

SUMMARY
Growing evidence suggests a major role for Src homology-2 domain-containing phosphatase 2 (SHP2/PTPN11) in MYCN-driven high-risk neuroblastoma, although biologic confirmation and a plausible mechanism for this contribution are lacking. Using a zebrafish model of MYCN-overexpressing neuroblastoma, we demonstrate that mutant ptpn11 expression in the adrenal gland analogue of MYCN transgenic fish promotes the proliferation of hyperplastic neuroblasts, accelerates neuroblastomagenesis and increases tumor penetrance. We identify a similar mechanism in tumors with wild-type ptpn11 and dysregulated Gab2, which encodes a Shp2 activator that is overexpressed in human neuroblastomas. In MYCN transgenic fish, Gab2 overexpression activated the Shp2-Ras-Erk pathway, enhanced neuroblastoma induction and increased tumor penetrance. We conclude that MYCN cooperates with either GAB2-activated or mutant SHP2 in human neuroblastomagenesis. Our findings further suggest that combined inhibition of MYCN and the SHP2-RAS-ERK pathway could provide effective targeted therapy for high-risk neuroblastoma patients with MYCN amplification and aberrant SHP2 activation.
Keywords: neuroblastoma, zebrafish, Gab2, Shp2, ptpn11, and MYCN
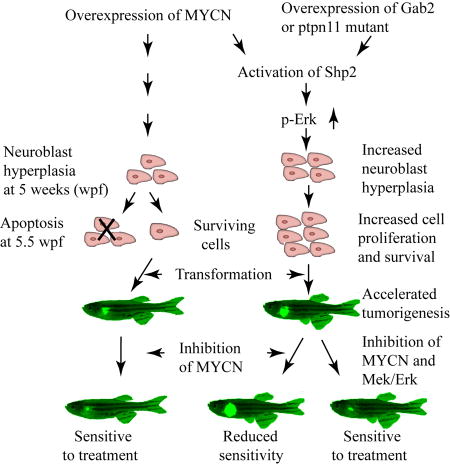
Graphical Abstract
INTRODUCTION
Neuroblastoma is the most common extracranial solid tumor in children, accounting for 10–13% of pediatric cancer deaths (Louis and Shohet, 2015; Park et al., 2013; Saito et al., 2012). This tumor arises from immature, primordial neural crest cells that develop into the adrenal medulla and other sympathetic neural ganglia in the peripheral sympathetic nervous system (PSNS) (Hoehner et al., 1996). Amplification of MYCN is found in 20% of neuroblastomas (Brodeur et al., 1984; Seeger et al., 1985), while mutations in ALK (anaplastic lymphoma kinase) account for the majority of hereditary cases (Bresler et al., 2014; Brodeur and Bagatell, 2014; De Brouwer et al., 2010; George et al., 2008; Janoueix-Lerosey et al., 2008; Mosse et al., 2008) and 8–10% of sporadic cases (Chen et al., 2008; George et al., 2008; Janoueix-Lerosey et al., 2008; Molenaar et al., 2012; Mosse et al., 2008; Pugh et al., 2013).
We and others have reported that PTPN11, which encodes SHP2 (Src homology-2 domain-containing phosphatase 2), is the third most frequently mutated gene in neuroblastoma, affecting up to 3.4% of cases. Interestingly, over half of PTPN11 mutant tumors also show MYCN amplification (Bentires-Alj et al., 2004; Pugh et al., 2013). PTPN11 is required for full activation of the RAS extracellular signal-regulated kinase (ERK) pathway in growth factor, cytokine, and integrin signaling, and mutant forms of SHP2 lower the threshold for activation of such pathways (Chan et al., 2008; Mohi and Neel, 2007). Very recently, PTPN11 and other genes encoding RAS-ERK pathway components were found to be mutated in relapsed cases (Eleveld et al., 2015), and activated Ras-Erk signaling in nf1-deficient zebrafish can accelerate MYCN-induced neuroblastomagenesis (He et al., 2016), suggesting that SHP2 could play a major role in neuroblastoma pathogenesis and treatment resistance, potentially through activation of the RAS-ERK pathway.
Despite growing evidence that SHP2 could play a critical role in neuroblastomagenesis and the emergence of relapse (Bentires-Alj et al., 2004; Pugh et al., 2013), neither direct biological evidence supporting an oncogenic role for PTPN11 mutations, nor the underlying mechanism of such activity has been provided. Because GAB2 (encoding GRB2-associated binding protein-2) and other upstream genetic regulators of SHP2 are highly expressed with PTPN11 in many neuroblastomas (http://r2.amc.nl), we hypothesized that dysregulation of one or more of their encoded proteins could also aberrantly activate SHP2. If so, inappropriate SHP2 activation leading to enhanced RAS-ERK signaling might play a much broader role in neuroblastoma pathogenesis than previously thought. To assess the significance of dysregulated SHP2 or GAB2 expression on neuroblastoma pathogenesis in the context of MYCN amplification, we generated transgenic zebrafish lines in which mutationally activated ptpn11 or wild-type Gab2 is overexpressed in the PSNS under control of the zebrafish dopamine-β-hydroxylase gene promoter. Concomitant overexpression of MYCN in these models reliably induces tumors that closely resemble human high-risk neuroblastoma (Zhu et al., 2012).
RESULTS
Mutant ptpn11 synergizes with overexpressed MYCN to induce neuroblastoma
We generated a stable transgenic zebrafish line that overexpresses EGFP-fused to ptpn11E69K (ptpn11mut). This mutation is the zebrafish homologue of a somatic activating mutation identified in human neuroblastoma (Bentires-Alj et al., 2004) and leukemia (Tartaglia et al., 2003). The ptpn11mut transgene is placed under control of the dopamine-beta-hydroxylase (dβh) promoter, which directs expression in sympathoadrenal cells in the interrenal gland (IRG), the analogue of human adrenal gland (Figure S1) (Hsu et al., 2004). Heterozygous ptpn11mut fish were bred to heterozygous transgenic fish overexpressing EGFP-MYCN in the PSNS (designated MYCN) (Zhu et al., 2012), and the offspring of three transgenic lines -- MYCN, ptpn11mut, and MYCN;ptpn11mut--were monitored over 6 months.
Expression of ptpn11mut or EGFP alone did not lead to tumor development (Figure 1A, 1B and 1E), while tumors arose in 2 of 68 MYCN fish between 5 and 9 weeks of age (Figure 1C and 1E). However, a much higher fraction of compound MYCN;ptpn11mut fish (16/70) developed tumors during the same period (Figure 1D and 1E). Tumors continued to arise after 9 weeks of age in both fish lines, but the rate of tumor development was much higher in the compound group over the entire monitoring period (Figure 1E). By 25 weeks of age, ~75% of the MYCN;ptpn11mut fish had tumors, compared with ~37% of the MYCN fish (Figure 1E, p<0.0001).
Figure 1. Mutant ptpn11 synergizes with MYCN in neuroblastomagenesis.

(A–D) Fluorescence micrographs of control Dβh (A), ptpn11mut (B), MYCN (C) and compound MYCN;ptpn11mut (D) transgenic fish. Note the EGFP-expressing tumor in the interrenal gland (IRG, arrowhead; panel D) at 5 weeks postfertilization (wpf). Scale bar, 1 mm.
(E) Kaplan-Meier analysis showing cumulative frequency of neuroblastoma induction over 6 months in the indicated stable transgenic zebrafish lines (p< 0.0001 for MYCN-only versus MYCN;ptpn11mut by log-rank test).
(F) Phospho-Erk1/2 immunoblots of lysates from individual MYCN-only (F1–F4) or MYCN;ptpn11mut tumors (F5–F8). Total Erk1/2 levels serve as a loading control.
(G) Kaplan-Meier analysis of cumulative frequency of neuroblastoma induction over 11 weeks in MYCN transgenic fish mosaically overexpressing mCherry alone (MYCN;mosaic mCherry), coexpressing mCherry with ptpn11wt (MYCN;mosaic ptpn11wt), or coexpressing mCherry with ptpn11mut (MYCN;mosaic ptpn11mut). MYCN;mosaic ptpn11mut versus MYCN;mosaic mCherry transgenic fish is significant by log-rank test (p=0.03).
See also Figure S1.
In both MYCN and compound MYCN;ptpn11mut transgenic fish, tumors arose in the IRG region (Figures 1D and S1B), and closely resembled high-risk human neuroblastoma histologically (Figure S1B, first two panels; also (Zhu et al., 2012)) as well as immunohistochemically (Figure S1B, middle two panels; also (Marusich et al., 1994; Teitelman et al., 1979)). Expression of Shp2 (the gene product of ptpn11) was confirmed by immunohistochemical analysis (Figure S1B, far-right panels). In addition, the phosphorylation of Erk1/2 was elevated in tumors coexpressing MYCN and ptpn11mut, as compared to those expressing MYCN alone, consistent with activation of the downstream Ras-Erk pathway in MYCN; ptpn11mut transgenic fish (Figure 1F).
We next asked whether overexpression of wild-type ptpn11 (ptpn11wt) can synergize with MYCN to promote neuroblastomagenesis. For these studies, we transiently co-overexpressed the ptpn11wt-EGFP fusion construct under control of the dβh promoter with a dβh-mCherry construct in MYCN transgenic fish. Expression of mCherry served as a marker for the transient coexpression of ptpn11wt or ptpn11mut (Langenau et al., 2008; Zhu et al., 2012). Consistent with the above finding in the stable transgenic fish lines (Figure 1E), we observed significantly increased fractions of MYCN transgenic fish mosaically overexpressing ptpn11mut and mCherry that developed tumors in the IRG during the 12 weeks of monitoring period (Figure 1F, p=0.03). By contrast, similar fractions of MYCN fish with either mosaic overexpression of ptpn11wt and mCherry or mCherry alone had tumors in the IRG during the observation period (Figure 1F), suggesting that mosaic overexpression of ptpn11wt at the levels driven by the dβh promoter is not adequate to collaborate with MYCN to potentiate neuroblastoma pathogenesis in this model system. These findings indicate that mutationally activated but not wild-type Shp2 cooperates with MYCN to increase tumor penetrance.
Transcriptome analysis reveals an association of GAB2 with PTPN11 expression in high-risk neuroblastoma with MYCN amplification
Having established a synergistic relationship between mutationally activated Shp2 and MYCN in our zebrafish model, we next investigated whether Shp2 might be activated by other means in MYCN-driven neuroblastomas that lack ptpn11 mutations. Analysis of transcriptome data from four publically available neuroblastoma data sets revealed that PTPN11 is overexpressed in most neuroblastoma cases (Figure S2A and http://r2.amc.nl). The available control data in those compilations were obtained from normal adrenal glands isolated from individuals of different ages, including adults (http://r2.amc.nl). Adult adrenal gland consists of adrenal cortex and adrenal medulla (containing mainly chromaffin cells), which differs from fetal adrenal gland (containing both neuroblasts and chromaffin cells) (Cooper et al., 1990; Crickard et al., 1982; Turkel and Itabashi, 1974). Thus, it is possible that high PTPN11 levels in neuroblastoma might be due to differences in the developmental or cellular origin of these tumors, or both. Nevertheless, we found that increased expression of PTPN11 is associated with a poorer overall survival rate among neuroblastoma patients with MYCN amplification (Figure S2B and S2C), suggesting an important role of PTPN11 in human neuroblastoma progression.
SHP2 is activated by binding to certain scaffolding adapters and receptors, including GRB2-associated binding proteins (GABs), fibroblast growth factor receptor substrate-2 (FRS2), insulin receptor substrates (IRSs), receptor tyrosine kinases (RTKs), cytokine receptors and other proteins (Chan et al., 2008; Mohi and Neel, 2007; Neel et al., 2003). Remarkably, we found that GAB2 is overexpressed in most neuroblastoma cases (Figure S2D and http://r2.amc.nl), and Pearson’s correlation analysis showed a significant association between the expression of PTPN11 and GAB2 in high-risk cases across different publically available neuroblastoma data sets, each comprising over 30 MYCN-amplified neuroblastoma cases (Figure 2A–C and Table S1). A number of other potential upstream regulators of SHP2, such as FRS2/3, IRS1/2/3, FGFR1/2, EGFR, PDGFRA/B, ALK, and NTRK1/2, also showed a significant correlation with PTPN11 expression, but this association did not extend to all data sets (Table S1). Notably, patients with an amplified MYCN gene and high level GAB2 expression had a significantly poorer overall survival in all three studies (Figure 2D–F and Table S1). Hence, these human genetic data suggest that GAB2 could be an important SHP2 activator contributing to high-risk neuroblastomagenesis.
Figure 2. Correlation of GAB2 and PTPN11 expression in high-risk human neuroblastomas with MYCN amplification.

(A–C) Relationship between PTPN11 and GAB2 mRNA levels in high-risk neuroblastomas with MYCN amplification, derived from three data sets, including GSE49710 (A), GSE73517 (B), and the TARGET HumanExon cohort for neuroblastoma -Asgharzadeh (C).
(D–F) Kaplan-Meier survival curves demonstrating the relationship between patient survival and GAB2 gene expression in high-risk cases with MYCN amplification. The difference between the curves for GAB2-low expression versus GAB2-high expression is significant by log-rank test at p=0.05 (D, GSE49710 data set), p=3.5e-03 (E, GSE73517 data set), and p=3.0e-04 (F, the TARGET HumanExon cohort for neuroblastoma-Asgharzadeh data set).
Gab2 potentiates MYCN-driven neuroblastomagenesis in vivo
To test whether GAB2 overexpression is important for neuroblastomagenesis, we generated a stable fish transgenic line overexpressing wild-type Gab2 in the PSNS (Gab2wt) by coinjecting dβh-Gab2wt and dβh-EGFP constructs, and bred this line to heterozygous MYCN transgenic fish. Monitoring of the offspring of this cross, including MYCN, Gab2wt and MYCN;Gab2wt fish, identified EGFP-positive masses as early as 4 weeks of age in the IRGs of transgenic fish co-expressing Gab2wt and MYCN (Figure 3B and 3C). The number of tumor-bearing fish increased rapidly: by 9 weeks of age, ~90 % of the compound transgenic animals had tumors (Figure 3C), a rate much higher than in the MYCN-only fish (~19%; Figure 3B and 3C, p<0.0001). To avoid the possible founder effect in our Gab2wt stable transgenic line, we transiently co-overexpressed the dβh-Gab2wt and dβh-mCherry constructs in MYCN transgenic fish. Over 11 weeks of observation, mCherry/GFP-positive tumors arose in 38% of MYCN transgenic fish co-overexpressing Gab2wt, compared with 13% of MYCN fish expressing mCherry alone (Figure 3D, p=0.003). Together, these results support our working hypothesis that overexpression of GAB2 collaborates with MYCN to promote neuroblastoma induction.
Figure 3. GAB2 and MYCN synergize in neuroblastomagenesis.

(A,B) Fluorescence micrographs of MYCN (A) and compound MYCN;Gab2wt (B) transgenic fish. Note EGFP-expressing tumor in the interrenal gland (arrow) of MYCN;Gab2wt compound transgenic fish at 4 weeks postfertilization (wpf). Scale bar, 1 mm.
(C) Kaplan-Meier analysis of the cumulative frequency of neuroblastoma induction over 9 weeks in the indicated stable transgenic zebrafish lines. (MYCN-only vs. MYCN;Gab2wt, p<0.0001by log-rank test).
(D) Kaplan-Meier analysis of the cumulative proportion of MYCN transgenic fish mosaically overexpressing mCherry alone (MYCN;mosaic mCherry) or co-expressing mCherry with Gab2wt (MYCN;mosaic Gab2wt) over 11 weeks. Tumor incidence in fish expressing Gab2wt and mCherry was increased over results for mCherry overexpression alone (p=0.003 by log-rank test).
(E) Immunoblot of Shp2-Erk pathway components in neuroblastomas isolated from MYCN-only or MYCN;Gab2wt transgenic fish. Total Erk1/2 levels serve as loading controls.
GAB2 contributes to activation of the SHP2-ERK pathway in multiple contexts (Gu et al., 1998; Gu et al., 2001; Nishida et al., 1999). To investigate the mechanism underlying Gab2wt-associated neuroblastomagenesis, we performed immunoblotting on tumors isolated from MYCN-only or MYCN;Gab2wt transgenic fish (Figure 3E). As expected, GAB2 was detected only in tumors from MYCN;Gab2wt fish. Phosphorylation of Erk1/2 in tumors overexpressing MYCN and Gab2wt was significantly increased over that in tumors overexpressing MYCN alone. Hence, these findings indicate that GAB2 signals through the SHP2-ERK pathway to potentiate MYCN activity during neuroblastoma pathogenesis.
Ptpn11mut/Gab2 synergy with MYCN promotes hyperplasia of sympathetic neuroblasts in the IRG
We have shown that overexpression of MYCN blocks chromaffin cell differentiation and induces sympathoadrenal neuroblastic hyperplasia in the zebrafish IRG at 5 weeks of age, which then often regresses due to apoptosis beginning at 5.5 weeks of age (Zhu et al., 2012). To explore how mutant Shp2 and Gab2 synergize with MYCN, we performed immunohistochemical analyses of sagittal sections through the IRG region of control Dβh (Zhu et al., 2012), ptpn11mut, MYCN, MYCN;ptpn11mut and MYCN;Gab2wt transgenic fish at 5 weeks of age. In transgenic fish expressing ptpn11mut alone, very few Hu+/GFP+ neuroblasts were detected in the IRG region, and the number of Hu+ neuroblasts was similar to that in control Dβh transgenic fish at 5 weeks of age (Figure 4A, first and second panels from top, and Figure 4B). These data indicate that overexpression of ptpn11mut alone does not affect the differentiation of sympathoadrenal cells. By contrast, transgenic fish overexpressing MYCN had significantly increased numbers of Hu+/GFP+ sympathoadrenal neuroblasts, compared with control Dβh fish (Figure 4A, third panel from top, and Figure 4B, p=0.0007), consistent with our previous report (Zhu et al., 2012). Importantly, the number of Hu+/GFP+ sympathoadrenal neuroblasts at 5 weeks of age was increased further in MYCN;ptpn11mut fish, compared with MYCN fish (Figure 4A, bottom panels, and Figure 4B, p=0.005), consistent with the large fraction of compound transgenic animals that developed neuroblastoma (Figure 1E).
Figure 4. Mutant ptpn11 enhances the proliferation of MYCN–induced hyperplastic neuroblasts in the interrenal gland (IRG).

(A) Sagittal sections through the IRGs of control Dβh, ptpn11mut, MYCN, and MYCN;ptpn11mut transgenic fish at 5 wpf (dorsal up, anterior left). GFP, green; Hu, red; DAPI, blue; merge, combined green, red and blue. Representative sections through the IRG of control Dβh or ptpn11mut fish contain 1–5 GFP+/Hu+ sympathetic neuroblasts (arrowheads). Hu+ cell numbers increase in MYCN and MYCN;ptpn11mut fish (brackets). Dotted lines indicate the head kidney (HK) boundary. Wpf, weeks postfertilization. Scale bar, 20 μm.
(B) Numbers of GFP+/Hu+ sympathetic neuroblasts in the IRG regions of control Dβh, ptpn11mut, MYCN, and MYCN;ptpn11mut transgenic fish at 5 wpf.
(C) Percentage of EdU+/Hu+ sympathetic neuroblasts in control Dβh, ptpn11mut, MYCN, and MYCN;ptpn11mut transgenic fish at 5 weeks. Mean values (horizontal bars) were compared by Welch t-test (two-tailed).
See also Figures S3 and S4.
Next, we performed EdU pulse-labeling experiments with the Dβh (control), ptpn11mut, MYCN, and MYCN; ptpn11mut fish. After 2 hours of labeling, the fraction of EdU-incorporating Hu+ neuroblasts was significantly increased in 5-week-old MYCN fish, compared with those in ptpn11mut or Dβh control fish (Figure 4C, p=0.003, and Figure S3, top three panels). Hence, MYCN overexpression not only blocks the differentiation of sympathoadrenal cells, but also increases the proliferative rate of neuroblasts in the IRG. The fraction of EdU-incorporating Hu+ neuroblasts in the IRG was increased even further in fish expressing MYCN and ptpn11mut (Figure 4C, p=0.014, and Figure S3, bottom panels), providing a cellular explanation for the increased rate of tumor induction in compound transgenic animals.
To determine whether overexpression of ptpn11mut can rescue the MYCN-induced, developmentally-timed apoptotic response in the IRG at 5.5 weeks of age, we assessed the expression of activated caspase-3, an indicator of apoptotic cell death. Consistent with our previous observations (Zhu et al., 2012), apoptotic sympathoadrenal cells coexpressing activated caspase-3 and GFP appeared in the IRG of MYCN-expressing fish (Figure 5A, top panels). The fraction of Caspase-3+/GFP+ neuroblasts to the total number of GFP+ neuroblasts was decreased significantly in the IRG region of fish overexpressing MYCN and ptpn11mut, compared with those in fish overexpressing MYCN alone (Figure 5A, lower panels, and 5B, p=0.03). Thus, coexpression of ptpn11mut with MYCN not only enhances the proliferation of hyperplastic neuroblasts in the IRG, but also promotes their survival, resulting in the continued accumulation of hyperplastic neuroblasts and culminating in the development of highly penetrant neuroblastoma.
Figure 5. Overexpression of Ptpn11mut inhibits the developmentally-timed apoptotic response triggered by MYCN overexpression in the IRG.

(A) Sagittal sections through the IRG of MYCN (top panels) and MYCN;ptpn11mut (bottom panels) transgenic fish at 5.5 wpf (dorsal up, anterior left). GFP, green; activated caspase-3, red; DAPI, blue. Arrowheads point to the activated caspase-3+ apoptotic cells. Dotted lines indicate the head kidney (HK) boundary. Scale bars, 10 μm.
(B) Percentage of activated caspase-3+ apoptotic cells to the total number of GFP+ sympathetic neuroblasts in the IRG of MYCN and MYCN;ptpn11mut fish at 5.5 wpf. Mean values (horizontal bars) were compared by the Welch t-test (two-tailed).
A similarly striking increase in the number of sympathoadrenal cells was detected in the IRGs of MYCN;Gab2wt transgenic fish at 5 weeks of age, as compared to MYCN-only fish (Figure S4, p=0.005), consistent with the profound acceleration of tumor onset and increased disease penetrance in the compound transgenic animals overexpressing both MYCN and Gab2.
Combined inhibition of MYCN signaling and Shp2-Erk activation enhances treatment-induced regression of MYCN-driven tumors
To assess the translational potential of our findings, we tested the effects of the small-molecule curaxin compound CBL0137. This inhibitor of histone chaperone FACT (facilitator of chromatin transcription) markedly reduces neuroblastoma initiation and progression in TH-MYCN transgenic mice by decreasing MYCN mRNA expression and protein stability (Carter et al., 2015). In our hands, CBL0137 inhibited neuroblastoma growth in the transgenic fish overexpressing dβh:EGFP-MYCN and dβh:EGFP (designated MYCN;GFP) when injected daily intraperitoneally for 2 weeks. At day-15 post-treatment, we detected larger GFP-positive tumor masses in all vehicle-treated fish as compared to tumors on day 1 of treatment (Figure 6A, 6B and 6E), suggesting that vehicle-treated MYCN tumors will continue to progress. By contrast, in 13 of 15 CBL0137-treated MYCN;GFP fish, GFP-positive tumor masses either regressed or remained essentially the same size as on day 1 (Figure 6C–E), suggesting that CBL0137 can effectively inhibit the growth of MYCN-driven tumors in zebrafish. A significant decrease in the number of PCNA-positive cells in CBL0137-treated compared with vehicle-treated tumors was detected by immunohistochemical analysis with an antibody against the proliferation marker PCNA (Kubben et al., 1994) (Figure 6F, right panels), but there was no detectable effect of the drug on tumor differentiation as assessed by H&E staining (Figure 6F, left panels). These effects correlated with a marked reduction of EGFP-MYCN levels in the CBL0137-treated tumors (Figure 6G). Levels of Ssrp1 and Spt16, two components of the FACT complex (Carter et al., 2015), were also decreased in CBL0137-treated tumors (Figure 6G), while the Erk1/2 and phosphorylated Erk1/2 levels remained unchanged. These results suggest that CBL0137 inhibits MYCN and FACT in zebrafish, leading to a significant inhibitory effect on neuroblastoma cell growth in MYCN-overexpressing animals.
Figure 6. Targeting MYCN with CBL0137 is effective against neuroblastoma in MYCN;GFP transgenic fish.

(A–D) EGFP-expressing tumors (arrowhead) in the MYCN;GFP compound transgenic fish, treated with vehicle (A and B) or CBL0137 (C and D) at days 1 or 15 of treatment. Scale bar, 1 mm.
(E) Differences in tumor volume in animals treated with vehicle or CBL0137 on day 15 vs day 1, as measured by the size of EGFP-positive tumor masses using Image J software. Mean values (horizontal bars) were compared by Welch t-test (two-tailed).
(F) Histopathologic and immunohistochemical analyses of MYCN;GFP tumors treated with vehicle or CBL0137. Left: H&E-stained sagittal sections. Right: immunohistochemical staining with PCNA antibody. Scale bar, 50 μm.
(G) Immunoblot of lysates from MYCN;GFP tumors treated with vehicle or CBL0137. Total levels of Erk1/2 serve as loading controls.
To test the efficacy of CBL0137 against MYCN;Gab2wt-overexpressing tumors, we administered this compound or vehicle for 2 weeks to MYCN;Gab2wt transgenic fish with established neuroblastomas. Rapid progression of tumors was observed in all vehicle-treated MYCN;Gab2wt fish (Figure 7A, 7B and 7G). Surprisingly, tumors grew continuously in 5 of 12 MYCN;Gab2wt fish treated with CBL0137 alone (Figure 7C, 7D and 7G, p=0.04, as compared to CBL0137-treated MYCN;GFP fish by Fisher’s exact test), suggesting that Gab2wt overexpression can dampen the inhibitory effect of CBL0137 on tumor growth. In earlier work, we showed that inactivation of the Ras-Erk pathway by trametinib, a MEK inhibitor approved by the FDA for use against BRAF V600E mutation-positive melanoma (Dhillon, 2016), can cooperate with retinoids (isotretinoin) to inhibit tumor growth in nf1-deficient fish with overexpression of MYCN (He et al., 2016). Thus, we asked whether inhibition of Erk activation could restore the sensitivity of resistant Gab2wt-expressing tumor cells to CBL0137. We first treated fish bearing MYCN;GFP or MYCN;Gab2wt tumors with trametinib. Tumor growth was not inhibited in all fish treated with trametinib alone (Figure S5), suggesting that Erk inhibition alone is not sufficient to inhibit the growth of tumors expressing MYCN or MYCN and Gab2wt. By contrast, combination treatment with CBL0137 and trametinib led to a significant reduction in tumor growth (Figure 7E, 7F, 7G and 7H). In addition, significant decreases in EGFP-MYCN, Ssrp1, Spt16 and pErk1/2 protein expression in tumors treated with the drug combination were detected by immunoblotting (Figure 7I). Although CBL0137 treatment alone or in combination with trametinib downregulated the expression of mycn at the RNA level, as expected (Carter et al., 2015), Gab2 levels were unaffected in CBL0137- or CBL0137 plus trametinib-treated Gab2wt-expressing embryos (Figure S6). These findings suggest that trametinib increases the efficacy of CBL0137 against established MYCN;Gab2wt tumors by cooperatively inhibiting MYCN signaling and Erk activation, respectively, further implicating cooperation between the Shp2-Ras-Erk pathway and MYCN in neuroblastoma maintenance.
Figure 7. Erk inhibition cooperates with CBL0137 to suppress MYCN;Gab2wt neuroblastoma.

(A–F) EGFP-expressing tumors (arrowhead) in the MYCN;Gab2wt transgenic fish treated with vehicle (A and B), CBL0137 alone (C and D), or CBL0137 plus trametinib (E and F) at day 1 or day 15 of treatment. Scale bar, 1 mm.
(G) Differences in tumor volume in animals treated with vehicle, CBL0137, or CBL0137 plus trametinib on day 15 vs day 1, as measured by the size of EGFP-positive tumor masses using Image J software. Mean values (horizontal bars) were compared by Welch t-test (two-tailed).
(H) Histopathologic and immunohistochemical analyses of MYCN;Gab2wt tumors treated with vehicle or combined CBL0137 and trametinib. Left: H&E-stained sagittal sections. Right: immunohistochemical staining with PCNA antibody. Scale bar, 50 μm.
(I) Immunoblot of lysates from MYCN;Gab2wt tumors treated with vehicle or CBL0137 and trametinib. Total levels of Erk1/2 serve as loading controls.
See also Figure S5 and S6.
DISCUSSION
We have demonstrated that aberrantly activated Shp2 cooperates with MYCN to accelerate tumor induction and increase disease penetrance in our zebrafish model of high-risk neuroblastoma. Mutant Shp2 fulfills this role by activating the Ras-Erk pathway, leading to increased proliferation and survival of sympathoadrenal neuroblasts in the IRG region of the fish. We also found that the pro-oncogenic role of Shp2 is not restricted to neuroblastoma with ptpn11 mutations, but extends to most high-risk cases. We attribute these effects to the overexpression of Gab2, an immediate upstream regulator of Shp2. In our zebrafish model, GAB2 activates the Shp2-Ras-Erk pathway, providing a means to potentiate MYCN activity in the absence of ptpn11 mutations. Inhibiting Erk activation with trametinib, and decreasing MYCN levels by treatment with CBL0137 effectively inhibits tumor growth in MYCN;Gab2wt transgenic fish. Hence, not only do MYCN amplification and stimulation of the Shp2-Ras-Erk pathway cooperate in neuroblastoma initiation, but both are required for tumor maintenance. These findings have clear implications for the treatment of most high-risk cases driven by amplified MYCN.
Disease-associated mutations of PTPN11 typically affect the interacting surfaces of the N-terminal SH2 and catalytic (PTP) domains of SHP2, leading to catalytic activation of SHP2 (Keilhack et al., 2005; Loh et al., 2004; Tartaglia et al., 2002; Tartaglia et al., 2001; Tartaglia et al., 2003). Indeed, Bentires-Alj et al. previously showed that the E69K neuroblastoma-associated mutation has ~15.5-fold higher catalytic activity in vitro than does wild-type SHP2 (Bentires-Alj et al., 2004). Consistent with these findings, overexpression of ptpn11mut (ptpn11E69K), but not mosaic overexpression of ptpn11wt, in MYCN-overexpressing transgenic fish resulted in accelerated neuroblastoma induction and increased tumor penetrance. Furthermore, biochemical analyses confirmed the predicted hyperactivation of Erk in ptpn11mut-expressing neuroblastoma cells. Hence, we conclude that ptpn11E69K acts as a gain-of-function mutation to activate Ras-Erk signaling during neuroblastomagenesis.
Importantly, Shp2-Ras-Erk pathway activation was also detected in the Gab2-overexpressing neuroblastomas, consistent with previous findings that recruitment of SHP2 to the GAB2-BCR/ABL complex and subsequent activation of RAS-ERK signaling is essential for BCR/ABL-induced transformation (Sattler et al., 2002) and leukemogenesis (Gu et al., 2016). Similarly, GAB2/SHP2 interaction is essential for GAB2 to enhance transformation of MCF10A mammary epithelial cells by HER2/Neu (Bentires-Alj et al., 2006) and to increase proliferation and altered growth factor dependence of MCF10A cells to promote mammary tumorigenesis (Brummer et al., 2006). Finally, GAB2 is required for the transformation of primary murine myeloid cells by SHP2E76K (Mohi et al., 2005). Although the mechanism underlying GAB2 overexpression in neuroblastoma remains unclear, amplification of 11q14.1, which harbors the GAB2 gene, has been identified as a recurrent alteration in breast (Bentires-Alj et al., 2006; Bocanegra et al., 2010) and ovarian (Brown et al., 2008) cancers, as well as metastatic melanoma (Horst et al., 2009). Amplification of 11q14.1 has not been identified in human neuroblastoma, while in our study high levels of human GAB2 expression in high-risk MYCN-amplified neuroblastoma cases correlated with high levels of PTPN11 expression. This emphasizes the need to clarify how GAB2 becomes aberrantly regulated in cases leading to aberrant SHP2 activation.
A recent study showed that mutations in genes encoding RAS-ERK pathway components, including PTPN11, account for more than three-fourths of all relapsed neuroblastoma cases and show moderate-to-high sensitivity to MEK inhibitors, both in vitro and in vivo (Eleveld et al., 2015). We find it interesting that the two MYCN-amplified neuroblastoma cell lines Kelly and IMR-5 did not respond to MEK inhibition, whether studied in vitro or in xenograft models (Eleveld et al., 2015). Similarly, our transgenic fish overexpressing MYCN only or MYCN and Gab2wt did not respond to MEK inhibitor treatment alone, suggesting that inhibition of Erk alone might not be sufficient to inhibit tumor growth in neuroblastomas with overexpression or amplification of MYCN. In support of this hypothesis, we observed a remarkable response of MYCN;Gab2wt-overexpressing neuroblastomas to combination treatment with a compound targeting MYCN and a MEK inhibitor. These findings provide a strong rationale for inhibiting both the RAS-ERK pathway and MYCN signaling in cases of MYCN-amplified relapsed neuroblastoma in which upregulation of GAB2 leads to aberrant activation of SHP2. This strategy could be readily extended to patients with MYCN-amplified primary neuroblastoma, most of whom show evidence of PTPN11 (SHP2) and/or GAB2 overexpression and thus hyperactivation of the SHP2-RAS-ERK pathway. In addition, given the success of recent preclinical efforts to target MYCN in neuroblastomas by using the FACT inhibitor CBL0137 (Carter et al., 2015) or the BET bromodomain inhibitor JQ1 (Puissant et al., 2013), the emerging challenge will likely be to forestall or prevent the development of resistance to these MYCN-directed agents. Our data suggest that by eliminating oncogenic signaling through the SHP2-RAS-ERK pathway, it may be possible to remove an important resistance mechanism. Very recently, a highly potent selective small-molecular SHP2 inhibitor, SHP099, developed by Novartis, was found to be efficacious in inhibiting the proliferation of receptor-tyrosine-kinase-driven human cancer cells both in vitro and in vivo (Chen et al., 2016). Whatever experimental approach is taken, our zebrafish model of neuroblastoma overexpressing ptpn11mut or Gab2wt should provide a valuable resource for evaluating new small-molecule inhibitors of the SHP2-RAS-ERK pathway.
EXPERIMENTAL PROCEDURES
Zebrafish
Zebrafish were all of the AB background strain. Embryos were staged according to Kimmel et al. (Kimmel et al., 1995). All zebrafish studies and maintenance of the animals were in accord with Mayo Clinic IACUC-approved protocol # A41213.
DNA constructs for transgenesis
Wild-type zebrafish ptpn11 (ptpn11wt), ptpn11E69K (ptpn11mut), and mouse Gab2wt were cloned into a modified destination vector downstream of a zebrafish dβh gene promoter by using the multisite Gateway system (Zhu et al., 2012). Wild-type embryos were injected with dβh:ptpn11mut-EGFP or co-injected with dβh:Gab2wt and dβh:EGFP constructs at the one-cell stage, and fish were allowed to grow to adulthood. Fin clips from the offspring were genotyped for stable integration and germline transmission of the transgenes. The Tg(dβh:ptpn11mut-EGFP) and Tg(dβh:Gab2wt;dβh:EGFP) zebrafish lines are designated “ptpn11mut “ and “Gab2wt” in this manuscript, respectively. To mosaically overexpress ptpn11wt or Gab2wt in MYCN-expressing transgenic fish, we co-injected dβh:ptpn11wt or dβh:Gab2wt DNA constructs with dβh:mCherry (3:1 ratio) into one-cell-stage MYCN transgenic embryos. The mCherry/EGFP-MYCN positive embryos were sorted at day 2–5 and monitored for tumor induction.
MYCN and ptpn11mut or Gab2wt heterozygous stable transgenic fish were crossed. Their offspring were grown under identical conditions and were screened every 2 weeks, beginning at 4–5 weeks postfertilization (wpf), for fluorescent EGFP-expressing cell masses indicative of tumors. Injected MYCN embryos with mosaic expression of mCherry and other genes (mentioned above) were monitored for tumors as described above. Fish with tumors were separated and analyzed further by H&E staining and immunohistochemical assays.
Cryosectioning, paraffin sectioning and immunostaining
Fish were fixed with 4% paraformaldehyde and embedded in agar/sucrose or paraffin blocks for cryosectioning or paraffin sectioning, respectively. Sections were immunostained by using conventional protocols (Macdonald, 1999) and antibodies against GFP, TH, Hu, P-Erk1/2, and Shp2.
Imaging
A Zeiss LSM 780 laser scanning confocal microscope was used to capture fluorescent images at high magnification, whereas a Leica MZ10F fluorescence microscope was used to capture bright field and fluorescent images of tumor-bearing fish. Images were processed with Zeiss ZEN 2012, Adobe Photoshop and Illustrator CS3 (Adobe) software.
Statistical analyses
The method of Kaplan and Meier was used to estimate the rate of tumor development. Fish that died without evidence of EGFP- or mCherry-positive masses were censored. The log-rank test (from the GraphPad Prism software) was used to assess differences in the cumulative frequency of neuroblastoma between MYCN-only and MYCN;ptpn11mut or MYCN;Gab2wt stable transgenic fish and between MYCN transgenic fish mosaically expressing mCherry alone and mCherry plus ptpn11mut, or Gab2wt at 11 wpf.
The number of EGFP+ or mCherry+ sympathoadrenal cells in the IRG of the Dβh-mCherry, ptpn11mut, MYCN, and MYCN;ptpn11mut transgenic fish at 5 wpf were compared between groups, by using the Welch t-test (from the GraphPad Prism software) to address the possibility of unequal variances. Means of the fraction of EdU-incorporating GFP+ sympathoadrenal cells in Dβh, ptpn11mut, MYCN, and MYCN;ptpn11mut transgenic fish at 5wpf and means of the percentage of activated caspase-3+ apoptotic cells in MYCN and MYCN;ptpn11mut fish at 5.5 wpf also were compared by the Welch t-test.
For the inhibitor treatment experiments, the means of changes in tumor volume between inhibitors-treated group and vehicle-treated group were compared by the Welch t-test (from GraphPad Prism 6). All p-values cited in the manuscript are two-sided.
Additional methods are presented in Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank J. R. Gilbert for editorial review of the manuscript and critical comments. This work was supported by grants K99/R00CA178189 (S.Z.) from the National Cancer Institute; young investigator awards from Alex’s Lemonade Stand Foundation (S.Z.) and the CureSearch for Children’s Cancer Foundation (S.Z.); a V Scholar award from the V Foundation for Cancer Research (S.Z.) and pilot project awards from the Fraternal Order of Eagles (S.Z.) and the Mayo Center for Biomedical Discovery (S.Z.); support from the Mayo Clinic Cancer Center, Center for Biomedical Discovery, and Center for Individualized Medicine (S.Z.); a R01 CA180692 (A.T.L.) from the National Cancer Institute; an Alex’s Lemonade Stand Foundation Innovation Award (A.T.L.); a fellowship from the Friends for Life and the Friends of DFCI (T.T.); a grant from the Children’s Neuroblastoma Cancer Foundation (T.T.); and R37 CA49152 (B.G.N.). During part of this work, B.G.N. was a Canada Research Chair, Tier 1, and was partially supported by the Princess Margaret Cancer Foundation. We also thank Incuron for providing the CBL0137 compound for our study.
Footnotes
Supplemental information includes 1 table, 6 figures, their legends and supplemental experimental procedures, which can be found with this article online at XXXX.
AUTHOR CONTRIBUTIONS
S.Z. designed the study and wrote the manuscript; A.T.L. and B.G.N. discussed the project and edited the manuscript; S.Z., X.Z., Z.D., S.H., and T.T. performed the zebrafish experiments; C.Z., C.Y.U and H.L. performed computational analyses; A.M. and B.J. performed immunohistochemical staining. A.M.O. performed pathology analysis.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bentires-Alj M, Gil SG, Chan R, Wang ZC, Wang Y, Imanaka N, Harris LN, Richardson A, Neel BG, Gu H. A role for the scaffolding adapter GAB2 in breast cancer. Nature medicine. 2006;12:114–121. doi: 10.1038/nm1341. [DOI] [PubMed] [Google Scholar]
- Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer research. 2004;64:8816–8820. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- Bocanegra M, Bergamaschi A, Kim YH, Miller MA, Rajput AB, Kao J, Langerod A, Han W, Noh DY, Jeffrey SS, et al. Focal amplification and oncogene dependency of GAB2 in breast cancer. Oncogene. 2010;29:774–779. doi: 10.1038/onc.2009.364. [DOI] [PubMed] [Google Scholar]
- Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady PW, et al. ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer cell. 2014;26:682–694. doi: 10.1016/j.ccell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nature reviews Clinical oncology. 2014;11:704–713. doi: 10.1038/nrclinonc.2014.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- Brown LA, Kalloger SE, Miller MA, Shih Ie M, McKinney SE, Santos JL, Swenerton K, Spellman PT, Gray J, Gilks CB, et al. Amplification of 11q13 in ovarian carcinoma. Genes, chromosomes & cancer. 2008;47:481–489. doi: 10.1002/gcc.20549. [DOI] [PubMed] [Google Scholar]
- Brummer T, Schramek D, Hayes VM, Bennett HL, Caldon CE, Musgrove EA, Daly RJ. Increased proliferation and altered growth factor dependence of human mammary epithelial cells overexpressing the Gab2 docking protein. The Journal of biological chemistry. 2006;281:626–637. doi: 10.1074/jbc.M509567200. [DOI] [PubMed] [Google Scholar]
- Carter DR, Murray J, Cheung BB, Gamble L, Koach J, Tsang J, Sutton S, Kalla H, Syed S, Gifford AJ, et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Science translational medicine. 2015;7:312ra176. doi: 10.1126/scitranslmed.aab1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer metastasis reviews. 2008;27:179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke VG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535:148–152. doi: 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- Cooper MJ, Hutchins GM, Israel MA. Histogenesis of the human adrenal medulla. An evaluation of the ontogeny of chromaffin and nonchromaffin lineages. The American journal of pathology. 1990;137:605–615. [PMC free article] [PubMed] [Google Scholar]
- Crickard K, Fujii DK, Jaffe RB. Isolation and identification of human fetal adrenal medullary cells in vitro. The Journal of clinical endocrinology and metabolism. 1982;55:1143–1148. doi: 10.1210/jcem-55-6-1143. [DOI] [PubMed] [Google Scholar]
- De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, Lakeman A, Vandesompele J, Hoebeeck J, Van Maerken T, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:4353–4362. doi: 10.1158/1078-0432.CCR-09-2660. [DOI] [PubMed] [Google Scholar]
- Dhillon S. Dabrafenib plus Trametinib: a Review in Advanced Melanoma with a BRAF (V600) Mutation. Targeted oncology. 2016;11:417–428. doi: 10.1007/s11523-016-0443-8. [DOI] [PubMed] [Google Scholar]
- Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, Schild L, Bentahar NB, Bellini A, Chicard M, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. 2015;47:864–871. doi: 10.1038/ng.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Pratt JC, Burakoff SJ, Neel BG. Cloning of p97/Gab2, the major SHP2-binding protein in hematopoietic cells, reveals a novel pathway for cytokine-induced gene activation. Molecular cell. 1998;2:729–740. doi: 10.1016/s1097-2765(00)80288-9. [DOI] [PubMed] [Google Scholar]
- Gu H, Saito K, Klaman LD, Shen J, Fleming T, Wang Y, Pratt JC, Lin G, Lim B, Kinet JP, et al. Essential role for Gab2 in the allergic response. Nature. 2001;412:186–190. doi: 10.1038/35084076. [DOI] [PubMed] [Google Scholar]
- Gu S, Chan WW, Mohi G, Rosenbaum J, Sayad A, Lu Z, Virtanen C, Li S, Neel BG, Van Etten RA. Distinct GAB2 signaling pathways are essential for myeloid and lymphoid transformation and leukemogenesis by BCR-ABL1. Blood. 2016 doi: 10.1182/blood-2015-06-653006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Mansour MR, Zimmerman MW, Ki DH, Layden HM, Akahane K, Gjini E, de Groh ED, Perez-Atayde AR, Zhu S, et al. Synergy between loss of NF1 and overexpression of MYCN in neuroblastoma is mediated by the GAP-related domain. eLife. 2016:5. doi: 10.7554/eLife.14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehner JC, Gestblom C, Hedborg F, Sandstedt B, Olsen L, Pahlman S. A developmental model of neuroblastoma: differentiating stroma-poor tumors’ progress along an extra-adrenal chromaffin lineage. Laboratory investigation; a journal of technical methods and pathology. 1996;75:659–675. [PubMed] [Google Scholar]
- Horst B, Gruvberger-Saal SK, Hopkins BD, Bordone L, Yang Y, Chernoff KA, Uzoma I, Schwipper V, Liebau J, Nowak NJ, et al. Gab2-mediated signaling promotes melanoma metastasis. The American journal of pathology. 2009;174:1524–1533. doi: 10.2353/ajpath.2009.080543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HJ, Lin G, Chung BC. Parallel early development of zebrafish interrenal glands and pronephros: differential control by wt1 and ff1b. Endocrine research. 2004;30:803. doi: 10.1081/erc-200044044. [DOI] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. The Journal of biological chemistry. 2005;280:30984–30993. doi: 10.1074/jbc.M504699200. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kubben FJ, Peeters-Haesevoets A, Engels LG, Baeten CG, Schutte B, Arends JW, Stockbrugger RW, Blijham GH. Proliferating cell nuclear antigen (PCNA): a new marker to study human colonic cell proliferation. Gut. 1994;35:530–535. doi: 10.1136/gut.35.4.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenau DM, Keefe MD, Storer NY, Jette CA, Smith AC, Ceol CJ, Bourque C, Look AT, Zon LI. Co-injection strategies to modify radiation sensitivity and tumor initiation in transgenic Zebrafish. Oncogene. 2008;27:4242–4248. doi: 10.1038/onc.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–2331. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- Louis CU, Shohet JM. Neuroblastoma: molecular pathogenesis and therapy. Annual review of medicine. 2015;66:49–63. doi: 10.1146/annurev-med-011514-023121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald R. Zebrafish immunohistochemistry. Methods Mol Biol. 1999;127:77–88. doi: 10.1385/1-59259-678-9:77. [DOI] [PubMed] [Google Scholar]
- Marusich MF, Furneaux HM, Henion PD, Weston JA. Hu neuronal proteins are expressed in proliferating neurogenic cells. Journal of neurobiology. 1994;25:143–155. doi: 10.1002/neu.480250206. [DOI] [PubMed] [Google Scholar]
- Mohi MG, Neel BG. The role of Shp2 (PTPN11) in cancer. Current opinion in genetics & development. 2007;17:23–30. doi: 10.1016/j.gde.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, Morgan K, Boulton C, Shigematsu H, Keilhack H, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer cell. 2005;7:179–191. doi: 10.1016/j.ccr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483:589–593. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends in biochemical sciences. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- Nishida K, Yoshida Y, Itoh M, Fukada T, Ohtani T, Shirogane T, Atsumi T, Takahashi-Tezuka M, Ishihara K, Hibi M, et al. Gab-family adapter proteins act downstream of cytokine and growth factor receptors and T- and B-cell antigen receptors. Blood. 1999;93:1809–1816. [PubMed] [Google Scholar]
- Park JR, Bagatell R, London WB, Maris JM, Cohn SL, Mattay KK, Hogarty M. Children’s Oncology Group’s 2013 blueprint for research: neuroblastoma. Pediatric blood & cancer. 2013;60:985–993. doi: 10.1002/pbc.24433. [DOI] [PubMed] [Google Scholar]
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013 doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer discovery. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito D, Takase Y, Murai H, Takahashi Y. The dorsal aorta initiates a molecular cascade that instructs sympatho-adrenal specification. Science. 2012;336:1578–1581. doi: 10.1126/science.1222369. [DOI] [PubMed] [Google Scholar]
- Sattler M, Mohi MG, Pride YB, Quinnan LR, Malouf NA, Podar K, Gesbert F, Iwasaki H, Li S, Van Etten RA, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer cell. 2002;1:479–492. doi: 10.1016/s1535-6108(02)00074-0. [DOI] [PubMed] [Google Scholar]
- Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. The New England journal of medicine. 1985;313:1111–1116. doi: 10.1056/NEJM198510313131802. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, Brunner HG, Bertola DR, Crosby A, Ion A, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. American journal of human genetics. 2002;70:1555–1563. doi: 10.1086/340847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- Teitelman G, Baker H, Joh TH, Reis DJ. Appearance of catecholamine-synthesizing enzymes during development of rat sympathetic nervous system: possible role of tissue environment. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:509–513. doi: 10.1073/pnas.76.1.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkel SB, Itabashi HH. The natural history of neuroblastic cells in the fetal adrenal gland. The American journal of pathology. 1974;76:225–244. [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, Kutok JL, Rodig SJ, Neuberg DS, Helman D, Feng H, et al. Activated ALK Collaborates with MYCN in Neuroblastoma Pathogenesis. Cancer cell. 2012;21:362–373. doi: 10.1016/j.ccr.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.