

Abstract
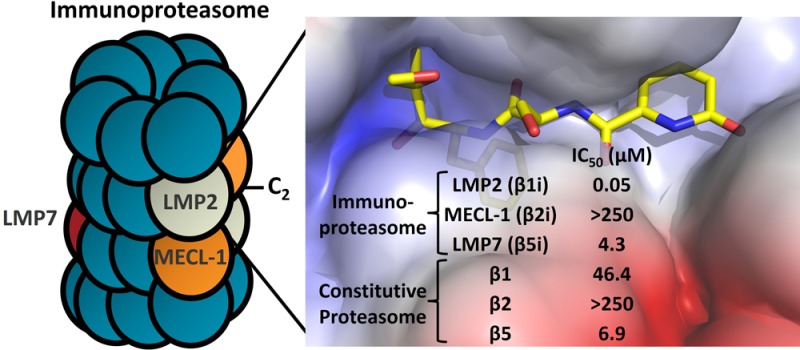
Building upon the success of bortezomib (VELCADE) and carfilzomib (KYPROLIS), the design of a next generation of inhibitors targeting specific subunits within the immunoproteasome is of interest for the treatment of autoimmune disease. There are three catalytic subunits within the immunoproteasome (low molecular mass polypeptide-7, -2, and multicatalytic endopeptidase complex subunit-1; LMP7, LMP2, and MECL-1), and a campaign was undertaken to design a potent and selective LMP2 inhibitor with sufficient properties to allow for sustained inhibition in vivo. Screening a focused library of epoxyketones revealed a series of potent dipeptides that were optimized to provide the highly selective inhibitor KZR-504 (12).
Keywords: LMP2, immunoproteasome, constitutive proteasome, β1i, β1c
With the Food and Drug Administration’s (FDA) approval of the peptide inhibitors bortezomib (1) and carfilzomib (2), inhibition of the proteasome has proven an effective therapeutic strategy to target the plasma cell neoplasm, multiple myeloma. In most cells, the constitutive proteasome is responsible for nonlysosomal intracellular protein degradation, mediated by the β1, β2, and β5 catalytic subunits.1 Cells of hematopoietic origin (e.g., lymphocytes) contain proteasomes expressing three distinct catalytic subunits, LMP7 (low-molecular mass polypeptide-7), LMP2, and MECL-1 (multicatalytic endopeptidase complex-like 1); referred to elsewhere as β5i, β1i, and β2i, respectively.2 This so-called immunoproteasome is also expressed in nonhematopoietic cells exposed to inflammatory cytokines and plays an important role in antigen presentation, influencing cytokine production, and T-cell differentiation and survival.3
Carfilzomib and bortezomib primarily inhibit with nearly equal potency the chymotrypsin-like β5 and LMP7 proteasome subunits.4 Building upon the positive clinical experience of these dual-inhibitors, it is quite intriguing to consider the effect of selectively targeting various subunits of the proteasome.5 Derivatization of the peptide backbone of epoxyketone analogues has allowed for the design of inhibitors that target specific subunits of the proteasome. For example, ONX 0914 (3, formerly PR-957)6 and PR-924 (4)7 selectively target immunoproteasome subunits, while PR-893 (5)7 and PR-825 (6)6 selectively target the β5 subunit of the constitutive proteasome (Figure 1). These compounds contain a epoxyketone functionality that covalently binds specifically to an N-terminal threonine residue in the active site of proteasome catalytic subunits.8 Administration of ONX 0914 reduces cytokine production in vitro and inflammation and tissue damage in mouse models of lupus nephritis,9 arthritis,6 experimental autoimmune encephalomyelitis (EAE), and colitis.10 Selective immunoproteasome inhibition also promotes cardiac allograft acceptance in mice.11 Thus, the immunoproteasome represents an intriguing target for the treatment of autoimmune disorders.
Figure 1.

Covalent proteasome inhibitors.
A first-generation, moderately selective covalent inhibitor of LMP2, UK-101 (7), induces cell death in prostate cancer cell lines at a high concentration and suppresses in vivo tumor growth in a xenograft mouse model of prostate cancer.12,13 Additional cell-permeable selective LMP2 inhibitors in the epoxyketone (az-NC-001 (8)14 and LU-001i (9)15) and boronate compound class have since been designed.16
LMP2–/– mice exhibit defects in B and T helper (Th) cell function and dendritic cell secretion of pro-inflammatory cytokines.17 These results, however, have not been confirmed with a selective LMP2 inhibitor. To gain a better understanding of the immunoregulatory effects of LMP2 inhibition both in vitro and in animal models, we sought to characterize a selective tool compound with suitable pharmaceutic properties to allow for sustained target inhibition in vivo.
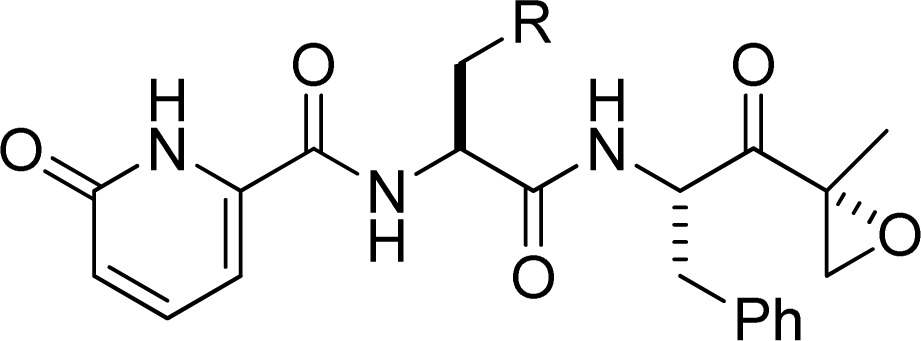
Primary screening of an in-house focused library of di- and tripeptide epoxyketones was performed utilizing the LMP2-selective fluorogenic substrate acetyl-Pro-Ala-Leu-7-amino-4-methylcoumarin (Ac-PAL-AMC, see Supplemental Table 1).18 Further characterization utilizing a subunit-specific ELISA (enzyme-linked immunosorbent assay), termed ProCISE, revealed that 10 (KZR-109) has excellent selectivity for LMP2 over other proteasomal subunits (Table 1). Although other reported dipeptide epoxyketone derivatives have demonstrated low affinity for β5 and LMP7, this motif is suitable for targeting LMP2.19
Table 1. Optimization of 10.

Utilizing MOLT-4 (human T cell leukemia) cellular lysate, a proteasome constitutive/immunoproteasome subunit ELISA (ProCISE) was employed for quantitative assessment of subunit-specific activity. Data are reported as the mean ± SD (n ≥ 2).
Liver S9 data collected at 90 min without NADPH present. Data are reported as the mean ± SD (n ≥ 3).
Solubility data collected at pH 7.2. Data are reported as the mean ± SD (n = 2).
The high degree of homology between proteasomal subunits and scarcity of structural data makes the design of subunit-selective inhibitors a challenge.20 Fortunately, a high degree of sequence homology is observed between mouse and human proteasome subunits around the respective catalytic sites. This allowed for an accurate human model to be built based upon the murine X-ray structure.21 In agreement with previous studies, modeling of the noncovalent complex accurately aligned with observed SAR within the epoxyketone series.22 Additionally, and as expected, IC50 values for all subunits are similar between species with the epoxyketone inhibitors tested herein (Table 1, see Supplemental Table 4 for mouse data). Previously, the selectivity of LMP7 inhibitors has been rationalized based on specific interactions within the S1–S3 pockets (Figure 2a).15,21,23 However, when examining the binding of 10, interactions with three residues (S48, S118, and H114) within the S3 pocket appears most critical for dictating LMP2 selectivity.
Figure 2.
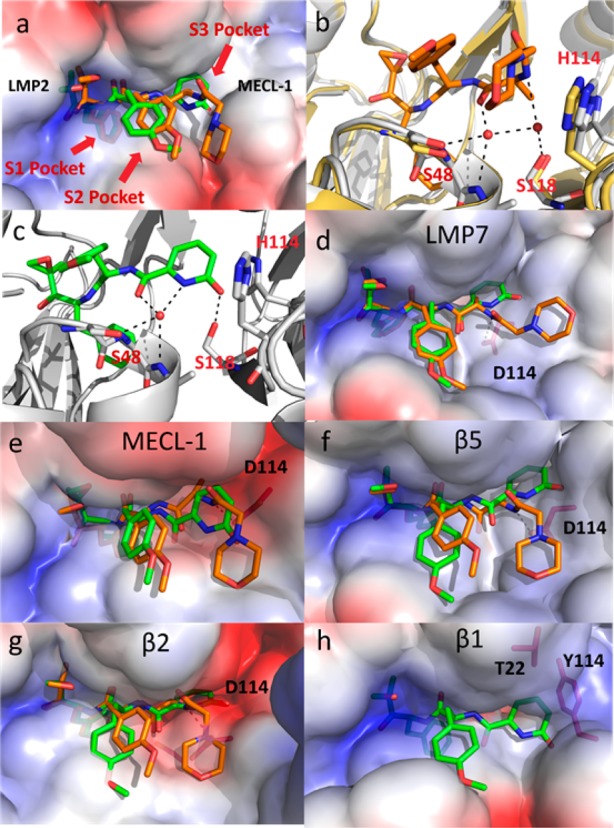
(a) ONX 0914 (orange) and 10 (green) modeled within the human LMP2 structure (van der Waals surfaces). The phenylalanine substituent of the peptide backbone of these inhibitors is buried below the surface within the S1 pocket. The shallow S3 pocket created at the interface of LMP2 and MECl-1 is occupied by the 2-pyridone of 10. (b) ONX 0914 (orange) modeled within the human LMP2 structure (gray) with mouse β1 (yellow, PDB code: 3UNF) overlaid. Based upon the high degree of sequence homology between mouse β1 and human LMP2, a similar water network is assumed to be present in both structures. (c) The 2-pyridone of 10 (green) displaces a water molecule within the S3 pocket of LMP2 (gray) while maintaining a hydrogen bonding network with S48 and S118. (d) Comparison of ONX 0914 (orange) and 10 (green) modeled in the human LMP7. (e) ONX 0914 (orange) and 10 (green) modeled in human MECL-1. (f) ONX0914 (orange) and 10 (green) modeled in human β5. (g) ONX 0914 (orange) and 10 (green) modeled in human β2. (h) Compound 10 (green) modeled in LMP2, overlaid on the human β1 model. The H114Y and A22T substitutions from LMP2 to β1 occupy the space in which the 2-pyridone would reside.
An overlay of ONX 0914 and 10 within LMP2 reveals a stark difference in how the 2-pyridone and morpholino tail are configured (Figure 2a). This was the first indication that the selectivity of 10 could be driven by interactions within the S3 pocket. The mouse cocrystal structure of ONX 0914 bound to β1 (PDB code: 3UNB) reveals that two water molecules sit within the S3 pocket forming an H-bonding network with the ligand (Figure 2b).24 Although these waters are not explicitly observed in the murine LMP2 (PDB code: 3UNF) structure, the two sequences are identical in this region, allowing us to deduce that a similar water network is also formed in LMP2. Modeling of 10 reveals that one water molecule in LMP2 is displaced with the 2-pyridone oxygen, while a hydrogen-bonding network including the remaining water, S48, and S118 is maintained (Figure 2c). A key structural difference between subunits in the S3 pocket is the D114H substitution from LMP7, MECL-1, β5, and β2 to LMP2. An H-bond is formed between D114 and the amide backbone of ONX 0914 that is absent with 10 (Figure 2d–g). The low affinity of 10 for LMP7, MECL-1, β5, and β2 is at least partially due to the 2-pyridone being unable to rotate within the S3 pocket to interact with D114. Poor binding of 10 to β1 is thought to come about from two amino acid substitutions from LMP2 to β1: H114Y and A22T. For 10 to adopt a similar binding mode in β1 as LMP2, these residues in the S3 pocket would clash with the 2-pyridone (Figure 2h). Collectively, this data suggests that the 2-pyridone motif of 10 is critical for favorable binding to LMP2 over other proteasomal subunits.
It is known that the high clearance of peptide epoxyketones is mediated predominantly by peptidase cleavage and epoxide hydrolysis.25,26 Therefore, in vitro stability data was routinely collected with liver mouse, rat, and human S9 fraction in the absence of NADPH (Table 1). Utilizing the ProCISE assay, inhibition of proteasome active sites in blood was measured 1 h after intravenous administration of compound. The low in vivo target inhibition (83% activity remaining) with 10 is likely related to its poor stability (Table 2). Therefore, another derivative that could address the shortcomings of 10 was sought.
Table 2. Mouse Pharmacodynamic Data for ONX 0914, 10, and 12a.
| analogue |
||||
|---|---|---|---|---|
| ONX 0914 | 10 | 12 | ||
| % activity remaining in blood relative to controla | LMP2 | 95.0 ± 10.6 | 83.0 ± 4.2 | 31.1 ± 6.3 |
| LMP7 | 65.3 ± 0.4 | 98.3 ± 2.5 | 102.5 ± 11.3 | |
| β5 | 99.8 ± 2.3 | 90.7 ± 3.9 | 101.7 ± 13.2 | |
Nominal doses: ONX 0914 = 1 mg/kg, 10 = 2 mg/kg, 12 = 1 mg/kg. Data are reported as the mean ± SD (n ≥ 3 animals).
A hydrophobic interaction with the p-methoxyphenyl group of 10 and the C48 residue leads to an increase in affinity to the antitarget LMP7 (Figure 3a). However, in LMP2 a more polar serine residue is present at this position (Figure 3b). Replacement of the p-methoxyphenylalanine residue in 10 with either alanine (11) or serine (12, KZR-504) diminishes this hydrophobic interaction and results in a substantial increase in solubility and stability while maintaining selectivity (Table 1).
Figure 3.

Comparison of ONX 0914 (orange) and 10 (green) within the human LMP7 and LMP2 structures (van der Waals surfaces). (a) In LMP7, a hydrophobic interaction between the p-methoxyphenyl group and C48 is a critical selectivity-driver for ONX 0914. (b) The LMP2 S2 pocket is more polar due to the C48S substitution. Replacement of the p-methoxyphenyl group on the ligand with less lipophilic functionality is well-tolerated.
Based on the attractive potency and selectivity profile of 12, it was evaluated in vivo. The greater stability of 12 vs 10, at least in part, contributes to greater target inhibition in blood (Table 2). This could also be due to differences in cell permeability/efflux; however, 12 exhibits significantly lower permeability than 10, and as further confirmation, activity in whole cells vs lysate is also compromised for 12 (see Supplemental Tables 2 and 3). Evaluating the inhibition of LMP2, and antitargets LMP7 and β5, in mouse tissues reveals that 12 is both selective and potent in vivo with >50% target inhibition achieved at >1 mg/kg in all tissues tested except brain (see Supplemental Figure 2).
Although broad immunoproteasome inhibition with ONX 0914 decreases pro-inflammatory cytokine production, the effect of selective LMP2 inhibition was unknown at the outset. A pro-inflammatory response in human peripheral blood mononuclear cells (PBMCs) stimulated with the endotoxin lipopolysaccharide (LPS) was observed with ONX 0914. However, selective inhibition of LMP2 with 10 or 12 appears to have little to no effect on cytokine production (Table 3).
Table 3. Cytokine Data for ONX 0914, 10, and 12a.
| analogue |
||||
|---|---|---|---|---|
| ONX 0914 | 10 | 12 | ||
| cytokine IC50 (μM)a | IL-12/23 p40 | 0.12 ± 0.07 | >5.6 | >9.7 |
| TNF-α | 0.56 ± 0.25 | >25 | >25 | |
| IL-6 | 0.24 ± 0.06 | >15.8 | >19.2 | |
| GMCSF | 0.22 ± 0.14 | >25 | >25 | |
| IL-8 | 1.11 ± 0.81 | >25 | >25 | |
| IL-1b | 1.47 ± 0.73 | >25 | >25 | |
| viability IC50 (μM)a,b | 3.4 ± 1.7 | >25 | >25 | |
Data are reported as the mean ± SD (n = 4).
Viability IC50 was measured using CellTiter-Glo.
Although the warhead is identical between ONX 0914 and the dipeptide structures we’ve described, exclusively changing the motif that binds within the S3 pocket is sufficient to alter the selectivity of the ligand from favoring LMP7, in the case of ONX 0914, to LMP2. Modeling was further leveraged to refine the pharmacophore of this dipeptide series and enable the design of derivatives with significantly improved in vivo stability. A quantitative measurement of proteasome active site occupancy was generated using ProCISE. Selectivity and potency data for 12 is comparable to literature data for LU-001i, the most selective analogue to date.15 Furthermore, 12 (KZR-504) can be administered at a selective dose in vivo. Collectively, the desirable properties of 12 make it an excellent addition to the armory of molecules available in the hunt for a more complete understanding of proteasome biology.
Acknowledgments
The authors would like to thank Joe Kansopon for collecting mouse ProCISE data.
Glossary
ABBREVIATIONS
- LMP2
low-molecular mass polypeptide-2
- LMP-7
low molecular mass polypeptide 7
- MECL-1
multicatalytic endopeptidase complex subunit 1
- ELISA
enzyme-linked immunosorbent assay
- PBMC
peripheral blood mononuclear cells
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00496.
Experimental procedures for the synthesis of all compounds, assay conditions, screening data for select hits, Michaelis–Menten saturation curves for characterization of the Ac-PAL-AMC substrate, full-tissue dose–response curves for 12, whole-cell ProCISE and permeability for select compounds, and modeling of 12 within LMP2 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Goldberg A. L. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem. Soc. Trans. 2007, 35, 12–17. 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- Groettrup M.; Khan S.; Schwarz K.; Schmidtke G. Interfeon-gamma inducible exchanges of 20S proteasome active site subunits: Why?. Biochimie 2001, 83, 367–372. 10.1016/S0300-9084(01)01251-2. [DOI] [PubMed] [Google Scholar]
- Groettrup M.; Kirk C. J.; Basler M. Proteasomes in immune cells: more than peptide producers?. Nat. Rev. Immunol. 2010, 10, 73–78. 10.1038/nri2687. [DOI] [PubMed] [Google Scholar]
- Huber E. M.; Groll M. Inhibitors for the Immuno- and Constitutive Proteasome: Current and Future Trends in Drug Development. Angew. Chem., Int. Ed. 2012, 51, 2–15. 10.1002/anie.201201616. [DOI] [PubMed] [Google Scholar]
- Miller Z.; Ao L.; Kim K.-B.; Lee W. Inhibitors of the immunoproteasome: current status and future directions. Curr. Pharm. Des. 2013, 19, 4140–4151. 10.2174/1381612811319220018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchamuel T.; Basler M.; Aujay M. A.; Suzuki E.; Kalim K. W.; Lauer C.; Sylvain C.; Ring E. R.; Shields J.; Jiang J.; Shwonek P.; Parlati F.; Demo S. D.; Bennett M. K.; Kirk C. J.; Groettrup M. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781. 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]
- Parlati F.; Lee S. J.; Aujay M.; Suzuki E.; Levitsky K.; Lorens J. B.; Micklem D. R.; Puurs P.; Sylvain C.; Lu Y.; Shenk K. D.; Bennett M. K. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. 10.1182/blood-2009-05-223677. [DOI] [PubMed] [Google Scholar]
- Schrader J.; Henneberg F.; Mata R. A.; Tittmann K.; Schneider T. R.; Stark H.; Gleb B.; Chari A. The inhibition mechanism of 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. 10.1126/science.aaf8993. [DOI] [PubMed] [Google Scholar]
- Ichikawa T. H.; Conley T.; Muchamuel T.; Jiang J.; Lee S.; Owen T.; Barnard J.; Nevarez S.; Goldman B. I.; Kirk C. J.; Looney J. R.; Anolik J. H. Beneficial Effect of Novel Proteasome Inhibitors in Murine Lupus via Dual Inhibition of Type I Interferon and Anutoantibody-Secreting Cells. Arthritis Rheum. 2012, 64, 493. 10.1002/art.33333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M.; Dajee M.; Moll C.; Groettrup M.; Kirk C. J. Prevention of Experimental Colitis by a Selective Inhibitor of the Immunoproteasome. J. Immunol. 2010, 185, 634–641. 10.4049/jimmunol.0903182. [DOI] [PubMed] [Google Scholar]
- Karreci E. S.; Fan H.; Uehara M.; Mihali A. B.; Singh P. K.; Kurdi A. T.; Solhjou Z.; Riella L. V.; Ghobrial I.; Laragione T.; Routray S.; Assaker J. P.; Wang R.; Sukenick G.; Shi L.; Barrat F. J.; Nathan C. F.; Lin G.; Azzi J. Brief Treatment with a Highly Selective Immunoproteasome Inhibitor Promotes Long-Term Cardiac Allograft Acceptance in Mice. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E8425–E8432. 10.1073/pnas.1618548114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y. K.; Bargagna-Mohan P.; Wehenkel M.; Mohan R.; Kim K.-B. LMP2-Specific Inhibitors: Chemical Genetic Tools for Proteasome Biology. Chem. Biol. 2007, 14, 419–430. 10.1016/j.chembiol.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehenkel M.; Ban J.-O.; Ho Y.-K.; Carmony K. C.; Hong J. T.; Kim K. B. A selective inhibitor of the immunoproteasome subunit LMP2 induces apoptosis in PC-3 cells and suppresses tumour growth in nude mice. Br. J. Cancer 2013, 107, 53–62. 10.1038/bjc.2012.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton M.; Lucas M. M.; Downey S. L.; Screen M.; Pletnev A. A.; Verdoes M.; Tokhunts R. A.; Amir O.; Goddard A. L.; Pelphrey P. M.; Wright D. L.; Overkleeft H. S.; Kisselev A. F. Selective inhibitor of proteasome’s caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem. Biol. 2009, 16, 1278–1289. 10.1016/j.chembiol.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin G.; Huber E. M.; Xin B.; van Rooden E. J.; Al-Ayed K.; Kim K.; Kisselev A. F.; Driessen C.; van der Stelt M.; van der Marel G. A.; Groll M.; Overkleeft H. S. Strucure-Based Design of β1i or β5i Specific Inhibitors of Human Immunoproteasomes. J. Med. Chem. 2014, 57, 6197–6209. 10.1021/jm500716s. [DOI] [PubMed] [Google Scholar]
- Basler M.; Lauer C.; Moebius J.; Weber R.; Przybylski M.; Kisselev A. F.; Tsu C.; Groettrup M. Why the Structure but Not the Activity of the Immunoproteasome Subunit Low Molecular Mass Polypeptide 2 Rescues Antigen Presentation. J. Immunol. 2012, 189, 1868–1877. 10.4049/jimmunol.1103592. [DOI] [PubMed] [Google Scholar]
- Hensley S. E.; Zanker D.; Dolan B. P.; David A.; Hickman H. D.; Embry A. C.; Skon C. N.; Grebe K. M.; Griffin T. A.; Chen W.; Bennink J. R.; Yewdell J. W. Unexpected role for the immunoproteasome subunit LMP2 in antiviral humoral and innate immune responses. J. Immunol. 2010, 184, 4115–4122. 10.4049/jimmunol.0903003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn C.; Gigstad K. M.; Hales P.; Garcia K.; Jones M.; Bruzzese F. J.; Barrett C.; Liu J. X.; Soucy T. A.; Sappal D. S.; Bump N.; Olhava E. J.; Fleming P.; Dick L. R.; Tsu C.; Sintchak M. D.; Blank J. L. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S β5-subunit. Biochem. J. 2010, 430, 461–476. 10.1042/BJ20100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.-J.; Aujay M. A.; Bennett M. K.; Dajee M.; Demo S. D.; Fang Y.; Ho M. N.; Jiang J.; Kirk C. J.; Laidig G. J.; Lewis E. R.; Lu Y.; Muchamuel T.; Parlati F.; Ring E.; Shenk K. D.; Shields J.; Shwonek P. J.; Stanton T.; Sun C. M.; Sylvain C.; Woo T. M.; Yang J. Design and Synthesis of an Orally Bioavailable and Selective Peptide Epoxyketone Proteasome Inhibitor (PR-047). J. Med. Chem. 2009, 52, 3028–3038. 10.1021/jm801329v. [DOI] [PubMed] [Google Scholar]
- Harshbarger W.; Miller C.; Diedrich C.; Sacchettini J. Human constitutive 20S proteasome in complex with carfilzomib. Structure 2015, 23, 418–424. 10.1016/j.str.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Huber E. M.; Basler M.; Schwab R.; Heinemeyer W.; Kirk C. J.; Groettrup M.; Groll M. Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity. Cell 2012, 148, 727–738. 10.1016/j.cell.2011.12.030. [DOI] [PubMed] [Google Scholar]
- Kawamura S.; Unno Y.; Tanaka M.; Sasaki T.; Yamano A.; Hirokawa T.; Kameda T.; Asai A.; Arisawa M.; Shuto S. Investigation of the Noncovalent Binding Mode of Covalent Proteasome Inhibitors around the Transition State by Combined Use of Cyclopropylic Strain-Based Conformational Restriction and Computational Modeling. J. Med. Chem. 2013, 56, 5829–5842. 10.1021/jm400542h. [DOI] [PubMed] [Google Scholar]
- McMinn D. M.Targeting the Immunoproteasome, 9th Annual Drug Discovery Conference, San Diego, 2014. [Google Scholar]
- For consistency, the human sequence and numbering from the published murine LMP2 X-ray structure (PDB code: 3UNF) are utilized throughout.
- Yang J.; Wang Z.; Fang Y.; Jiang J.; Zhao F.; Wong H.; Bennett M. K.; Molineaux C. J.; Kirk C. J. Pharmacokinetics, pharmacodynamics, metabolism, distribution, and excretion of carfilzomib in rats. Drug Metabolism & Disposition 2011, 39, 1873–1882. 10.1124/dmd.111.039164. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Yang J.; Kirk C.; Fang Y.; Alsina M.; Badros A.; Papadopoulos K.; Wong A.; Woo T.; Bomba D.; Li J.; Infante J. R. Clinical pharmacokinetics, metabolism, and drug-drug interaction of carfilzomib. Drug Metab. Dispos. 2013, 41, 230–237. 10.1124/dmd.112.047662. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.