. E., Fields F. O., Kunisawa R., Bishop J. M., and Thorner J.. 1990. A candidate protein kinase C gene, PKC1, is required for the S. cerevisiae cell cycle. Cell. 62: 213–224. [DOI] [PubMed] [Google Scholar]
- 12.Levin D. E., and Bartlett-Heubusch E.. 1992. Mutants in the S. cerevisiae PKC1 gene display a cell cycle- specific osmotic stability defect. J. Cell Biol. 116: 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamada Y., Jung U. S., Piotrowski J., and Levin D. E.. 1995. The protein kinase C-activated MAP kinase pathway of Saccharomyces cerevisiae mediates a novel aspect of the heat shock response. Genes Dev. 9: 1559–1571. [DOI] [PubMed] [Google Scholar]
- 14.Perez P., and Calonge T. M.. 2002. Yeast protein kinase C. J. Biochem. 132: 513–517. [DOI] [PubMed] [Google Scholar]
- 15.Schmitz H. P., and Heinisch J. J.. 2003. Evolution, biochemistry and genetics of protein kinase C in fungi. Curr. Genet. 43: 245–254. [DOI] [PubMed] [Google Scholar]
- 16.Mellor H., and Parker P. J.. 1998. The extended protein kinase C superfamily. Biochem. J. 332: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newton A. C., Antal C. E., and Steinberg S. F.. 2016. Protein kinase C mechanisms that contribute to cardiac remodelling. Clin. Sci. (Lond.). 130: 1499–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosse C., Linch M., Kermorgant S., Cameron A. J., Boeckeler K., and Parker P. J.. 2010. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11: 103–112. [DOI] [PubMed] [Google Scholar]
- 19.Newton A. C. 2010. Protein kinase C: poised to signal. Am. J. Physiol. Endocrinol. Metab. 298: E395–E402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antonsson B., Montessuit S., Friedli L., Payton M. A., and Paravicini G.. 1994. Protein kinase C in yeast. Characteristics of the Saccharomyces cerevisiae PKC1 gene product. J. Biol. Chem. 269: 16821–16828. [PubMed] [Google Scholar]
- 21.Watanabe M., Chen C-Y., and Levin D. E.. 1994. Saccharomyces cerevisiae PKC1 encodes a protein kinase C (PKC) homolog with a substrate specificity similar to that of mammalian PKC. J. Biol. Chem. 269: 16829–16836. [PubMed] [Google Scholar]
- 22.Kamada Y., Qadota H., Python C. P., Anraku Y., Ohya Y., and Levin D. E.. 1996. Activation of yeast protein kinase C by Rho1 GTPase. J. Biol. Chem. 271: 9193–9196. [DOI] [PubMed] [Google Scholar]
- 23.Xu Z., Su W-M., and Carman G. M.. 2012. Fluorescence spectroscopy measures yeast PAH1-encoded phosphatidate phosphatase interaction with liposome membranes. J. Lipid Res. 53: 522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishizuka Y. 1984. The role of protein kinase C in cell surface signal transduction and tumor promotion. Nature. 308: 693–698. [DOI] [PubMed] [Google Scholar]
- 25.Nishizuka Y. 1988. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 334: 661–665. [DOI] [PubMed] [Google Scholar]
- 26.Ogita K., Miyamoto S., Koide H., Iwai T., Oka M., Ando K., Kishimoto A., Ikeda K., Fukami Y., and Nishizuka Y.. 1990. Protein kinase C in Saccharomyces cerevisiae: Comparison with the mammalian enzyme. Proc. Natl. Acad. Sci. USA. 87: 5011–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon A. J., Milner Y., Saville S. P., Dvir A., Mochly-Rosen D., and Orr E.. 1991. The identification and purification of a mammalian-like protein kinase C in the yeast Saccharomyces cerevisiae. Proc. Biol. Sci. 243: 165–171. [DOI] [PubMed] [Google Scholar]
- 28.Siniossoglou S., Santos-Rosa H., Rappsilber J., Mann M., and Hurt E.. 1998. A novel complex of membrane proteins required for formation of a spherical nucleus. EMBO J. 17: 6449–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santos-Rosa H., Leung J., Grimsey N., Peak-Chew S., and Siniossoglou S.. 2005. The yeast lipin Smp2 couples phospholipid biosynthesis to nuclear membrane growth. EMBO J. 24: 1931–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Hara L., Han G-S., Peak-Chew S., Grimsey N., Carman G. M., and Siniossoglou S.. 2006. Control of phospholipid synthesis by phosphorylation of the yeast lipin Pah1p/Smp2p Mg2+-dependent phosphatidate phosphatase. J. Biol. Chem. 281: 34537–34548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su W-M., Han G-S., and Carman G. M.. 2014. Yeast Nem1-Spo7 protein phosphatase activity on Pah1 phosphatidate phosphatase is specific for the Pho85-Pho80 protein kinase phosphorylation sites. J. Biol. Chem. 289: 34699–34708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schenk P. M., Baumann S., Mattes R., and Steinbiss H. H.. 1995. Improved high-level expression system for eukaryotic genes in Escherichia coli using T7 RNA polymerase and rare ArgtRNAs. Biotechniques. 19: 196–198, 200. [PubMed] [Google Scholar]
- 33.Sambrook J., Fritsch E. F., and Maniatis T.. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 34.Innis M. A., and Gelfand D. H.. 1990. Optimization of PCRs. In PCR Protocols: A Guide to Methods and Applications. M. A. Innis, D. H. Gelfand, J. J. Sninsky, et al., editors. Academic Press, Inc., San Diego. 3–12. [Google Scholar]
- 35.Thomas B. J., and Rothstein R.. 1989. Elevated recombination rates in transcriptionally active DNA. Cell. 56: 619–630. [DOI] [PubMed] [Google Scholar]
- 36.Choi H-S., Han G-S., and Carman G. M.. 2010. Phosphorylation of yeast phosphatidylserine synthase by protein kinase A: identification of Ser46 and Ser47 as major sites of phosphorylation. J. Biol. Chem. 285: 11526–11536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rose M. D., Winston F., and Heiter P.. 1990. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 38.Atkinson K. D., Jensen B., Kolat A. I., Storm E. M., Henry S. A., and Fogel S.. 1980. Yeast mutants auxotropic for choline or ethanolamine. J. Bacteriol. 141: 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laemmli U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227: 680–685. [DOI] [PubMed] [Google Scholar]
- 40.Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O’Shea E. K., and Weissman J. S.. 2003. Global analysis of protein expression in yeast. Nature. 425: 737–741. [DOI] [PubMed] [Google Scholar]
- 41.Han G-S., Wu W-I., and Carman G. M.. 2006. The Saccharomyces cerevisiae lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J. Biol. Chem. 281: 9210–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang W-L., McDonough V. M., Ozier-Kalogeropoulos O., Adeline M-T., Flocco M. T., and Carman G. M.. 1994. Purification and characterization of CTP synthetase, product of the URA7 gene in Saccharomyces cerevisiae. Biochemistry. 33: 10785–10793. [DOI] [PubMed] [Google Scholar]
- 43.Bradford M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- 44.Guengerich F. P., Wang P., and Davidson N. K.. 1982. Estimation of isozymes of microsomal cytochrome P-450 in rats, rabbits, and humans using immunochemical staining coupled with sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Biochemistry. 21: 1698–1706. [DOI] [PubMed] [Google Scholar]
- 45.Burnette W. N. 1981. Western blotting: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112: 195–203. [DOI] [PubMed] [Google Scholar]
- 46.Haid A., and Suissa M.. 1983. Immunochemical identification of membrane proteins after sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Methods Enzymol. 96: 192–205. [DOI] [PubMed] [Google Scholar]
- 47.Choi H-S., Su W-M., Morgan J. M., Han G-S., Xu Z., Karanasios E., Siniossoglou S., and Carman G. M.. 2011. Phosphorylation of phosphatidate phosphatase regulates its membrane association and physiological functions in Saccharomyces cerevisiae: identification of Ser602, Thr723, and Ser744 as the sites phosphorylated by CDC28 (CDK1)-encoded cyclin-dependent kinase. J. Biol. Chem. 286: 1486–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyle W. J., Van der Geer P., and Hunter T.. 1991. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 201: 110–149. [DOI] [PubMed] [Google Scholar]
- 49.MacDonald J. I. S., and Kent C.. 1994. Identification of phosphorylation sites in rat liver CTP:phosphocholine cytidylyltransferase. J. Biol. Chem. 269: 10529–10537. [PubMed] [Google Scholar]
- 50.MacDonald R. C., MacDonald R. I., Menco B. P., Takeshita K., Subbarao N. K., and Hu L. R.. 1991. Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim. Biophys. Acta. 1061: 297–303. [DOI] [PubMed] [Google Scholar]
- 51.Smith S. W., Weiss S. B., and Kennedy E. P.. 1957. The enzymatic dephosphorylation of phosphatidic acids. J. Biol. Chem. 228: 915–922. [PubMed] [Google Scholar]
- 52.Choi H-S., Su W-M., Han G-S., Plote D., Xu Z., and Carman G. M.. 2012. Pho85p-Pho80p phosphorylation of yeast Pah1p phosphatidate phosphatase regulates its activity, location, abundance, and function in lipid metabolism. J. Biol. Chem. 287: 11290–11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holt L. J., Tuch B. B., Villen J., Johnson A. D., Gygi S. P., and Morgan D. O.. 2009. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 325: 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swaney D. L., Beltrao P., Starita L., Guo A., Rush J., Fields S., Krogan N. J., and Villén J.. 2013. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods. 10: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wittenberg J., and Kornberg A.. 1953. Choline phosphokinase. J. Biol. Chem. 202: 431–444. [PubMed] [Google Scholar]
- 56.Lieberman I. 1956. Enzymatic amination of uridine triphosphate to cytidine triphosphate. J. Biol. Chem. 222: 765–775. [PubMed] [Google Scholar]
- 57.Long C. W., and Pardee A. B.. 1967. Cytidine triphosphate synthetase of Escherichia coli B. Purification and kinetics. J. Biol. Chem. 242: 4715–4721. [PubMed] [Google Scholar]
- 58.Pascual F., Hsieh L-S., Soto-Cardalda A., and Carman G. M.. 2014. Yeast Pah1p phosphatidate phosphatase is regulated by proteasome-mediated degradation. J. Biol. Chem. 289: 9811–9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsieh L-S., Su W-M., Han G-S., and Carman G. M.. 2015. Phosphorylation regulates the ubiquitin-independent degradation of yeast Pah1 phosphatidate phosphatase by the 20S proteasome. J. Biol. Chem. 290: 11467–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Atkinson K., Fogel S., and Henry S. A.. 1980. Yeast mutant defective in phosphatidylserine synthesis. J. Biol. Chem. 255: 6653–6661. [PubMed] [Google Scholar]
- 61.Letts V. A., Klig L. S., Bae-Lee M., Carman G. M., and Henry S. A.. 1983. Isolation of the yeast structural gene for the membrane-associated enzyme phosphatidylserine synthase. Proc. Natl. Acad. Sci. USA. 80: 7279–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nomura W., Ito Y., and Inoue Y.. 2017. Role of phosphatidylserine in the activation of Rho1-related Pkc1 signaling in Saccharomyces cerevisiae. Cell. Signal. 31: 146–153. [DOI] [PubMed] [Google Scholar]
- 63.Levin D. E. 2011. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics. 189: 1145–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Levin D. E. 2005. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 69: 262–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carman G. M., and Han G-S.. 2009. Phosphatidic acid phosphatase, a key enzyme in the regulation of lipid synthesis. J. Biol. Chem. 284: 2593–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pascual F., and Carman G. M.. 2013. Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim. Biophys. Acta. 1831: 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Su W-M., Han G-S., Casciano J., and Carman G. M.. 2012. Protein kinase A-mediated phosphorylation of Pah1p phosphatidate phosphatase functions in conjunction with the Pho85p-Pho80p and Cdc28p-cyclin B kinases to regulate lipid synthesis in yeast. J. Biol. Chem. 287: 33364–33376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nomura W., and Inoue Y.. 2015. Methylglyoxal activates the target of rapamycin complex 2-protein kinase C signaling pathway in Saccharomyces cerevisiae. Mol. Cell. Biol. 35: 1269–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]