

Abstract
Background
Progressive supranuclear palsy (PSP) with a frontal presentation, characterized by cognitive deficits and behavioral changes, has been recognized as an early clinical picture, distinct from the classical so-called Richardson and parkinsonism presentations. The midcingulate cortex is associated with executive and attention tasks and has consistently been found to be impaired in imaging studies of patients with PSP. The aim of the present study was to determine alterations in neurotransmission underlying the pathophysiology of PSP, as well as their significance for clinically identifiable PSP subgroups.
Methods
In vitro receptor autoradiography was used to quantify densities of 20 different receptors in the caudate nucleus and midcingulate area 24' of patients with PSP (n = 16) and age- and sex-matched control subjects (n = 14).
Results
Densities of γ-aminobutyric acid type B, peripheral benzodiazepine, serotonin receptor type 2, and N-methyl-d-aspartate receptors were significantly higher in area 24′ of patients with PSP, where tau impairment was stronger than in the caudate nucleus. Kainate and nicotinic cholinergic receptor densities were significantly lower, and adenosine receptor type 1 (A1) receptors significantly higher, in the caudate nucleus of patients with PSP. Receptor fingerprints also segregated PSP subgroups when clinical parameters such as occurrence of frontal presentation and tau pathology severity were taken into consideration.
Conclusions
We demonstrate, for the first time to our knowledge, that kainate and A1 receptors are altered in PSP and that clinically identifiable PSP subgroups differ at the neurochemical level. Numerous receptors were altered in the midcingulate cortex, further suggesting that it may prove to be a key region in PSP. Finally, we add to the evidence that nondopaminergic systems play a role in the pathophysiology of PSP, thus highlighting potential novel treatment strategies.
Keywords: Progressive supranuclear palsy, Frontal presentation, Neurotransmitter receptors, Midcingulate cortex, Caudate nucleus
Background
Progressive supranuclear palsy (PSP) is a neurodegenerative disorder clinically characterized by early postural instability, supranuclear gaze palsy, parkinsonism, and cognitive decline [1]. Frontal presentation characterized by cognitive deficits and behavioral changes has recently been recognized as an early clinical picture [2, 3], distinct from the classical so-called Richardson and parkinsonism presentations [4]. Accordingly, the focus of research in PSP has expanded over the years from neuropathological studies of subcortical structures to investigations of the disease as a more diffuse condition with varying cortical involvement [5].
The midcingulate cortex, comprising areas 24′ and 32′ [6], is associated with executive and attention tasks [7] and has consistently been found to be impaired in patients with PSP [3]. A recent perfusion single-photon emission computed tomography study confirmed and extended these findings; the degree of midcingulate cortical hypoperfusion correlated with the extent of executive dysfunction in patients [8], the cardinal feature of cognitive dysfunction in PSP [9]. Therefore, understanding the neurochemical changes in this region may prove crucial to finding a treatment for cognitive symptoms.
Previous neurotransmitter studies in PSP have been focused mainly on nigrostriatal dopaminergic and cholinergic systems. Authors of a comprehensive review of in vivo imaging studies addressing PSP-associated alterations of synaptic transmission revealed that most existing studies showed decreased dopamine transporter and dopamine receptor type 2 (D2) binding densities in the striatum, whereas dopamine receptor type 1 (D1) densities were demonstrated to be unaltered [10]. In studies focused on the cholinergic system, researchers reported significant reductions of muscarinic and nicotinic receptors in the striatum [11, 12]. Despite these findings, dopamine and cholinergic replacement therapies in PSP have not proven to be effective [13]. Therefore, other neurotransmitter systems may be involved.
An intriguing question is whether the midcingulate hypometabolism found in PSP is accompanied by alterations in the densities of specific neurotransmitter receptors. Therefore, we applied quantitative in vitro receptor autoradiography on unfixed brain tissue from patients with PSP and control subjects to quantify the densities of 20 different receptor binding sites and determine PSP-related alterations in the “receptor fingerprints” [14] of midcingulate area 24′. Furthermore, because the midcingulate cortex and caudate nucleus differ considerably in their neurochemical composition in the healthy brain [6, 14], and because different brain regions are not necessarily affected in the same way by disease [15], we also examined caudate nucleus tissue obtained from the same patients with PSP.
Methods
Subjects
Brains were obtained from patients with PSP (aged 72 ± 7 years, 9 male, 7 female) recruited in a nationwide study on PSP between 2000 and 2009. Brain autopsy was conducted by The Netherlands Brain Bank according to its Legal and Ethical Code of Conduct. Control subjects consisted of age- and sex-matched subjects (aged 76 ± 10 years old, 9 male, 5 female) without a history of neurological or psychiatric diseases.
Patients were examined after referral to the outpatient department of the Erasmus University Medical Center and by visiting patients in nursing homes as part of a large longitudinal study [2, 16] approved by the medical ethics committee of the Erasmus University Medical Center. All participants or their first-degree relatives signed informed consent forms. All patients were examined by a research physician (W.Z.C. or L.D.K.) or a neurologist (A.J.W.B. or J.C.v.S.). A detailed clinical history was obtained from patients and their family members and by reviewing medical records. The neurological examination was videotaped according to a standardized protocol. Structural neuroimaging of patients was reviewed to exclude other disease causes. Family history was considered positive when at least one first-degree relative had dementia or parkinsonism. The possibility of postmortem examination was discussed with patients and their relatives. Relevant medication used in the last 3 months of life was recorded. Clinical diagnosis of patients was established in a consensus meeting according to the National Institute of Neurological Diseases and Stroke/Society for Progressive Supranuclear Palsy criteria [1]. Neuropathological diagnosis of PSP was established according to international criteria [17].
Standard neuropathology
At The Netherlands Brain Bank, the right hemispheres of all brains are processed for routine staining and immunohistochemistry against several antibodies: AT8 (1:40; Innogenetics, Ghent, Belgium), ubiquitin (1:500; Dako, Glostrup, Denmark), three-repeat tau isoform (1:3000; Upstate Biotechnology, Charlottesville, VA, USA), four-repeat tau isoform (1:100; Upstate Biotechnology), p62 (1:200, following 80 °C antigen retrieval; BD Biosciences Pharmingen, San Diego, CA, USA), trans-activation response DNA-binding protein 43 (TDP-43, 1:100 following pressure-cooking; Proteintech, Chicago, IL, USA), β-amyloid (anti-β-amyloid, 1:100 following formic acid pretreatment; Dako), and α-synuclein (anti-α-synuclein, undiluted following formic acid pretreatment; Zymed Laboratories, South San Francisco, CA, USA). Slides were incubated overnight at 4 °C. Endogenous peroxidase activity was inhibited by 30-minute incubation in a PBS-hydrogen peroxide-sodium azide solution (100 ml of 0.1 M PBS, 2 ml of 30% H2O2, 1 ml of NaN3). The Histostain-Plus broad-spectrum immunohistochemistry kit with 3,3′-diaminobenzidine (Zymed Laboratories) was used as a detection system. Slides were counterstained with Mayer’s hematoxylin and mounted in Entellan medium (EMD Millipore, Billerica, MA, USA).
A separate semiquantitative assessment of tau pathology for area 24′ and the caudate nucleus was carried out by two raters (W.Z.C. and J.C.v.S.) using a 2-point grading scale, which is an adaptation of the visual guide proposed by Williams et al. [18]. We defined Williams’s grades 1 and 2 as mild and grades 3 and 4 as moderate to severe.
In vitro receptor autoradiography
Probes from midcingulate area 24′ and the caudate putamen were taken from the left hemisphere, frozen in isopentane at −40 °C with a postmortem delay of 6 ± 1 h (patients with PSP) and 8 ± 1 h (control subjects), and serially sectioned at −20 °C in 10-μm-thick sections with a cryostat. Alternating sections were processed for the visualization of 20 transmitter receptors according to standard protocols (Table 1) comprising a preincubation to remove endogenous ligands and external substances such as medication, a main incubation to label binding sites with a tritiated ligand in the presence (nonspecific binding) or absence (total binding) of a nonlabeled displacer, and a rinsing step to eliminate unbound radioactivity [14, 19]. Nonspecific binding was less than 5% of total binding for all examined binding sites and thus was ignored in the present study. All sections intended for the visualization of a given receptor type were incubated in the same radioactive solution.
Table 1.
Ligands and binding protocols used for receptor autoradiography
| Transmitter | Receptor | [3H]-Ligand | Displacer | Incubation buffer | Preincubation | Main incubation | Final rinsea |
|---|---|---|---|---|---|---|---|
| Glutamate | AMPA | AMPA [10 nM] |
Quisqualate [10 μM] |
50 mM Tris-acetate (pH 7.2) +100 mM KSCNb |
3 × 10 minutes, 4 °C | 45 minutes, 4 °C | 1. 4 × 4 seconds 2. Acetone/glutaraldehyde (100 ml/2.5 ml), 2 × 2 seconds, 4 °C |
| Kainate | Kainate [9.4 nM] |
SYM 2081 [100 μM] |
50 mM Tris-acetate (pH 7.1) + 10 mM Ca2+-acetate |
3 × 10 minutes, 4 °C | 45 minutes, 4 °C | 1. 4 × 4 seconds, 4 °C 2. Acetone/glutaraldehyde (100 ml/2.5 ml), 2 × 2 seconds, 4 °C |
|
| NMDA | MK-801 [3.3 nM] |
(+)MK-801 [100 μM] |
50 mM Tris-acetate (pH 7.2) + 50 μM glutamate + 30 μM glycineb + 50 μM spermidineb |
15 minutes, 4 °C | 60 minutes, 22 °C | 2 × 5 minutes | |
| mGlu2/3 |
LY341495 [1 nM] |
l-Glutamate [1 mM] |
10 mM phosphate buffer (pH 7.6) + 100 mM KBrb |
2 × 5 minutes, 22 °C | 60 minutes, 4 °C | 2 × 5 minutes | |
| GABA | GABAA | Muscimol [7.7nM] |
GABA [10 μM] |
50 mM Tris-citrate (pH 7.0) | 3 × 5 minutes, 4 °C | 40 minutes, 4 °C | 3 × 3 seconds |
| GABAB | CGP 54626 [2 nM] |
CGP 55845 [100 μM] |
50 mM Tris-HCl (pH 7.2) + 2.5 mM CaCl2 |
3 × 5 minutes, 4 °C | 60 minutes, 4 °C | 3 × 2 seconds | |
| BZ | Flumazenil [1 nM] |
Clonazepam [2 μM] |
170 mM Tris-HCl (pH 7.4) | 15 minutes, 4 °C | 60 minutes, 22 °C | 2 × 1 seconds | |
| pBZ | PK 11195 [0.1 nM] |
PK 11195 [10 μM] |
50 mM Tris-HCl (pH 7.4) | 15 minutes, 22 °C | 60 minutes, 22 °C | 3 × 5 minutes | |
| Acetylcholine | M1 | Pirenzepine [1 nM] |
Pirenzepine [2 μM] |
Krebs buffer (pH 7.4) + 4 mM KCl + 120 mM NaCl |
15 minutes, 22 °C | 60 minutes, 22 °C | 3 × 4 minutes |
| M2 | Oxotremorine-M [1.7 nM] |
Carbachol [10 μM] |
20 mM HEPES-Tris (pH 7.5) + 10 mM MgCl2 + 300 nM pirenzepine |
20 minutes, 22 °C | 60 minutes, 22 °C | 2 × 2 minutes | |
| M3 | 4-DAMP [1 nM] |
Atropine sulfate [10 μM] |
50 mM Tris-HCl (pH 7.4) + 0.1 mM PMSF + 1 mM EDTA |
15 minutes, 22 °C | 45 minutes, 22 °C | 2 × 5 minutes | |
| nACh | Epibatidine [0.5 nM] |
Nicotine [100 μM] |
1 mM HEPES (pH 7.5) + 120 mM NaCl + 5.4 mM KCl + 0.8 mM MgCl2 + 1.8 mM CaCl2 |
20 minutes, 22 °C | 90 minutes, 22 °C | 5 minutes | |
| Noradrenaline | α1 | Prazosin [0.2 nM] |
Phentolamine mesylate [10 μM] |
50 mM Na+/K+-phosphate buffer (pH 7.4) | 15 minutes, 22 °C | 60 minutes, 22 °C | 2 × 5 minutes |
| α2 | UK 14,304 [0.64 nM] |
Phentolamine mesylate [10 μM] |
50 mM Tris-HCl (pH 7.7) + 100 μM MnCl2 |
15 minutes, 22 °C | 90 minutes, 22 °C | 5 minutes | |
| Serotonin | 5-HT1A | 8-OH-DPAT [1 nM] |
5-Hydroxytryptamine [1 μM] |
170 mM Tris-HCl (pH 7.4) + 4 mM CaCl2 b + 0.01% ascorbateb |
30 minutes, 22 °C | 60 minutes, 22 °C | 5 minutes |
| 5-HT2 | Ketanserin [1.14 nM] |
Mianserin [10 μM] |
170 mM Tris-HCl (pH 7.7) | 30 minutes, 22 °C | 120 minutes, 22 °C | 2 × 10 minutes | |
| Dopamine | D1 | SCH 23390 [1.67 nM] |
SKF 83566 [1 μM] |
50 mM Tris-HCl (pH 7.4) + 120 mM NaCl + 5 mM KCl + 2 mM CaCl2 + 1 mM MgCl2 |
20 minutes, 22 °C | 90 minutes, 22 °C | 2 × 10 minutes |
| D2 | Raclopride [0.55 nM] |
Butaclamol [1 μM] |
50 mM Tris-HCl (pH 7.4) + 0.1% ascorbate + 150 mM NaCl |
20 minutes, 22 °C | 45 minutes, 22 °C | 6 × 1 minutes | |
| Adenosine | A1 | DPCPX [1 nM] |
R-PIA [100 μM] |
170 mM Tris-HCl (pH 7.4) + 2 U/L adenosine deaminase + 100 μM Gpp(NH)pb |
15 minutes, 4 °C | 120 minutes, 22 °C | 2 × 5 minutes |
| A2A | ZM 241385 [0.42 nM] |
2-Chloroadenosine [20 μM] |
170 mM Tris-HCl (pH 7,4) + 2 U/L adenosine deaminase + 10 mM MgCl2 |
2 × 10 minutes, 22 °C | 120 minutes, 22 °C | 2 × 5 minutes |
Abbreviations: A 1 Adenosine receptor type 1, A 2A Adenosine receptor type 2A, α 1 Adrenoceptor type 1, α 2 Adrenoceptor type 2, AMPA α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, BZ γ-Aminobutyric acid type A-associated benzodiazepine-binding site, D1 Dopamine receptor type 1, D2 Dopamine receptor type 2, 4-DAMP 1,1-Dimethyl-4-diphenylacetoxypiperidinium iodide, EDTA Ethylenediaminetetraacetic acid, GABA A γ-Aminobutyric acid receptor type A, GABA B γ-Aminobutyric acid receptor type B, Gpp(NH)p 5′-guanylimidodiphosphate, 5-HT 1A Serotonin receptor type 1A, 5-HT 2 Serotonin receptor type 2, KBr Potassium bromide, KSCN Potassium thiocyanate, M 1, M 2 , and M 3 Muscarinic cholinergic receptor types 1, 2, and 3, mGlu2/3 Metabotropic glutamate receptor type 2/3, MK-801 Dizocilpine, nACh Nicotinic cholinergic receptor of the α4/β2 type, NMDA N-methyl-d-aspartate, 8-OH-DPAT 8-Hydroxy-2-(dipropylamino)tetralin, pBZ Peripheral benzodiazepine receptor, PMSF Phenylmethylsulfonyl fluoride, R-PIA N 6-R-phenylisopropyladenosine, SYM 2081 (2S,4R)-4-Methylglutamic acid
a Final rinsing was carried out in incubation buffer at 4 °C, followed by one to three dips in distilled water at room temperature, unless otherwise specified
b Substance included only in the main incubation buffer solution
Radioactively labeled sections were coexposed against tritium-sensitive films (Amersham Hyperfilm®; GE Healthcare Life Sciences, Braunschweig, Germany) with plastic [3H]-standards of known concentrations of radioactivity (Amersham Microscales®; GE Healthcare Life Sciences). Upon purchase, Microscales® were calibrated with the aid of brain homogenate standards for which total protein content had been determined by means of the Bradford assay [20]. Resulting autoradiographs were processed by densitometry with a video-based image-analyzing technique [19]. The Microscales® were used to compute a calibration curve, which, together with the parameters specific for each binding experiment (i.e., specific activity, dissociation constant, and concentration of the ligand), enabled transformation of grayscale values in the autoradiographs of samples into a binding site density per unit of protein (femtomoles per milligram of protein). Mean densities were thus obtained for a series of three or four sections per receptor type in the area 24′ and caudate nucleus probe of each case.
Statistical analysis
IBM SPSS Statistics version 21.0 for Windows software (IBM, Armonk, NY, USA) was used for analysis. Demographic features were analyzed by independent samples t test or chi-square test. Discriminant analyses were performed separately for data from area 24′ and the caudate nucleus to visualize the multivariate distance between control subjects and patients with PSP and between the two groups into which the patients could be subdivided on the basis of tau pathology severity or frontal versus nonfrontal presentation. Only in the case of a significant result did we perform post hoc tests (univariate F-tests) to reveal which receptor types differed between control subjects and patients with PSP or PSP subgroups. These p values were not corrected for multiple comparisons. Significance levels were set at p < 0.05 for the omnibus tests and p < 0.01 for the post hoc tests. The discriminant analysis was chosen as a global test because it offers several advantages over the procedures classically used to test group differences [21], the most important of which are that it is nonparametric and that it supports the analysis of multivariate datasets with more dependent variables (receptor densities, comprising 20 in this study) than cases (individuals in this study, comprising 14 control subjects and 16 patients with PSP). This is accomplished by reducing the receptor densities to a smaller number of discriminant scores (two in this study) for statistical testing and graphing.
Results
No cases of PSP-parkinsonism were identified in the present cohort, and relevant clinical data are summarized in Tables 2 and 3.
Table 2.
Demographic features of patients with progressive supranuclear palsy and healthy control subjects
| Patients | Control subjects | p Value | |
|---|---|---|---|
| n | 16 | 14 | |
| Age at death, years | 72.5 (6.92)a | 75.7 (10.07) | 0.34 |
| Male sex, n (%) | 9 (56) | 9 (64) | 0.8 |
| Disease duration, years | 8.2 (2.26)a | – | – |
| Postmortem delay, h, mean (SD) | 6.29 (1.21) | 7.53 (1.31) | 0.01 |
aThree cases that underwent euthanasia were not included
Table 3.
Detailed clinical features of patients with progressive supranuclear palsy
| Patient | Age at onset (years) | Age at death (years) | NINDS-SPSP criteria during life | Frontal presentation | Family history | Relevant medication in the last 3 months of life | Brain weight (g) | Tau pathology area 24' | Tau pathology caudate |
|---|---|---|---|---|---|---|---|---|---|
| 1a | 71 | 73 | Probable | Nonfrontal | Negative | Clonazepam, temazepam, piracetam | 1398 | Grade 3 | Grade 2 |
| 2 | 66 | 76 | Probable | Frontal | Negative | Oxybutynin, acetylcysteine, thiopental, pancuronium | 1060 | Grade 3 | Grade 2 |
| 3 | 63 | 68 | Probable | Nonfrontal | Positive | Temazepam, oxybutynin | 1405 | Grade 3 | Grade 4 |
| 4 | 70 | 79 | Possible | Frontal | Positive | Tolterodine | 1069 | Grade 2 | Grade 2 |
| 5 | 51 | 60 | Probable | Frontal | Negative | Temazepam | 1256 | Grade 3 | Grade 4 |
| 6a | 74 | 80 | Possible | Nonfrontal | Negative | Thiopental, pancuronium | 1100 | Grade 2 | Grade 1 |
| 7 | 66 | 75 | Possible | Frontal | Negative | Levomeprazine | 1253 | Grade 3 | Grade 1 |
| 8 | 54 | 64 | Possible | Frontal | Positive | Amantadine | 1290 | Grade 3 | Grade 1 |
| 9 | 79 | 85 | Possible | Nonfrontal | Negative | Oxazepam, nitrazepam, acetylcysteine | 1175 | Grade 2 | Grade 2 |
| 10 | 68 | 79 | Probable | Nonfrontal | Negative | Amitriptyline | 922 | Grade 2 | Grade 1 |
| 11 | 60 | 72 | Possible | Nonfrontal | Positive | Levodopa/carbidopa, amitriptyline | 1045 | Grade 2 | Grade 2 |
| 12 | 60 | 67 | Probable | Frontal | Positive | Levodopa/carbidopa, lormetazepam, nortriptyline | 1013 | Grade 4 | Grade 2 |
| 13 | 62 | 70 | Probable | Nonfrontal | Negative | Amantadine, temazepam, diazepam, amitriptyline | 1160 | Grade 2 | Grade 3 |
| 14a | 79 | 85 | Possible | Nonfrontal | Negative | Alprazolam, tamsulosin, thiopental, pancuronium | 1305 | Grade 3 | Grade 3 |
| 15 | 67 | 72 | Possible | Nonfrontal | Positive | Levodopa/carbidopa | 1525 | Grade 2 | Grade 4 |
| 16 | 61 | 69 | Probable | Frontal | Positive | Midazolam, clozapine | 1270 | Grade 3 | Grade 3 |
NINDS-SPSP National Institute of Neurological Disorders and Stroke/Society for Progressive Supranuclear Palsy
aPatients who underwent euthanasia by sodium thiopental and pancuronium bromide
Progressive supranuclear palsy cohort vs control subjects
On the basis of discriminant analyses of receptor densities, classification of patients with PSP and control subjects was significant in the caudate nucleus (Wilks’ lambda = 0.103, chi-square = 34.127, p = 0.025) and in area 24′ (Wilks’ lambda = 0.071, chi-square = 47.592, p < 0.001). Post hoc univariate F-tests revealed significantly higher densities of peripheral benzodiazepine (pBZ) and adenosine receptor type 1 (A1) receptors, but lower densities of kainate receptors and of nicotinic cholinergic receptors of the α4/β2 type (nACh) in the caudate nucleus of PSP brains than in brains of control subjects (Table 4, Fig 1a). In contrast to this, significantly higher N-methyl-d-aspartate (NMDA), γ-aminobutyric acid receptor type B (GABAB), pBZ, and serotonin receptor type 2 (5-HT2) receptor densities were found in area 24′ of PSP patient brains than in control brains (Table 5, Fig. 1d).
Table 4.
Mean receptor densities in the caudate nucleus of control subjects and patients with progressive supranuclear palsy
| Progressive supranuclear palsy vs. control subjects | Progressive supranuclear palsy, frontal vs. nonfrontal presentation | Progressive supranuclear palsy, mild vs. severe tau pathology | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Receptor | Control subjects | Patients | p Value | Frontal | Nonfrontal | p Value | Mild | Severe | p Value |
| AMPA | 607 (108) | 583 (228) | 0.749 | 477 (248) | 665 (184) | 0.102 | 631 (220) | 503 (238) | 0.290 |
| Kainate | 879 (61) | 800 (73) | 0.007 | 783 (70) | 813 (76) | 0.426 | 793 (68) | 812 (85) | 0.628 |
| NMDA | 1216 (42) | 1218 (115) | 0.951 | 1210 (122) | 1224 (118) | 0.823 | 1251 (116) | 1163 (99) | 0.413 |
| mGlu2/3 | 9295 (625) | 9416 (1455) | 0.799 | 8735 (1746) | 9945 (980) | 0.100 | 9502 (1272) | 9272 (1843) | 0.771 |
| GABAA | 1273 (164) | 1091 (203) | 0.021 | 986 (214) | 1173 (160) | 0.065 | 1133 (178) | 1021 (239) | 0.304 |
| GABAB | 2477 (299) | 2684 (397) | 0.155 | 2529 (402) | 2805 (370) | 0.177 | 2788 (382) | 2511 (389) | 0.184 |
| BZ | 1684 (136) | 1458 (330) | 0.023 | 1463 (422) | 1454 (266) | 0.956 | 1635 (284) | 1163 (121) | 0.002 |
| pBZ | 1759 (89) | 2050 (347) | 0.005 | 2127 (392) | 1989 (317) | 0.450 | 2154 (365) | 1876 (251) | 0.123 |
| M1 | 1174 (35) | 922 (379) | 0.018 | 968 (351) | 886 (418) | 0.686 | 904 (412) | 952 (353) | 0.815 |
| M2 | 567 (56) | 497 (127) | 0.063 | 467 (140) | 520 (119) | 0.422 | 501 (158) | 490 (54) | 0.049 |
| M3 | 1755 (36) | 1740 (255) | 0.829 | 1753 (309) | 1731 (223) | 0.869 | 1751 (299) | 1723 (183) | 0.843 |
| nACh | 200 (42) | 105 (43) | <0.001 | 97 (30) | 111 (52) | 0.522 | 119 (42) | 82 (38) | 0.094 |
| α1 | 339 (28) | 338 (53) | 0.945 | 314 (51) | 356 (50) | 0.123 | 331 (47) | 349 (65) | 0.543 |
| α2 | 467 (65) | 510 (92) | 0.199 | 502 (98) | 516 (94) | 0.767 | 498 (91) | 531 (99) | 0.500 |
| 5-HT1A | 129 (16) | 135 (27) | 0.543 | 139 (28) | 132 (28) | 0.646 | 139 (33) | 128 (12) | 0.428 |
| 5-HT2 | 1069 (100) | 976 (142) | 0.074 | 976 (138) | 976 (153) | 0.996 | 1048 (105) | 856 (115) | 0.004 |
| D1 | 291 (8) | 332 (49) | 0.010 | 341 (30) | 326 (61) | 0.456 | 349 (53) | 305 (25) | 0.085 |
| D2 | 885 (50) | 818 (103) | 0.057 | 765 (108) | 860 (82) | 0.066 | 845 (99) | 774 (102) | 0.188 |
| A1 | 1522 (239) | 1952 (288) | <0.001 | 1966 (327) | 1942 (274) | 0.786 | 1938 (349) | 1977 (167) | 0.806 |
| A2A | 1814 (195) | 2105 (327) | 0.014 | 2112 (409) | 2100 (273) | 0.944 | 2093 (391) | 2124 (212) | 0.863 |
Abbreviations: A 1 Adenosine receptor type 1, A 2A Adenosine receptor type 2A, α 1 Adrenoceptor type 1, α 2 Adrenoceptor type 2, AMPA α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, BZ γ-Aminobutyric acid type A-associated benzodiazepine-binding site, D1 Dopamine receptor type 1, D2 Dopamine receptor type 2, GABA A γ-Aminobutyric acid receptor type A, GABA B γ-Aminobutyric acid receptor type B, 5-HT 1A Serotonin receptor type 1A, 5-HT 2 Serotonin receptor type 2, M 1, M 2, M 3 Muscarinic cholinergic receptor types 1, 2, 3, mGlu2/3 Metabotropic glutamate receptor type 2/3, nACh Nicotinic cholinergic receptor of the α4/β2 type, NMDA N-methyl-d-aspartate, pBZ Peripheral benzodiazepine receptor
Absolute densities (SD) in femtomoles per milligram of protein as well as p values for the post hoc tests (significant values are highlighted in boldface type) are provided for each receptor type
Fig. 1.
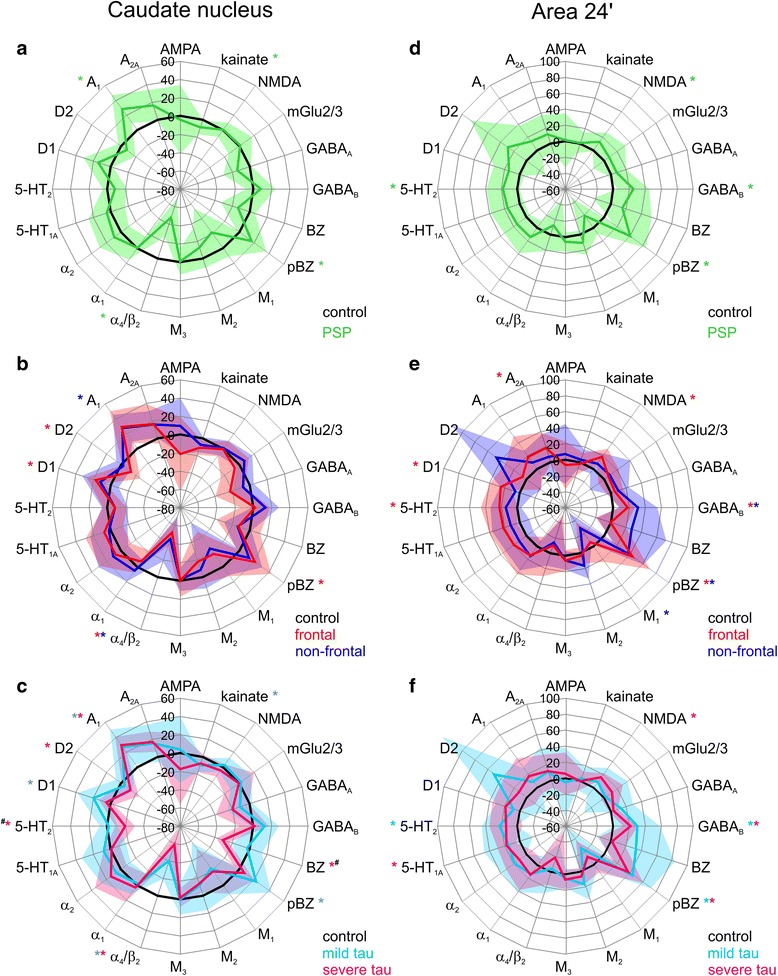
Receptor fingerprints of progressive supranuclear palsy (PSP)-related receptor density alterations in the caudate nucleus (a–c) and area 24′ (d–f). Polar plots showing the mean relative changes (in percent) in binding density of tissue obtained from the caudate nucleus (a) and area 24′ (d) of patients with PSP (mean value coded by green lines, SD given by transparent surface) with respect to control subjects (0%, in black). Polar plots showing the mean relative changes (in percent) in binding density of tissue obtained from the caudate nucleus (b) and area 24′ (e) of patients with PSP with frontal presentation (mean value coded by red lines, SD given by red transparent surface) or with nonfrontal presentation (mean value coded by blue lines, SD given by blue transparent surface) with respect to control subjects (0%, in black). Polar plots showing the mean relative changes (in percent) in binding density of tissue obtained from the caudate nucleus (c) and area 24′ (f) of patients with PSP with no to mild tau pathology (mean value coded by turquoise lines, SD given by turquoise transparent surface) or with moderate to severe tau pathology (mean value coded by pink lines, SD given by pink transparent surface) with respect to control subjects (0%, in black). Colored asterisks highlight receptors that were significantly altered in a given PSP cohort compared with control subjects. Hashtags indicate receptors significantly different when comparing PSP cases with no to mild tau pathology and PSP cases with moderate to severe tau pathology. α 4 /β 2 Nicotinic cholinergic receptors of the α4/β2 type, BZ γ-Aminobutyric acid receptor type A-associated benzodiazepine binding sites, pBZ Peripheral benzodiazepine receptors
Table 5.
Mean receptor densities in midcingulate area 24' of control subjects and progressive supranuclear palsy patients
| Progressive supranuclear palsy vs. control subjects | Progressive supranuclear palsy, frontal vs. nonfrontal presentation | Progressive supranuclear palsy, mild vs. severe tau pathology | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Receptor | Control subjects | Patients | p Value | Frontal | Nonfrontal | p Value | Mild | Severe | p Value |
| AMPA | 731 (39) | 739 (246) | 0.899 | 681 (216) | 785 (271) | 0.421 | 695 (314) | 773 (191) | 0.546 |
| Kainate | 1129 (182) | 1138 (150) | 0.880 | 1102 (162) | 1166 (144) | 0.422 | 1161 (166) | 1120 (145) | 0.602 |
| NMDA | 1290 (102) | 1455 (188) | 0.007 | 1531 (155) | 1397 (198) | 0.164 | 1404 (272) | 1495 (79) | 0.350 |
| mGlu2/3 | 7932 (1816) | 8576 (1960) | 0.361 | 8462 (1215) | 8665 (2464) | 0.845 | 8136 (2647) | 8918 (1283) | 0.448 |
| GABAA | 1975 (188) | 2125 (241) | 0.070 | 2078 (204) | 2162 (272) | 0.506 | 2232 (323) | 2042 (112) | 0.181 |
| GABAB | 4089 (484) | 5126 (801) | <0.001 | 4826 (360) | 5360 (982) | 0.176 | 5322 (1155) | 4974 (376) | 0.406 |
| BZ | 2686 (296) | 3094 (1028) | 0.164 | 2676 (525) | 3419 (1226) | 0.158 | 3567 (1373) | 2726 (468) | 0.165 |
| pBZ | 1698 (227) | 2380 (416) | <0.001 | 2444 (257) | 2330 (402) | 0.605 | 2452 (464) | 2325 (394) | 0.564 |
| M1 | 621 (62) | 505 (175) | 0.023 | 581 (186) | 446 (150) | 0.131 | 458 (204) | 542 (150) | 0.358 |
| M2 | 277 (43) | 306 (43) | 0.076 | 285 (29) | 322 (47) | 0.094 | 323 (54) | 293 (29) | 0.185 |
| M3 | 1071 (84) | 1135 (127) | 0.117 | 1137 (142) | 1133 (122) | 0.959 | 1123 (129) | 1144 (132) | 0.752 |
| nACh | 116 (40) | 102 (40) | 0.325 | 104 (47) | 100 (35) | 0.833 | 111 (38) | 94 (41) | 0.425 |
| α1 | 705 (57) | 805 (174) | 0.042 | 803 (204) | 807 (159) | 0.963 | 802 (195) | 808 (167) | 0.949 |
| α2 | 1124 (190) | 1288 (220) | 0.039 | 1342 (201) | 1246 (237) | 0.407 | 1337 (221) | 1250 (225) | 0.453 |
| 5-HT1A | 542 (53) | 630 (119) | 0.017 | 660 (99) | 606 (133) | 0.380 | 621 (172) | 636 (65) | 0.817 |
| 5-HT2 | 817 (74) | 963 (170) | 0.006 | 999 (192) | 934 (158) | 0.471 | 1001 (177) | 933 (169) | 0.446 |
| D1 | 125 (22) | 143 (25) | 0.050 | 157 (17) | 132 (26) | 0.050 | 136 (29) | 149 (21) | 0.307 |
| D2 | 71 (17) | 92 (41) | 0.092 | 75 (19) | 104 (49) | 0.165 | 107 (57) | 80 (18) | 0.191 |
| A1 | 1300 (151) | 1476 (318) | 0.069 | 1542 (407) | 1424 (241) | 0.479 | 1413 (281) | 1525 (352) | 0.504 |
| A2A | 120 (9) | 134 (30) | 0.089 | 142 (24) | 128 (35) | 0.392 | 133 (38) | 134 (26) | 0.941 |
Abbreviations: A 1 Adenosine receptor type 1, A 2A Adenosine receptor type 2A, α 1 Adrenoceptor type 1, α 2 Adrenoceptor type 2, AMPA α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, BZ γ-Aminobutyric acid type A-associated benzodiazepine-binding site, D1 Dopamine receptor type 1, D2 Dopamine receptor type 2, GABA A γ-Aminobutyric acid receptor type A, GABA B γ-Aminobutyric acid receptor type B, 5-HT 1A Serotonin receptor type 1A, 5-HT 2 Serotonin receptor type 2, M 1, M 2, M 3 Muscarinic cholinergic receptor types 1, 2, 3, mGlu2/3 Metabotropic glutamate receptor type 2/3, nACh Nicotinic cholinergic receptor of the α4/β2 type, NMDA N-methyl-d-aspartate, pBZ Peripheral benzodiazepine receptor
Absolute densities (SD) in femtomoles per milligram of protein as well as p values for the post hoc tests (significant values are highlighted in boldface type) are provided for each receptor type. Note that although comparison of mild versus severe tau pathology resulted in a nonsignificant omnibus test, results of the post hoc tests are displayed
Because patients with PSP displayed high interindividual variability in receptor density alterations (see large SD in Fig. 1a and d), we subdivided the cohort on the basis of the clinical parameters presence of frontal presentation and severity of tau pathology in the examined regions, and we tested separately for data obtained from the caudate nucleus and area 24′ whether these factors were associated with receptor density alterations in patients with PSP.
Progressive supranuclear palsy subgroups
Frontal presentation versus nonfrontal presentation
Discriminant analyses of receptor densities when the PSP cohort was divided into cases with frontal (n = 7) and cases with nonfrontal (n = 9) presentation resulted in a significant segregation of these two clinically relevant pictures in both the caudate nucleus (Wilks’ lambda = 0.016, chi-square = 29.107, p = 0.01) and area 24′ (Wilks’ lambda = 0.029, chi-square = 24.737, p = 0.037). However, this result revealed by the omnibus test could not be attributed to distinct receptors in the subsequent post hoc tests, because none of them reached significance in either brain structure. The significant omnibus test in the case of the caudate nucleus can be explained by the fact that 11 of 20 receptors presented lower densities in cases with frontal presentation than in those with nonfrontal presentation, and the opposite situation was found for only 8 receptors (5-HT2 receptor densities were identical in both groups). The significant omnibus test in the case of area 24′ is due to the fact that 11 of 20 receptors presented higher densities in cases with frontal presentation than in those with nonfrontal presentation, and the opposite situation was found for only 9 receptors. However, relatively large SDs resulted for both regions in a lack of significance at the post hoc test level.
Interestingly, discriminant analyses and subsequent post hoc tests revealed that patients with PSP with and without frontal presentation also had different variations from control subjects in both the caudate nucleus (Fig. 1b) and area 24′ (Fig. 1e). In the caudate nucleus (Fig. 1b), patients with frontal presentation PSP (Wilks’ lambda = 0.009, chi-square = 37.711, p = 0.002 by omnibus test) had significantly higher pBZ and D1 but lower nACh and D2 receptor densities than did control subjects, whereas patients with nonfrontal PSP (Wilks’ lambda = 0.008, chi-square = 43.257, p = 0.001 by omnibus test) presented significantly lower nACh and higher A1 receptor densities than did control subjects. In area 24′ (Fig. 1e), patients with frontal PSP (Wilks’ lambda = 0.035, chi-square = 31.799, p = 0.033 by omnibus test) showed significantly higher NMDA, GABAB, pBZ, 5-HT2, D1, and adenosine receptor type 2A receptor densities than control subjects, whereas patients with nonfrontal PSP (Wilks’ lambda = 0.005, chi-square = 58.876, p < 0.001 by omnibus test) presented higher GABAB and pBZ densities, but lower muscarinic cholinergic receptor type 1 (M1) densities, than control subjects.
Mild versus moderate to severe tau burden
The degree of tau pathology in both area 24′ and the caudate nucleus varied in our series of PSP brains. Tau pathology in area 24′ was mild in seven brains. In five of these cases, tau pathology in the caudate nucleus was also mild, but in two of them, it was moderate to severe. Tau pathology in area 24′ was moderate to severe in nine brains. In four of these cases, tau pathology in the caudate nucleus was also moderate to severe, but in five of them, it was only mild. Discriminant analyses of receptor densities resulted in a significant segregation of PSP cases into patients with mild tau burden and patients with moderate to severe tau burden in the caudate nucleus (Wilks’ lambda = 0.019, chi-square = 27.751, p = 0.015), but not in area 24′ (Wilks’ lambda = 0.098, chi-square = 16.259, p = 0.298). Post hoc univariate F-tests revealed significantly lower γ-aminobutyric acid type A-associated benzodiazepine (BZ) binding site (p = 0.002) and 5-HT2 (p = 0.004) receptor densities in the caudate nucleus of PSP brains with moderate to severe tau pathology than in PSP brains with mild tau pathology.
Discriminant analyses and subsequent post hoc tests revealed that PSP cases with no to mild and moderate to severe tau pathology also compared differently from control subjects in both the caudate nucleus (Fig. 1c) and area 24′ (Fig. 1f). In the caudate nucleus (Fig. 1c), patients with PSP with no to mild tau pathology (Wilks’ lambda = 0.016, chi-square = 39.477, p = 0.004 by omnibus test) presented higher pBZ, D1, and A1 densities, but lower kainate and nACh receptor densities, than did control subjects, whereas patients with PSP with moderate to severe tau pathology (Wilks’ lambda = 0.012, chi-square = 33.177, p = 0.004 by omnibus test) showed higher A1 densities, but lower benzodiazepine binding site as well as nACh, 5-HT2, and D2 receptor densities, than control subjects.
In area 24′ (Fig. 1f), patients with PSP with no to mild tau pathology (Wilks’ lambda = 0.003, chi-square = 55.124, p < 0.001 by omnibus test) showed higher GABAB, pBZ, and 5-HT2 receptor densities than did control subjects, whereas patients with PSP with moderate to severe tau pathology (Wilks’ lambda = 0.017, chi-square = 44.973, p = 0.001 omnibus test) presented higher NMDA, GABAB, pBZ, and serotonin receptor type 1A densities than control subjects.
Discussion
The present study shows a divergence in the severity of tau pathology between area 24′ and the caudate nucleus of patients with PSP, as well as significant PSP-related alterations in the densities of multiple receptors from different neurotransmitter systems that differentially affected both structures. In the caudate nucleus of PSP brains, densities of pBZ and A1 receptors were higher, and those of kainate and nACh receptors were lower, than in control subjects. In area 24′, NMDA, GABAB, pBZ, and 5-HT2 receptor densities were higher in PSP than in control tissue. Furthermore, clinically relevant PSP subgroups could be differentiated on the basis of their receptor fingerprints.
To our knowledge, this is the first study to show that patients with PSP with frontal and nonfrontal presentations can be differentiated postmortem with a high degree of accuracy on the basis of differences in receptor densities in both the caudate nucleus and area 24′. Receptor fingerprints also segregate mild from moderate to severe tau cases. We are aware that a drawback of our study is the fact that we were not able to assess the effect of medication on receptor densities, owing to the variability in drug therapy among patients.
Tau pathology is the histological hallmark of PSP, though the severity and distribution of tau pathology may differ between PSP subgroups [4]. The cerebral cortex and caudate nucleus are among the regions where the differences in severity are greatest [4]. This divergence in severity of pathology between the cingulate cortex and the caudate nucleus is supported by the present semiquantitative evaluation and is reflected by our receptor data.
The widespread alterations in the GABAergic system highlight its importance in the pathophysiology of PSP. GABAB receptor densities were increased in area 24′ of patients with PSP, but they were unaltered in the caudate nucleus. Because the increased density of GABAB receptors occurred in all PSP subgroups (frontal/nonfrontal and mild/severe tau), they seem to be the most vulnerable receptor type in PSP. Furthermore, the GABAB receptor increase in the midcingulate cortex is of particular interest because its activation is associated with the induction of long-term potentiation [22] and results in amelioration of the cognitive impairment associated with chronic cerebral hypoperfusion [23].
BZ binding sites were decreased in the caudate nucleus of patients with PSP only in cases of moderate to severe tau pathology. This decrease may be caused by a loss of GABAergic projection neurons in this PSP subgroup, leading to a reduction of pre- and postsynaptic γ-aminobutyric acid receptor type A (GABAA) and could explain the therapeutic effectivity of BZ agonists [24]. The GABAA receptor density demonstrated by the binding with the agonist [3H]muscimol also showed a decrease, but this did not reach significance (Tables 4 and 5). Because the agonistic binding prefers high-affinity binding sites of the receptor, these data may indicate a shift of the ratio between low- and high-affinity binding sites of the GABAA receptor in PSP.
Densities of pBZ receptors were higher in area 24′ and the caudate nucleus of patients with PSP than in control subjects. This is in line with the increased PK11195 binding in these regions revealed by a positron emission tomography (PET) study [25] and reflects microglial activation. However, when the cohort was subdivided into frontal/nonfrontal cases or mild/severe tau pathology, consistent alterations were found only in area 24′. Taken together, receptors of the GABAergic system are more affected in area 24′ than in the caudate nucleus, and impairment does not depend on the severity of tau pathology and frontal or nonfrontal clinical type.
Our findings of widespread PSP-related changes in the glutamatergic system may be relevant for potential future treatment strategies in PSP, similar to recent studies in Parkinson’s disease [26, 27]. The divergence in the severity of receptor impairments between cortical and subcortical sites is further supported by our findings regarding NMDA receptors, which were altered only in area 24′. This increase in NMDA receptor densities is probably due to region-specific disease-induced alterations, and not caused by the long-term administration of amantadine, because patients treated with this NMDA receptor antagonist (patients 8 and 12; see Table 3) presented normal NMDA receptor densities. The unchanged NMDA receptor density in the caudate nucleus is in accordance with the one other study investigating NMDA receptors in patients with PSP [28]. Furthermore, we found a decrease of kainate receptor densities but unaltered α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and metabotropic glutamate receptor type 2/3 densities, where up to now no information was available in patients with PSP.
Drugs targeting the cholinergic system have failed to relieve the cognitive and motor impairments of PSP [29]. Interestingly, of the four cholinergic receptor types examined here, only the nACh receptors were found to be altered in the PSP cohort (as a whole and in all subgroups), though only in the caudate nucleus. Our results for nACh and M1 receptors in the caudate nucleus are in line with previous findings [11]. The unaltered caudate nucleus muscarinic cholinergic receptor type 2 (M2) receptor densities, however, contrast with the findings in another postmortem study in which researchers reported reduced M2 receptor densities in the posterior caudate nucleus [12]. The discrepancy may be explained by differences in postmortem delay times (45 h versus 6 h in our study); ligands used (the antagonist [3H]-AFDX 384 versus the agonist [3H]oxotremorine-M in our study); or the rostrocaudal anatomical, neurochemical, and functional differences that characterize the caudate nucleus [30].
Interestingly, the nACh receptor plays a major role in the control of dopamine release in the caudate nucleus [31]. Consequently, the remarkably strong decrease in nACh receptor densities leads to a reduction of dopamine release, which results in a global impairment of dopaminergic effects in the caudate nucleus of patients with PSP.
The normal density of adrenoceptors in the caudate nucleus and area 24′ in our cases, together with the normal adrenaline levels in various brain regions of patients with PSP as found by Kish et al. [32], as well as the ineffectiveness of noradrenergic replacement therapies [33], suggests that this neurotransmitter system does not contribute significantly to the symptomatology of PSP. It must be noted, however, that researchers in the single other autoradiographic study on adrenoceptors in PSP to date found a generalized reduction of adrenoceptor type 2 receptors [34], though their findings were based on a case report.
The 5-HT2 receptor also emphasizes the divergent severity of alterations between area 24′ and the caudate nucleus in PSP, because it was increased only in the former structure, preferentially in the frontal group. Our results are difficult to compare with those of an in vivo PET imaging study in which investigators reported normal densities of 5-HT2 receptors in the neocortex, but higher concentrations in the putamen [35], because different regions were examined and different ligands ([18F]altanserin versus [3H]ketanserin in our cases) were used. Furthermore, [18F]altanserin PET does not directly reflect 5-HT2 receptor density, because it is confounded by the uptake of blood-brain barrier-penetrating metabolites and nonspecific binding of [18F]altanserin itself [36].
The decrease of 5-HT2 receptor densities in the caudate nucleus of PSP brains with moderate to severe tau pathology compared with those with mild tau pathology cannot be explained merely by a more severe neurodegeneration in the former group, because we did not observe an association between tau pathology and 5-HT2 receptor alterations in area 24′. Interestingly, although a differential effect of serotonergic denervation on tau pathology in various brain regions has been described previously, the underlying explanation for this selective vulnerability remains unclear [37].
Dopaminergic receptors are localized on medium spiny stellate cells. Cells expressing D1 receptors, or D1 colocalized with D2 receptors, preferentially project to the substantia nigra and the internal segment of the globus pallidus, whereas those expressing D2 receptors target the external segment of the globus pallidus [38]. Our finding of unaltered D1 receptor densities is in accordance with previous reports [10, 39], and the unchanged D2 receptor densities described here add to the controversial data concerning this receptor type [10, 11, 39].
A1 receptors are frequently localized presynaptically and control glutamate release. Thus, the significant PSP-related increase of receptor densities in the caudate nucleus may be a plastic reaction to (1) decreased inhibition resulting from BZ binding site downregulation and (2) increased excitation resulting from higher NMDA and lower kainate receptor densities, because the latter can also control glutamate release. The PSP-related increase in A1 receptor densities in the caudate nucleus may be the result of an ongoing inflammatory process because these receptors are expressed in microglia [40]. Furthermore, it could be a compensatory mechanism to counteract the decreased concentrations of the adenosine precursors adenosine diphosphate and adenosine triphosphate measured in the basal ganglia of patients with PSP [41]. Therefore, an intriguing question is whether modulation targeting the adenosine receptors may represent a therapeutic strategy in PSP.
Conclusions
We have demonstrated the involvement of multiple nondopaminergic neurotransmitter systems in the pathophysiology of PSP, which may be relevant for potential novel treatment strategies. We provide further evidence that the midcingulate cortex may prove to be a key region in this disease. GABAergic, glutamatergic, and serotonergic receptors in patients with PSP deviated most from those of control subjects in area 24′, where the highest frequency of tau pathology was found. This is in sharp contrast to dopaminergic, cholinergic, and adenosine receptors, which were preferentially impaired in the caudate nucleus. Finally, “receptor fingerprints” not only differentiated patients with PSP from control subjects neurochemically but also segregated PSP subgroups when clinical parameters such as presence of frontal presentation and severity of tau pathology were taken into consideration.
Acknowledgements
The authors thank Angelika Hallenberger for excellent technical assistance.
Funding
This study was supported by Parkinson Vereniging grant number 2010-V02 and by the ’Horizon 2020 European Union Framework Programme for Research and Innovation under grant agreement 7202070 (HBP SGA1).
Availability of data and materials
The datasets generated during the present study are available from the corresponding author on reasonable request.
Authors’ contributions
JCvS and NPG conceived of and designed the study. WZC, LDK, WK, AS, and NPG acquired and analyzed data. WZC, JCvS, KZ, and NPG drafted the manuscript or figures. WZC, LDK, AJWB, WK, AS, KZ, JCvS, and NPG critically revised the manuscript for important intellectual content. WZC, AS, and NPG performed statistical analysis. JCvS and KZ obtained funding. WZC, JCvS, and NPG supervised the study. All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study was approved by the medical ethics committee of the Erasmus University Medical Center, and all participants or their first-degree relatives signed informed consent forms. Brain autopsies were conducted by The Netherlands Brain Bank according to their legal and ethical code of conduct.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- 4-DAMP
1,1-Dimethyl-4-diphenylacetoxypiperidinium iodide
- 5-HT1A
Serotonin receptor type 1A
- 5-HT2
Serotonin receptor type 2
- 8-OH-DPAT
8-Hydroxy-2-(dipropylamino)tetralin
- A1
Adenosine receptor type 1
- A2A
Adenosine receptor type 2A
- AF-DX 384
(±)-5,11-dihydro-11-{[(2-{2-[(dipropylamino)methyl]-1-piperidinyl}ethyl)amino]carbonyl}-6H-pyrido(2,3-b)(1,4)-benzodiazepine-6-one
- AMPA
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- BZ
γ-Aminobutyric acid type A-associated benzodiazepine-binding site
- D1
Dopamine receptor type 1
- D2
Dopamine receptor type 2
- DCPCX
8-Cyclopentyl-1,3-dipropylxanthine
- EDTA
Ethylenediaminetetraacetic acid
- GABAA
γ-Aminobutyric acid receptor type A
- GABAB
γ-Aminobutyric acid receptor type B
- Gpp(NH)p
5′-guanylimidodiphosphate
- KBr
Potassium bromide
- KSCN
Potassium thiocyanate
- M1
M2, M3, Muscarinic cholinergic receptor types 1, 2, 3
- mGlu2/3
Metabotropic glutamate receptor type 2/3
- MK-801
Dizocilpine
- nACh
Nicotinic cholinergic receptor of the α4/β2 type
- NINDS-SPSP
National Institute of Neurological Disorders and Stroke and Society for Progressive Supranuclear Palsy
- NMDA
N-methyl-d-aspartate
- pBZ
Peripheral benzodiazepine receptor
- PET
Positron emission tomography
- PMSF
Phenylmethylsulfonyl fluoride
- PSP
Progressive supranuclear palsy
- R-PIA
N 6-R-phenylisopropyladenosine
- SYM 2081
(2S,4R)-4-Methylglutamic acid
- α1
Adrenoceptor type 1
- α2
Adrenoceptor type 2
Contributor Information
Wang Zheng Chiu, Email: w.chiu@erasmusmc.nl.
Laura Donker Kaat, Email: l.donker_kaat@lumc.nl.
Agnita J. W. Boon, Email: a.j.w.boon@erasmusmc.nl
Wouter Kamphorst, Email: w.kamphorst@online.nl.
Axel Schleicher, Email: abschleicher@gmail.com.
Karl Zilles, Email: k.zilles@fz-juelich.de.
John C. van Swieten, Email: j.c.vanswieten@erasmusmc.nl
Nicola Palomero-Gallagher, Email: n.palomero-gallagher@fz-juelich.de.
References
- 1.Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9. doi: 10.1212/WNL.47.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Donker Kaat L, Boon AJ, Kamphorst W, Ravid R, Duivenvoorden HJ, van Swieten JC. Frontal presentation in progressive supranuclear palsy. Neurology. 2007;69:723–9. doi: 10.1212/01.wnl.0000267643.24870.26. [DOI] [PubMed] [Google Scholar]
- 3.Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014;29:1758–66. doi: 10.1002/mds.26054. [DOI] [PubMed] [Google Scholar]
- 4.Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8:270–9. doi: 10.1016/S1474-4422(09)70042-0. [DOI] [PubMed] [Google Scholar]
- 5.Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. 2010;23:394–400. doi: 10.1097/WCO.0b013e32833be924. [DOI] [PubMed] [Google Scholar]
- 6.Palomero-Gallagher N, Vogt BA, Schleicher A, Mayberg HS, Schleicher A, Zilles K. Receptor architecture of human cingulate cortex: evaluation of the four-region neurobiological model. Hum Brain Mapp. 2009;30:2336–55. doi: 10.1002/hbm.20667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torta DM, Cauda F. Different functions in the cingulate cortex, a meta-analytic connectivity modeling study. Neuroimage. 2011;56:2157–72. doi: 10.1016/j.neuroimage.2011.03.066. [DOI] [PubMed] [Google Scholar]
- 8.Chiu WZ, Papma JM, de Koning I, Donker Kaat L, Seelaar H, Reijs AE, et al. Midcingulate involvement in progressive supranuclear palsy and tau positive frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2012;83:910–5. doi: 10.1136/jnnp-2011-302035. [DOI] [PubMed] [Google Scholar]
- 9.Gerstenecker A, Mast B, Duff K, Ferman TJ, Litvan I. Executive dysfunction is the primary cognitive impairment in progressive supranuclear palsy. Arch Clin Neuropsychol. 2013;28:104–13. doi: 10.1093/arclin/acs098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikolaus S, Antke C, Müller HW. In vivo imaging of synaptic function in the central nervous system: I. Movement disorders and dementia. Behav Brain Res. 2009;204:1–31. doi: 10.1016/j.bbr.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Warren NM, Piggott MA, Greally E, Lake M, Lees AJ, Burn DJ. Basal ganglia cholinergic and dopaminergic function in progressive supranuclear palsy. Mov Disord. 2007;22:1594–600. doi: 10.1002/mds.21573. [DOI] [PubMed] [Google Scholar]
- 12.Warren NM, Piggott MA, Lees AJ, Burn DJ. The basal ganglia cholinergic neurochemistry of progressive supranuclear palsy and other neurodegenerative diseases. J Neurol Neurosurg Psychiatry. 2007;78:571–5. doi: 10.1136/jnnp.2006.099937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burn DJ, Warren NM. Toward future therapies in progressive supranuclear palsy. Mov Disord. 2005;20(Suppl 12):S92–8. doi: 10.1002/mds.20546. [DOI] [PubMed] [Google Scholar]
- 14.Zilles K, Palomero-Gallagher N, Grefkes C, Scheperjans F, Boy C, Amunts K, et al. Architectonics of the human cerebral cortex and transmitter receptor fingerprints: reconciling functional neuroanatomy and neurochemistry. Eur Neuropsychopharmacol. 2002;12:587–99. doi: 10.1016/S0924-977X(02)00108-6. [DOI] [PubMed] [Google Scholar]
- 15.Hammers A, Koepp MJ, Labbe C, Brooks DJ, Thom M, Cunningham VJ, et al. Neocortical abnormalities of [11C]-flumazenil PET in mesial temporal lobe epilepsy. Neurology. 2001;56:897–906. doi: 10.1212/WNL.56.7.897. [DOI] [PubMed] [Google Scholar]
- 16.Chiu WZ, Kaat LD, Seelaar H, Rosso SM, Boon AJ, Kamphorst W, et al. Survival in progressive supranuclear palsy and frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2010;81:441–5. doi: 10.1136/jnnp.2009.195719. [DOI] [PubMed] [Google Scholar]
- 17.Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol. 1996;55:97–105. doi: 10.1097/00005072-199601000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ, et al. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain. 2007;130:1566–76. doi: 10.1093/brain/awm104. [DOI] [PubMed] [Google Scholar]
- 19.Zilles K, Schleicher A, Palomero-Gallagher N, Amunts K. Quantitative analysis of cyto- and receptor architecture of the human brain. In: Toga AW, Mazziotta JC, editors. Brain mapping: the methods. 2. Amsterdam: Academic Press/Elsevier Science; 2002. pp. 573–602. [Google Scholar]
- 20.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Cleophas TJ. Clinical trials with large numbers of variables: important advantages of canonical analysis. Am J Ther. 2016;23:e825–36. doi: 10.1097/MJT.0b013e31827e9765. [DOI] [PubMed] [Google Scholar]
- 22.Dutar P, Nicoll RA. A physiological role for GABAB receptors in the central nervous system. Nature. 1988;332:156–8. doi: 10.1038/332156a0. [DOI] [PubMed] [Google Scholar]
- 23.Li CJ, Lu Y, Zhou M, Zong XG, Li C, Xu XL, et al. Activation of GABAB receptors ameliorates cognitive impairment via restoring the balance of HCN1/HCN2 surface expression in the hippocampal CA1 area in rats with chronic cerebral hypoperfusion. Mol Neurobiol. 2014;50:704–20. doi: 10.1007/s12035-014-8736-3. [DOI] [PubMed] [Google Scholar]
- 24.Chang AY, Weirich E. Trial of zolpidem, eszopiclone, and other GABA agonists in a patient with progressive supranuclear palsy. Case Rep Med. 2014;2014:107064. doi: 10.1155/2014/107064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord. 2006;21:89–93. doi: 10.1002/mds.20668. [DOI] [PubMed] [Google Scholar]
- 26.Johnson KA, Conn PJ, Niswender CM. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol Disord Drug Targets. 2009;8:475–91. doi: 10.2174/187152709789824606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duty S. Targeting glutamate receptors to tackle the pathogenesis, clinical symptoms and levodopa-induced dyskinesia associated with Parkinson’s disease. CNS Drugs. 2012;26:1017–32. doi: 10.1007/s40263-012-0016-z. [DOI] [PubMed] [Google Scholar]
- 28.Holemans S, Javoy F, Agid Y, Laterre EC, Maloteaux JM. [3H]MK-801 binding to NMDA glutamatergic receptors in Parkinson’s disease and progressive supranuclear palsy. Brain Res. 1991;565:154–7. doi: 10.1016/0006-8993(91)91747-O. [DOI] [PubMed] [Google Scholar]
- 29.Warren NM, Piggott MA, Perry EK, Burn DJ. Cholinergic systems in progressive supranuclear palsy. Brain. 2005;128:239–49. doi: 10.1093/brain/awh391. [DOI] [PubMed] [Google Scholar]
- 30.Grahn JA, Parkinson JA, Owen AM. The cognitive functions of the caudate nucleus. Prog Neurobiol. 2008;86:141–55. doi: 10.1016/j.pneurobio.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 31.Cao YJ, Surowy CS, Puttfarcken PS. Different nicotinic acetylcholine receptor subtypes mediating striatal and prefrontal cortical [3H]dopamine release. Neuropharmacology. 2005;48:72–9. doi: 10.1016/j.neuropharm.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Kish SJ, Chang LJ, Mirchandani L, Shannak K, Hornykiewicz O. Progressive supranuclear palsy: relationship between extrapyramidal disturbances, dementia, and brain neurotransmitter markers. Ann Neurol. 1985;18:530–6. doi: 10.1002/ana.410180504. [DOI] [PubMed] [Google Scholar]
- 33.Rascol O, Sieradzan K, Peyro-Saint-Paul H, Thalamas C, Brefel-Courbon C, Senard JM, et al. Efaroxan, an alpha-2 antagonist, in the treatment of progressive supranuclear palsy. Mov Disord. 1998;13:673–6. doi: 10.1002/mds.870130411. [DOI] [PubMed] [Google Scholar]
- 34.Pascual J, Berciano J, González AM, Grijalba B, Figols J, Pazos A. Autoradiographic demonstration of loss of α2-adrenoceptors in progressive supranuclear palsy: preliminary report. J Neurol Sci. 1993;114:165–9. doi: 10.1016/0022-510X(93)90293-8. [DOI] [PubMed] [Google Scholar]
- 35.Stamelou M, Matusch A, Elmenhorst D, Hurlemann R, Eggert KM, Zilles K, et al. Nigrostriatal upregulation of 5-HT2A receptors correlates with motor dysfunction in progressive supranuclear palsy. Mov Disord. 2009;24:1170–5. doi: 10.1002/mds.22533. [DOI] [PubMed] [Google Scholar]
- 36.Hurlemann R, Matusch A, Eickhoff SB, Palomero-Gallagher N, Meyer PT, Boy C, et al. Analysis of neuroreceptor PET-data based on cytoarchitectonic maximum probability maps: a feasibility study. Anat Embryol. 2005;210:447–53. doi: 10.1007/s00429-005-0034-4. [DOI] [PubMed] [Google Scholar]
- 37.Ramos-Rodriguez JJ, Molina-Gil S, Rey-Brea R, Berrocoso E, Garcia-Alloza M. Specific serotonergic denervation affects tau pathology and cognition without altering senile plaques deposition in APP/PS1 mice. PLoS One. 2013;8:e79947. doi: 10.1371/journal.pone.0079947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–91. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pascual J, Berciano J, Grijalba B, del Olmo E, González AM, Figols J, et al. Dopamine D1 and D2 receptors in progressive supranuclear palsy: an autoradiographic study. Ann Neurol. 1992;32:703–7. doi: 10.1002/ana.410320516. [DOI] [PubMed] [Google Scholar]
- 40.Luongo L, Guida F, Imperatore R, Napolitano F, Gatta L, Cristino L, et al. The A1 adenosine receptor as a new player in microglia physiology. Glia. 2014;62:122–32. doi: 10.1002/glia.22592. [DOI] [PubMed] [Google Scholar]
- 41.Stamelou M, Pilatus U, Reuss A, Magerkurth J, Eggert KM, Knake S, et al. In vivo evidence for cerebral depletion in high-energy phosphates in progressive supranuclear palsy. J Cereb Blood Flow Metab. 2009;29:861–70. doi: 10.1038/jcbfm.2009.2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during the present study are available from the corresponding author on reasonable request.