

Abstract
Background
The genetic basis of virulence in Mycobacterium tuberculosis has been investigated through genome comparisons of virulent (H37Rv) and attenuated (H37Ra) sister strains. Such analysis, however, relies heavily on the accuracy of the sequences. While the H37Rv reference genome has had several corrections to date, that of H37Ra is unmodified since its original publication.
Results
Here, we report the assembly and finishing of the H37Ra genome from single-molecule, real-time (SMRT) sequencing. Our assembly reveals that the number of H37Ra-specific variants is less than half of what the Sanger-based H37Ra reference sequence indicates, undermining and, in some cases, invalidating the conclusions of several studies. PE_PPE family genes, which are intractable to commonly-used sequencing platforms because of their repetitive and GC-rich nature, are overrepresented in the set of genes in which all reported H37Ra-specific variants are contradicted. Further, one of the sequencing errors in H37Ra masks a true variant in common with the clinical strain CDC1551 which, when considered in the context of previous work, corresponds to a sequencing error in the H37Rv reference genome.
Conclusions
Our results constrain the set of genomic differences possibly affecting virulence by more than half, which focuses laboratory investigation on pertinent targets and demonstrates the power of SMRT sequencing for producing high-quality reference genomes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3687-5) contains supplementary material, which is available to authorized users.
Keywords: Mycobacteria, Tuberculosis, H37Rv, H37Ra, Virulence, Single-molecule sequencing, De novo assembly, Comparative genomics, Reference genomes, Sequencing errors
Background
Tuberculosis is a serious and pervasive public health problem [1]. It is a disease caused by infection of bacteria from the Mycobacterium tuberculosis complex (MTBC). The reference strain, Mycobacterium tuberculosis H37Rv, has an attenuated counterpart known as H37Ra that is available for studies where facilities to handle virulent samples are lacking. H37Ra exhibits a distinct colony morphology, an absence of cord formation, decreased resistance to stress and hypoxia, and attenuated virulence in mammalian models [2–4]. The H37Ra genome was assembled by Zheng and colleagues in 2008 and compared to H37Rv for the purpose of identifying the genetic basis of virulence attenuation [5]. The resulting sequence has been used as the primary avirulent reference genome for M. tuberculosis since its publication in 2008.
As genome sequencing technology has significantly improved [6], we sought to assess the ability of single-molecule, real-time (SMRT) sequencing for finishing mycobacterial genomes. In addition to a high overall GC-content, these genomes have GC-rich repetitive sequences, a source of systematic error for many sequencing protocols. Even sample preparation methods commonly used for shotgun Sanger sequencing are prone to such bias [7]. Sequencing errors in the H37Rv reference have been sought out, with some corrected, others remaining to be discovered, and still others discovered and remaining to be corrected [8, 9]. The Pacific Biosciences RS II platform has been shown to produce finished-grade assemblies of microbial genomes exceeding the quality of Sanger sequencing [10–12].
In this study, we sequenced and assembled the genome of M. tuberculosis H37Ra and compared it to the reference sequence. We further compared both sequences against the reference sequence for M. tuberculosis H37Rv and re-evaluated the conclusions of Zheng and colleagues with respect to the genetic basis of virulence attenuation.
Results
Genome assembly and methylation motif detection
Using the data from two sequencing runs (SMRTCells), the genome assembled with 217x average coverage into a single contig containing 4426109 base pairs after circularization and polishing. Applying the same protocol using data from only one of the two SMRTCells (103x average coverage) resulted in an identical sequence. Figure 1 shows sequencing coverage and GC-content as a function of genome position.
Fig. 1.
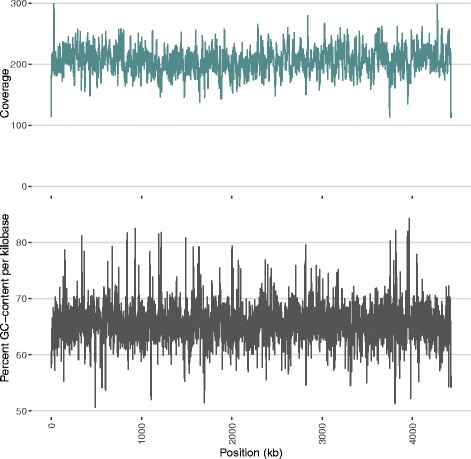
Sequencing Coverage and GC-content by Genome Position. GC-content and coverage are shown in 1kb windows. The coverage plot refers to reads mapped to our assembly during the final polishing round. Reads with mapping quality values less than 10 were not used in polishing and are not counted here. Imposing linearity in the contig despite circularity of the genome creates mapping difficulties at the contig edges, resulting in irregularities in apparent sequencing coverage at these sites
Circularization was impeded by discrepancies in the edges of the contig, where an IS6110 insertion was present in only one of the two edges. It appears heterogeneously in our sample, as aligning our reads against our assembly shows that a minority of reads have interrupted mapping to this segment while the majority do not. With regard to base modifications, N6-methyladenine was detected in 99.67% of the instances of the partner sequence motifs CTGGAG and CTCCAG. The methylation of these motifs in both H37Ra and H37Rv was previously reported by Zhu and colleagues in H37Ra as part of their study of mycobacterial methylomes [13].
Direct comparison with the hitherto H37Ra reference genome
Comparison of our assembly with the H37Ra reference sequence (NC_009525.1, hereafter referred to as H37RaJH, for Johns Hopkins) showed significant variation. We found 33 single nucleotide polymorphisms (SNPs), and 77 insertions and deletions in our assembly with respect to H37RaJH (Additional file 1).
Structural variations
Two of the insertions with respect to H37RaJH were substantial structural variations: one was an insertion of IS6110 into the gene corresponding to Rv1764 and the other was an in-frame insertion of 3456bp into the PPE54 gene.
The insertion of IS6110 into Rv1764 (an IS6110 transposase) is unsurprising, as IS6110 insert frequently into that general region of the genome, as well as within their transposase [14, 15]. This insertion was the heterogeneous insertion responsible for the discrepant contig ends in our raw genome assembly. Such heterogeneity implies either a lack of selection pressure on the insertion in culture, a recent emergence of the insertion, or both.
The 3456bp insertion in ppe54 with respect to H37RaJH incidentally corresponds to a tandem duplication of a 1728bp sequence at the same site in H37Rv with 100% identity. The complete absence of this tandem repeat at this site in H37RaJH, however, is not necessarily an assembly error, as this is also observed in several clinical isolates (unpublished data). This, along with the 100% identity between each 1728bp duplicate of the tandem repeat with respect to H37Rv, lead us to believe that both the duplication in our sequence and the deletion observed in H37RaJH are instances of in vitro evolution, following the divergence of the lineages from which H37RaJH and our assembly were drawn. These two structural variations, or, at least, very similar structural variations, have been observed previously in virulent strains of M. tuberculosis, and therefore likely do not contribute to virulence attenuation in H37Rv (unpublished data) [14, 16], but shed light on in vitro evolution of this strain [8, 17].
Analysis of motif variants in H37Ra and H37Rv
With the knowledge that the CTGGAG/CTCCAG motifs are methylated in both H37Ra and H37Rv [13], we determined the motif variants, or sequence polymorphisms that create or destroy motifs, between H37Rv and H37Ra. By first comparing H37RaJH to H37Rv, we see that all but two motif variants were due to structural variations. Both of these variants instantiate the CTGGAG motif in H37Ra where it is absent in the H37Rv reference sequence. The first is due to the G→T polymorphism at H37Rv position 2043284 (upstream of PPE30) in H37RaJH, but this variant is contradicted by our H37Ra assembly. The second is due to the T→G polymorphism at H37Rv position 2718852 (upstream of nadD) and confirmed by our H37Ra assembly, yet also appears in CDC1551 and is a previously reported sequencing error in H37Rv [8] that has not been applied to the current reference. Based on these results, DNA methylation and motif variants do not play a role in the attenuation of virulence in H37Ra.
Status of previously reported “H37Ra-specific” polymorphisms
With our assembly, we aimed to replicate the study performed by Zheng and colleagues when they first assembled the H37Ra genome [5]. In their study, they compared their assembly with H37Rv, then filtered out variants also present in CDC1551 (NC_002755.2) to find mutations likely specific to H37Ra [5]. Zheng and colleagues identified a set of mutations in H37Ra unique with respect to H37Rv and CDC1551 as “H37Ra-specific”. These mutations fall within or adjacent to (which we term “affecting”) 56 genes in H37Rv, which we refer to as the high-confidence (HC) gene set. While comparing the variants, Zheng and colleagues also discovered sequencing errors in the H37Rv reference sequence [5], a number of which were corrected in NC_000962.3 [9], the version used in our study.
To see how well the HC genes are supported by our assembly of H37Ra, we determined variants with respect to H37Rv for our assembly and H37RaJH and performed set comparisons after excluding mutations shared with CDC1551 and other finished assemblies for H37Rv (H37RvBroad: NC_018143.2; H37RvSiena: NZ_CP007027.1; H37RvTMC102: NZ_CP009480.1) (Additional files 1 and 2). We then categorized the HC genes as follows. We labeled a gene “contradicted” if all mutations affecting it were observed only in H37RaJH. We labeled a gene “supported” if all mutations affecting it were observed in both H37Ra assemblies. Otherwise, we labeled a gene “adjusted” if it had a different variant profile between H37RaJH and our assembly in a manner distinct from the two categories defined above. Figure 2 shows example classifications based on these criteria.
Fig. 2.
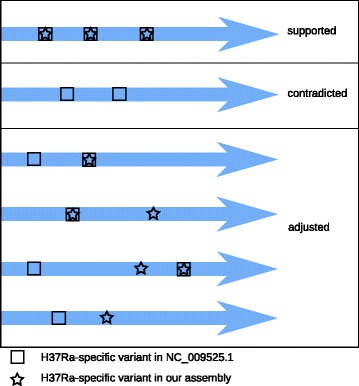
Example Classification of Genes Based on Variant Comparisons. Considering the profile of H37Ra-specific variants (those with respect to H37Rv not also appearing in CDC1551), a given gene (blue arrow) is categorized as “supported”, “contradicted”, or “adjusted” by our H37Ra assembly as a result of comparison with the hitherto reference sequence NC_009525.1. The illustration shows examples of the different variant profiles a gene could have and their resulting classifications. Genes in the “supported” and “contradicted” categories are strictly those where our assembly either fully matches the H37Ra reference (supported) or the H37Rv reference (contradicted). Multiple factors may cause a gene to be classified as “adjusted”. Such genes may have variant profiles not fully meeting the criteria of “supported” or “contradicted”, or they may have novel H37Ra-specific variants observed only in our assembly
We first noted that two of the HC variants reported by Zheng and colleagues, those affecting nadD (Rv2421c) and nrdH (Rv3053), were included erroneously (Table 1 d). These variants were a T→G mutation 44 bases upstream of nadD, at H37Rv position 2718852, and a 14bp deletion in the promoter of nrdH. These mutations, although confirmed by our assembly, also appear in CDC1551 and thus cannot be considered H37Ra-specific.
Table 1.
Status of Genes Previously Reported as Affected by H37Ra-specific Mutations
| Locus Tag | Gene name | Description | Notes | Citation |
|---|---|---|---|---|
| (a) Genes with all High-Confidence Variants Contradicted by our Assembly | ||||
| Rv0037c | Rv0037c | Probable conserved integral membrane protein | ||
| Rv0124 | PE_PGRS2 | PE-PGRS family protein PE_PGRS2 | ||
| Rv0189c | ilvD | Probable dihydroxy-acid dehydratase IlvD (dad) | [48, 58] | |
| Rv0279c | PE_PGRS4 | PE-PGRS family protein PE_PGRS4 | ∙ | |
| Rv0383c | Rv0383c | Possible conserved secreted protein | masks sequencing error in H37Rv | [8] |
| Rv0578c | PE_PGRS7 | PE-PGRS family protein PE_PGRS7 | ∙ | |
| Rv0880 | Rv0880 | Possible MarR-family transcriptional regulatory protein | [48] | |
| Rv0977 | PE_PGRS16 | PE-PGRS family protein PE_PGRS16 | ∙ | |
| Rv1068c | PE_PGRS20 | PE-PGRS family protein PE_PGRS20 | ∙ | |
| Rv1091 | PE_PGRS22 | PE-PGRS family protein PE_PGRS22 | [36, 38] ∙ | |
| Rv1095 | phoH2 | Probable PHOH-like protein PhoH2 | ||
| Rv1196 | PPE18 | PPE family protein PPE18 | [25, 36] ∙ | |
| Rv1386 | PE15 | PE family protein PE15 | ∙ | |
| Rv1450c | PE_PGRS27 | PE-PGRS family protein PE_PGRS27 | ∙ | |
| Rv1802 | PPE30 | PPE family protein PPE30 | SNV instantiates CTGGAG motif | ∙ |
| Rv1929c | Rv1929c | Conserved hypothetical protein | ||
| Rv2048c | pks12 | Polyketide synthase Pks12 | ||
| Rv2068c | blaC | Class a beta-lactamase BlaC | ||
| Rv2069 | sigC | RNA polymerase sigma factor, ECF subfamily, SigC | [18, 19] | |
| Rv2098c | PE_PGRS36 | PE-PGRS family protein PE_PGRS36 | Likely pseudogene | ∙ |
| Rv2202c | adoK | Adenosine kinase | Synonymous mutation | |
| Rv2396 | PE_PGRS41 | PE-PGRS family protein PE_PGRS41 | ∙ | |
| Rv2649 | Rv2649 | Probable transposase for IS6110 | ||
| Rv2733c | Rv2733c | Conserved hypothetical alanine, arginine-rich protein | ||
| Rv2734 | Rv2734 | Conserved hypothetical protein | ||
| Rv2825c | Rv2825c | Conserved hypothetical protein | ||
| Rv3031 | Rv3031 | Conserved protein | Synonymous mutation | |
| Rv3191c | Rv3191c | Probable transposase | labeled intergenic in H37RaJH | |
| Rv3192 | Rv3192 | Conserved hypothetical alanine and proline-rich protein | labeled intergenic in H37RaJH | |
| Rv3303c | lpdA | NAD(P)H quinone reductase LpdA | tandem repeat copy number variation | [48] |
| Rv3350c | PPE56 | PPE family protein | ∙ | |
| Rv3388 | PE_PGRS52 | PE-PGRS family protein PE_PGRS52 | [36] ∙ | |
| Rv3389c | htdY | Probable 3-hydroxyacyl-thioester dehydratase HtdY | ||
| Rv3507 | PE_PGRS53 | PE-PGRS family protein PE_PGRS53 | [37] ∙ | |
| Rv3595c | PE_PGRS59 | PE-PGRS family protein PE_PGRS59 | [36, 38] ∙ | |
| Rv3611 | Rv3611 | Hypothetical arginine and proline rich protein | One deletion also at ftsH -57bp | |
| (b) Genes with Different H37Ra-specific Variant Profiles in our Assembly | ||||
| Rv1764 | Rv1764 | Putative transposase of insertion element IS6110 | disrupted by IS6110 in our assembly | |
| Rv3343c | PPE54 | PPE family protein | tandem repeat copy number variation | [36] ∙ |
| Rv3508 | PE_PGRS54 | PE-PGRS family protein PE_PGRS54 | [36–38] ∙ | |
| Rv3514 | PE_PGRS57 | PE-PGRS family protein PE_PGRS57 | [36, 37] ∙ | |
| (c) Genes with High-Confidence Variant Profiles Fully Confirmed by our Assembly | ||||
| Rv0010c | Rv0010c | Probable conserved membrane protein | ||
| Rv0039c | Rv0039c | Possible conserved transmembrane protein | ||
| Rv0101 | nrp | Probable peptide synthetase Nrp (peptide synthase) | [9, 24] | |
| Rv0635 | hadA | (3R)-hydroxyacyl-ACP dehydratase subunit HadA | [33, 38] | |
| Rv0637 | hadC | (3R)-hydroxyacyl-ACP dehydratase subunit HadC | [25, 33] | |
| Rv0757 | phoP | Member of Two-component response complex PhoPR | [18, 23, 25, 49, 58] | |
| Rv0878c | PPE13 | PPE family protein PPE13 | [36, 38] ∙ | |
| Rv1005c | pabB | Probable para-aminobenzoate synthase component I | [48] | |
| Rv1006 | Rv1006 | Unknown protein | ||
| Rv1021 | mazG | NTP Pyrophosphohydrolase, MazG | [31, 32, 48, 58, 60] | |
| Rv1755c | plcD | Probable phospholipase C 4 (fragment) PlcD | [8, 61] | |
| Rv1759c | wag22 | PE-PGRS family protein Wag22 | [36] ∙ | |
| Rv2352c | PPE38 | PPE family protein PPE38 | exact, adjacent duplication of PPE38 | [25, 40] ∙ |
| Rv3879c | espK | ESX-1 secretion-associated protein EspK. | ||
| (d) Genes with Variant Profiles Erroneously Declared as H37Ra-specific | ||||
| Rv2421c† | nadD | Probable nicotinate-nucleotide adenylyltransferase NadD | SNV instantiates CTGGAG motif | [48] |
| Rv3053c | nrdH | Probable glutaredoxin electron transport component of NRDEF NrdH | [48] | |
Of the variants in the remaining 54 HC genes, our assembly contradicts 36 (Table 1 a), adjusts 4 (Table 1 b), and confirms 14 (Table 1 c). We then considered how these results affect the picture of how the genotypic differences between H37Rv and H37Ra give rise to the phenotypic differences observed between the two strains, which are discussed below and depicted graphically in Fig. 3. As our analysis focused on the HC gene set reported by Zheng and colleagues [5], we did not re-evaluate whether additional genes and variants should belong to this grouping. We did, however, carefully consider all variants unique to our assembly (Table 2, Additional file 2) and their potential effect on the organism’s phenotype.
Fig. 3.
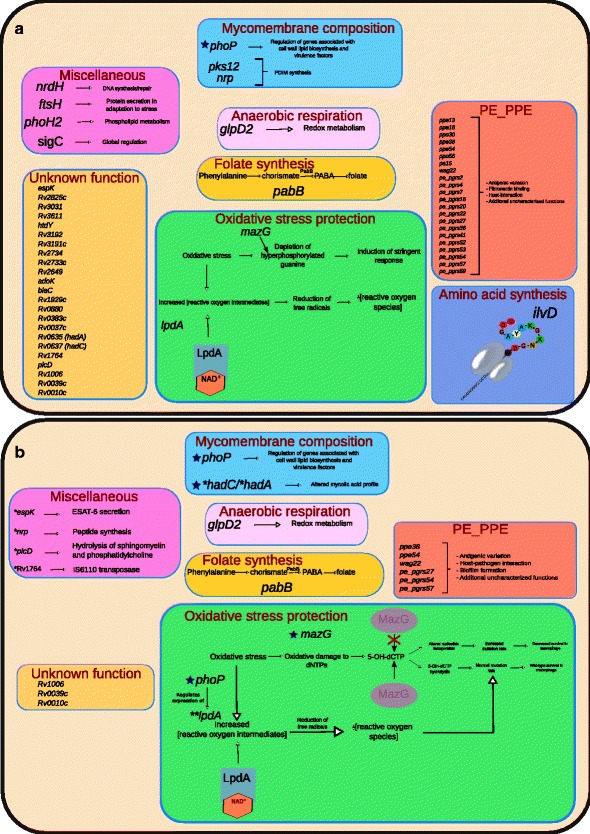
Visualization of the Reduced Set of H37Ra-specific Variants and Their Effect on Phenotype. Our assembly contradicts many variants previously thought to be H37Ra-specific, reducing the number of genes that may contribute to H37Ra’s virulence attenuation. Several of these genes have been reassigned function since the first published assembly of the H37Ra genome [5], which is reflected in the figure. a The set of genes identified to carry H37Ra-specific polymorphisms in the original H37Ra genome publication [5] and their contribution to phenotype as understood at that time. 56 genes are affected, the majority of which were PE_PPE genes or were of unknown function. b The set of genes with H37Ra-specific variants confirmed by our assembly is reduced markedly, particularly in PE_PPE genes, highlighting the strength of single-molecule sequencing in resolving GC-rich and repetitive stretches of DNA. Genes with functions not yet characterized were also reduced significantly.*Though in a few instances this was because these genes’ function was characterized between 2008 and now, most were due to our assembly showing that they matched that of H37Rv and, therefore, are not H37Ra-specific. **For lpdA, the altered copy number in H37Ra was found not to be specific to the avirulent phenotype. However, the observed altered expression of lpdA in H37Ra may be due to altered regulation from PhoP. blueThe H37Ra-specific variant(s) in these genes have been shown to confer a phenotypic change in H37Ra relative to H37Rv in wet-lab studies. For these genes, the mechanisms affected by the H37Ra-specific variant are illustrated in detail (see Fig. 4 for hadC and phoP). For other genes, their general function is described or briefly illustrated
Table 2.
Genes with Variants in H37Ra Unique to our Assembly
| Locus Tag | Gene name | Variant | Notes |
|---|---|---|---|
| Rv0279ca, b | PE_PGRS4 | Two substitutions | Both mutations are not specific to H37Ra |
| Rv0383ca, b | Rv0383c | A459399C - 84bp upstream of Rv0383c | Potential sequencing error in H37Rv [8] |
| Rv1450ca, b | PE_PGRS27 | 208bp inframe insertion | |
| Rv1764 | Rv1764 | insertion of IS6110 | |
| Rv3303ca, b | lpdA | 174bp insertion 12bp upstream | Tandem repeat CNV |
| Rv3343ca | PPE54 | 1728bp insertion | Tandem duplication with respect to H37Rv |
| Rv3508a | PE_PGRS54 | multiple variants | |
| Rv3514b | PE_PGRS57 | multiple variants | Only two are H37Ra-specific |
The mutations in this table are with respect to the H37Rv reference (NC_000962.3), so variants with respect to the current H37Ra reference sequence (NC_009525.1) that cause agreement with the H37Rv sequence do not appear here.
agene previously implicated as affected by H37Ra-specific mutations [5].
bone or more mutations affecting this gene are also present in at least one of the sequences CDC1551 (NC_002755.2), H37RvBroad (NC_018143.2), H37RvSiena (NZ_CP007027.1), and H37RvTMC102 (NZ_CP009480.1)
Accuracy of the H37Rv reference sequence
Ioerger and colleagues listed 73 polymorphisms (excluding those in PE_PPE genes) with respect to the H37Rv reference shared between six H37Rv strains from different laboratories, but considered all but one of them as errors in the reference sequence because they also appeared in the H37Ra reference [8]. The remaining polymorphism was a A→C transversion at position 459399, a position upstream of Rv0383c masked by a 55bp deletion in H37RaJH. Interestingly, our assembly contradicts this 55bp deletion, but is in perfect concordance with the transversion at position 459399. The revelation that H37Ra is in fact the same as all H37Rv strains at this position invalidates the maximum parsimony tree in Fig. 1 of their publication [8]. Thus, through our improved assembly of the H37Ra genome, we have identified an additional error in H37Rv, the standard reference genome of M. tuberculosis.
SNPs previously reported to cause expression changes in H37Ra are contradicted by our assembly
Interestingly, SNPs in the putative promoter regions of two genes, phoH2 and sigC, found by Zheng and colleagues to be up-regulated in vitro and down-regulated in macrophage in H37Ra relative to H37Rv, were contradicted by our assembly [5]. Zheng and colleagues attributed this differential expression to these (now contradicted) SNPs, but it appears there instead must be a distal causative factor driving the observed expression changes of both genes. The SNP affecting sigC has been cited as the cause of the differential expression of SigC in macrophages relative to H37Rv [18, 19], illustrating how incorrect sequences can propagate through the literature.
SNPs previously thought to affect polyketide synthesis in H37Ra are contradicted by our assembly
Altered polyketide synthesis has been proposed as one of the primary mechanisms attenuating virulence in H37Ra, through disrupting phthiocerol dimycocerosate (PDIM) production, which has shown to manifest deleteriously in H37Ra [20, 21]. Our assembly contradicts both reported SNPs in pks12 (polyketide synthase 12) of H37RaJH. This means that some factor other than disruption of pks12 causes the observed lowered PDIM production in H37Ra. Thus, it remains unclear which (epi)genomic factor(s) underlie the observed reduction in PDIM synthesis in H37Ra, as supported variants (those in phoP and nrp) once considered to cause this reduction [22] have been shown not to [23, 24]. However, it is possible the decreased production of PDIMs is merely an artifact of repeated subculturing in vitro [17].
Variants in phoP, mazG, and hadC account for much of the virulence attenuation in H37Ra
Of all the HC genes, only variants in phoP, mazG, and hadC have been connected strongly with virulence attenuation in H37Ra through wet-lab work, each of which our assembly supports.
Of these, the most thoroughly studied is the nsSNP (S219L) in the DNA-binding region of phoP, part of the two component PhoPR regulatory system. There is an abundance of literature linking phoP to virulence attenuation in H37Ra, through several mechanisms, including disrupted sulfolipid and trehalose synthesis (Fig. 4), diminished ESAT-6 secretion, and additional downstream effects from altered expression of other genes under its regulon [5, 18, 23, 25–30]. However, several of these studies also show that phoP alone [23, 29] is not responsible for virulence attenuation in H37Ra, but rather that the genomic cause behind virulence attenuation in H37Ra is multifactorial.
Fig. 4.
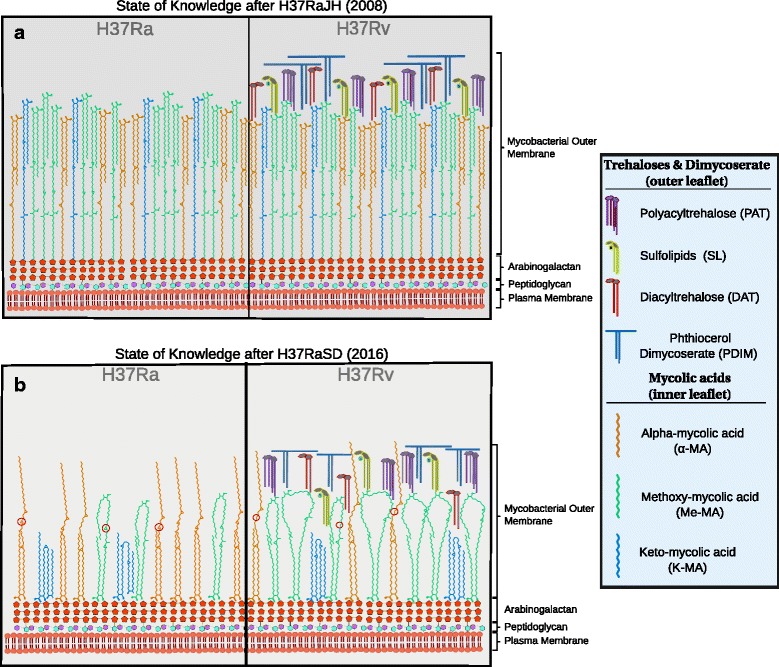
Cell Wall Differences in H37Ra and H37Rv. a State of knowledge following publication of H37RaJH. At this time it was known that the SNP in the DNA-binding site of phoP abrogated synthesis of sulfolipids (yellow) and acyltrehaloses (purple and red) of the mycomembrane outer leaflet, while two SNPs in pks12, both of which were refuted in our assembly, were thought to cause the observed lack of phthiocerol dimycocerosates (blue) in H37Ra. b Current state of knowledge. Advances were made in understanding the inner leaflet. A single nucleotide, frameshift deletion in the now annotated hadC gene was shown by Slama and colleagues [33] to alter the mycolic acid profile in three distinct ways: i. Lower proportion of oxygenated mycolic acids (K-MA and Me-MA; green and blue carbon skeletons, respectively) to α-MAs (orange carbon skeleton). There are seven Me-MAs depicted in H37Rv compared to three in H37Ra, reflecting the proportions reported by Slama and colleagues [33]. ii. Extra degree of unsaturation (red circles) in H37Ra mycolic acids due to truncation of the HadC protein in H37Ra. iii. Shorter chain lengths of mycolic acids in H37Ra. Note that Me-MAs have larger loops in H37Rv than in H37Ra, and that the height of the α-MAs is shorter in H37Ra than H37Rv. Carbon chain lengths are based on results reported by Slama and colleagues. The folding geometry of the mycolic acids is depicted in panel B, as described by Groenewald and colleagues [56], and inspired by the illustration style of Minnikin and colleagues [57]
The second gene, mazG, has a nsSNP (A219E) in a region coding for a highly conserved alpha-helix residue in its protein product, a nucleoside triphosphate (NTP) pyrophosphohydrolase [5]. MazG exhibits diminished hydrolysis activity in H37Ra relative to both MazG in H37Rv and MazG of the fast-growing Mycobacterium smegmatis. Wild-type MazG hydrolyzes all NTPs, including those that are mutagenic and appear more frequently with oxidative stress (Fig. 3 b), which is experienced by the bacterium inside activated macrophages [31]. This decreased ability to hydrolyze mutagenic NTPs contributes to virulence attenuation in H37Ra [32].
In the third gene, hadC, there is a frameshift-inducing 1-bp insertion, which creates a premature stop codon and results in truncation of HadC. hadC is a member of the essential hadA-hadB-hadC gene cluster, which forms two hydratases (HadAB and HadBC) of the M. tuberculosis fatty acid synthase II system. Our assembly and H37RaJH both show a 5-bp insertion in hadA which, along with hadC, are the only genes with variants in H37Ra [33] that encode proteins known to be necessary for mycolic acid synthesis.
Recent complementation and knockout studies using hadC from H37Ra and H37Rv showed that intact HadC is necessary for cord formation, and that the truncated form H37R a/h a d C affects length and oxygenation of mycolic acids (Fig. 4 b). Furthermore, when tested in murine lung and spleen, H37R a/h a d C Rv grew an intermediate amount of colony forming units, between that of H37Ra and H37Rv, at a level commensurate with H37R v Δ h a d C which suggests that the H37Ra hadC variant underlies some of its virulence attenuation [33].
Interestingly, while both our assembly and H37RaJH harbor a 5-bp insertion in hadA, sequences obtained by Lee, Slama, and their respective colleagues do not [29, 33]. These two sequences were both derived from a culture from Institut Pasteur, while ours and that of Zheng and colleagues [5] were acquired directly from ATCC, which suggests that the two cultures diverged in vitro prior to sequencing despite sharing the same ATCC identifier. We expect the deleterious effects of h a d C Ra shown by Slama and colleagues would be exacerbated by the 5bp insertion in our assembly, as it results in an abnormal HadAB enzyme which, when normal, has been posited to compensate for faulty HadBC [33]. However, the experiments discussed above indicate that the hadC variant alone is sufficient to attenuate virulence, and is one of the primary sources of attenuation in H37Ra.
Copy number variation in lpdA promoter
The polymorphism reported in H37RaJH that affects lpdA (NAD(P)H quinone reductase) is a third (as opposed to the two in H37Rv) 58bp repeat in its promoter region. Our assembly reveals an additional two copies of this 58bp region, resulting in a total of five copies of the repeat. LpdA has been shown to protect bacilli from oxidative stress and improve M. tuberculosis survival in a mouse model [34]. However the copy number of this tandem repeat in our assembly matched two of the H37Rv assemblies—H37RvBroad and H37RvTMC102—meaning this copy number variation is not specific to the avirulent strain and does not contribute to the phenotype of H37Ra. Despite the contradiction of this copy number variation being H37Ra-specific, the observed differential expression of lpdA with respect to H37Rv [5] may contribute to virulence, perhaps through altered regulation by PhoP, as lpdA is under its regulon [30].
Variants affecting uncharacterized hypothetical genes
Several genes classified with unknown or hypothetical functions were among the HC genes of H37RaJH (Table 1). Our assembly contradicts all variants in the majority of these, leaving three which we supported in full.
Though none of these genes have an implicated role in virulence in the literature, they may in reality. These genes should be investigated, as they are three of the few supported HC genes yet to be explored. The value of exploring hypothetical genes is evidenced by the recent discovery of a significant contribution of HadC [33]—the function of which was unknown when H37RaJH was published—to virulence attenuation in H37Ra (Fig. 4).
Significant reduction of H37Ra-specific variants in PE_PPE genes
The PE_PPE family of genes is unique to mycobacteria but poorly characterized, both functionally and genomically, in M. tuberculosis, the latter owing to the family’s high-GC content and repetitive nature [35]. Evidence for contribution from PE_PPE family members to virulence has amassed support since 2008[36–39], but this gene family was the most drastically altered by our assembly: while PE_PPE genes comprise approximately 10% of the genome, they account for nearly half (16/36) of the contradicted genes. It is likely that the majority of these are errors in H37RaJH rather than manifestations of hypervariability, as few PE_PPE genes fell into the adjusted or novel categories, as one would expect if they were due to hypervariability.
Consequently, some extant work examining polymorphic PE_PPE genes between H37RaJH and H37Rv is invalidated by our assembly. For example, our assembly contradicts or changes the variant profile of all four PE_PPE genes reported to be positively selected for in H37Ra in an evolutionary genomics study by Zhang and colleagues [38] using H37RaJH.
Another study affected profoundly by our results is that of Kohli and colleagues [36], which used H37RaJH and H37Rv in an in silico genomic and proteomic comparison of PE_PPE family genes. Though our assembly renders much of the results from their analyses invalid, applying their methodology to our updated assembly would yield interesting results.
Our assembly contains polymorphisms in 6 of 22 genes that encode PE_PPE family members reported as unique to H37RaJH (Table 1, Fig. 3 b). Of the three PE_PPE family members fully corroborated by our sequence, one was the duplication of ppe38, which McEvoy and colleagues have also identified in 3 different samples of H37Rv, suggesting this duplication likely plays no role in virulence [40]. All 3 of the adjusted PE_PPE family members, as well as the supported Wag22 and PPE13, belong to PE_PPE sublineage V. Sublineage V members comprise the majority of PE_PPE proteins that interact with the host, and are overrepresented in proteomic studies of in vivo infection [35]. This enrichment of subfamily V PE_PPE family members in the set of supported or adjusted genes suggests they may be more integral to virulence attenuation in H37Ra than other PE_PPE family members. The role of PE_PPE family members in virulence should become better understood as more genomes are sequenced using third-generation platforms.
In addition to the differences due to sequence alterations in PE_PPE family genes, the corroborated polymorphism in phoP may confer altered expression of many PE_PPE family proteins, as at least 13 are under its regulon [35], which could mediate some virulence attenuation.
The precise roles of PE_PPE family members have yet to be elucidated in full. It is difficult to evaluate rigorously the effect of each PE_PPE variant, as their function in wild-type M. tuberculosis is poorly characterized [35]. Moreover, their contribution to virulence may well require complexities of the native host environment beyond what can be replicated in vitro or ex vivo with current technology. Thus, the role the polymorphisms in this family play in the phenotype of H37Ra compel further research, which our reduction of variants has made more tractable.
Discussion
Since its publication in 2008 [5], several studies have used the whole genome [8, 36, 41–46] of H37RaJH, or the reported differences between H37RaJH and H37Rv [47] in their analyses. Our improved assembly changes the implications of several of these in silico analyses. Additionally, several studies have used the set of genes with variants in H37RaJH with respect to H37Rv to guide wet-lab experiments [48, 49]. Re-examining these studies with our assembly of H37Ra may yield novel insights, as contradicted variants can serve as a retroactive control.
Our de novo assembly using single-molecule sequencing has reduced the set of genes polymorphic to H37Rv by more than half, clarifying which genomic factors most likely give rise to virulence attenuation and other H37Ra-specific phenotypes. For an expanded discussion of genes affected and their ties to virulence, see Additional file 3. Supported variants affecting PhoP, MazG, and HadC have been experimentally affirmed [23, 32, 33], gaining insight into how they manifest in the phenotype of H37Ra, but basic mechanisms for their contributions are not fully elucidated. Two other supported or adjusted genes (pabB and nrp) have been indirectly connected to the avirulence of H37Ra through experiments on other mycobacterial species [22] or H37Ra complementation studies measuring proxies of virulence [48].
It is clear that the nsSNP in phoP remains a potent mediator of virulence of M. tuberculosis through affecting SL and ATHL activity (Fig. 4), while the truncation of HadC enfeebles the mycomembrane (Fig. 4 b). The polymorphism in mazG may compromise stress response mechanisms in H37Ra (Fig. 3), which are critical to enduring the intramacrophage environment of the host [32, 34]. Variants affecting genes with regulatory functions—phoP and others with roles in regulation not yet known—may also cause downstream effects on H37Ra phenotype, which may prove difficult to characterize. The variants in genes of the PE_PPE family and hypothetical genes (Rv0010c, Rv0039c, and Rv1006) potentially contribute to virulence attenuation through mechanisms not yet identified. Thus, with the greater accuracy of our assembly, wet-lab studies can focus on the true differences between H37Ra and H37Rv, and computational studies will be in greater concordance with reality, yielding more useful results.
The advantages of single-molecule sequencing are readily apparent in our results. The random error profile of this technology allows for consensus accuracy to increase as a function of sequencing depth [10]. The coverage depth of our assembly corresponds to a Phred quality value greater than 60 (QV >60), which translates to fewer than four expected errors [11]. If such errors exist, they would most likely appear as single-base insertions or deletions unique to our assembly. The fact that performing the assembly with half of our available data resulted in an identical sequence indicates that a single sequencing run is sufficient for accurate assembly of M. tuberculosis genomes with our methodology. The long reads produced by this technology allowed us to easily and unambiguously capture known structural variants in H37Ra, as well as two novel to the strain. We were also able to fully resolve the GC-rich and repetitive PE_PPE genes, sequences which compound the weaknesses of most sequencing technologies. As a result, our assembly demonstrates that H37Ra is significantly more similar to H37Rv than indicated by the Sanger-based reference sequence H37RaJH, with contradicted variants overrepresented in the difficult sequences of the PE_PPE genes. While in vitro evolution may underlie some of the differences between our assembly and H37RaJH, we believe that most of the contradicted variants (Table 1a) reflect sequencing errors in H37RaJH due to the disparity in sequencing quality. Regardless, the contradicted variants should not be considered as characteristic of H37Ra or its attenuated virulence. These sites were concordant with H37Rv and we did not find additional polymorphic PE_PPE genes with respect to H37Rv (Table 2), indicating a disparity in sequencing accuracy even among the Sanger-based references.
Conclusions
Studies that have relied on the accuracy of PE_PPE sequences in the H37Ra reference sequence were the most severely impacted by our study. We consequently advise caution when analyzing GC-rich and repetitive sequences among reference genomes, not to mention draft genomes. As de novo assembly can be routinely performed for microbes using single-molecule sequencing, we strongly recommend this for mycobacteria, especially because of their PE_PPE genes.
Methods
Sample preparation and whole-genome sequencing
M. tuberculosis H37Ra (ATCC25177) was obtained from ATCC and cultured on Lowenstein-Jenson slants and Middlebrook 7H11 plates. Cultures were incubated until growth of a full bacterial lawn. DNA was extracted using Genomic-tips (Qiagen Inc.) following the manufacturer’s sample preparation and lysis protocol for bacteria with the following modifications. Culture was harvested directly into buffer B1/RNAse solution, homogenized by vigorous vortex mixing and inactivated at 80°C for 15 minutes. Lysozyme was added and incubated at 37°C for 30 minutes followed by the addition of proteinase K and further incubation at 37°C for an additional 60 minutes. Buffer B2 was added and the mixture was incubated overnight at 50°C. Wide-bore pipet tips were used to optimize recovery of large DNA fragments. The remainder of the Genomic-tip protocol was carried out exactly as described by the manufacturer. DNA purity and concentration was analyzed on a Nanodrop 1000 (Thermo Scientific). The DNA was then sequenced using two SMRTCells on the Pacific Biosciences RS II instrument with the P6-C4 chemistry and a 20kb insert library preparation.
Genome assembly and methylome determination
The genome was assembled using Pacific Biosciences’ Hierarchical Genome Assembly Process [12] (HGAP) as implemented in SMRTAnalysis 2.3.0. This version of SMRTAnalysis provides two implementations of HGAP: HGAP.2 and the newer HGAP.3. HGAP.3 differs from HGAP.2 by replacing the Celera Assembler’s assembly consensus step with Pacific Biosciences’ speed-optimized implementation. We used HGAP.2 because, in our experiments, we found that HGAP.3 consistently produced spurious contigs while HGAP.2 did not.
The overlapping ends of the contig, an artifact of the assembly due to the circularity of the chromosome, were trimmed and joined using the minimus2 program from AMOS (http://amos.sourceforge.net). Discrepancies between the contig ends were resolved manually by selecting an authoritative sequence and trimming the discrepant one. The circularization was also performed with Circlator [50] to confirm the minimus2 results.
We validated the assembly structure using PBHoney [51], a structural variation detection tool, by using our assembled genome as input. Any structural variations detected would indicate potential misassemblies.
Circularization was followed by iterative assembly polishing using Quiver in SMRTAnalysis until the consensus sequence converged, which amounted to three rounds. Quiver was used with the maximum coverage parameter set to 1000 and otherwise default settings.
The methylome was determined using the base modification and motif analysis protocol in SMRTAnalysis.
Sequence selection and comparative genomics
Multiple finished assemblies exist for H37Rv and H37Ra (Table 3). While the unconventionally named samples “F1” (H37Rv) and “F28” (H37Ra) were sequenced on the Pacific Biosciences platform by Zhu and colleagues [13], “F28” had substantial differences from both our assembly and H37RaJH, leading us to suspect reduced sequencing accuracy in their data, potentially due to sample overloading. Because the authors did not provide the sequencing summary statistics, we were unable to verify this and establish a reasonable consensus sequence accuracy, so the “F1” and “F28” assemblies were excluded from our analysis.
Table 3.
Available Finished Assemblies for the Reference Strains M. tuberculosis H37Rv and H37Ra
| Strain | Name | ATCC identifier | Accession | Technology | Last updated |
|---|---|---|---|---|---|
| H37Rv | F1a[13] | 27294 | CP010329.1 | Pacific Biosciences (P4-C2) | 02/2016 |
| H37RvSiena | unspecified | NZ_CP007027.1/CP007027.1 | Illumina | 01/2015 | |
| H37RvTMC102 | 27294D-2 | NZ_CP009480.1/CP009480.1 | Illumina | 09/2014 | |
| H37RvBroad | unspecified | NC_018143.2/CP003248.2 | 454/Sanger/Illumina | 10/2013 | |
| H37Rv [62] | 25618b | NC_000962.3/AL123456.3 | Sanger | 02/2013 | |
| H37Ra | H37RaSD [present study] | 25177 | CP016972.1 | Pacific Biosciences (P6-C4) | 08/2016 |
| F28a[13] | 25177 | NZ_CP010330.1/CP010330.1 | Pacific Biosciences (P4-C2) | 02/2016 | |
| H37RaJH [5] | 25177 | NC_009525.1/CP000611.1 | Sanger | 05/2007 |
Unreferenced entries were direct database submissions and do not have an associated publication
aThe unconventional names for these samples were not explained by Zhu and colleagues [13]. The name F28 in particular is already known from the literature to refer to a family of clinical isolates [63]
bThe ATCC number was unspecified by Cole and colleagues [62]. However, the ATCC catalog entry for this strain identifies it as the source for the sequence
For genome comparison, we initially used MUMmer [52], but it did not precisely specify structural variations, making their resolution and comparison difficult. For example, while it identified the lpdA tandem repeat variants, it could not properly resolve the actual sequence differences. Therefore, in our study, variants were identified using GNU diff (http://www.gnu.org/software/diffutils), an implementation of Myers’ algorithm for solving the longest-common-subsequence problem [53, 54] and converted to the Variant Call Format for analysis. This process is implemented in our custom tool, biodiff (https://www.gitlab.com/LPCDRP/biodiff). Biodiff has been tested by applying variants called for a given query sequence to the reference and ensuring that the result is identical to the original query sequence. In particular, the variants that biodiff called in H37RaJH with respect to H37Rv were manually compared with those reported by Zheng and colleagues [5] to ensure that the variants referred to in our analysis correspond to what they discussed in their study.
Because insertions and deletions in repetitive regions can be represented equivalently in multiple ways, we normalized the variants using the “norm” function of bcftools (http://samtools.github.io/bcftools), giving every mutation a standard representation to facilitate a proper comparison. Variants were then compared using bcftools isec and annotated using the Ensembl Variant Effect Predictor [55]. Motif variants were analyzed using in villa code.
Literature review
In order to gain a holistic view of the research built off of and conclusions drawn from the unique variants of H37RaJH with respect to H37Rv, we performed an exhaustive literature review. Common names and Rv numbers were searched using Google scholar within all publications which cited Zheng et al, 2008 [5] as of March 14th, 2016, for all genes with H37RaJH specific variants within CDS or potential promoter regions, according to Table 2 of [5]. All mentions of these genes were compiled and evaluated to illustrate how our assembly alters the picture of how the genomic differences between the reference strains contribute to the observed virulence attenuation of H37Ra (Discussion). Genes are discussed in the present study according to the H37Rv annotation (as opposed to H37Ra’s own annotation), as this convention relates to extant publications most readily.
Additional files
Raw Variants. Zip archive containing the following data in Variant Call Format (VCF): H37RaSD-H37RaJH.vcf. Variants in our H37Ra assembly with respect to the H37Ra reference sequence (NC_009525.1). H37RaSD-H37Rv.vcf. Variants in our H37Ra assembly with respect to the H37Rv reference sequence (NC_000962.3). H37RaJH-H37Rv.vcf. Variants in the H37Ra reference sequence (NC_009525.1) with respect to the H37Rv reference sequence (NC_000962.3). (ZIP 16 kb)
Annotated Variants with Respect to H37Rv. Spreadsheet containing annotated variants in our assembly and the H37Ra reference sequence with respect to the H37Rv reference sequence. The sheets separate variants that are common to the two H37Ra assemblies and those that are unique to each. (XLSX 28 kb)
Expanded Discussion of Virulence Attenuation Mechanisms in M. tuberculosis H37Ra. (PDF 136 kb)
Computer Code used for Analyses. Workflow and scripts to run variant comparisons and related analyses. May be extracted using 7zip (http://www.7-zip.org/). (TAR 50 kb)
Acknowledgements
We would like to thank Jason Chin and Richard Hall from Pacific Biosciences for discussions on de novo assembly methodology and quality assessment. Logan Fink also provided some assistance with quality assessment of the assembly. We would also like to thank Antonino Catanzaro, Timothy Rodwell, and their staff for bacterial culture and DNA extraction. Jonas Korlach, Anthony Baughn, Sarah Ramirez-Busby, Ragavi Shanmugam, Carmela Chan, Amy Goodmanson, Daeheon Oh, and Logan Fink reviewed and provided helpful feedback on drafts of the manuscript.
Funding
This work was funded by a grant from National Institute of Allergy and Infectious Diseases (NIAID Grant No. R01AI105185). A.E., S.J.M., and F.V. were supported by this grant. S.J.M. was also supported by scholarships from a National Science Foundation Grant (no. 0966391). The funding bodies had no role in the design of the study or collection, analysis, and interpretation of data or in writing the manuscript.
Availability of data and materials
Data for this project have been submitted to Genbank and the NCBI Sequence Read Archive under Bioproject PRJNA329548. The accession number for the assembly is CP016972. Our motif variants detection tool is available from https://gitlab.com/LPCDRP/motif-variants. Analysis code for this study is provided in Additional file 4. All figures were created using Inkscape (http://inkscape.org).
Authors’ contributions
F.V., A.E., and S.J.M. designed the study. A.E. performed the de novo assembly, methylation analysis, and comparative genomics analyses. S.J.M. performed the literature review, interpreted the results, and wrote Additional file 3. A.E. and S.J.M. prepared the manuscript, which was reviewed and approved by all authors.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- H37RaJH
H37Ra Johns Hopkins (The assembly of H37Ra performed there)
- HC
High-confidence
- HGAP
Hierarchical genome assembly process
- MTBC
Mycobacterium tuberculosis complex
- nsSNP
Non-synonymous single nucleotide polymorphism
- NTP
Nucleoside triphosphate
- PDIM
Phthiocerol dimycocerosate
- QV
Phred quality value (a metric for base-call accuracy)
- SMRT
Single-molecule, real-time
- SNP
Single nucleotide polymorphism
References
- 1.World Health Organization . Global Tuberculosis Report. Geneva: WHO Press, World Health Organization; 2015. [Google Scholar]
- 2.Middlebrook G, Dubos RJ, Pierce C. Virulence and morphological characteristics of mammalian tubercle bacilli. J Exp Med. 1947;86(2):175–84. doi: 10.1084/jem.86.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heplar JQ, Clifton CE, Raffel S, Futrelle CM. Virulence of the tubercle bacillus: I, Effect of oxygen tension upon respiration of virulent and avirulent bacilli. J Infect Dis. 1954;94(1):90–8. doi: 10.1093/infdis/94.1.90. [DOI] [PubMed] [Google Scholar]
- 4.Alsaadi AI, Smith DW. The fate of virulent and attenuated mycobacteria in guinea pigs infected by the respiratory route. Am Rev Respir Dis. 1973;107(6):1041–6. doi: 10.1164/arrd.1973.107.6.1041. [DOI] [PubMed] [Google Scholar]
- 5.Zheng H, Lu L, Wang B, Pu S, Zhang X, Zhu G, Shi W, Zhang L, Wang H, Wang S, Zhao G, Zhang Y. Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra versus H37Rv. PLoS ONE. 2008;3(6):2375. doi: 10.1371/journal.pone.0002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koren S, Phillippy AM. One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr Opin Microbiol. 2015;23:110–20. doi: 10.1016/j.mib.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 7.Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14(5):51. doi: 10.1186/gb-2013-14-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ioerger TR, Feng Y, Ganesula K, Chen X, Dobos KM, Fortune S, Jacobs WR, Mizrahi V, Parish T, Rubin E, Sassetti C, Sacchettini JC. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J Bacteriol. 2010;192(14):3645–53. doi: 10.1128/JB.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Köser CU, Niemann S, Summers DK, Archer JAC. Overview of errors in the reference sequence and annotation of Mycobacterium tuberculosis H37Rv, and variation amongst its isolates. Infect Genet Evol. 2012;12(4):807–10. doi: 10.1016/j.meegid.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 10.Roberts RJ, Carneiro MO, Schatz MC. The advantages of SMRT sequencing. Genome Biol. 2013;14(6):405. doi: 10.1186/gb-2013-14-6-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koren S, Harhay GP, Smith TP, Bono JL, Harhay DM, Mcvey SD, Radune D, Bergman NH, Phillippy AM. Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol. 2013;14(9):1–16. doi: 10.1186/gb-2013-14-9-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10(6):563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 13.Zhu L, Zhong J, Jia X, Liu G, Kang Y, Dong M, Zhang X, Li Q, Yue L, Li C, Fu J, Xiao J, Yan J, Zhang B, Lei M, Chen S, Lv L, Zhu B, Huang H, Chen F. Precision methylome characterization of Mycobacterium tuberculosis complex (MTBC) using PacBio single-molecule real-time (SMRT) technology. Nucleic Acids Res. 2016;44(2):730–43. doi: 10.1093/nar/gkv1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roychowdhury T, Mandal S, Bhattacharya A. Analysis of IS6110 insertion sites provide a glimpse into genome evolution of Mycobacterium tuberculosis. Sci Rep. 2015;5:12567. doi: 10.1038/srep12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alonso H, Samper S, Martín C, Otal I. Mapping IS6110 in high-copy number Mycobacterium tuberculosis strains shows specific insertion points in the Beijing genotype. BMC Genomics. 2013;14(1):422. doi: 10.1186/1471-2164-14-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lari N, Rindi L, Garzelli C. Identification of one insertion site of IS6110 in Mycobacterium tuberculosis H37Ra and analysis of the RvD2 deletion in M, tuberculosis clinical isolates. J Med Microbiol. 2001;50(9):805–11. doi: 10.1099/0022-1317-50-9-805. [DOI] [PubMed] [Google Scholar]
- 17.Andreu N, Gibert I. Cell population heterogeneity in Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinburgh, Scotland) 2008;88(6):553–9. doi: 10.1016/j.tube.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 18.Dokladda K, Billamas P, Palittapongarnpim P. Different behaviours of promoters in Mycobacterium tuberculosis H37Rv and H37Ra. World J Microbiol Biotechnol. 2015;31(2):407–13. doi: 10.1007/s11274-014-1794-x. [DOI] [PubMed] [Google Scholar]
- 19.Malhotra V, Tyagi JS, Clark-Curtiss JE. DevR-mediated adaptive response in Mycobacterium tuberculosis H37Ra: links to asparagine metabolism. Tuberculosis (Edinburgh, Scotland) 2009;89(2):169–74. doi: 10.1016/j.tube.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daffé M, Lacave C, Lanéelle MA, Gillois M, Lanéelle G. Polyphthienoyl trehalose, glycolipids specific for virulent strains of the tubercle bacillus. Eur J Biochem FEBS. 1988;172(3):579–84. doi: 10.1111/j.1432-1033.1988.tb13928.x. [DOI] [PubMed] [Google Scholar]
- 21.Middlebrook G, Coleman CM, Schaefer WB. Sulfolipid from virulent tubercle bacilli. Proc Natl Acad Sci U S A. 1959;45(12):1801–4. doi: 10.1073/pnas.45.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hotter GS, Wards BJ, Mouat P, Besra GS, Gomes J, Singh M, Bassett S, Kawakami P, Wheeler PR, de Lisle GW, Collins DM. Transposon mutagenesis of Mb0100 at the ppe1-nrp locus in Mycobacterium bovis disrupts phthiocerol dimycocerosate (PDIM) and glycosylphenol-PDIM biosynthesis, producing an avirulent strain with vaccine properties at least equal to those of M. bovis BCG. J Bacteriol. 2005;187(7):2267–77. doi: 10.1128/JB.187.7.2267-2277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chesne-Seck ML, Barilone N, Boudou F, Asensio JG, Kolattukudy PE, Martín C, Cole ST, Gicquel B, Gopaul DN, Jackson M. A point mutation in the two-component regulator PhoP-PhoR accounts for the absence of polyketide-derived acyltrehaloses but not that of phthiocerol dimycocerosates in Mycobacterium tuberculosis H37Ra. J Bacteriol. 2008;190(4):1329–34. doi: 10.1128/JB.01465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hotter GS, Collins DM. Mycobacterium bovis lipids: Virulence and vaccines. Vet Microbiol. 2011;151(1-2):91–8. doi: 10.1016/j.vetmic.2011.02.030. [DOI] [PubMed] [Google Scholar]
- 25.Li AH, Waddell SJ, Hinds J, Malloff CA, Bains M, Hancock RE, Lam WL, Butcher PD, Stokes RW. Contrasting transcriptional responses of a virulent and an attenuated strain of Mycobacterium tuberculosis infecting macrophages. PLoS One. 2010; 5(6). doi:10.1371/journal.pone.0011066. [DOI] [PMC free article] [PubMed]
- 26.Asensio JG, Maia C, Ferrer NL, Barilone N, Laval F, Soto CY, Winter N, Daffé M, Gicquel B, Martín C, Jackson M. The virulence-associated two-component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J Biol Chem. 2006;281(3):1313–1316. doi: 10.1074/jbc.C500388200. [DOI] [PubMed] [Google Scholar]
- 27.Frigui W, Bottai D, Majlessi L, Monot M, Josselin E, Brodin P, Garnier T, Gicquel B, Martin C, Leclerc C, Cole ST, Brosch R. Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS Pathogens. 2008; 4(2). doi:10.1371/journal.ppat.0040033. [DOI] [PMC free article] [PubMed]
- 28.Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol. 2006;60(2):312–30. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 29.Lee JS, Krause R, Schreiber J, Mollenkopf HJ, Kowall J, Stein R, Jeon BY, Kwak JY, Song MK, Patron JP, Jorg S, Roh K, Cho SN, Kaufmann SHE. Mutation in the transcriptional regulator PhoP contributes to avirulence of Mycobacterium tuberculosis H37Ra strain. Cell Host Microbe. 2008;3(2):97–103. doi: 10.1016/j.chom.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Solans L, Gonzalo-Asensio J, Sala C, Benjak A, Uplekar S, Rougemont J, Guilhot C, Malaga W, Martín C, Cole ST. The PhoP-dependent ncRNA Mcr7 modulates the TAT secretion system in Mycobacterium tuberculosis. PLoS Pathogens. 2014; 10(5). doi:10.1371/journal.ppat.1004183. [DOI] [PMC free article] [PubMed]
- 31.Lyu LD, Tang BK, Fan XY, Ma H, Zhao GP. Mycobacterial MazG safeguards genetic stability via housecleaning of 5-OH-dCTP. PLoS Pathogens. 2013; 9(12). doi:10.1371/journal.ppat.1003814. [DOI] [PMC free article] [PubMed]
- 32.Lu LD, Sun Q, Fan XY, Zhong Y, Yao YF, Zhao GP. Mycobacterial MazG is a novel NTP pyrophosphohydrolase involved in oxidative stress response. J Biol Chem. 2010;285(36):28076–85. doi: 10.1074/jbc.M109.088872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slama N, Jamet S, Frigui W, Pawlik A, Bottai D, Laval F, Constant P, Lemassu A, Cam K, Daffé M, Brosch R, Eynard N, Quémard A. The changes in mycolic acid structures caused by hadC mutation have a dramatic effect on the virulence of Mycobacterium tuberculosis. Mol Microbiol. 2015. doi:10.1111/mmi.13266. [DOI] [PubMed]
- 34.Akhtar P, Singh S, Bifani P, Kaur S, Srivastava BS, Srivastava R. Variable-number tandem repeat 3690 polymorphism in Indian clinical isolates of Mycobacterium tuberculosis and its influence on transcription. J Med Microbiol. 2009;58(6):798–805. doi: 10.1099/jmm.0.002550-0. [DOI] [PubMed] [Google Scholar]
- 35.Fishbein S, van Wyk N, Warren RM, Sampson SL. Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol Microbiol. 2015;96(5):901–16. doi: 10.1111/mmi.12981. [DOI] [PubMed] [Google Scholar]
- 36.Kohli S, Singh Y, Sharma K, Mittal A, Ehtesham NZ, Hasnain SE. Comparative genomic and proteomic analyses of PE/PPE multigene family of Mycobacterium tuberculosis H37Rv and H37Ra reveal novel and interesting differences with implications in virulence. Nucleic Acids Res. 2012;40(15):7113–22. doi: 10.1093/nar/gks465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu G, Fu X, Jin K, Zhang L, Wu W, Cui Z, Hu Z, Li Y. Integrative analysis of transcriptome and genome indicates two potential genomic islands are associated with pathogenesis of Mycobacterium tuberculosis. Gene. 2011;489(1):21–9. doi: 10.1016/j.gene.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Zhang H, Zhou T, Zhong Y, Jin Q. Genes under positive selection in Mycobacterium tuberculosis. Comput Biol Chem. 2011;35(5):319–22. doi: 10.1016/j.compbiolchem.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 39.Ahmed A, Das A, Mukhopadhyay S. Immunoregulatory functions and expression patterns of PE/PPE family members: Roles in pathogenicity and impact on anti-tuberculosis vaccine and drug design. IUBMB Life. 2015;67(6):414–27. doi: 10.1002/iub.1387. [DOI] [PubMed] [Google Scholar]
- 40.McEvoy CR, Helden PD, Warren RM, Pittius NCG. Evidence for a rapid rate of molecular evolution at the hypervariable and immunogenic Mycobacterium tuberculosis PPE38 gene region. BMC Evol Biol. 2009;9:237. doi: 10.1186/1471-2148-9-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu W, Xiao S, Li M, Guo S, Li S, Luo R, Feng Z, Li B, Zhou Z, Shao G, Chen H, Fang L. Comparative genomic analyses of Mycoplasma hyopneumoniae pathogenic 168 strain and its high-passaged attenuated strain. BMC Genomics. 2013;14:80. doi: 10.1186/1471-2164-14-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Zhang Y, Zheng H, Pan Y, Liu H, Du P, Wan L, Liu J, Zhu B, Zhao G, Chen C, Wan K. Genome sequencing and analysis of BCG vaccine strains. PLoS ONE. 2013; 8(8). doi:10.1371/journal.pone.0071243. [DOI] [PMC free article] [PubMed]
- 43.Zhang S, Chen J, Shi W, Liu W, Zhang W, Zhang Y. Mutations in panD encoding aspartate decarboxylase are associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerg Microbes Infect. 2013;2(6):34. doi: 10.1038/emi.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song T, Park Y, Shamputa IC, Seo S, Lee SY, Jeon HS, Choi H, Lee M, Glynne RJ, Barnes SW, Walker JR, Batalov S, Yusim K, Feng S, Tung CS, Theiler J, Via LE, Boshoff HIM, Murakami KS, Korber B, Barry CE, Cho SN. Fitness costs of rifampicin-resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β’ subunit of RNA polymerase. Mol Microbiol. 2014;91(6):1106–19. doi: 10.1111/mmi.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freidlin PJ, Goldblatt D, Kaidar-Shwartz H, Rorman E. Polymorphic Exact Tandem Repeat A (PETRA): a newly defined lineage of Mycobacterium tuberculosis in Israel originating predominantly in Sub-Saharan Africa. J Clin Microbiol. 2009;47(12):4006–20. doi: 10.1128/JCM.01270-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Namouchi A, Karboul A, Fabre M, Gutierrez MC, Mardassi H. Evolution of smooth tubercle bacilli PE and PE_PGRS genes: Evidence for a prominent role of recombination and imprint of positive selection. PLoS ONE. 2013; 8(5). doi:10.1371/journal.pone.0064718. [DOI] [PMC free article] [PubMed]
- 47.Banerjee R, Vats P, Dahale S, Kasibhatla SM, Joshi R. Comparative genomics of cell envelope components in mycobacteria. PLoS ONE. 2011; 6(5). doi:10.1371/journal.pone.0019280. [DOI] [PMC free article] [PubMed]
- 48.Zhang G, Zhu B, Shi W, Wang M, Da Z, Zhang Y. Evaluation of mycobacterial virulence using rabbit skin liquefaction model. Virulence. 2010;1(3):156–63. doi: 10.4161/viru.1.3.11748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Målen H, De Souza GA, Pathak S, Søfteland T, Wiker HG. Comparison of membrane proteins of Mycobacterium tuberculosis H37Rv and H37Ra strains. BMC Microbiol. 2011;11:18. doi: 10.1186/1471-2180-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunt M, Silva ND, Otto TD, Parkhill J, Keane JA, Harris SR. Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015; 16(1). doi:10.1186/s13059-015-0849-0. [DOI] [PMC free article] [PubMed]
- 51.English AC, Salerno WJ, Reid JG. PBHoney: identifying genomic variants via long-read discordance and interrupted mapping. BMC Bioinformatics. 2014;15(1):180. doi: 10.1186/1471-2105-15-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Myers EW. An O(ND) difference algorithm and its variations. Algorithmica. 1986;1(1):251–66. doi: 10.1007/BF01840446. [DOI] [Google Scholar]
- 54.Miller W, Myers EW. A file comparison program. Softw Pract Experience. 1985;15(11):1025–40. doi: 10.1002/spe.4380151102. [DOI] [Google Scholar]
- 55.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Groenewald W, Baird MS, Verschoor JA, Minnikin DE, Croft AK. Differential spontaneous folding of mycolic acids from Mycobacterium tuberculosis. Chem Phys Lipids. 2014;180:15–22. doi: 10.1016/j.chemphyslip.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 57.Minnikin DE, Lee OY, Wu HHT, Nataraj V, Donoghue HD, Ridell M, Watanabe M, Alderwick L, Bhatt A, Besra GS. Pathophysiological implications of cell envelope structure in Mycobacterium tuberculosis and related taxa In: Ribon W, editor. Tuberculosis - Expanding Knowledge: 2015. doi:10.5772/59585.
- 58.Jena L, Kashikar S, Kumar S, Harinath BC. Comparative proteomic analysis of Mycobacterium tuberculosis strain H37Rv versus H37Ra. Int J Mycobacteriology. 2013;2(4):220–6. doi: 10.1016/j.ijmyco.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 59.Mohareer K, Tundup S, Hasnain SE. Transcriptional regulation of Mycobacterium tuberculosis PE/PPE Genes: A molecular switch to virulence? J Mol Microbiol Biotechnol. 2011;21(3-4):97–109. doi: 10.1159/000329489. [DOI] [PubMed] [Google Scholar]
- 60.Squires AH, Atas E, Meller A. Genomic pathogen typing using solid-state nanopores. PLoS One. 2015;10(11):0142944. doi: 10.1371/journal.pone.0142944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Velayati AA, Abeel T, Shea T, Konstantinovich Zhavnerko G, Birren B, Cassell GH, Earl AM, Hoffner S, Farnia P. Populations of latent Mycobacterium tuberculosis lack a cell wall: isolation, visualization, and whole-genome characterization. Int J Mycobacteriol. 2016;5(1):66–73. doi: 10.1016/j.ijmyco.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 63.Streicher EM, Warren RM, Kewley C, Simpson J, Rastogi N, Sola C, van der Spuy GD, van Helden PD, Victor TC. Genotypic and phenotypic characterization of drug-resistant Mycobacterium tuberculosis isolates from rural districts of the Western Cape province of South Africa. J Clin Microbiol. 2004;42(2):891–4. doi: 10.1128/JCM.42.2.891-894.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Raw Variants. Zip archive containing the following data in Variant Call Format (VCF): H37RaSD-H37RaJH.vcf. Variants in our H37Ra assembly with respect to the H37Ra reference sequence (NC_009525.1). H37RaSD-H37Rv.vcf. Variants in our H37Ra assembly with respect to the H37Rv reference sequence (NC_000962.3). H37RaJH-H37Rv.vcf. Variants in the H37Ra reference sequence (NC_009525.1) with respect to the H37Rv reference sequence (NC_000962.3). (ZIP 16 kb)
Annotated Variants with Respect to H37Rv. Spreadsheet containing annotated variants in our assembly and the H37Ra reference sequence with respect to the H37Rv reference sequence. The sheets separate variants that are common to the two H37Ra assemblies and those that are unique to each. (XLSX 28 kb)
Expanded Discussion of Virulence Attenuation Mechanisms in M. tuberculosis H37Ra. (PDF 136 kb)
Computer Code used for Analyses. Workflow and scripts to run variant comparisons and related analyses. May be extracted using 7zip (http://www.7-zip.org/). (TAR 50 kb)
Data Availability Statement
Data for this project have been submitted to Genbank and the NCBI Sequence Read Archive under Bioproject PRJNA329548. The accession number for the assembly is CP016972. Our motif variants detection tool is available from https://gitlab.com/LPCDRP/motif-variants. Analysis code for this study is provided in Additional file 4. All figures were created using Inkscape (http://inkscape.org).