

Abstract
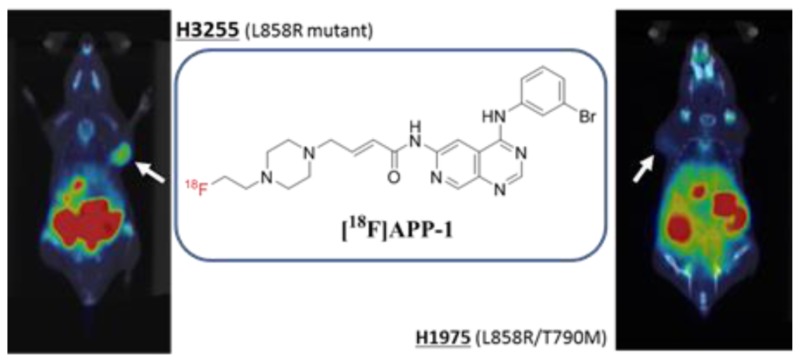
In nonsmall-cell lung carcinoma patients, L858R mutation of epidermal growth factor receptor (EGFR) is often found, and molecular target therapy using EGFR tyrosine kinase inhibitors is effective for the patients. However, the treatment frequently develops drug resistance by secondary mutation, of which approximately 50% is T790M mutation. Therefore, the ability to predict whether EGFR will undergo secondary mutation is extremely important. We synthesized a novel radiofluorinated 4-(anilino)pyrido[3,4-d]pyrimidine derivative ([18F]APP-1) and evaluated its potential as a positron emission tomography (PET) imaging probe to discriminate the difference in mutations of tumors. EGFR inhibition assay, cell uptake, and biodistribution study showed that [18F]APP-1 binds specifically to the L858R mutant EGFR but not to the L858R/T790M mutant. Finally, on PET imaging study using [18F]APP-1 with tumor-bearing mice, the H3255 tumor (L858R mutant) was more clearly visualized than the H1975 tumor (L858R/T790M mutant).
Keywords: Epidermal growth factor receptor tyrosine kinase (EGFR-TK); L858R mutant EGFR; positron emission tomography; fluorin-18; 4-(anilino)pyrido[3,4-d]pyrimidine
Nonsmall-cell lung carcinoma (NSCLC) is a class of lung cancer that accounts for 80% of lung cancer cases and is a major cause of cancer-related deaths. In approximately 10–30% of patients with NSCLC, a somatic activating mutation occurs in the tyrosine kinase (TK) domain of the epidermal growth factor receptor (EGFR) and causes the exon 21 mutation, resulting in leucine being replaced by arginine at position 858 (L858R mutant) or exon 19 deletions in the EGFR gene.1 The increased kinase activity of mutant EGFR plays a key role in cell proliferation and angiogenesis in cancer cells. Therefore, EGFR-TK inhibitors (EGFR-TKIs), such as gefitinib (Iressa), erlotinib (Tarceva), and afatinib (Giotriff), are used in NSCLC therapy to bind to the ATP domain in mutant EGFR-TK.2
However, patients who initially respond to EGFR-TKIs typically develop drug resistance within 6–12 months after the start of therapy, thereby inevitably disrupting the therapy.3 The most common factor is the occurrence of secondary mutation of the mutant EGFR gene, which leads to methionine replacing threonine at position 790 of EGFR (T790M mutation). The T790M mutation occurs in approximately 50% of patients with resistance to EGFR-TKIs.3,4 Therefore, predicting whether EGFR undergoes this secondary mutation is extremely important for planning the treatment of EGFR-TKIs. For clinical settings, genetic testing is conducted via biopsy to find mutations and provide a definitive cancer diagnosis before starting the therapy.5 However, because of the invasiveness of this procedure, such testing is done only when the effect of molecular targeting drugs decreases over the course of treatment. Therefore, developing noninvasive diagnostics techniques that can determine the therapeutic efficiency of EFGR-TKIs has become necessary.
Positron emission tomography (PET) is a molecular imaging technique that enables noninvasive detection of specific molecules in living systems and is expected, in particular, to detect the presence of mutant EGFR. Furthermore, some radiolabeled EGFR-TKIs, including [18F]gefitinib, [18F]afatinib, and [11C]erlotinib, have been investigated using PET probes for EGFR-TK-positive tumor imaging.6−12 However, the use of these compounds does not permit visualization of an EGFR-TK-positive tumor. In addition, the use of [11C]erlotinib reportedly allows primary and secondary mutant tumors to be differentiated by visualizing the former but not the latter,12 and it leads to unclear images of L858R mutant tumors, similar to the results for other radiolabeled EGFR-TKIs.
Previous investigations of a series of 4-(anilino)pyrido[3,4-d]pyrimidine derivatives as EGFR-TKIs claim that these compounds show lower half-maximal inhibitory concentration (IC50) to EGFR-TK compared with 4-(anilino)quinazoline.13−15 Therefore, the radiolabeled 4-(anilino)pyrido[3,4-d]pyrimidine derivative is also expected to become an imaging-probe candidate for tumors targeted by EGFR-TKIs. In addition, we aimed drug design on the grounds that incorporating a Michael acceptor unit at position 6 of pyrido[3,4-d]pyrimidine and quinazoline scaffolds leads to irreversible inhibition of EGFR-TK through a covalent modification of cysteine 797 in the ATP binding domain16−18 and that the piperazinyl group is useful to afford suitable water solubility and induce the radiolabeling site. Moreover, if the accumulation of the derivative differs between the primary and secondary mutant tumor, we hope to use 4-(anilino)pyrido[3,4-d]pyrimidine as an imaging probe to identify whether EGFR mutations are present. In the present study, we synthesize a novel radiofluorinated 4-(anilino)pyrido[3,4-d]pyrimidine derivative APP-1 (Figure 1) and evaluate its ability to discriminate between primary and secondary mutations in in vitro and in vivo experiments.
Figure 1.

Chemical structure of EGFR-targeting PET imaging probes.
The nonradioactive APP-1 was synthesized as indicated in Scheme 1. First, 6-aminopyrido[3,4-d]pyrimidine 1 was prepared from 2-fluoro-5-aminopyridine via the synthetic route reported by Rewcastle et al.14 Compound 1 was coupled with 4-bromocrotonic acid by using 1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride as the condensing agent followed by the addition of 1-(tert-butyloxycarbonyl)piperazine to obtain amide 2 at a 91% yield. The deprotection of the tert-butyloxycarbonyl group of compound 2 gave amine 3. Finally, compound 3 was reacted with 2-fluoroethyl tosylate to obtain APP-1.
Scheme 1. Synthesis of Nonradioactive APP-1 and Its Precursor 3.

Reagents and conditions: (a) 4-bromocrotonic acid, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, DIPEA, DMF, rt, 1 h followed by 1-(tert-butyloxycarbonyl)piperazine, rt, 30 min, 91%; (b) TFA, dichloromethane, rt, 85%; (c) 2-fluoroethyl tosylate, Et3N, DMF, rt, 45%.
The inhibition assay of APP-1 involved using erlotinib and AZD9291 to inhibit the L858R mutant and L858R/T790M mutant EGFR-TK; the results for IC50 appear in Table 1. Erlotinib inhibits the L858R mutant EGFR kinase but not the L858R/T790M mutant,12 and AZD9291 (osimertinib, Tagrisso), developed by AstraZeneca to inhibit the L858R/T790M mutant EGFR-TK and recently approved by the US Food and Drug Administration, inhibits both the L858R and L858R/T790M mutants of EGFR-TK.19 As a result, both APP-1 and erlotinib inhibit L858R mutant EGFR-TK somewhat more than L858R/T790M mutant of EGFR-TK (15.6 ± 0.8 nM vs 326 ± 64 nM and 12.5 ± 6.0 nM vs 4040 ± 1270 nM, respectively), and AZD9291 potently inhibits mutant EGFR-TK (12.3 ± 3.1 nM vs 14.5 ± 5.3 nM). Thus, radioactive APP-1 is also expected to become a probe candidate for imaging EGER-TK-positive tumors. In addition, APP-1 is found to be selective, although the range of selectivity is smaller than that of erlotinib. However, whether its selectivity suffices to visually discriminate between L858R- and L858R/T790M mutant tumors remains to be determined. Therefore, we continue evaluating the potential of APP-1 for use in PET imaging.
Table 1. IC50 for APP-1, Erlotinib, and AZD9291.
| IC50a (nM) |
||
|---|---|---|
| compd | L858R | L858R/T790M |
| APP-1 | 15.6 ± 0.8 | 326 ± 64 |
| Erlotinib | 12.5 ± 6.0 | 4040 ± 1270 |
| AZD9291 | 12.3 ± 3.1 | 14.5 ± 5.3 |
The values represent the mean ± SE (n = 3).
We synthesized 18F-labeled [18F]APP-1 from precursor 3 in a two-step reaction (Scheme 2). First, we prepared radioactive 2-[18F]fluoroethyl tosylate by a nucleophilic displacement reaction of ethylene glycol-1,2-ditosylate with the [18F]fluoride anion. After purification by preparative high-performance liquid chromatography (HPLC) and solid-phase extraction, 2-[18F]fluoroethyl tosylate was reacted with compound 3 for 20 min at 110 °C to yield [18F]APP-1 in a radiochemical yield of 3.2 ± 0.94% [n = 5, end of synthesis (EOS), from potassium [18F]fluoride] after preparative HPLC. The total operation took less than 110 min. The isolated [18F]APP-1 was identified by a HPLC analysis with coinjection of APP-1 (Figure 2). The radiochemical purity and the specific activity exceeded 95% and 40.4 GBq/μmol, respectively.
Scheme 2. Radiosynthesis of [18F]APP-1.

Reagent and conditions: (d) potassium [18F]fluoride, Kryptofix 2.2.2, MeCN, 90 °C, 5 min; and (e) Et3N, DMF, 110 °C, 20 min, 3.2 ± 0.94% radiochemical yield (n = 5, EOS from potassium [18F]fluoride).
Figure 2.

HPLC analysis of [18F]APP-1 coinjected with nonradioactive APP-1. HPLC conditions: the column was a Cosmosil 5C18-AR-II 10 mm × 250 mm; flow rate was 5.0 mL/min; UV excitation at 280 nm; and mobile phase systems were MeCN (0.1% TFA)/H2O (0.1% TFA) = 20:80 (0 min) to 40:60 (20 min).
We next investigated the cellular uptake of [18F]APP-1 by two human NSCLC cells: H3255 cells expressing the L858R mutant EGFR and H1975 cells expressing the L858R/T790M mutant. The results appear in Figure 3. The uptake of [18F]APP-1 in H3255 cells was twice as much as that in the H1975 cells. Furthermore, upon adding AZD9291 as inhibitor, the H3255-cell uptake decreases (104% ± 8.6% dose/mg protein to 46.8% ± 7.6% dose/mg protein, P < 0.01), whereas the uptake of H1975 cells remains stable. This result suggests that the different uptake is caused by the specific binding of [18F]APP-1 to L858R mutant EGFR-TK. Because the inhibition rate of H3255 did not change even when 5 μM or more of AZD9291 was added (data not shown), incomplete blockade of H3255 cells may represent nonspecific binding.
Figure 3.

Accumulation of [18F]APP-1 in H3255 and H1975 cells after incubation with and without AZD9291. The values represent the mean ± standard deviation (n = 3; *P < 0.01; “n.s.” means “not significant”).
We studied the biodistribution of [18F]APP-1 in H3255-tumor-bearing mice (Table 2). The highest accumulation of [18F]APP-1 occurred in the intestines [small intestine, 44.94% injected dose per gram (ID/g) at 1 h postinjection; colon, 59.65% ID/g at 3 h postinjection] and was excreted over time. The accumulation of [18F]APP-1 in bone was low. Therefore, [18F]APP-1 was stable in vivo. The accumulation of [18F]APP-1 in tumors was retained for at least 3 h (3.62% ID/g at 1 h postinjection, 3.80% ID/g at 3 h postinjection). Because the accumulation of [11C]erlotinib for 1 h in H3255 tumors is reported to be decreasing over time,9 we assume that [18F]APP-1 strongly binds to EGFR-TK in H3255 tumors by irreversible binding associated with a covalent bond via the Michael acceptor group. However, the slow clearance from normal tissue must be improved. At each time point, Table 2 also gives the tumor-to-blood, tumor-to-muscle, and tumor-to-lung ratios. All ratios increase over time and exceed three at 3 h postinjection. Therefore, we expect the use of [18F]APP-1 for visualization of H3255 tumors in mice.
Table 2. Biodistribution of [18F]APP-1 in H3255-Tumor-Bearing Micea.
| Injected
dose/g (%) |
||
|---|---|---|
| tissue | 1 h after injection | 3 h after injection |
| blood | 1.60 ± 0.33 | 1.12 ± 0.08 |
| heart | 1.61 ± 0.38 | 0.72 ± 0.12 |
| lung | 3.48 ± 0.51 | 1.18 ± 0.14 |
| stomach | 17.93 ± 7.12 | 3.64 ± 1.68 |
| small intestine | 44.94 ± 5.51 | 14.70 ± 4.31 |
| colon | 6.96 ± 0.61 | 59.65 ± 11.68 |
| liver | 7.86 ± 1.23 | 2.78 ± 0.38 |
| pancreas | 5.80 ± 0.78 | 1.12 ± 0.12 |
| spleen | 4.37 ± 0.50 | 1.08 ± 0.17 |
| kidney | 8.58 ± 1.25 | 2.05 ± 0.22 |
| bone | 0.81 ± 0.46 | 1.32 ± 0.40 |
| muscle | 0.63 ± 0.21 | 0.29 ± 0.06 |
| tumor (H3255) | 3.62 ± 0.87 | 3.80 ± 0.88 |
| ratios | ||
|---|---|---|
| tumor/blood | 2.25 ± 0.18 | 3.35 ± 0.66 |
| tumor/muscle | 6.15 ± 1.88 | 13.37 ± 4.02 |
| tumor/lung | 1.04 ± 0.20 | 3.21 ± 0.54 |
The values represent the mean ± SD (n = 5).
Furthermore, we studied in vivo blocking to determine the ability of [18F] to discriminate between L858R and L858R/T790M mutant EGFRs (Figure 4). Coadministration of excess AZD9291 significantly reduced the accumulation of [18F]APP-1 in H3255 tumors (54% inhibition) at 3 h postinjection. However, accumulation in H1975 tumors was not blocked by excess AZD9291. These results correspond to those of the cell uptake study (Figure 3). In other words, these results confirm that, in mice with mutant-EGFR-TK tumors, [18F]APP-1 binds specifically to L858R mutant EGFR-TK but not to L858R/T790M mutant EGFR-TK.
Figure 4.

Effect of coadministration of AZD9291 on biodistribution of [18F]APP-1 (3 h postinjection). The graphs show the mean %ID/g of four mice with the error bars giving the standard deviation (*P < 0.01).
Furthermore, we performed PET imaging of [18F]APP-1 in H3255- or H1975-tumor-bearing mice (Figure 5). The images at 3 h postinjection of [18F]APP-1 show that H3255 tumors are more clearly visualized than H1975 tumors. In addition, we measured the radioactivity in each organ and tissue after image acquisition and calculated the tumor-to-blood, tumor-to-muscle, and tumor-to-lung ratios (Table 3). The contrast between H3255 tumors and surrounding tissue is higher than that when using [11C]erlotinib.13 These results suggest that [18F]APP-1 is effective as an imaging probe that targets L858R mutant EGFR.
Figure 5.
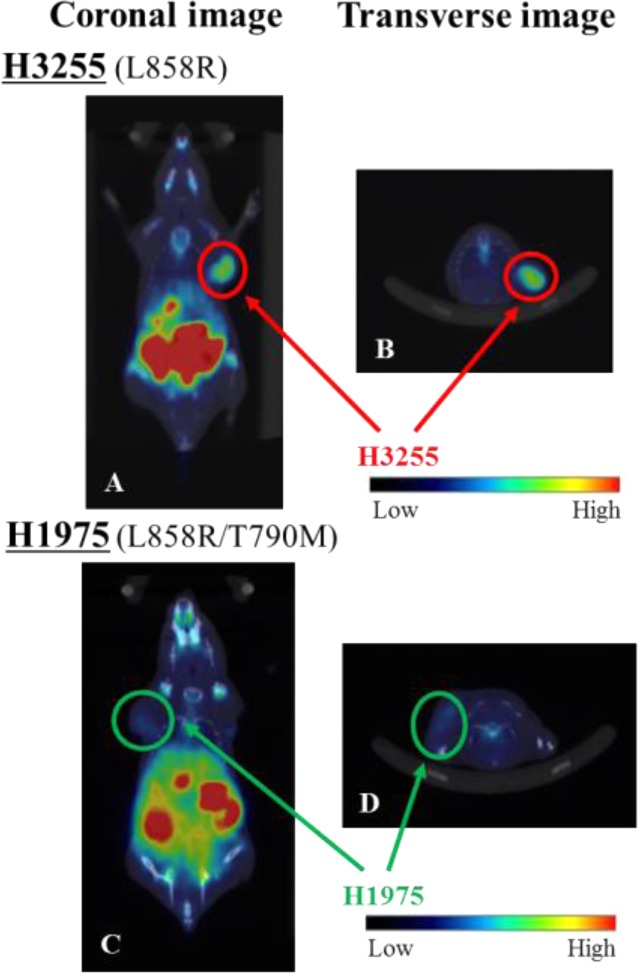
PET-CT image of [18F]APP-1 (A,B) in H3255- or (C,D) in H1975-tumor-bearing mice at 3 h postinjection. Panels (A) and (C) show coronal images, while panels (B) and (D) show transverse images.
Table 3. Ratios of Accumulated Radioactivity in H3255 and H1975 Tumor-Bearing Mice after PET-CT Imaging.
| ratio |
|||
|---|---|---|---|
| tumor | tumor/blood | tumor/muscle | tumor/lung |
| H3255 | 3.12 | 6.80 | 3.25 |
| H1975 | 0.74 | 2.95 | 0.74 |
We designed and synthesized a radiofluorinated 4-(anilino)pyrido[3,4-d]pyrimidine derivative, APP-1, as a PET imaging agent to discriminate between L858R and L858R/T790M mutant EGFRs. When used in an EGFR-TK inhibition assay, APP-1 was the strongest inhibitor of the L858R mutant EGFR-TK, and a weak inhibitor of the L858R/T790M mutant EGFR-TK. The cell-uptake study shows that [18F]APP-1 binds specifically to the L858R mutant EGFR-TK but not to the L858R/T790M mutant. Further assessment of the biodistribution revealed that [18F]APP-1 results in a high tumor-to-tissue ratio in H3255-tumor-bearing mice, while coinjection of AZD9291 suggests that the accumulation of [18F]APP-1 in tumors is specific to EGFR-TK. In addition, for PET imaging, the H3255 tumor is more clearly visualized than the H1975 tumor. Based on these results, we conclude that [18F]APP-1 has a potential to be used as a PET imaging probe to discriminate between L858R and L858R/T790M mutant EGFRs in NSCLC.
Glossary
ABBREVIATIONS
- ATP
adenosine triphosphate
- DIPEA
N,N-diisopropylethylamine
- Boc
tert-butyloxycarbonyl
- DMF
N,N-dimethylformamide
- EGFR
epidermal growth factor receptor
- EGFR-TK
epidermal growth factor receptor tyrosine kinase
- EGFR-TKIs
epidermal growth factor receptor tyrosine kinase inhibitors
- ID
injected dose
- MeCN
acetonitrile
- NSCLC
nonsmall-cell lung carcinoma
- PET
positron emission tomography
- HPLC
high-performance liquid chromatography
- rt
room temperature
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00520.
Full experimental procedures and characterization data for all new compounds described in this study (PDF)
Author Contributions
# These authors have contributed equally. The manuscript was written through contributions of all authors, and all the authors approve the final version of the manuscript.
This work was supported by the Practical Research for Innovative Cancer Control from the Japan Agency for Medical Research and Development (AMED) and Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science.
The authors declare no competing financial interest.
Supplementary Material
References
- Janku F.; Garrido-Laguna I.; Petruzelka L. B.; Stewart D. J.; Kurzrock R. Novel therapeutic targets in non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1601–1612. 10.1097/JTO.0b013e31822944b3. [DOI] [PubMed] [Google Scholar]
- Ellis P. M.; Coakley N.; Feld R.; Kuruvilla S.; Ung Y. C. Use of the epidermal growth factor receptor inhibitors gefitinib, erlotinib, afatinib, dacomitinib, and icotinib in the treatment of non-small-cell lung cancer: A systematic review. Curr. Oncol. 2015, 22, e183–e215. 10.3747/co.22.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. V.; Bell D. W.; Settleman J.; Haber D. A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Boggon T. J.; Dayaram T.; Jänne P. A.; Kocher O.; Meyerson M.; Johnson B. E.; Eck M. J.; Tenen D. G.; Halmos B. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Jackman D.; Pao W.; Riely G. J.; Engelman J. A.; Kris M. G.; Jänne P. A.; Lynch T.; Johnson B. E.; Miller V. A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360. 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H.; Seimbille Y.; Ferl G. Z.; Bodenstein C.; Fueger B.; Kim K. J.; Hsu Y. T.; Dubinett S. M.; Phelps M. E.; Czernin J.; Weber W. A. Evaluation of [18F]gefitinib as a molecular imaging probe for the assessment of the epidermal growth factor receptor status in malignant tumors. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1089–1099. 10.1007/s00259-007-0636-6. [DOI] [PubMed] [Google Scholar]
- Slobbe P.; Windhorst A. D.; Walsum M. S.; Schuit R. C.; Smit E. F.; Niessen H. G.; Solca F.; Stehle G.; van Dongen G. A. M. S.; Poot A. J. Development of [18F]afatinib as new TKI-PET tracer for EGFR positive tumors. Nucl. Med. Biol. 2014, 41, 749–757. 10.1016/j.nucmedbio.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Bonasera T. A.; Ortua G.; Rozena Y.; Kraisa R.; Freedmanc N. M. T.; Chisina R.; Gazitd A.; Levitzkib A.; Mishani E. Potential 18F-labeled biomarkers for epidermal growth factor receptor tyrosine kinase. Nucl. Med. Biol. 2001, 28, 359–374. 10.1016/S0969-8051(01)00200-1. [DOI] [PubMed] [Google Scholar]
- Dissoki S.; Eshet R.; Billauer H.; Mishani E. Modified PEG- anilinoquinazoline derivatives as potential EGFR PET agents. J. Labelled Compd. Radiopharm. 2009, 52, 41–52. 10.1002/jlcr.1569. [DOI] [Google Scholar]
- Memon A. A.; Jakobsen S.; Hansen F. D.; Sorensen B. S.; Keiding S.; Nexo E. Positron emission tomography (PET) imaging with [11C]-labeled erlotinib: A micro-PET study on mice with lung tumor xenografts. Cancer Res. 2009, 69, 873–878. 10.1158/0008-5472.CAN-08-3118. [DOI] [PubMed] [Google Scholar]
- Bahce I.; Smit E. F.; Lubberink M.; van der Veldt A. A. M.; Yaqub M.; Windhorst A. D.; Schuit R. C.; Thunnissen E.; Heideman D. A. M.; Postmus P. E.; Lammertsma A. A.; Hendrikse N. H. Development of [11C]erlotinib positron emission tomography for in vivo evaluation of EGF receptor mutational status. Clin. Cancer Res. 2013, 19, 183–193. 10.1158/1078-0432.CCR-12-0289. [DOI] [PubMed] [Google Scholar]
- Abourbeh G.; Itamar B.; Salnikov O.; Beltsov S.; Mishani E. Identifying erlotinib-sensitive non-small cell lung carcinoma tumors in mice using [11C]erlotinib PET. EJNMMI Res. 2015, 5, 4. 10.1186/s13550-014-0080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges A. J.; Zhou H.; Cody D. R.; Rewcastle G. W.; McMichael A.; Showalter H. D. H.; Fry D. W.; Kraker A. J.; Denny W. A. Tyrosine kinase inhibitors. 8. An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor. J. Med. Chem. 1996, 39, 267–276. 10.1021/jm9503613. [DOI] [PubMed] [Google Scholar]
- Rewcastle G. W.; Palmer B. D.; Thompson A. M.; Bridges A. J.; Cody D. R.; Zhou H.; Fry D. W.; McMichael A.; Denny W. A. Tyrosine kinase inhibitors. 10. isomeric 4-[(3-bromophenyl)amino]pyrido[d]pyrimidines are potent ATP binding site inhibitors of the tyrosine kinase function of the epidermal growth factor receptor. J. Med. Chem. 1996, 39, 1823–1835. 10.1021/jm9508651. [DOI] [PubMed] [Google Scholar]
- Rewcastle G. W.; Murray D. K.; Elliott W. L.; Fry D. W.; Howard C. T.; Nelson J. M.; Roberts B. J.; Vincent P. W.; Showalter H. D. H.; Winters R. T.; Denny W. A. Tyrosine kinase inhibitors. 14. structure-activity relationships for methylamino-substituted derivatives of 4-[(3-bromophenyl)amino]-6-(methylamino)pyrido[3,4-d]pyrimidine (PD 158780), a potent and specific inhibitor of the tyrosine kinase activity of receptors for the EGF family of growth factors. J. Med. Chem. 1998, 41, 742–751. 10.1021/jm970641d. [DOI] [PubMed] [Google Scholar]
- Smaill J. B.; Showalter H. D. H.; Zhou H.; Bridges A. J.; McNamara D. J.; Fry D. W.; Nelson J. M.; Sherwood V.; Vincent P. W.; Roberts B. J.; Elliott W. L.; Denny W. A. Tyrosine kinase inhibitors. 18. 6-substituted 4-anilinoquinazolines and 4-anilinopyrido[3,4-d]pyrimidines as soluble, irreversible inhibitors of the epidermal growth factor receptor. J. Med. Chem. 2001, 44, 429–440. 10.1021/jm000372i. [DOI] [PubMed] [Google Scholar]
- Tsou H. R.; Mamuya N.; Johnson B. D.; Reich M. F.; Gruber B. C.; Ye F.; Nilakantan R.; Shen R.; Discafani C.; DeBlanc R.; Davis R.; Koehn F. E.; Greenberger L. M.; Wang Y. F.; Wissner A. 6-Substituted-4-(3-bromophenylamino) quinazolines as putative irreversible inhibitors of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor (HER-2) tyrosine kinases with enhanced antitumor activity. J. Med. Chem. 2001, 44, 2719–2734. 10.1021/jm0005555. [DOI] [PubMed] [Google Scholar]
- Klutchko S. R.; Zhou H.; Winters R. T.; Tran T. P.; Bridges A. J.; Althaus I. W.; Amato D. M.; Elliott W. L.; Ellis P. A.; Meade M. A.; Roberts B. J.; Fry D. W.; Gonzales A. J.; Harvey P. J.; Nelson J. M.; Sherwood V.; Han H. K.; Pace G.; Smaill J. B.; Denny W. A.; Showalter H. D. H. Tyrosine kinase inhibitors. 19. 6-alkynamides of 4-anilinoquinazolines and 4-anilinopyrido[3,4-d]pyrimidines as irreversible inhibitors of the erbB family of tyrosine kinase receptors. J. Med. Chem. 2006, 49, 1475–1485. 10.1021/jm050936o. [DOI] [PubMed] [Google Scholar]
- Cross D. A. E.; Ashton S. E.; Ghiorghiu S.; Eberlein C.; Nebhan C. A.; Spitzler P. J.; Orme J. P.; Finlay M. R. V.; Ward R. A.; Mellor M. J.; Hughes G.; Rahi A.; Jacobs V. N.; Brewer M. R.; Ichihara E.; Sun J.; Jin H.; Ballard P.; Al-Kadhimi K.; Rowlinson R.; Klinowska T.; Richmond G. H. P.; Cantarini M.; Kim D. W.; Ranson M. R.; Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery 2014, 4, 1046–1061. 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.