

Abstract

Photoremovable protecting groups added to bioactive molecules provide spatial and temporal control of the biological effects. We present synthesis and characterization of the first photoactivatable small-molecule tubulin inhibitor. By blocking the pharmacophoric OH group on compound 1 with photoremovable 4,5-dimethoxy-2-nitrobenzyl moiety we developed the photocaged prodrug 2 that had no effect in biological assays. Short UV light exposure of the derivative 2 or UV-irradiation of cells treated with 2 resulted in fast and potent inhibition of tubulin polymerization, attenuation of cell viability, and apoptotic cell death, implicating release of the parent active compound. This study validates for the first time the photoactivatable prodrug concept in the field of small molecule tubulin inhibitors. The caged derivative 2 represents a novel tool in antitubulin approaches.
Keywords: Photoactivatable caged prodrug, tubulin inhibitor, glioblastoma
Delivering bioactive molecules to cells with temporal and spatial precision is useful for elucidating complex biological processes. One method for regulating the action of bioactive molecules employs photolabile-protecting groups (PPGs).1 The PPG is a chromophore covalently attached to the pharmacophoric moiety of the bioactive molecule, thus blocking its biological activity, a concept known as “caging”. The covalent bond between the bioactive molecule and the PPG is cleaved by irradiation with ultraviolet (UV) light, leading to the release of the parent bioactive molecule (“uncaging”). A number of PPGs have been developed for this purpose, including p-nitrobenzyl, 4,5-dimethoxy-2-nitrobenzyl (DMNB), and 6-bromo-7-hydroxycoumarine-4-ylmethyl,2 and the caging concept has been successfully applied to phototrigger calcium, neurotransmitters, nucleic acids, and antibiotics.3,4 For example, photocaged rapamycin has been used to induce controlled activity of the small GTPase Rac in the cellular context,5 and photocaged anisomycin has been employed to locally inhibit protein synthesis.6 Photocaged puromycin was effectively applied for spatiotemporal monitoring of mRNA translation.7 We have recently developed a number of caged kinase inhibitors that serve as valuable molecular probes to delineate signaling pathways.8−10 Furthermore, photocaging has been applied for delivery of molecules across membranes and for the control of side effects.11,12
Microtubule-targeting agents (MTAs) disrupt polymerization of α- and β-tubulin to form microtubules. Microtubules are crucial for cell division in mitosis, and this explains why compounds that bind to tubulin and interfere with tubulin polymerization are highly effective in killing rapidly proliferating cancer cells. Importantly, tubulin has a crucial role also in nonmitotic cells, which underlies the overall success of MTAs in cancer therapy.13−15
MTAs are structurally diverse and very often structurally complex molecules as the vast majority of these agents are natural products isolated from bacteria, plants, and marine sponges.16 With the long history of clinical efficacy, MTAs remain to date the most classical yet reliable chemotherapeutics. Microtubules targeting Vinca alkaloids (vinblastine, vincristine) and taxanes (paclitaxel, cabazitaxel) are frontline treatments for breast, ovarian, and hormone-refractory prostate cancers. However, the acquired resistance developed over the time of treatment has plagued the success of these drugs.
Mechanisms of MTA resistance are manifold, including overexpression of efflux proteins, point mutations at the paclitaxel-binding site, or polymorphism resulting in the overexpression of various β-tubulin isotypes.13 Another major limitation in the use of MTAs is the high rate of neuropathy induced by these compounds. This effect manifests itself as a painful peripheral axonal pain for which there is currently no effective symptomatic treatment.17 Myeloid toxicity and neutropenia is also frequently observed with MTAs, with subtle differences between compounds within the same family.
Clinically approved MTAs are ineffective for treatment of brain tumors as their large molecular weight (>800 g/mol) renders them unable to cross the blood–brain barrier. Hence, there has been increasing research interest toward the development of effective MTA delivery methods18−20 or identification of small-molecule tubulin inhibitors able to cross the blood–brain barrier.21,22 We discovered that a small-molecule known as CMPD1 and initially developed to inhibit p38 MAPK-MK2 signaling pathway,23 primarily inhibits tubulin polymerization.24 CMPD1 showed potent antimitotic and apoptotic activity in a panel of cancer cells. This cytotoxic activity and the small molecular weight (349 g/mol) made CMPD1 an attractive lead for the development of potential chemotherapeutic agents for brain tumors. Recently, we reported synthesis of CMPD1 analogues with improved molecular properties and demonstrated their anticancer efficacy in patient-derived glioblastoma cells.25
Herein, we present a novel concept in the class of small-molecule tubulin inhibitors. In order to reduce side effects associated with tubulin inhibitors, we developed a caged tubulin inhibitor by addition of a photoactivatable protecting group and describe its pharmacology in glioblastoma cells. We have chosen glioblastoma cell-based models as this heterogeneous brain cancer represents a major unmet medical need. Although glioblastoma was one of the first cancers to be profiled through The Cancer Genome Atlas project, making it genomically a well-characterized cancer,26 the results of glioblastoma trials using inhibitors of oncogenic drivers have been disappointing so far.27 Importantly, glioblastoma cells are sensitive to MTAs,22,25 suggesting that MTAs able to cross the blood–brain barrier could be effective in glioblastoma therapy. The photoactivatable approach presented in this work opens a new avenue to reduce side effects of MTAs as the active drug may be locally released at the tumor site.
To synthesize the photoactivatable tubulin inhibitor, the tubulin inhibitor 1 was converted into a photocaged derivative 2 (Scheme 1). We have chosen the 4,5-dimethoxy-2-nitrobenzyl (DMNB) as the PPG because of its excellent quantitative cleavage by UV irradiation.1,9,10 DMNB was attached to the phenol moiety of compound 1, as the SAR study demonstrated that the removal of the OH group resulted in significant loss of cellular efficacy.25 DMNB-caged derivative 2 was obtained in 60% yield in the final synthetic step using DMNB-bromide as a reagent, followed by reversed-phase chromatography purification. UV spectra of compounds 1 and 2 (Figure S1) revealed that inhibitor 1 shows no absorption at 365 nm, whereas the photoprodrug 2 possesses an absorption maximum around 350 nm. As the high absorption of photocaged molecules at the irradiation wavelength is crucial to trigger the PPG cleavage, we identified 365 nm as suitable irradiation wavelength.
Scheme 1. Synthesis of the Photoactivatable Tubulin Inhibitor 2.

Reagents and conditions: (a) AlCl3, HCl, N2, 96 h; (b) Zn/Hg, HCl, toluene, 24 h; (c) PYBOP, DIPEA, DMF; (d) Pd(PPh3)4, Na2CO3, EtOH/H2O, MW 130°C, 20 min; (e) K2CO3, DMF, RT.
In the photocaging concept it is essential that the parent molecule is sufficiently stable under the conditions used for uncaging by UV irradiation. Otherwise, the irradiation would degrade the released active drug immediately after the PPG cleavage. Initially, in an analytical setup to examine the UV stability of the tubulin inhibitor 1, we used a light-emitting diode (LED) reactor and a wavelength of 365 nm (5400 mW, Figure S2, lamp A) to irradiate compound 1 (1 mM). HPLC and LC–MS analysis of samples collected over the period of 20 min confirmed that inhibitor 1 was stable under these conditions (Figure S3).
To determine the kinetics of the photorelease, caged analogue 2 was UV irradiated at 365 nm (2700 mW, Figure S2, lamp A), and samples were collected at indicated time points for quantitative HPLC analysis (Figure 1). After 1 min of irradiation, approximately 80% of the bioactive inhibitor 1 was released from the photoprodrug 2. The reaction progress curve excellently fitted (R2 = 1) to the exponential one-phase decay kinetics with a time constant (τ) of 0.605 min. The maximum measured concentration of parent inhibitor 1 was reached after 2 min of irradiation. This data confirm that DMNB-caged compound 2 possesses suitable uncaging kinetics.
Figure 1.
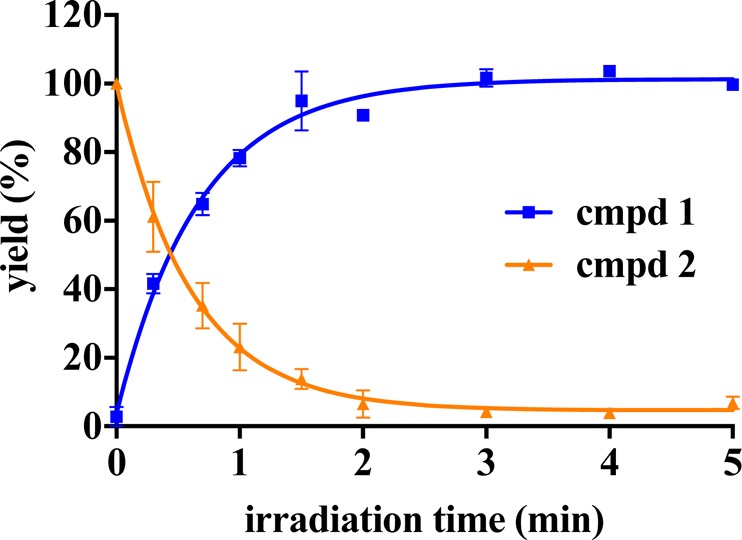
Photolytic characterization of the caged derivative 2. A solution of compound 2 (1 mM) was irradiated at 365 nM, and samples collected at indicated time points were analyzed by HPLC. Compound 2 was photolyzed to produce compound 1 in one-phase decay kinetics (τ = 0.605 min, R2 = 1). Data represent mean ± SEM from two independent experiments performed in duplicate.
To assess the inactivity of caged prodrug 2, as well as reactivation and recovery of the cytotoxic effects, we performed a series of cell viability assays using U251 and patient-derived RN1 glioblastoma cells. The U251 cell line was established prior to genome analysis of glioblastoma tumors and is not assigned to any glioblastoma subtype. The patient-derived RN1 cell line was established in our laboratories28,29 and represents the most common (>60%) classical subtype of glioblastomas. RN1 cells were grown as stem cells under defined conditions in order to maintain the phenotype and genotype of the primary resected tumor.30
First, we investigated the effects of UV light on cell viability in order to determine tolerable levels of UV exposure. We exposed U251 and RN1 cells to UV light up to 5 min (lamp B, 1800 mW, Figure S2) and after 24 h of incubation performed viability assay using Cell TiterBlue reagent. We found that U251 and RN1 cells tolerated a continuous UV light exposure of 1 min and 30s, respectively (Figure S4).
To assess the cytotoxic effects, cells were treated with uncaged inhibitor 1 and caged derivative 2 for 72 h, and cell viability assays were performed to evaluate the number of viable cells. The parent compound 1 decreased the viability of U251 and RN1 cells with EC50 values of 1.3 and 0.3 μM, respectively (Figure 2), which is in good agreement with previously published data.24,25 UV irradiation of cells treated with compound 1 did not affect the efficacy of the uncaged derivative (Figure 2). In contrast to the bioactive compound 1 and as expected by our PPG design, caged derivative 2 had no significant cytotoxicity up to high micromolar concentrations (EC50 = 72 and 37.4 μM for U251 and RN1, respectively), providing evidence that addition of the PPG to compound 1 resulted in the loss of cytotoxic activity. UV irradiation (30 s of RN1 and 1 min of U251 cells) restored the activity of compound 2 and efficacy in both cell lines was equivalent to the efficacy of the uncaged compound (EC50 = 2.1 and 1.2 μM for U251 and RN1, respectively), suggesting that a recovery of the cytotoxic activity is achieved with a short period of UV irradiation.
Figure 2.
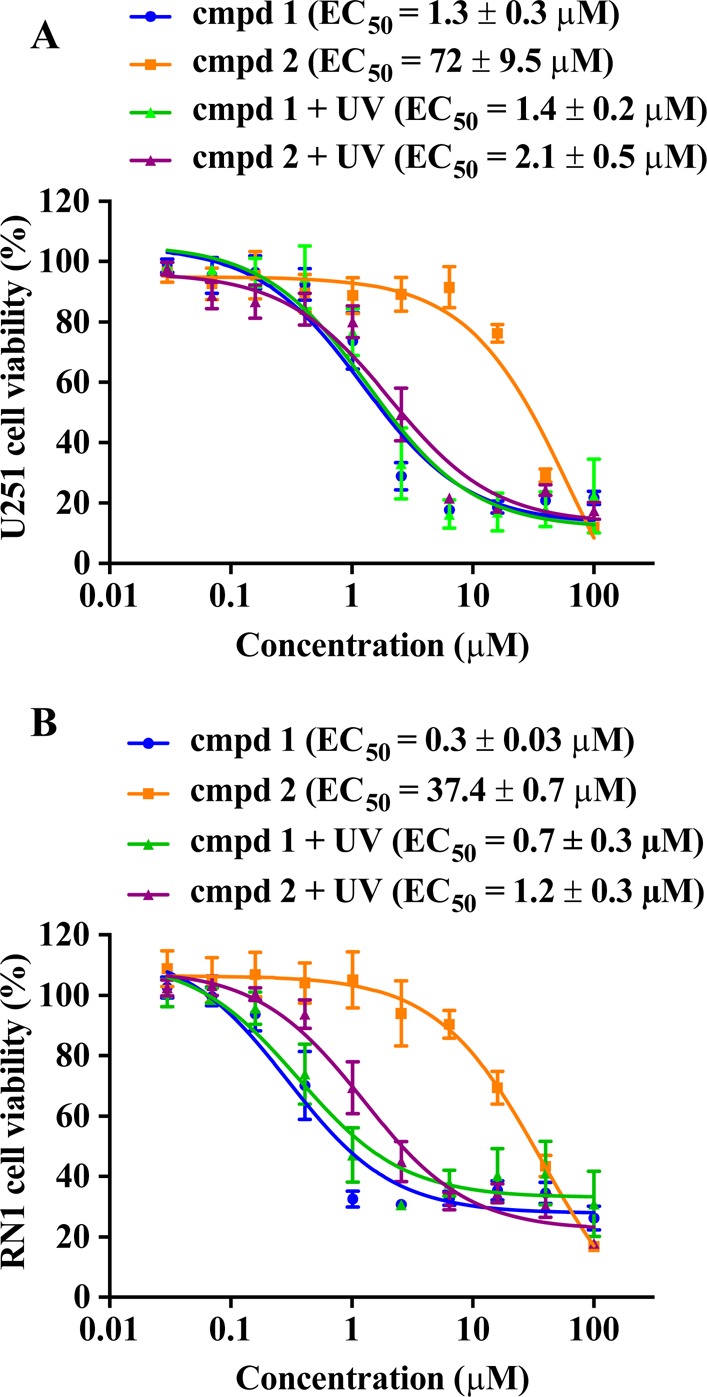
Evaluation of the uncaging protocol in a cell viability assay. (A) U251 and (B) patient-derived RN1 glioblastoma cells were grown as adherent cultures, treated with uncaged inhibitor 1 and caged derivative 2. U251 cells were UV irradiated (365 nm, 1800 mW) for 1 min, RN1 cells were irradiated for 30 s. Cellular efficacy (EC50) values were determined using Cell TiterBlue viability assay after 72 h of drug treatment. Data represent mean ± SEM from three independent experiments performed in triplicate.
To confirm that addition of the bulky PPG was detrimental to the cytotoxicity of compound 1, compound 3 containing a benzyl moiety on the phenolic OH group was synthesized and its cytotoxicity tested using U251 glioblastoma cells (Figure 3). Compound 3 affected U251 cell viability only at concentrations higher than 40 μM. We also synthesized 4,5-dimethoxy-2-nitrobenzyl (tert-butoxycarbonyl)alaninate 4 to investigate if the DMNB moiety released via irradiation of the caged derivative 2 could be cytotoxic to the cells. Importantly, compound 4 when UV irradiated to release DMNB did not change the viability of U251 cells up to 100 μM concentration (Figure 3). Together, these data indicate that (i) the unsubstituted phenolic group is crucial for the biological activity of 1 and that (ii) cytotoxicity after UV irradiation results from uncaging the inhibitor 1 and not from the released DMNB.
Figure 3.
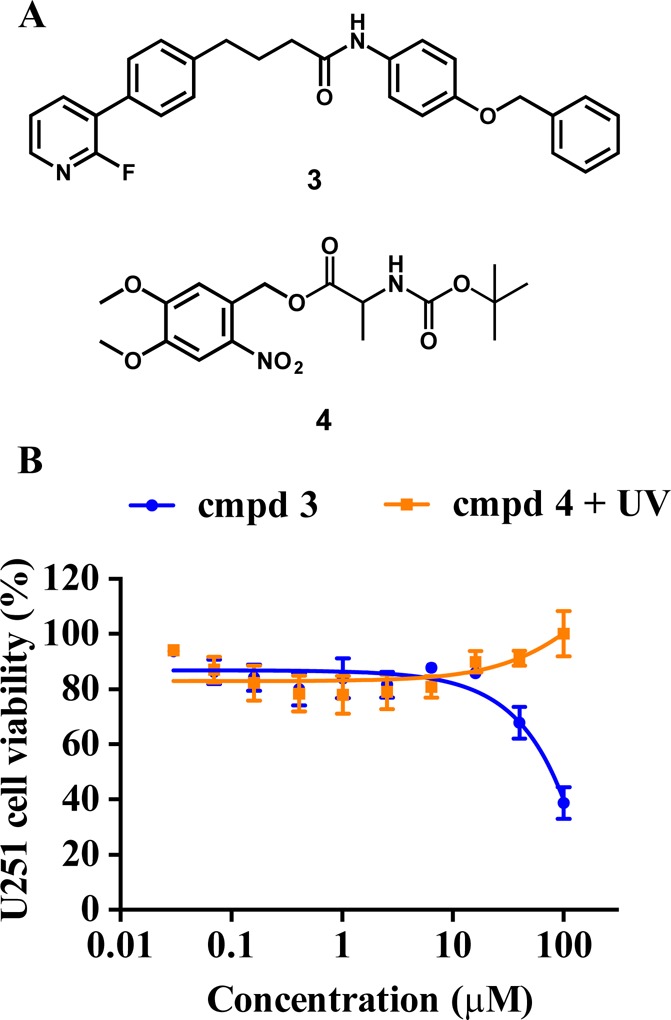
Efficacy of negative control compounds 3 and 4 in the cell viability assay. (A) Chemical structures of compounds 3 and 4. (B) Cellular efficacy of compound 3 and UV-irradiated (365 nm, 1800 mW, 1 min) compound 4 in U251 glioblastoma cells was determined using Cell TiterBlue viability assay after 72 h of drug treatment. Data represent mean ± SEM from three independent experiments performed in triplicate.
To further validate that the caging with DMNB caused loss of biological activities determined for compound 1 in cells, we conducted an in vitro tubulin polymerization and tubulin binding assays using uncaged and caged analogues 1 and 2, respectively (Figure 4). Purified β-tubulin was incubated with clinical MTAs paclitaxel and vinblastine, as well as with compounds 1 and 2. Compared to control, paclitaxel enhanced tubulin polymerization, whereas vinblastine and compound 1 inhibited tubulin polymerization (Figure 4A), which is in agreement with their established mechanism of action.24 In contrast, caged derivative 2 did not exhibit any effect on the kinetics of tubulin polymerization. However, if the same assay was performed with derivative 2 exposed to UV irradiation, the inhibition of tubulin polymerization was indistinguishable from that obtained with the uncaged compound 1. Thus, the uncaging of 2 by UV irradiation produced a bioactive molecule inhibiting tubulin polymerization with the same efficacy as the unmodified tubulin inhibitor 1 (Figure 4A).
Figure 4.
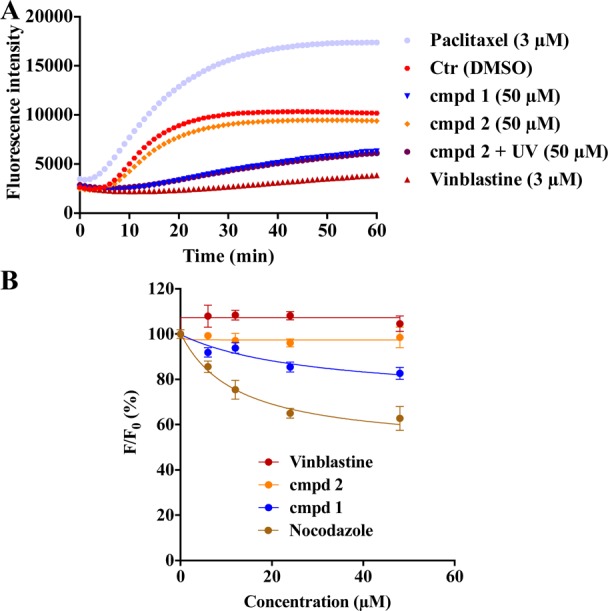
Tubulin polymerization and colchicine binding assay. (A) Porcine brain tubulin was incubated with paclitaxel, vinblastine, and compounds 1 and 2 ± UV irradiation (365 nm, 1800 mW, 5 min). Assembly of microtubules was monitored by an increase in fluorescence. Data represent the mean from three independent experiments; each data point was performed in triplicate. (B) Colchicine binding assay. Porcine tubulin was incubated with colchicine and tested compounds. Fluorescence intensity (F) was normalized to the fluorescence of the colchicine–tubulin complex (F0). Data represent mean ± SEM from four independent experiments.
Small molecules inhibiting tubulin polymerization predominantly bind into the colchicine binding site on tubulin.31 To investigate the binding site of compound 1, we performed fluorescence-based colchicine binding assay.32,33 Competition of the inhibitor and colchicine for the binding site will decrease the intrinsic fluorescence of colchicine-tubulin complex by reducing the amount of colchicine bound. With this assay, we confirmed that compound 1, but not the caged derivative 2, decreased the intrinsic colchicine fluorescence in a dose-dependent manner (Figure 4B). Nocodazole, tubulin inhibitor binding to the colchicine site (positive control) also efficiently decreased the fluorescence, whereas vinblastine (negative control) had no effect on the fluorescence. The observation that compound 1 caused a weaker decrease in the fluorescence compared to nocodazole suggests that compound 1 is likely to bind in the vicinity or allosterically to colchicine.
We next examined in greater detail how addition of the PPG to compound 1 alters the microtubule network in cells. For this, U251 glioblastoma cells were treated with 1 and 2, and the effect on the microtubules was investigated via immunofluorescence staining of β-tubulin. Treatment of U251 cells with compound 1 (5 μM) led to a disassembly of microtubules and pronounced changes in cell morphology (Figure 5). However, treatment with compound 2 had no effect on cell morphology and the tubulin network; the images of cells treated with 2 resembled the images of untreated control cells. Importantly, if cells were treated with UV irradiated (365 nm, 1800 mW, 5 min) compound 2, cells rounded up and lost their star-shaped structure. Furthermore, tubulin filaments lost their organization, suggesting that UV irradiation of compound 2 released a compound that acts as tubulin inhibitor and disrupts the highly organized tubulin network in cells.
Figure 5.
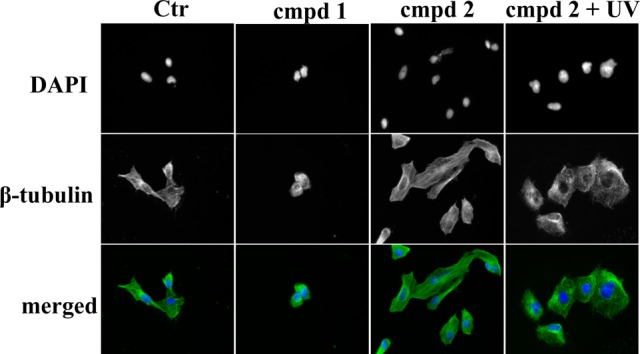
Immunofluorescence imaging of treated cells. U251 cells treated with compounds 1 and 2 and UV-irradiated (365 nm, 1800 mW, 5 min) compound 2. All treatments were done with 5 μM concentration for 24 h. Cells were fixed and stained with Alexa488-labeled anti-β-tubulin antibody (green) or DAPI (blue). Representative images of two independent experiments are shown.
Microtubule targeting agents not only characteristically disrupt the tubulin network and cell morphology as demonstrated in Figure 5, they also induce apoptosis through the intrinsic (mitochondrial) apoptotic pathway.34 In order to determine whether this mechanism contributes to the cellular efficacy of the caged derivative 2 after UV irradiation, we quantified apoptosis in drug-treated RN1 cells by Annexin V staining (Figure 6). UV irradiation (365 nm, 1800 mW, 30 s) of the patient-derived RN1 cells did not increase the basal level of Annexin V-positive cells (8.1% and 10.1% for Ctr and UV treated cells, respectively; Figure 6). Treatment of RN1 cells with the parent bioactive compound 1 (5 μM, 48 h) increased the amount of apoptotic cells to 47.8%. In agreement with previous data, compound 2 was ineffective in inducing apoptosis (12.6% of Annexin V-positive cells). However, the quantity (55.9%) of apoptotic RN1 cells when treated with compound 2 combined with UV irradiation (365 nm, 1800 mW, 30 s) was comparable to the quantity of apoptotic cells after treatment with compound 1 (47.8%), further confirming that the uncaging with UV light released an active compound.
Figure 6.
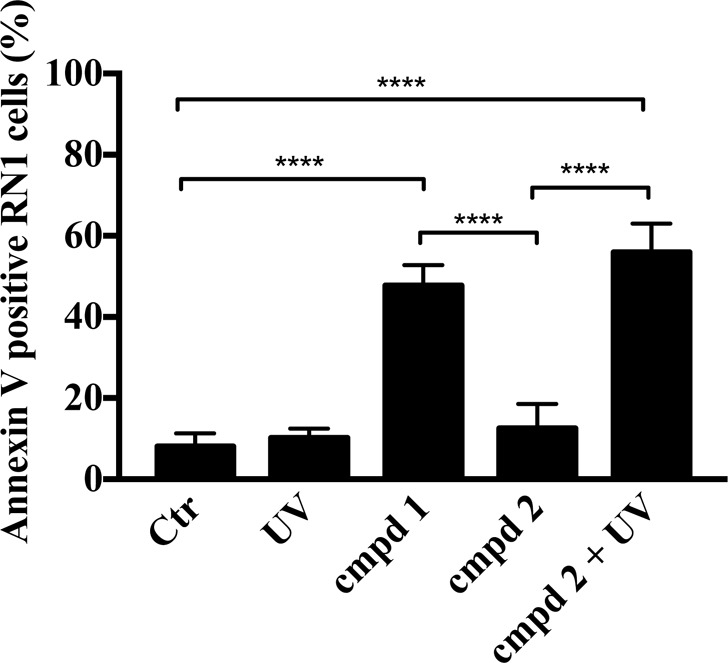
Tubulin inhibitor 1 and caged derivative 2 combined with UV irradiation induce apoptosis in RN1 glioblastoma cells. Cells were treated with 1 and 2 (5 μM ± UV irradiation) for 48 h. Control cells received an equivalent amount of DMSO or UV irradiation (365 nm, 1800 mW, 30 s). Cells were stained with Annexin V and analyzed using the MUSE Cell Analyzer. Data represent mean ± SEM from three independent experiments (****P < 0.0001, one-way ANOVA followed by Tukey’s multiple comparison test).
In summary, we have described the synthesis of the novel photocaged tubulin inhibitor 2, as well as its photolytic and pharmacological characterization. By using DMNB as photolabile protecting group to cage a small molecule tubulin inhibitor, we demonstrate spatial and temporal photoinducible toxicity to glioblastoma cells, inhibition of tubulin polymerization, and induction of apoptotic cell death. Collectively, these data show for the first time that caging concept combined with UV irradiation can be used to control the activity of small molecule tubulin inhibitors. This concept offers a novel tool for pharmacological studies and potentially a novel therapeutic approach to reduce the side effects of microtubule-targeting agents.
Glossary
ABBREVIATIONS
- DMNB
4,5-dimethoxy-2-nitrobenzyl
- MAPK
mitogen-activated protein kinase
- MK2
MAPK-activated protein kinase 2
- MTA
microtubule-targeting agent
- PPG
photolabile-protecting group
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00483.
Experimental details, compounds characterization, and Figures S1–S5 (PDF)
Author Present Address
# College of Pharmacy, Taibah University, Almadinah Almonawarrah 41477, Saudi Arabia.
Author Contributions
A.D. and M.A. synthesized and characterized compounds presented in this study. A.D. and A.F.P. designed, performed, and analyzed all biological assays, except for the colchicine binding assay, which was designed, performed, and analyzed by R.H.A. B.W.S., B.W.D., and T.J.G. derived and characterized RN1 patient-derived glioblastoma cells. L.M. and P.F. designed the study concept and supervised the study. All authors have given approval to the final version of the manuscript.
L.M. is a Cancer Institute NSW Career Development Fellow (grant reference 15/CDF/1-07). This study was partially funded by the National Foundation for Medical Research and Innovation (NFMRI) and Sydney Medical School Foundation grants to L.M.; and DFG (German Research Society) grant PE 1605/2-1 to C.P. A.F.P. is supported by The University of Sydney Australian Postgraduate Award scholarship. M.A. was funded by the German Academic Exchange Service (DAAD) and A.D. via the DAAD/PROMOS scholarship funds from the Federal German Ministry for Education and Research. T.G.J. was supported by the NH&MRC (Project Grant #1028552) and Cure Brain Cancer Foundation. T.G.J., B.W.S., and B.W.D. are members of the Brain Cancer Discovery Collaborative, which is supported by the Cure Brain Cancer Foundation. This work was partly supported by the Victorian Government’s Operational and Infrastructure Support Program (to T.G.J.).
The authors declare no competing financial interest.
Supplementary Material
References
- Klán P.; Šolomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Photoremovable protecting groups in chemistry and biology: Reaction mechanisms and efficacy. Chem. Rev. 2013, 113 (1), 119–191. 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens R. S.; Rubina M.; Wirz J. Applications of p-hydroxyphenacyl (pHP) and coumarin-4-ylmethyl photoremovable protecting groups. Photochem. Photobiol. Sci. 2012, 11 (3), 472–488. 10.1039/c2pp05399c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G.; Heckel A. Biologically active molecules with a “Light Switch. Angew. Chem., Int. Ed. 2006, 45 (30), 4900–4921. 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]
- Cui J.; Gropeanu R. A.; Stevens D. R.; Rettig J.; Campo A. d. New photolabile BAPTA-based Ca2+ cages with improved photorelease. J. Am. Chem. Soc. 2012, 134 (18), 7733–7740. 10.1021/ja2115184. [DOI] [PubMed] [Google Scholar]
- Umeda N.; Ueno T.; Pohlmeyer C.; Nagano T.; Inoue T. A photocleavable rapamycin conjugate for spatiotemporal control of small GTPase activity. J. Am. Chem. Soc. 2011, 133 (1), 12–14. 10.1021/ja108258d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goard M.; Aakalu G.; Fedoryak O. D.; Quinonez C.; St. Julien J.; Poteet S. J.; Schuman E. M.; Dore T. M. Light-mediated inhibition of protein synthesis. Chem. Biol. 2005, 12 (6), 685–693. 10.1016/j.chembiol.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Buhr F.; Kohl-Landgraf J.; tom Dieck S.; Hanus C.; Chatterjee D.; Hegelein A.; Schuman E. M.; Wachtveitl J.; Schwalbe H. Design of photocaged puromycin for nascent polypeptide release and spatiotemporal monitoring of translation. Angew. Chem., Int. Ed. 2015, 54 (12), 3717–3721. 10.1002/anie.201410940. [DOI] [PubMed] [Google Scholar]
- Gropeanu R. A.; Baumann H.; Ritz S.; Mailänder V.; Surrey T.; del Campo A. Phototriggerable 2′,7-caged paclitaxel. PLoS One 2012, 7 (9), e43657. 10.1371/journal.pone.0043657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horbert R.; Pinchuk B.; Davies P.; Alessi D.; Peifer C. Photoactivatable prodrugs of antimelanoma agent vemurafenib. ACS Chem. Biol. 2015, 10 (9), 2099–2107. 10.1021/acschembio.5b00174. [DOI] [PubMed] [Google Scholar]
- Zindler M.; Pinchuk B.; Renn C.; Horbert R.; Döbber A.; Peifer C. Design, synthesis, and characterization of a photoactivatable caged prodrug of imatinib. ChemMedChem 2015, 10 (8), 1335–1338. 10.1002/cmdc.201500163. [DOI] [PubMed] [Google Scholar]
- Fan N.-C.; Cheng F.-Y.; Ho J.-A. A.; Yeh C.-S. Photocontrolled targeted drug delivery: Photocaged biologically active folic acid as a light-responsive tumor-targeting molecule. Angew. Chem., Int. Ed. 2012, 51 (35), 8806–8810. 10.1002/anie.201203339. [DOI] [PubMed] [Google Scholar]
- Dcona M. M.; Mitra D.; Goehe R. W.; Gewirtz D. A.; Lebman D. A.; Hartman M. C. T. Photocaged permeability: a new strategy for controlled drug release. Chem. Commun. 2012, 48 (39), 4755–4757. 10.1039/c2cc30819c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10 (3), 194–204. 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- Mccarroll J.; Parker A.; Kavallaris M. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. 10.3389/fonc.2014.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E.; Sackett D.; Wilkerson J.; Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 2011, 8 (4), 244–250. 10.1038/nrclinonc.2010.228. [DOI] [PubMed] [Google Scholar]
- Dumontet C.; Jordan M. A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discovery 2010, 9 (10), 790–803. 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudlowitz D.; Muggia F. Defining risks of taxane neuropathy: Insights from randomized clinical trials. Clin. Cancer Res. 2013, 19 (17), 4570–4577. 10.1158/1078-0432.CCR-13-0572. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ran R.; Chen J.; Kuang Q.; Tang J.; Mei L.; Zhang Q.; Gao H.; Zhang Z.; He Q. Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials 2014, 35 (17), 4835–4847. 10.1016/j.biomaterials.2014.02.031. [DOI] [PubMed] [Google Scholar]
- Kang T.; Gao X.; Hu Q.; Jiang D.; Feng X.; Zhang X.; Song Q.; Yao L.; Huang M.; Jiang X.; Pang Z.; Chen H.; Chen J. iNGR-modified PEG-PLGA nanoparticles that recognize tumor vasculature and penetrate gliomas. Biomaterials 2014, 35 (14), 4319–4332. 10.1016/j.biomaterials.2014.01.082. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Zhang Y.; Liao Z.; Jiang T.; Zhao J.; Tuo Y.; She X.; Shen S.; Chen J.; Zhang Q.; Jiang X.; Hu Y.; Pang Z. UPA-sensitive ACPP-conjugated nanoparticles for multi-targeting therapy of brain glioma. Biomaterials 2015, 36, 98–109. 10.1016/j.biomaterials.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Senese S.; Lo Y. C.; Huang D.; Zangle T. A.; Gholkar A. A.; Robert L.; Homet B.; Ribas A.; Summers M. K.; Teitell M. A.; Damoiseaux R.; Torres J. Z. Chemical dissection of the cell cycle: probes for cell biology and anti-cancer drug development. Cell Death Dis. 2014, 5 (10), e1462. 10.1038/cddis.2014.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu S.; Harris F.; Lea R.; Snape T. J. Small-molecule clinical trial candidates for the treatment of glioma. Drug Discovery Today 2014, 19 (9), 1298–1308. 10.1016/j.drudis.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Davidson W.; Frego L.; Peet G. W.; Kroe R. R.; Labadia M. E.; Lukas S. M.; Snow R. J.; Jakes S.; Grygon C. A.; Pargellis C.; Werneburg B. G. Discovery and characterization of a substrate selective p38α inhibitor. Biochemistry 2004, 43 (37), 11658–11671. 10.1021/bi0495073. [DOI] [PubMed] [Google Scholar]
- Gurgis F.; Åkerfeldt M. C.; Heng B.; Wong C.; Adams S.; Guillemin G. J.; Johns T. G.; Chircop M.; Munoz L. Cytotoxic activity of the MK2 inhibitor CMPD1 in glioblastoma cells is independent of MK2. Cell Death Discovery 2015, 1, 15028. 10.1038/cddiscovery.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoa A. F.; Browne S.; Gurgis F. M. S.; Åkerfeldt M. C.; Döbber A.; Renn C.; Peifer C.; Stringer B. W.; Day B. W.; Wong C.; Chircop M.; Johns T. G.; Kassiou M.; Munoz L. Pharmacology of novel small-molecule tubulin inhibitors in glioblastoma cells with enhanced EGFR signalling. Biochem. Pharmacol. 2015, 98 (4), 587–601. 10.1016/j.bcp.2015.10.014. [DOI] [PubMed] [Google Scholar]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455 (7216), 1061–1068. 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon D. A.; Wen P. Y.; Mellinghoff I. K. Targeted molecular therapies against epidermal growth factor receptor: Past experiences and challenges. Neuro-Oncology 2014, 16 (suppl 8), viii7–viii13. 10.1093/neuonc/nou232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day B.; Stringer B.; Wilson J.; Jeffree R.; Jamieson P.; Ensbey K.; Bruce Z.; Inglis P.; Allan S.; Winter C.; Tollesson G.; Campbell S.; Lucas P.; Findlay W.; Kadrian D.; Johnson D.; Robertson T.; Johns T.; Bartlett P.; Osborne G.; Boyd A. Glioma Surgical Aspirate: A Viable Source of Tumor Tissue for Experimental Research. Cancers 2013, 5 (2), 357–371. 10.3390/cancers5020357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day B. W.; Stringer B. W.; Al-Ejeh F.; Ting M. J.; Wilson J.; Ensbey K. S.; Jamieson P. R.; Bruce Z. C.; Lim Y. C.; Offenhäuser C.; Charmsaz S.; Cooper L. T.; Ellacott J. K.; Harding A.; Leveque L.; Inglis P.; Allan S.; Walker D. G.; Lackmann M.; Osborne G.; Khanna K. K.; Reynolds B. A.; Lickliter J. D.; Boyd A. W. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell 2013, 23 (2), 238–248. 10.1016/j.ccr.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Pollard S. M.; Yoshikawa K.; Clarke I. D.; Danovi D.; Stricker S.; Russell R.; Bayani J.; Head R.; Lee M.; Bernstein M.; Squire J. A.; Smith A.; Dirks P. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4 (6), 568–580. 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Chen J.; Xiao M.; Li W.; Miller D. D. An Overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.-B.; Gong J.-H.; Liu X.-J.; Wu S.-Y.; Li Y.; Xu X.-D.; Shang B.-Y.; Zhou J.-M.; Zhu Z.-L.; Si S.-Y.; Zhen Y.-S. A novel nitrobenzoate microtubule inhibitor that overcomes multidrug resistance exhibits antitumor activity. Sci. Rep. 2016, 6, 31472. 10.1038/srep31472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibbeson B. M.; Laraia L.; Alza E.; O’Connor C. J.; Tan Y. S.; davies H. M. L.; Mckenzie G.; Venkitaraman A. R.; Spring D. R. Diversity-oriented synthesis as a tool for identifying new modulators of mitosis. Nat. Commun. 2014, 5, 3155. 10.1038/ncomms4155. [DOI] [PubMed] [Google Scholar]
- Gan P. P.; McCarroll J. A.; Po’uha S. T.; Kamath K.; Jordan M. A.; Kavallaris M. Microtubule dynamics, mitotic arrest, and apoptosis: Drug-induced differential effects of βIII-tubulin. Mol. Cancer Ther. 2010, 9 (5), 1339–1348. 10.1158/1535-7163.MCT-09-0679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.