

Abstract
Dihydroorotate dehydrogenase (DHODH) is an enzyme necessary for pyrimidine biosynthesis in protozoan parasites of the genus Plasmodium, the causative agents of malaria. We recently reported the identification of novel compounds derived from diversity-oriented synthesis with activity in multiple stages of the malaria parasite life cycle. Here, we report the optimization of a potent series of antimalarial inhibitors consisting of azetidine-2-carbonitriles, which we had previously shown to target P. falciparum DHODH in a biochemical assay. Optimized compound BRD9185 (27) has in vitro activity against multidrug-resistant blood-stage parasites (EC50 = 0.016 μM) and is curative after just three doses in a P. berghei mouse model. BRD9185 has a long half-life (15 h) and low clearance in mice and represents a new structural class of DHODH inhibitors with potential as antimalarial drugs.
Keywords: BRD7539, BRD9185, DHODH, malaria, diversity-oriented synthesis, Plasmodium falciparum
Malaria is a global health concern with nearly 200 million cases annually, many of which occur in sub-Saharan Africa. The disease is caused by parasitic protozoans of the genus Plasmodium and transmitted by female Anopheles mosquitos.1 Malaria is treatable using chemotherapy, but reduced efficacy of first-line treatments artemisinin and its derivatives at the Cambodia-Thailand border underscores the need for new, safe, and effective antimalarial therapies.2−5 Moreover, while most current antimalarial drugs target asexual blood-stage parasites, next-generation antimalarials should ideally also target the liver- and/or sexual blood-stage parasites to impede parasite replication and transmission from host-to-vector, respectively.6 New antimalarial candidates have entered clinical trials in this regard, including one that targets dihydroorotate dehydrogenase (DHODH).
DHODH catalyzes the flavin mononucleotide (FMN)-dependent oxidation of l-dihydroorotate to orotate as the fourth step in de novo pyrimidine biosynthesis. While most organisms use both de novo and salvage pathways to generate pyrimidines, Plasmodium parasites lack the necessary genes for the latter, making de novo pyrimidine synthesis an essential pathway for the parasite.7 One compound in the antimalarial pipeline, DSM265, has progressed to phase-II clinical trials and has activity against both asexual blood-stage and liver-stage parasites.8 DSM265 and secondary candidate DSM421 (Chart 1) comprise a class of selective and potent antimalarial DHODH inhibitors.9−15 These triazolopyrimidines remain the most well-studied and clinically relevant antimalarial DHODH inhibitors to date, but 5-benzimidazolyl-thiophene-2-carboxamides16 and 7-arylaminopyrazolo[1,5-α]pyrimidines have also been reported.17
Chart 1. Structures of PfDHODH Inhibitors in Clinical or Preclinical Development.

We recently identified numerous compounds with multistage activity by growth-inhibition phenotypic screening of 100,000 compounds prepared in advance using diversity-oriented synthesis (DOS).18 The DOS collection was synthesized using modern asymmetric organic chemistry to impart three-dimensional topographical features using the build–couple–pair strategy.19 The success of this strategy in revealing novel therapeutic targets is illustrated by the discovery of small-molecule antimalarial inhibitors of phenylalanyl-tRNA synthetase,18 PI4K,18 and cytochrome bc1 Qi,20,21 as well as others with as-yet unidentified targets.22 While our initial efforts focused on small-molecule inhibitors of phenylalanyl–tRNA synthetase, we became intrigued in BRD7539, which we had previously shown to target PfDHODH (IC50 = 0.033 μM) selectively over human (Hs) DHODH (IC50 > 50 μM). BRD7539 was reported to have potent activity against both multidrug-resistant asexual blood-stage (P. falciparum, Dd2 strain, EC50 = 0.010 μM) and liver-stage (P. berghei, EC50 = 0.015 μM) parasites but no activity against sexual blood-stage (P. falciparum, stages IV–V, EC50 > 20 μM) parasites. BRD7539 is an azetidine carbonitrile with three contiguous stereocenters (2S,3S,4S), and stereochemistry-based structure–activity relationships (SSARs) showed that only two of eight possible stereoisomers are active.18 The clinical relevance of PfDHODH inhibitors and the selectivity and potency of BRD7539 arising directly from a high-throughput screen encouraged us to pursue this series further. Here, we report our efforts to optimize this compound and to evaluate this series in vivo.
To confirm the biological activity of BRD7539 and to explore structure–activity relationships (SARs) of the scaffold, we resynthesized core structure 1 (Scheme 1) as reported.23 Deallylation of the protected azetidine core and sequential capping of the nitrogen with propyl isocyanate gave urea 2. Trityl deprotection followed by a Heck alkynylation or Suzuki reaction diversified the para-Br position and served as a route to most analogues. Biological activity of BRD7539 was confirmed in 20-point dose (n ≥ 2 independent experiments in triplicate) against a multidrug-resistant strain (P. falciparum, Dd2 strain) using a phenotypic blood-stage growth inhibition assay that models a human blood-stage infection. Additionally, in vitro stability against mice and human microsomes for 1 h was used as a guide to identify analogues with potential in vivo stability.
Scheme 1. Elaboration of the Azetidine-2-carbonitrile Scaffold.

Reagents and conditions: (a) Pd(PPh3)4, 1,3-dimethylbarbituric acid, 2:1 EtOH/DCM, 40 °C, 16 h, 92%; (b) propylisocyanate, DIPEA, DCM, 23 °C, 1 h, 96%; (c) trifluoroacetic acid, Et3SiH, DCM, 23 °C, 1 h, 87%; (d) 1-ethynyl-3-fluorobenzene, XPhos Pd-G3, Et3N, MeCN, 70 °C, 6 h, 91%.
Our initial SAR focused primarily on the acetylene (R1) region of BRD7539 as this was the most facile point of diversification to explore and a possible toxicity concern (Table 1).24 Activity was assessed using the phenotypic blood-stage growth inhibition assay. The activity of BRD7539 was reconfirmed in dose, and in vitro activity was maintained with a wide variety of hydrophobic acetylenes (4–6, 9, 10). Heteroaromatic 2- and 3-pyridyl analogues (7–8) showed significant loss in activity compared to aromatic analogues. Interestingly, cis-alkene (11) and alkane (12) derivatives of BRD7539 showed only a slight loss in activity, suggesting that the acetylene was not necessary. Indeed, unsubstituted biaryl 13 is essentially equipotent to BRD7539 while removing the distal ring (17, Scheme S1) abolished activity, indicating the need for a large hydrophobic region in the scaffold. Having removed the acetylenic toxicity concern, we decided to use this region to modulate mouse and human microsomal stability while maintaining activity. Improved stability should correlate with favorable pharmacokinetic (PK) properties. Analogues bearing a 4-pyridyl (22) and 4-methanesulfonyl (23) distal aryl were synthesized in an effort to improve solubility but were inactive in vitro. CF3-substitution (24–26) was found to impart greater in vitro microsomal stability than methoxy substituents (28–30). Ultimately, we found that the addition of two −CF3 groups on the distal phenyl ring (BRD9185, 27) to be comparable in activity and microsomal stability to BRD7539.
Table 1. Activity of BRD7539 Analogues at R1 Position against Dd2 Parasitesa.

EC50 values are the mean of at least two independent experiments.
Mouse (M) and human (H) microsomal stability (% remaining after 1 h). Data were obtained from a single experiment performed in duplicate and calculated from a six-point curve over 1 h.
We briefly sought to evaluate the role of the primary alcohol (R2) and secondary nitrile (R3) on activity (Table 2). Analogues were synthesized from BRD7539 in 1–3 steps or from commercially available starting materials (Schemes S2–S7). Modifying the nitrile to a methyl ester (32) or alcohol (33) abolished activity. This is unsurprising given our previous result that the 2S,3S,4R diastereomer, which only differs from BRD7539 at the nitrile-bearing stereocenter, is inactive, hinting at the importance of this functional group. This also illustrates the subtle but significant role stereochemistry can have on small molecule–protein interactions and highlights the strength of diversity-oriented synthesis in identifying these key interactions. Any modification of the primary alcohol, including methylation (34) or conversion to a primary amine (36), resulted in large loss in activity.
Table 2. Activity of BRD7539 Analogues at R2 and R3 Positions against Dd2 Parasitesa.
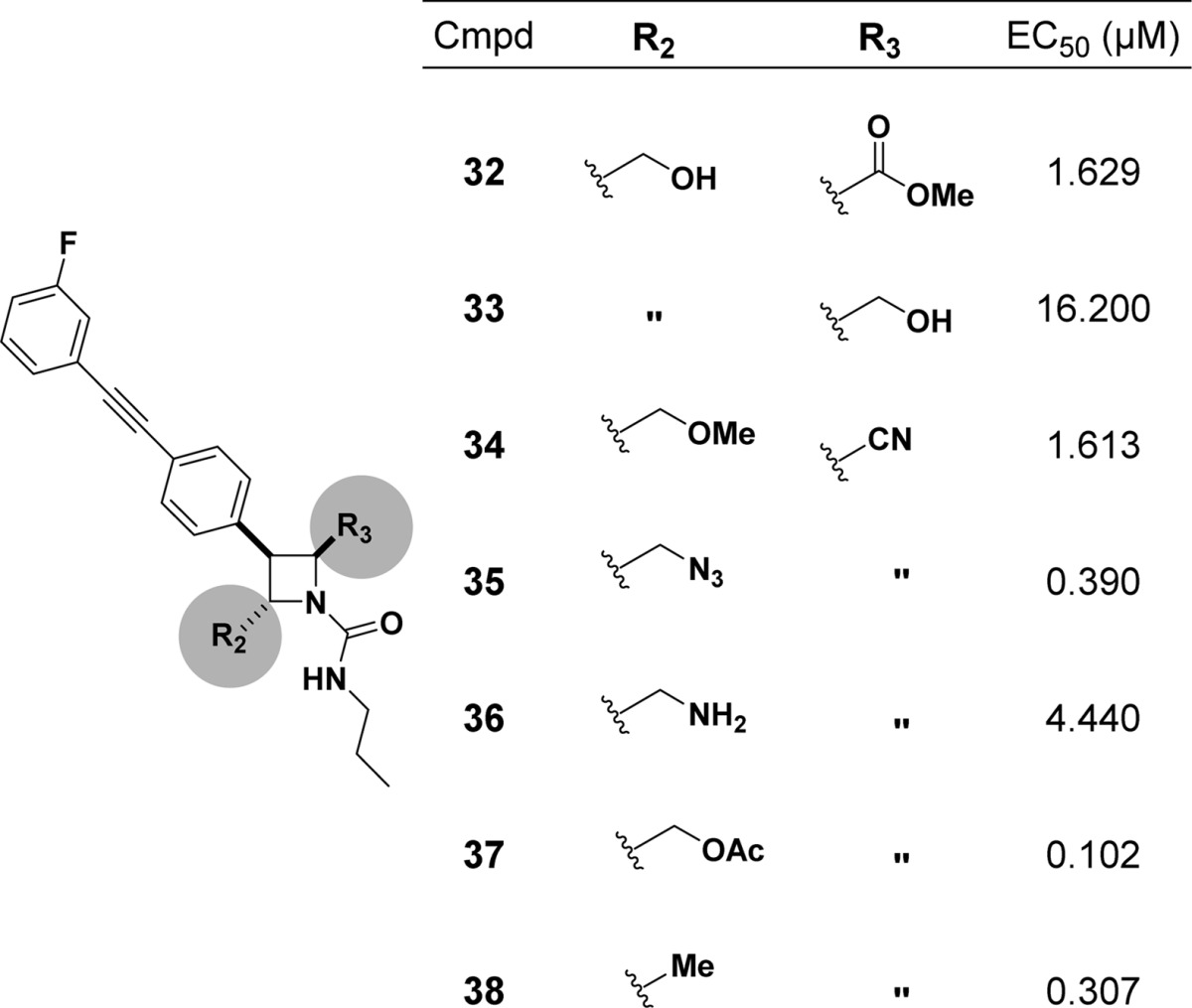
EC50 values are the mean of at least two independent experiments.
To confirm that our lead compound 27 inhibits PfDHODH despite removal of the acetylene motif, we performed biochemical assays against both recombinant P. falciparum and human DHODH enzyme (Table 3, Figure S1). Similar to hit compound BRD7539, 27 is a potent inhibitor of PfDHODH (IC50 = 0.012 μM) but not HsDHODH (IC50 > 50 μM), suggesting that this class of DHODH inhibitors provides selectivity between orthologues. The IC50 of selected analogues against PfDHODH was also shown to track well with Dd2 EC50 (Table S1). To assess the suitability of BRD9185 further for in vivo use, we measured plasma protein binding and obtained mouse PK data. Lead compound 27 is highly protein bound in both mouse and human plasma (>99%) and is a highly bioavailable (94%), long half-life (15 h) compound in mice (PO 5 mg/kg; IV 1 mg/kg) with low clearance (0.40 mL/min/kg). Notably, the DNAUC0-inf of 27 is >54.3 μM, higher than the EC50in vitro.
Table 3. Key Properties of Lead Compound 27.
| in vitro enzyme inhibition, IC50a | |
|---|---|
| PfDHODH (μM) | 0.012 |
| HsDHODH (μM) | >50 |
| plasma protein bindingb | |
|---|---|
| mouse (%) | 99.3 |
| human (%) | >99.0 |
| mouse PKc | |
|---|---|
| t1/2 (h) | 15.2 |
| C0 (μM) | 4.9 |
| Cmax (μM) | 16.9 |
| DNAUC0-inf (μM·h) | >54.3 |
| Vss (L/kg) | 0.37 |
| CL (mL/min/kg) | 0.40 |
| F (%) | 94 |
Mean of a single experiment in triplicate.
Single experiment, calculated from a six-point curve over 1 h.
t1/2, terminal half-life; C0, initial serum concentration at t = 0; Cmax, peak serum concentration; DNAUC0-inf, dose-normalized area under the plasma concentration vs time curve following PO dosing; Vss, volume of distribution at steady state; CL, plasma clearance; F%, bioavailability. IV dosing in 5% DMSO/10% cremophor/85% H2O at 0.25 mg/mL (1 mg/kg). PO dosing in 70% PEG300/30% (5% dextrose in H2O) at 0.50 mg/mL (5 mg/kg). n = 3 mice in both IV and PO groups.
Based on the promising PK properties, we were interested in evaluating the efficacy of 27in vivo (Figure 1). We used a blood-stage model with the rodent malaria parasite P. berghei that expresses luciferase and treated infected CD-1 mice for 3 days with 66.6 mg/kg of 27 or vehicle (70% PEG300, 30% solution of 5% dextrose in H2O). Bioluminescence intensity was used to measure parasite growth, and artesunate was used as a positive control. No parasites were detected after 30 days in mice treated with 27, suggesting that the analogue achieved a sterile cure for P. berghei. These results are particularly interesting in light of the properties of DSM265 and the triazolopyrimidine series, which are not effective in the P. berghei (Pb) model due to poor binding to the PbDHODH enzyme.9,14 This raises the possibility that 27 has a different mechanism of action from DSM265 on PfDHODH. However, 27 binds competitively with decylubiquinone (Figure S2), the same proposed binding site as DSM265.8 X-ray crystallography studies are underway to gain insights into binding features of these compounds.
Figure 1.
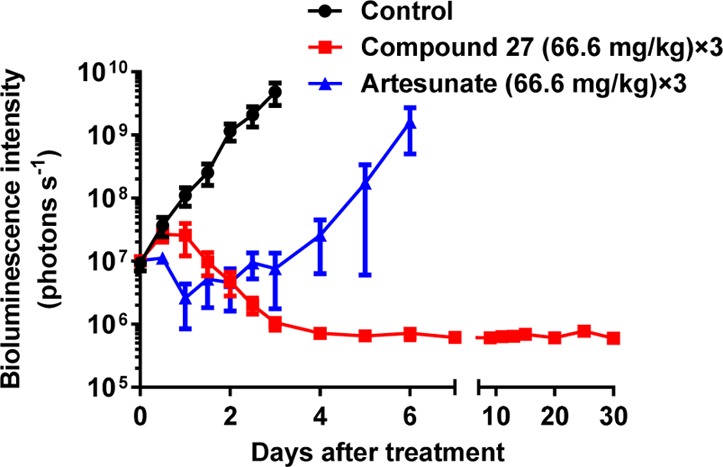
In vivo efficacy studies of BRD9185 (27) using P. berghei mouse model. CD-1 mice were infected with P. berghei (ANKA GFP-luc) at −24 h (Day −1) and dosed orally at 0, 24, and 48 h with compound 27, artesunate, or vehicle solution. Infections were monitored using the in vivo imaging system (IVIS), and bioluminescence intensity was quantified from each mouse and plotted against time. Animals with parasitemia exceeding 25% were humanely euthanized. No recrudescence was observed after 3 × 66.6 mg/kg doses of 27 (n = 4). By contrast, recrudescence is observed quickly after treatment with 3 × 66.6 mg/kg doses of artesunate (n = 2).
These data collectively show that azetidine-2-carbonitriles comprise a promising, potent, and selective new class of inhibitors of PfDHODH. In contrast to other antimalarial DHODH inhibitors to date, compound 27 exhibits a sterile cure in an in vivo P. berghei model after just three doses. Additional efforts assessing the inhibition of azetidine-2-carbonitriles against DHODH from other Plasmodium species and evaluating efficacy of this series in the humanized NSG mouse P. falciparum model are underway.
Acknowledgments
We thank Leslie N. Aldrich, Jennifer A. Beaudoin, Christopher J. Gerry, and Jingqiang Wei (Broad Institute) for advice regarding compound synthesis; Eamon Comer and Marshall Morningstar (Broad Institute) for helpful discussions; and Broad Institute Compound Management and analytical teams for assistance with compound access and characterization.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00030.
Supplementary figures, experimental details, compound characterization, and abbreviations (PDF)
This work was supported in part by the Bill & Melinda Gates Foundation (grant OPP1032518 awarded to S.L.S.). S.L.S. is an Investigator at the Howard Hughes Medical Institute. M.M. was supported by a fellowship from the National Science Foundation (DGE1144152) and from Harvard University’s Graduate Prize Fellowship. Biochemical and mechanistic studies for DHODH were supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), grant number 2012/25075-0 (to M.C.N.).
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. World Malaria Report 2015. http://who.int/malaria/publications/world-malaria-report-2015/report/en/.
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P. J.; Lindegardh N.; Socheat D.; White N. J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien C.; Henrich P. P.; Passi N.; Fidock D. A. Recent clinical and molecular insights into emerging artemisinin resistance in Plasmodium falciparum. Curr. Opin. Infect. Dis. 2011, 24, 570–577. 10.1097/QCO.0b013e32834cd3ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery E. L.; Chatterjee A. K.; Winzeler E. A. Antimalarial drug discovery: approaches and progress towards new medicines. Nat. Rev. Microbiol. 2013, 11, 849–862. 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto O.; Almagro-Garcia J.; Manske M.; MacInnis B.; Campino S.; Rockett K. A.; Amaratunga C.; Lim P.; Suon S.; Sreng S.; Anderson J. M.; Duong S.; Nguon C.; Chuor C. M.; Saunders D.; Se Y.; Lon C.; Fukuda M. M.; Amenga-Etego L.; Hodgson A. V.; Asoala V.; Imwong M.; Takala-Harrison S.; Nosten F.; Su X.-Z.; Ringwald P.; Ariey F.; Dolecek C.; Hien T. T.; Boni M. F.; Thai C. Q.; Amambua-Ngwa A.; Conway D. J.; Djimde A. A.; Doumbo O. K.; Zongo I.; Ouedraogo J. B.; Alcock D.; Drury E.; Auburn S.; Koch O.; Sanders M.; Hubbart C.; Maslen G.; Ruano-Rubio V.; Jyothi D.; Miles A.; O’Brien J.; Gamble C.; Oyola S. O.; Rayner J. C.; Newbold C. I.; Berriman M.; Spencer C. C.; McVean G.; Day N. P.; White N. J.; Bethell D.; Dondorp A. M.; Plowe C. V.; Fairhurst R. M.; Kwiatkowski D. P. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat. Genet. 2013, 45, 648–655. 10.1038/ng.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony M. P.; Burrows J. N.; Duparc S.; Moehrle J. J.; Wells T. N. The global pipeline of new medicines for the control and elimination of malaria. Malar. J. 2012, 11, 316. 10.1186/1475-2875-11-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Rathod P. K. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect. Disord.: Drug Targets 2010, 10, 226–239. 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Lotharius J.; Marsh K.; White J.; Dayan A.; White K. L.; Njoroge J. W.; El Mazouni F.; Lao Y.; Kokkonda S.; Tomchick D. R.; Deng X.; Laird T.; Bhatia S. N.; March S.; Ng C. L.; Fidock D. A.; Wittlin S.; Lafuente-Monasterio M.; Benito F. J.; Alonso L. M.; Martinez M. S.; Jimenez-Diaz M. B.; Bazaga S. F.; Angulo-Barturen I.; Haselden J. N.; Louttit J.; Cui Y.; Sridhar A.; Zeeman A. M.; Kocken C.; Sauerwein R.; Dechering K.; Avery V. M.; Duffy S.; Delves M.; Sinden R.; Ruecker A.; Wickham K. S.; Rochford R.; Gahagen J.; Iyer L.; Riccio E.; Mirsalis J.; Bathhurst I.; Rueckle T.; Ding X.; Campo B.; Leroy D.; Rogers M. J.; Rathod P. K.; Burrows J. N.; Charman S. A. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci. Transl. Med. 2015, 7, 296ra111. 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coteron J. M.; Marco M.; Esquivias J.; Deng X.; White K. L.; White J.; Koltun M.; El Mazouni F.; Kokkonda S.; Katneni K.; Bhamidipati R.; Shackleford D. M.; Angulo-Barturen I.; Ferrer S. B.; Jimenez-Diaz M. B.; Gamo F. J.; Goldsmith E. J.; Charman W. N.; Bathurst I.; Floyd D.; Matthews D.; Burrows J. N.; Rathod P. K.; Charman S. A.; Phillips M. A. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 2011, 54, 5540–5561. 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujjar R.; El Mazouni F.; White K. L.; White J.; Creason S.; Shackleford D. M.; Deng X.; Charman W. N.; Bathurst I.; Burrows J.; Floyd D. M.; Matthews D.; Buckner F. S.; Charman S. A.; Phillips M. A.; Rathod P. K. Lead-optimization of aryl and aralkyl amine based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in mice. J. Med. Chem. 2011, 54, 3935–3949. 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Gujjar R.; Malmquist N. A.; White J.; El Mazouni F.; Baldwin J.; Rathod P. K. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite, Plasmodium falciparum. J. Med. Chem. 2008, 51, 3649–3653. 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Kokkonda S.; El Mazouni F.; White J.; Burrows J. N.; Kaminsky W.; Charman S. A.; Matthews D.; Rathod P. K.; Phillips M. A. Fluorine modulates species selectivity in the triazolopyrimidine class of Plasmodium falciparum dihydroorotate dehydrogenase inhibitors. J. Med. Chem. 2014, 57, 5381–5394. 10.1021/jm500481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkonda S.; Deng X.; White K. L.; Coteron J. M.; Marco M.; de Las Heras L.; White J.; El Mazouni F.; Tomchick D. R.; Manjalanagara K.; Rudra K. R.; Chen G.; Morizzi J.; Kaminsky W.; Leroy D.; Martinez-Martinez M. S.; Jimenez-Diaz M. B.; Bazaga S. F.; Angulo-Barturen I.; Waterson D.; Burrows J. N.; Matthews D.; Charman S. A.; Phillips M. A.; Rathod P. K. Tetrahydro-2-naphthyl and 2-indanyl triazolopyrimidines targeting Plasmodium falciparum dihydroorotate dehydrogenase display potent and selective anti-malarial activity. J. Med. Chem. 2016, 59, 5416–5431. 10.1021/acs.jmedchem.6b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; White K. L.; Kokkonda S.; Deng X.; White J.; El Mazouni F.; Marsh K.; Tomchick D. R.; Manjalanagara K.; Rudra K. R.; Wirjanata G.; Noviyanti R.; Price R. N.; Marfurt J.; Shackleford D. M.; Chiu F. C; Campbell M.; Jimenez-Diaz M. B.; Bazaga S. F.; Angulo-Barturen I.; Martinez M. S.; Lafuente-Monasterio M.; Kaminsky W.; Silue K.; Zeeman A.-M.; Kocken C.; Leroy D.; Blasco B.; Rossignol E.; Rueckle T.; Matthews D.; Burrows J. N.; Waterson D.; Palmer M. J.; Rathod P. K.; Charman S. A. A triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor (DSM421) with improved drug-like properties for treatment and prevention of malaria. ACS Infect. Dis. 2016, 2, 945–957. 10.1021/acsinfecdis.6b00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Gujjar R.; El Mazouni F.; Kaminsky W.; Malmquist N. A.; Goldsmith E. J.; Rathod P. K.; Phillips M. A. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J. Biol. Chem. 2009, 284, 26999–27009. 10.1074/jbc.M109.028589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker M. L.; Bastos C. M.; Kramer M. L.; Barker R. H. Jr; Skerlj R.; Sidhu A. B.; Deng X.; Celatka C.; Cortese J. F.; Guerrero-Bravo J. E.; Crespo-Llado K. N.; Serrano A. E.; Angulo-Barturen I.; Jimenez-Diaz M. B.; Viera S.; Garuti H.; Wittlin S.; Papastogiannidis P.; Lin J. W.; Janse C. J.; Khan S. M.; Duraisingh M.; Coleman B.; Goldsmith E. J.; Phillips M. A.; Munoz B.; Wirth D. F.; Klinger J. D.; Wiegand R.; Sybertz E. Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J. Biol. Chem. 2010, 285, 33054–33064. 10.1074/jbc.M110.162081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azeredo L. F.; Coutinho J. P.; Jabor V. A.; Feliciano P. R.; Nonato M. C.; Kaiser C. R.; Menezes C. M.; Hammes A. S.; Caffarena E. R.; Hoelz L. V.; de Souza N. B.; Pereira G. A.; Ceravolo J. P.; Krettli A. U.; Boechat N. Evaluation of 7-arylaminopyrazolo[1,5-α]pyrimidines as anti-Plasmodium falciparum, antimalarial, and Pf-dihydroorotate dehydrogenase inhibitors. Eur. J. Med. Chem. 2017, 126, 72–83. 10.1016/j.ejmech.2016.09.073. [DOI] [PubMed] [Google Scholar]
- Kato N.; Comer E.; Sakata-Kato T.; Sharma A.; Sharma M.; Maetani M.; Bastien J.; Brancucci N. M.; Bittker J. A.; Corey V.; Clarke D.; Derbyshire E. R.; Dornan G. L.; Duffy S.; Eckley S.; Itoe M. A.; Koolen K. M.; Lewis T. A.; Lui P. S.; Lukens A. K.; Lund E.; March S.; Meibalan E.; Meier B. C.; McPhail J. A.; Mitasev B.; Moss E. L.; Sayes M.; VanGessel Y.; Wawer M. J.; Yoshinaga T.; Zeeman A.-M.; Avery V. M.; Bhatia S. N.; Burke J. E.; Catteruccia F.; Clardy J. C.; Clemons P. A.; Dechering K. J.; Duvall J. R.; Foley M. A.; Gusovsky F.; Kocken C. H.; Marti M.; Morningstar M. L.; Munoz B.; Neafsey D. E.; Sharma A.; Winzeler E. A.; Wirth D. F.; Scherer C. A.; Schreiber S. L. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 2016, 538, 344–349. 10.1038/nature19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancík V.; Seiler K. P.; Young D. W.; Schreiber S. L.; Clemons P. A. Distinct biological network properties between the targets of natural products and disease genes. J. Am. Chem. Soc. 2010, 132, 9259–9261. 10.1021/ja102798t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidebrecht R. W. Jr.; Mulrooney C.; Austin C. P.; Barker R. H. Jr.; Beaudoin J. A.; Cheng K. C.; Comer E.; Dandapani S.; Dick J.; Duvall J. R.; Ekland E. H.; Fidock D. A.; Fitzgerald M. E.; Foley M.; Guha R.; Hinkson P.; Kramer M.; Lukens A. K.; Masi D.; Marcaurelle L. A.; Su X.-Z.; Thomas C. J.; Weïwer M.; Wiegand R. C.; Wirth D.; Xia M.; Yuan J.; Zhao J.; Palmer M.; Munoz B.; Schreiber S. L. Diversity-oriented synthesis yields a novel lead for the treatment of malaria. ACS Med. Chem. Lett. 2012, 3, 112–117. 10.1021/ml200244k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens A. K.; Heidebrecht R. W. Jr.; Mulrooney C.; Beaudoin J. A.; Comer E.; Duvall J. R.; Fitzgerald M. E.; Masi D.; Galinsky K.; Scherer C. A.; Palmer M.; Munoz B.; Foley M.; Schreiber S. L.; Wiegand R. C.; Wirth D. F. Diversity-oriented synthesis probe targets Plasmodium falciparum cytochrome b ubiquinone reduction site and synergizes with oxidation site inhibitors. J. Infect. Dis. 2015, 211, 1097–1103. 10.1093/infdis/jiu565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann J.; Corey V.; Scherer C. A.; Kato N.; Comer E.; Maetani M.; Antonova-Koch Y.; Reimer C.; Gagaring K.; Ibanez M.; Plouffe D.; Zeeman A.-M.; Kocken C. H.; McNamara C. W.; Schreiber S. L.; Campo B.; Winzeler E. A.; Meister S. High-throughput luciferase-based assay for the discovery of therapeutics that prevent malaria. ACS Infect. Dis. 2016, 2, 281–293. 10.1021/acsinfecdis.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J. T.; Lee M. D. IV; Akella L. B.; Davoine E.; Donckele E. J.; Durak L.; Duvall J. R.; Gerard B.; Holson E. B.; Joliton A.; Kesavan S.; Lemercier B. C.; Liu H.; Marie J.-C.; Mulrooney C. A.; Muncipinto G.; Welzel-O’Shea M.; Panko L. M.; Rowley A.; Suh B.-C.; Thomas M.; Wagner F. F.; Wei J.; Foley M. A.; Marcaurelle L. A. Synthesis and profiling of a diverse collection of azetidine-based scaffolds for the development of CNS-focused lead-like libraries. J. Org. Chem. 2012, 77, 7187–7211. 10.1021/jo300974j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peukert S.; Nunez J.; He F.; Dai M.; Yusuff N.; DiPesa A.; Miller-Moslin K.; Karki R.; Lagu B.; Harwell C.; Zhang Y.; Bauer D.; Kelleher J. F.; Egan W. A method for estimating the risk of drug-induced phototoxicity and its application to smoothened inhibitors. MedChemComm 2011, 2, 973–976. 10.1039/c1md00144b. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.