

Abstract

Lysine specific demethylase 1 (LSD1) plays a pivotal role in regulating the lysine methylation. The aberrant overexpression of LSD1 has been reported to be involved in the progression of certain human malignant tumors. Abrogation of LSD1 with RNAi or small molecule inhibitors may lead to the inhibition of cancer proliferation and migration. Herein, a series of [1,2,3]triazolo[4,5-d]pyrimidine derivatives were synthesized and evaluated for their LSD1 inhibitory effects. The structure–activity relationship studies (SARs) were conducted by exploring three regions of this scaffold, leading to the discovery of compound 27 as potent LSD1 inhibitor (IC50 = 0.564 μM). Compound 27 was identified as a reversible LSD1 inhibitor and showed certain selectivity to LSD1 over monoamine oxidase A/B (MAO-A/B). When MGC-803 cells were treated with compound 27, the activity of LSD1 can be significantly inhibited, and the cell migration ability was also suppressed. Docking studies indicated that the hydrogen interaction between the nitrogen atom in the pyridine ring and Met332 could be responsible for the improved activity of 2-thiopyridine series. The [1,2,3]triazolo[4,5-d]pyrimidine scaffold can be used as the template for designing new LSD1 inhibitors.
Keywords: [1,2,3]Triazolo[4,5-d]pyrimidine; LSD1 inhibitor; migration inhibition; molecular docking
Unlike histone acetylation, phosphorylation, and ubiquitination, which were identified as a dynamic process, histone methylation has long been considered as an irreversible modification until the identification of lysine specific demethylase 1 (LSD1) in 2004.1 LSD1 can demethylate mono- and dimethylated K4 of histone 3 (H3K4) via flavin adenine dinucleotide (FAD)-dependent enzymatic oxidation, as well as remove mono- and dimethylated K9 of histone 3 (H3K9) through the interaction with other nuclear receptors.2,3 In addition, LSD1 could also demethylate nonhistone proteins such as p53, E2F transcription factor, and DNA methyltransferases (DNMTs) and further modulate their activity.4−6 By regulating the expression of target genes, LSD1 is closely associated with tumorigenesis, pluripotent stem cells, and neurodegenerative disorders.7−9 Particularly, aberrant expression of LSD1 has been associated with malignant tumors such as prostate, gastric, breast, lung cancers, and acute myelocytic leukemia.10−12 Downregulation of LSD1 expression by RNAi or inhibition of its activity by small molecules can inhibit the proliferation of cancer cells.13−15 Thus, LSD1 is considered as a promising epigenetic target for anticancer drug discovery.16
Since the discovery of LSD1, a large number of inhibitors with different chemotypes have been reported and are mainly classified into two types: (A) Irreversible inhibitors such as trans-2-phenylcyclopropylamine (2-PCPA) based LSD1 inhibitor.16−19 (B) Reversible inhibitors including (bis)thiourea, amidoxime, hydrazone, pyridine–piperidine hybrids, and other heterocyclic based LSD1 inhibitors.20−23 To date, ORY-1001, GSK2879552, and INCB059872 have advanced into clinical trials for the treatment of acute myeloid leukemia.9,24,25 Our group has previously reported several classes of LSD1 inhibitors based on different skeletons including dithiocarbamate-triazoles, pyrimidine-thioureas, steroide-triazoles, and [1,2,4]triazolo[1,5-a]pyrimidines, and some of these inhibitors exhibited potent inhibitory activity against LSD1 and were proved to be orally active.26−28 Recently, a series of [1,2,3]triazolo[4,5-d]pyrimidine derivatives possessing hydrazone moiety were reported by our lab as potent antiproliferative agents. Among them compound A (Figure 1) displayed the most potent antiproliferative ability, albeit with weak inhibitory activity toward LSD1 (only 26.77% inhibition at 10 μM).29 As for this weak affinity toward LSD1, docking studies revealed that compound A failed to occupy the active pocket of LSD1 due to the rigid hydrazone moiety. Based on the [1,2,3]triazolo[4,5-d]pyrimidine scaffold, further modification by replacing the hydrazone moiety with flexible groups (R3, X = N, O, S), along with alterations in R1 and R2, were then carried out in this work, aiming to identify more potent LSD1 inhibitors as shown in Figure 1.
Figure 1.
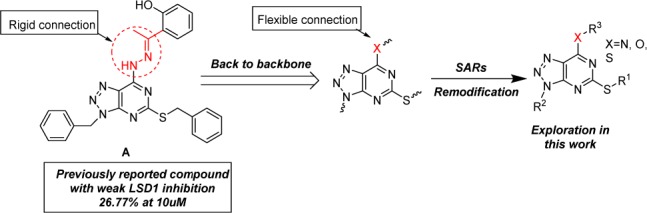
Compound A with weak anti-LSD1 activity and strategy employed for further optimization described in this work.
The synthetic route of designed compounds was illustrated in Scheme 1. The intermediates 1a–c and 1f–h were previously reported,29 and the intermediates 1d–e were synthesized following the same procedure as that for compounds 1a–c and 1f–h. The target compounds 3–27 were prepared in 50–85% yield by reacting 1a–h with 2a–q (amines, phenols, thiophenols, and 2-thiopyridine) under alkaline conditions.
Scheme 1. Synthesis of [1,2,3]Triazolo[4,5-d]pyrimidine Derivatives.
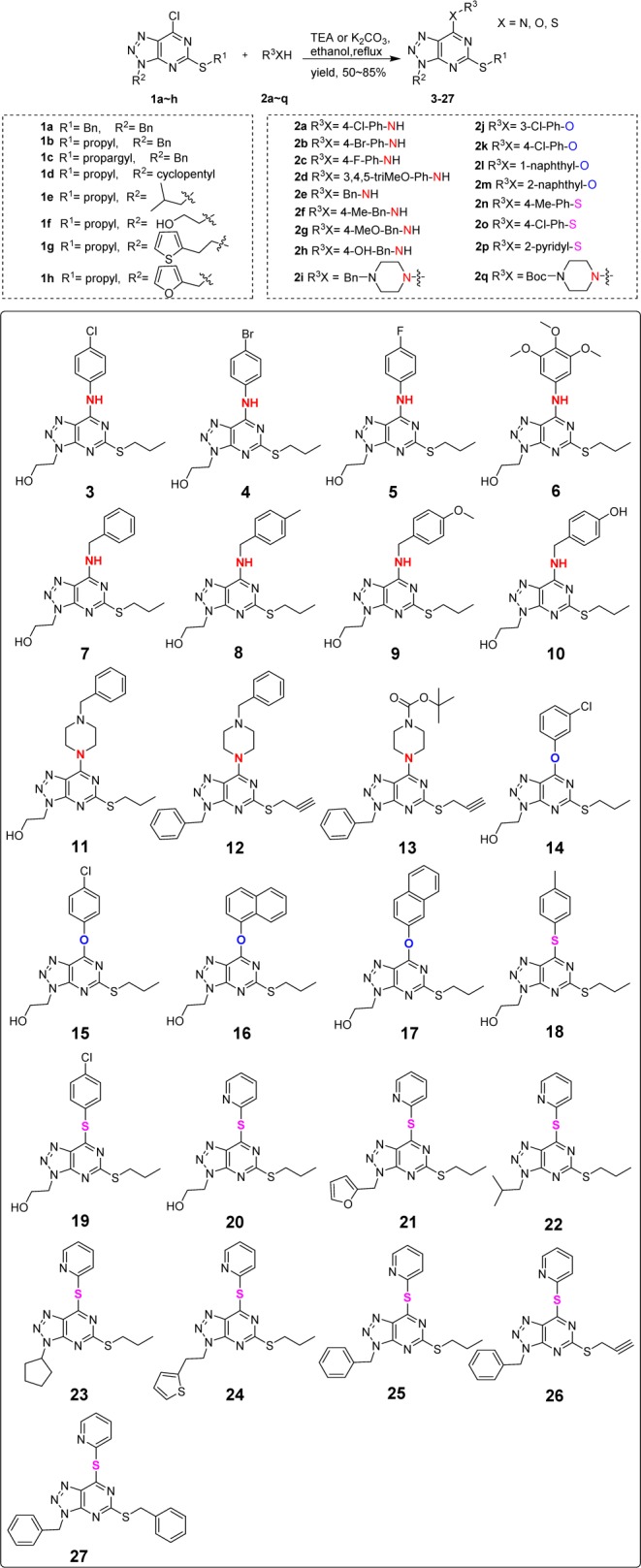
All the target compounds were screened for their in vitro inhibitory activity toward LSD1,26 and GSK2879552 was chosen as the positive control.30 Results were summarized in Tables 1 and 2. As shown in Table 1, with the exception of compound 5 having weak inhibition, the other aniline derivatives 3, 4, and 6 were found to be inactive toward LSD1 regardless of the substituent groups attached. Among the benzyl amine substituted series, compounds 7 and 8 also showed weak activities. However, compounds 9 and 10 had around 45% of inhibition rate toward LSD1, possibly due to the electron-donating property of methoxyl and hydroxyl groups on the phenyl ring. For piperazine substituted compounds 11–13, the inhibitory effects were not satisfactory as well. Phenol derivatives 14 and 15 with X = O also displayed poor inhibition against LSD1, while compounds 16 and 17 bearing the bulky naphthol groups showed significantly increased potency against LSD1 with an inhibitory rate of 57.52% and 51.73% at 10 μM, respectively. As for compounds 18 and 19, the introduction of sulfur atom led to a slight improvement in comparison with compounds 14 and 15. Noticeably, the introduction of 2-thiopyridine into the scaffold resulted in a remarkable improvement of activity (compound 20) with inhibitory rate reaching 88.24% at 10 μM.
Table 1. Inhibition of Compounds 3–20 toward LSD1.
| compd | inhibition at 10 μMa (%) | compd | inhibition at 10 μMa (%) |
|---|---|---|---|
| 3 | inactive | 12 | 27.16 |
| 4 | inactive | 13 | inactive |
| 5 | 10.18 | 14 | 15.70 |
| 6 | inactive | 15 | 11.38 |
| 7 | 23.25 | 16 | 57.52 |
| 8 | 14.31 | 17 | 51.73 |
| 9 | 45.45 | 18 | 28.29 |
| 10 | 44.86 | 19 | 38.16 |
| 11 | inactive | 20 | 88.24 |
| GSK2879552 IC50 = 0.041 ± 0.006 μM | |||
Data are represented as inhibition % at 10 μM. All experiments were independently carried out at least three times.
Table 2. Inhibition of Compounds 20–27 toward LSD1.
| compd | inhibition at 10 μMa (%) | IC50 (μM) | Log Pb |
|---|---|---|---|
| 20 | 88.24 | 2.633 ± 0.401 | 2.38 |
| 21 | 97.17 | 0.758 ± 0.003 | 3.86 |
| 22 | 94.11 | 0.960 ± 0.003 | 4.34 |
| 23 | 95.63 | 1.153 ± 0.062 | 4.69 |
| 24 | 90.79 | 0.717 ± 0.025 | 4.58 |
| 25 | 99.98 | 0.685 ± 0.062 | 4.70 |
| 26 | 95.91 | 0.678 ± 0.250 | 3.82 |
| 27 | 98.40 | 0.564 ± 0.003 | 5.30 |
| GSK2879552 | 0.041 ± 0.006 |
Data are represented as mean ± SD. All experiments were independently carried out at least three times.
Log P values were predicted at http://molsoft.com/mprop/.
Inspired by these results, we then performed further structural modifications by altering R1 and R2 groups while keeping the R3X as 2-thiopyridine unchanged. The results were listed in Table 2. It is evident that compounds 21–27 demonstrated significantly improved but comparable inhibition against LSD1 (IC50 < 2 μM), regardless of their substitution patterns. Among these compounds, the most hydrophobic compound 27 (Log P = 5.30) exerted the best activity with an IC50 value of 0.564 μM. Generally, the hydrophobic groups for R1 and R2 are preferred for the inhibitory activity against LSD1. Also, the Log P values are given in Table 2.
Next, we examined inhibitory effects of compound 27 against MAO-A/B as LSD1 shares similar amine acid sequence with MAO-A/B (Figure 2).16,26 We found that compound 27 may inactivate MAO-A 59% at 10 μM and 34% at 1 μM. Meanwhile, we also observed that compound 27 may inactivate MAO-B 39% at 10 μM and 11% at 1 μM. These results indicated that compound 27 had certain selectivity to LSD1 over MAO-A/B. To evaluate the reversibility of compound 27 for LSD1, the dilution assay were performed (Figure 3). Our results suggested that 80-fold dilution of the LSD1/compound 27 mixture resulted in the recovery of LSD1 activity, indicating that compound 27 may interact noncovalently with the enzyme. However, in the presence of the covalently binding inhibitor GSK2879552,30 LSD1 activity cannot be recovered after dilution. These results indicated the reversibility of compound 27.
Figure 2.

Inhibitory activities of compound 27 against LSD1 and MAO-A/B. Data are mean ± SD. All experiments were independently carried out at least three times.
Figure 3.

Reversibility of compound 27 was determined by dilution assay, and GSK2879552 was used as control. Data are mean ± SD. p < 0.01 was considered statistically highly significant. All experiments were independently carried out at least three times.
Besides, we also evaluated its enzymatic activity in LSD1 overexpressed gastric cancer cell line MGC-803.26 As indicated in Figure 4, MGC-803 cells were seeded in a 96-well plate and treated with compound 27 for 5 days. Then the cells were subjected to immunofluorescence with H3K4me2 antibody, which indicated the activity of LSD1 in cells. Meanwhile, 4′,6-diamidino-2-phenylindole (DAPI) was also applied for cell counting. By analyzing each well with 12 sights of immunofluorescence with high content analysis, intensity of H3K4me2 was evaluated, and the ratio of the intensity of H3K4me2 to cell number indicates the activity of LSD1. As can be seen from Figure 4A, treatment with compound 27 dose-dependently led to the significant accumulation of H3K4me2, the substrate of LSD1, without impact on the expression of LSD1 (Figure 4B). To further support its cellular activity, proteins of cells treated with compound 27 for 5 days were extracted and subjected to Western blot. As indicated in Figure 4C, both H3K4me1/2 were accumulated after treatment with compound 27. Nevertheless, the amount of H3K9me1/2 and H3K4me3 was unchanged, indicating that inactivation of LSD1 by compound 27 had no impact on the activity of other histone methyltransferase or histone demethylase. As CD86 is a surrogate cellular biomarker for LSD1 activity,31 RT-qPCR analysis of CD86 was also performed when cells were treated with compound 27, and significant induction of CD86 mRNA was observed (Figure 4D). Hence, we confirmed the cellular inhibitory effect of compound 27 against LSD1 in MGC-803 cells.
Figure 4.

Inhibitory effect of compound 27 against LSD1 in MGC-803 cells. (A) High content analysis of the LSD1 inactivation status at indicated concentration using H3K4me2 as an indicator. (B) High content analysis of LSD1 expression with indicated treatment. (C) Western blot analysis of different histone modifications at indicated concentration. (D) Relative mRNA level of CD86 in MGC-803 cells with 4 μM compound 27 treatment. Data are mean ± SD. *p < 0.05 was considered significant; **p < 0.01 was considered statistically significant. All experiments were carried out at least three times.
Inactivation of LSD1 may abrogate the gastric cancer cell proliferation and migration.26 Antiproliferative evaluation of compound 27 against MGC-803 cells was performed with MTT assay.26 When cells were treated with compound 27 for 5 days, the IC50 value of compound 27 was 7.64 ± 0.59 μM, while GSK2879552 with high potency against LSD1 (IC50 = 41 nM) presented weak cytotoxic activity toward MGC-803 (IC50 > 125 μM). As reported, the majority of cell lines were insensitive to growth inhibition by GSK2879552, and the cytotoxicity of LSD1 inhibitors is closely associated with the levels of DNA methylation, as observed in small cell lung carcinoma (SCLC).30 Cell migration ability was evaluated by transwell experiment coupled with high content analysis. For this experiment, three low concentrations of compound 27 were applied to MGC-803 cells for 24 h treatment. As shown in Figure 5A, such lower concentration of compound 27 can inhibit the migration of MGC-803 cells significantly, and further Western blot analysis also indicated the accumulation of epithelial cell marker E-Cadherin and decreasing amount of mesenchymal cell marker N-Cadherin induced by compound 27 (Figure 5B). All these results indicated that compound 27 may be a promising scaffold for developing new LSD1 inhibitor.
Figure 5.

Inhibitory effect of compound 27 against MGC-803 cells migration. (A) Treatment with compound 27 at indicated concentrations resulted in the decreased migration ability of MGC-803 cells. (B) Expression of E-Cadherin and N-Cadherin when cells were treated with compound 27 at indicated concentrations. **p < 0.01 was considered statistically significant. Data are mean ± SD. All experiments were carried out at least three times.
In order to understand the important role of 2-thiopyridine in determining the inhibitory activity toward LSD1, we chose structurally similar compounds 18 and 20 for the docking studies to rationalize the remarkable difference in the activity toward LSD1 using the MOE2015 software (PDB: 2V1D).32 With the lowest docking energy in the active site of LSD1, the triazole ring and hydroxy group in compound 20 formed hydrogen bonds with Arg316 (Figure 6A,B). In addition to the H-bond, Tyr761 had an arene–H interaction with the triazole ring. Besides, the nitrogen atom in the pyridine ring formed hydrogen interaction with Met332, which also had a hydrogen interaction with the flavin ring of FAD. For the docking prediction of compound 18 with benzene ring substitution, the results (Figure 6C,D) showed that there were only interactions between the triazole ring and residues Arg316 and Tyr761 and that no interaction was observed around the benzene ring attached to the 2-position of pyrimidine ring. Therefore, the increased activities of 2-thiopyridine series may be due to the favorable interaction of the pyridine ring with Met332.
Figure 6.
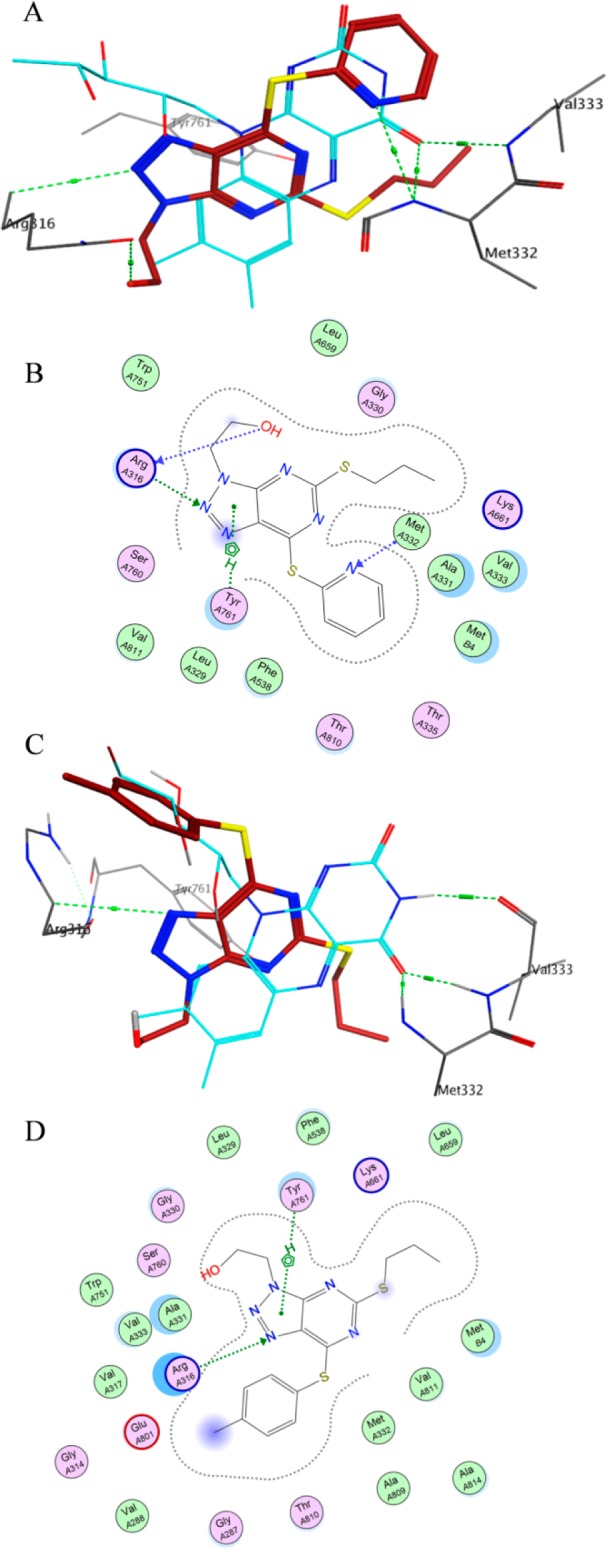
Binding modes of compounds 18 and 20 in the active site of LSD1/CoREST complex (PDB: 2V1D). For clarity, only key residues (in gray) in the active site of LSD1 are shown, together with the cofactor FAD (light blue stick model). Compounds 18 and 20 are highlighted in brown tube model, and hydrogen bonds are shown as green dashed lines. (A,B) Three- and two-dimensional binding modes of compound 20 in LSD1 active site. (C,D) Three- and two-dimensional binding modes of compound 18 in LSD1 active site.
In summary, we have designed and synthesized a novel class of [1,2,3]triazolo[4,5-d]pyrimidine derivatives as potent LSD1 inhibitors. Among them, compound 27 exhibited the most anti-LSD1 potency, as well as modulated cancer cell growth and migration in vitro at lower concentrations. SAR investigation suggested that R3X group was important for the LSD1 inhibition, and the replacement of the C atom in phenyl ring with the N atom significantly affected the inhibitory activity against LSD1. In addition, the potency of 2-thiopyridine series was also rationally explained by docking studies through forming interactions with residues Arg316 and Tyr761, especially with Met332. Thus, [1,2,3]triazolo[4,5-d]pyrimidine core may serve as an attractive structure point for the discovery of potent LSD1 inhibitors.
Glossary
ABBREVIATIONS
- LSD1
lysine specific demethylase 1
- MAO
monoamine oxidase
- FAD
flavin adenine dinucleotide
- DNMTs
DNA methyltransferases
- 2-PCPA
trans-2-phenylcyclopropylamine
- SAR
structure–activity relationship
- DAPI
4′,6-diamidino-2-phenylindole
- SCLC
small cell lung carcinoma
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00423.
Experimental sections, characterization and 1H and 13C NMR spectra of the synthesized compounds (PDF)
Author Contributions
# Z.H.L. and X.Q.L. contributed equally to this work.
This work was supported by National Key Research Programs of Proteins(2016YFA0501800); National Natural Science Foundation of China (Project No. 81430085 and No. 21372206, to H.-M.L.); Ph.D. Educational Award from Ministry of Education (No. 20134101130001, to H.-M.L.); The 2013 Young Teacher Foundation from Zhengzhou University (No. 000001307615, for Y.-C.Z.); Key Scientific Research Project for Higher Education by Department of Education of Henan Province (No. 15A350018, to Y.-C.Z.); Research on Foundation and Frontier Technology of Science and Technology Department of Henan Province (No. 162300410236, to Y.-C.Z.); National Natural Science Foundation of Youth Fund (No. 81602961, to Y.-C.Z.).
The authors declare no competing financial interest.
Supplementary Material
References
- Shi Y.; Lan F.; Matson C.; Mulligan P.; Whetstine J. R.; Cole P. A.; Casero R. A.; Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–53. 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Metzger E.; Wissmann M.; Yin N.; Muller J. M.; Schneider R.; Peters A. H. F. M.; Gunther T.; Buettner R.; Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Garcia-Bassets I.; Kwon Y. S.; Telese F.; Prefontaine G. G.; Hutt K. R.; Cheng C. S.; Ju B. G.; Ohgi K. A.; Wang J.; Escoubet-Lozach L.; Rose D. W.; Glass C. K.; Fu X. D.; Rosenfeld M. G. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 2007, 128, 505–518. 10.1016/j.cell.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Hevi S.; Kurash J. K.; Lei H.; Gay F.; Bajko J.; Su H.; Sun W. T.; Chang H.; Xu G. L.; Gaudet F.; Li E.; Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- Ooi S. K. T.; Qiu C.; Bernstein E.; Li K. Q.; Jia D.; Yang Z.; Erdjument-Bromage H.; Tempst P.; Lin S. P.; Allis C. D.; Cheng X. D.; Bestor T. H. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–U13. 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccone D. N.; Su H.; Hevi S.; Gay F.; Lei H.; Bajko J.; Xu G. L.; Li E.; Chen T. P. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009, 461, 415–U115. 10.1038/nature08315. [DOI] [PubMed] [Google Scholar]
- Wang G. G.; Allis C. D.; Chi P. Chromatin remodeling and cancer, part I: covalent histone modifications. Trends Mol. Med. 2007, 13, 363–372. 10.1016/j.molmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Di Stefano B.; Collombet S.; Jakobsen J. S.; Wierer M.; Sardina J. L.; Lackner A.; Stadhouders R.; Segura-Morales C.; Francesconi M.; Limone F.; Mann M.; Porse B.; Thieffry D.; Graf T. C/EBP alpha creates elite cells for iPSC reprogramming by upregulating Klf4 and increasing the levels of Lsd1 and Brd4. Nat. Cell Biol. 2016, 18, 371. 10.1038/ncb3326. [DOI] [PubMed] [Google Scholar]
- Maes T.; Mascaro C.; Ortega A.; Lunardi S.; Ciceri F.; Somervaille T. C. P.; Buesa C. KDM1 histone lysine demethylases as targets for treatments of oncological and neurodegenerative disease. Epigenomics 2015, 7, 609–626. 10.2217/epi.15.9. [DOI] [PubMed] [Google Scholar]
- Kahl P.; Gullotti L.; Heukamp L. C.; Wolf S.; Friedrichs N.; Vorreuther R.; Solleder G.; Bastian P. J.; Ellinger J.; Metzger E.; Schule R.; Buettner R. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006, 66, 11341–7. 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- Gao L.; Alumkal J. Epigenetic regulation of androgen receptor signaling in prostate cancer. Epigenetics 2010, 5, 100–4. 10.4161/epi.5.2.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magerl C.; Ellinger J.; Braunschweig T.; Kremmer E.; Koch L. K.; Holler T.; Buttner R.; Luscher B.; Gutgemann I. H3K4 dimethylation in hepatocellular carcinoma is rare compared with other hepatobiliary and gastrointestinal carcinomas and correlates with expression of the methylase Ash2 and the demethylase LSD1. Hum. Pathol. 2010, 41, 181–9. 10.1016/j.humpath.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Pollock J. A.; Larrea M. D.; Jasper J. S.; McDonnell D. P.; McCafferty D. G. Lysine-Specific Histone Demethylase 1 Inhibitors Control Breast Cancer Proliferation in ER alpha-Dependent and -Independent Manners. ACS Chem. Biol. 2012, 7, 1221–1231. 10.1021/cb300108c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q. S.; Huang Y.; Marton L. J.; Woster P. M.; Davidson N. E.; Casero R. A. Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino Acids 2012, 42, 887–898. 10.1007/s00726-011-1004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv T. F.; Yuan D. M.; Miao X. H.; Lv Y. L.; Zhan P.; Shen X. K.; Song Y. Over-Expression of LSD1 Promotes Proliferation, Migration and Invasion in Non-Small Cell Lung Cancer. PLoS One 2012, 7, e35065. 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. C.; Ma J. L.; Wang Z. R.; Li J.; Jiang B.; Zhou W.; Shi X.; Wang X.; Zhao W.; Liu H. M. A Systematic Review of Histone Lysine-Specific Demethylase 1 and Its Inhibitors. Med. Res. Rev. 2015, 35, 1032–71. 10.1002/med.21350. [DOI] [PubMed] [Google Scholar]
- Vianello P.; Botrugno O. A.; Cappa A.; Ciossani G.; Dessanti P.; Mai A.; Mattevi A.; Meroni G.; Minucci S.; Thaler F.; Tortorici M.; Trifiro P.; Valente S.; Villa M.; Varasi M.; Mercurio C. Synthesis, biological activity and mechanistic insights of 1-substituted cyclopropylamine derivatives: a novel class of irreversible inhibitors of histone demethylase KDM1A. Eur. J. Med. Chem. 2014, 86, 352–63. 10.1016/j.ejmech.2014.08.068. [DOI] [PubMed] [Google Scholar]
- Schmidt D. M.; McCafferty D. G. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–16. 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- Vianello P.; Botrugno O. A.; Cappa A.; Dal Zuffo R.; Dessanti P.; Mai A.; Marrocco B.; Mattevi A.; Meroni G.; Minucci S.; Stazi G.; Thaler F.; Trifiro P.; Valente S.; Villa M.; Varasi M.; Mercurio C. Discovery of a Novel Inhibitor of Histone Lysine-Specific Demethylase 1A (KDM1A/LSD1) as Orally Active Antitumor Agent. J. Med. Chem. 2016, 59, 1501–17. 10.1021/acs.jmedchem.5b01209. [DOI] [PubMed] [Google Scholar]
- Sharma S. K.; Wu Y.; Steinbergs N.; Crowley M. L.; Hanson A. S.; Casero R. A.; Woster P. M. (Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. J. Med. Chem. 2010, 53, 5197–212. 10.1021/jm100217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazeldine S.; Pachaiyappan B.; Steinbergs N.; Nowotarski S.; Hanson A. S.; Casero R. A. Jr.; Woster P. M. Low molecular weight amidoximes that act as potent inhibitors of lysine-specific demethylase 1. J. Med. Chem. 2012, 55, 7378–91. 10.1021/jm3002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorna V.; Theisen E. R.; Stephens B.; Warner S. L.; Bearss D. J.; Vankayalapati H.; Sharma S. High-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J. Med. Chem. 2013, 56, 9496–508. 10.1021/jm400870h. [DOI] [PubMed] [Google Scholar]
- Wu F.; Zhou C.; Yao Y.; Wei L.; Feng Z.; Deng L.; Song Y. 3-(Piperidin-4-ylmethoxy)pyridine Containing Compounds Are Potent Inhibitors of Lysine Specific Demethylase 1. J. Med. Chem. 2016, 59, 253–63. 10.1021/acs.jmedchem.5b01361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. C.; Yu B.; Chen Z. S.; Liu Y.; Liu H. M. TCPs: privileged scaffolds for identifying potent LSD1 inhibitors for cancer therapy. Epigenomics 2016, 8, 651–66. 10.2217/epi-2015-0002. [DOI] [PubMed] [Google Scholar]
- Wu L. C.; Konkol L. C.; Lajkiewicz Neil.; Lu L.; Xu M. Z.; Yao W. Q.; Yu Z. Y.; Zhang C.; He C. H.. Imidazopyridines and imidazopyrazines as LSD1 inhibitors. US 2016/0009712 A1.
- Zheng Y. C.; Duan Y. C.; Ma J. L.; Xu R. M.; Zi X.; Lv W. L.; Wang M. M.; Ye X. W.; Zhu S.; Mobley D.; Zhu Y. Y.; Wang J. W.; Li J. F.; Wang Z. R.; Zhao W.; Liu H. M. Triazole-dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration. J. Med. Chem. 2013, 56, 8543–60. 10.1021/jm401002r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L. Y.; Zheng Y. C.; Wang S. Q.; Wang B.; Wang Z. R.; Pang L. P.; Zhang M.; Wang J. W.; Ding L.; Li J.; Wang C.; Hu B.; Liu Y.; Zhang X. D.; Wang J. J.; Wang Z. J.; Zhao W.; Liu H. M. Design, synthesis, and structure-activity relationship of novel LSD1 inhibitors based on pyrimidine-thiourea hybrids as potent, orally active antitumor agents. J. Med. Chem. 2015, 58, 1705–16. 10.1021/acs.jmedchem.5b00037. [DOI] [PubMed] [Google Scholar]
- Yu B.; Qi P. P.; Shi X. J.; Huang R. L.; Guo H.; Zheng Y. C.; Yu D. Q.; Liu H. M. Efficient synthesis of new antiproliferative steroidal hybrids using the molecular hybridization approach. Eur. J. Med. Chem. 2016, 117, 241–255. 10.1016/j.ejmech.2016.04.024. [DOI] [PubMed] [Google Scholar]
- Li Z. H.; Yang D. X.; Geng P. F.; Zhang J.; Wei H. M.; Hu B.; Guo Q.; Zhang X. H.; Guo W. G.; Zhao B.; Yu B.; Ma L. Y.; Liu H. M. Design, synthesis and biological evaluation of [1,2,3]triazolo[4,5-d]pyrimidine derivatives possessing a hydrazone moiety as antiproliferative agents. Eur. J. Med. Chem. 2016, 124, 967–980. 10.1016/j.ejmech.2016.10.022. [DOI] [PubMed] [Google Scholar]
- Mohammad H. P.; Smitheman K. N.; Kamat C. D.; Soong D.; Federowicz K. E.; Van Aller G. S.; Schneck J. L.; Carson J. D.; Liu Y.; Butticello M.; Bonnette W. G.; Gorman S. A.; Degenhardt Y.; Bai Y.; McCabe M. T.; Pappalardi M. B.; Kasparec J.; Tian X.; McNulty K. C.; Rouse M.; McDevitt P.; Ho T.; Crouthamel M. T.; Hart K.; Concha N. O.; McHugh C. F.; Miller W. H.; Dhanak D.; Tummino P. J.; Carpenter C. L.; Johnson N. W.; Hann C. L.; Kruger R. G. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Lynch J. T.; Cockerill M. J.; Hitchin J. R.; Wiseman D. H.; Somervaille T. C. CD86 expression as a surrogate cellular biomarker for pharmacological inhibition of the histone demethylase lysine-specific demethylase 1. Anal. Biochem. 2013, 442, 104–6. 10.1016/j.ab.2013.07.032. [DOI] [PubMed] [Google Scholar]
- Forneris F.; Binda C.; Adamo A.; Battaglioli E.; Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J. Biol. Chem. 2007, 282, 20070–4. 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.