

Here, Levikova et al. show that the motor activity of both recombinant yeast and human DNA2 promotes efficient degradation of long stretches of ssDNA, particularly in the presence of the replication protein A. Their results support a model of DNA2 and the RecQ family helicase partner forming a bidirectional motor machine in which the RecQ family helicase is the lead helicase, and the motor of DNA2 functions as a ssDNA translocase to promote degradation of 5′-terminated DNA.
Keywords: DNA helicase, DNA nuclease, yDna2, hDNA2, ssDNA translocase, DNA end resection
Abstract
DNA2 nuclease–helicase functions in DNA replication and recombination. This requires the nuclease of DNA2, while, in contrast, the role of the helicase activity has been unclear. We now show that the motor activity of both recombinant yeast and human DNA2 promotes efficient degradation of long stretches of ssDNA, particularly in the presence of the replication protein A. This degradation is further stimulated by a direct interaction with a cognate RecQ family helicase, which functions with DNA2 in DNA end resection to initiate homologous recombination. Consequently, helicase-deficient yeast dna2 K1080E cells display reduced resection speed of HO-induced DNA double-strand breaks. These results support a model of DNA2 and the RecQ family helicase partner forming a bidirectional motor machine, where the RecQ family helicase is the lead helicase, and the motor of DNA2 functions as a ssDNA translocase to promote degradation of 5′-terminated DNA.
ssDNA translocases are motor proteins that couple ATP hydrolysis to translocation on ssDNA with either 3′ → 5′ or 5′ → 3′ polarity (Lohman et al. 2008). Some but not all ssDNA translocases are DNA helicases capable of unwinding dsDNA (Lohman et al. 2008). The DNA replication-dependent helicase/nuclease 2 (yDna2 from Saccharomyces cerevisiae, hDNA2 from humans) possesses a RecB family nuclease domain and a superfamily I helicase domain (Budd et al. 1995, 2000; Bae et al. 1998). The nuclease of both yeast and human DNA2 is specific for ssDNA and can degrade both 3′- and 5′-terminated ssDNA strands (Bae et al. 1998; Masuda-Sasa et al. 2006). The cognate ssDNA-binding replication protein A (RPA) specifically inhibits 3′ → 5′ DNA degradation by DNA2 (Cejka et al. 2010; Niu et al. 2010; Nimonkar et al. 2011), suggesting that the DNA2 nuclease degrades ssDNA exclusively with a 5′ → 3′ directionality under physiological conditions.
The helicase activity of both yeast and human DNA2 is paradoxically cryptic, as it unwinds dsDNA only when the nuclease activity is inactivated (Levikova et al. 2013; Pinto et al. 2016). The structure of mouse DNA2 revealed that the nuclease active site is located inside a narrow tunnel that accommodates only ssDNA (Zhou et al. 2015). Importantly, the nuclease tunnel is positioned ahead of the superfamily I helicase domain. This supported a model in which the DNA2 nuclease cleaves ssDNA overhangs that are required for loading of the DNA2 helicase domain on the DNA substrate (Levikova et al. 2013; Pinto et al. 2016). Overhang cleavage by the DNA2 nuclease thus prevents DNA unwinding by the DNA2 helicase, but the reason why the helicase domain appears to compete with the nuclease domain for DNA substrate in a physiological context has remained unclear. Genetic and cell biological experiments offer few insights into the function of the DNA2 helicase. These experiments are limited by the fact that DNA2 and its nuclease activity are required for viability in yeast cells, while both helicase and nuclease activities are essential in human cells, making phenotypic analysis associated with DNA2 defects challenging (Formosa and Nittis 1999; Budd et al. 2000; Duxin et al. 2012; Wanrooij and Burgers 2015; Olmezer et al. 2016).
To repair a DNA double-strand break (DSB) by homologous recombination, the 5′-terminated strand of the DSB must be first nucleolytically resected to reveal a 3′-terminated ssDNA overhang (Cejka 2015). This serves as a substrate for the strand exchange protein RAD51 and primes DNA synthesis in later steps of the recombination pathway. There are two main pathways capable of resecting long lengths of dsDNA, catalyzed by either the EXO1 or DNA2 nuclease (Gravel et al. 2008; Mimitou and Symington 2008; Zhu et al. 2008). While EXO1 is able to resect a 5′-terminated DNA strand of a DNA duplex (Tran et al. 2002; Cannavo et al. 2013), research from several laboratories established that DNA2 functions in complex with a cognate RecQ family helicase. This includes Sgs1 in S. cerevisiae and the Werner syndrome protein (WRN) or Bloom syndrome protein (BLM) in human cells (Zhu et al. 2008; Nimonkar et al. 2011; Sturzenegger et al. 2014). The helicase activity of the respective RecQ family member and the nuclease of DNA2 are essential in DNA end resection (Zhu et al. 2008; Nimonkar et al. 2011; Sturzenegger et al. 2014). All studies to date failed to explain the function of the DNA2 helicase. Here we report that the motor activity of both yeast and human DNA2 proteins greatly stimulates degradation of long ssDNA molecules by acting as a ssDNA translocase with a 5′ → 3′ polarity. The motor activity within wild-type DNA2 is incapable of unwinding and degrading dsDNA (Levikova et al. 2013; Pinto et al. 2016) but, in contrast, is required to efficiently degrade long ssDNA. These results infer a model in which the cognate RecQ helicase partner is the lead helicase that provides DNA2 with ssDNA, and DNA2 is using its motor coupled with nuclease activity to efficiently degrade the unwound 5′-terminated ssDNA strand. Yeast cells expressing physiological levels of the helicase-deficient Dna2 K1080E variant from its natural locus in genomic DNA display an impaired DNA end resection phenotype, suggesting that the helicase activity of DNA2 functions to promote DNA end resection in vivo.
Results
The motor of yeast and human DNA2 promotes degradation of long ssDNA
Both yeast and human DNA2 possess the capacity to unwind dsDNA, yet this is paradoxically revealed only when the nuclease of DNA2 is inactivated (Levikova et al. 2013; Pinto et al. 2016). In accord with these observations, we show here that nuclease-deficient yDna2 E675A efficiently unwound Y-structured DNA, producing ssDNA. In contrast, the processing of this substrate by wild-type and helicase-deficient yDna2 K1080E variants was indistinguishable and resulted only in DNA degradation (Fig. 1A–C; Supplemental Fig. S1A). The helicase activity of yDna2 thus had no apparent effect on the degradation of the oligonucleotide-based DNA by wild-type yDna2 (Fig. 1A–C; Supplemental Fig. S1A). The nuclease of yDna2 masks its unwinding activity, in agreement with previous data in both yeast and human systems (Levikova et al. 2013; Pinto et al. 2016). To elucidate the function of the DNA2 motor, we investigated the helicase-proficient and helicase-deficient yeast Dna2 variants in the degradation of long ssDNA. As DNA2 proteins in all eukaryotic organisms function in conjunction with a DNA helicase, ssDNA may better mimic the structure on which DNA2 acts in vivo. To this point, we digested λDNA with HindIII, 32P-labeled the restricted fragments at the 3′ end, and heat-denatured the dsDNA to prepare ssDNA fragments of ∼100 to ∼23,000 nucleotides (nt) in length. Strikingly, wild-type yDna2 was dramatically faster in ssDNA degradation than the helicase-deficient K1080E variant (Fig. 1D) despite equivalent levels of nuclease activity in both preparations when assayed on oligonucleotide-based DNA (Fig. 1A–C; Supplemental Fig. S1A). We detected no endonuclease activity of yDna2 under our experimental conditions when yRPA was present (Supplemental Fig. S1B), which suggested that the ssDNA degradation occurs from an open end. The gradual DNA degradation and the presence of smear below the 3′-labeled substrate bands in Figure 1D clearly indicate that the ssDNA degradation by yDna2 occurred with a 5′ → 3′ polarity, as expected for yDna2 in the presence of yRPA (Cejka et al. 2010; Niu et al. 2010). The motor activity of yDna2 also promoted ssDNA degradation when using a yDna2 mutant lacking the N-terminal regulatory domain (yDna2 ΔN), showing that the first 405 residues of yDna2 are dispensable for the motor-assisted ssDNA degradation (Supplemental Fig. S1C,D). These results could be recapitulated with recombinant human DNA2, which lacks the N-terminal regulatory domain found in yeast (Fig. 1E). In accord with the slower rate of DNA unwinding by hDNA2 compared with yDna2 (Levikova et al. 2013; Pinto et al. 2016), the overall rate of ssDNA degradation was slower by the human protein than by the yeast homolog. Importantly, however, the helicase-deficient hDNA2 K654R was clearly less efficient in ssDNA degradation than wild-type hDNA2, showing that the involvement of the DNA2 motor in accelerating ssDNA degradation is conserved in evolution (Fig. 1E). In summary, we show that, unlike in the processing of duplex DNA, where the helicase of yeast and human DNA2 is entirely masked by its nuclease, the motor activity of DNA2 promotes degradation of long ssDNA.
Figure 1.

The helicase activity of yeast and human DNA2 promotes ssDNA degradation. (A) Processing of 5′ 32P-labeled Y-structured DNA substrate by wild-type, helicase-deficient K1080E, and nuclease-deficient E675A yDna2 variants. The reactions contained 22.5 nM yeast RPA. (Heat) Heat-denatured substrate. The panel shows representative native 10% polyacrylamide gels. (B) Quantitation of the assays shown in A. Averages are shown for n = 2. Error bars indicate range. (C) Quantitation of experiments that are shown in Supplemental Figure S1A. Various concentrations of the yDna2 variants were used with the Y-structured DNA substrate. (D) Representative 1% agarose gel showing degradation kinetics of 3′ 32P-labeled ssDNA fragments (derived from λDNA) of various lengths by wild-type or helicase-deficient K1080E yDna2 in the presence of 1.08 µM yRPA. The sizes of the corresponding dsDNA fragments are indicated at the left. (E) Similar experiment as in D, showing the degradation kinetics of ssDNA by 20 nM human wild-type and helicase-deficient K654R hDNA2 in the presence of 576 nM hRPA.
To gain further insights into the role of the yDna2 motor in DNA degradation, we prepared a 2200-nt-long ssDNA randomly labeled with [α-32P] dATP. We analyzed the degradation of this substrate by wild-type yDna2 and the helicase-deficient yDna2 K1080E variant in a kinetic setup and separated the reaction products on agarose gels (Fig. 2A). Wild-type yDna2 degraded ssDNA rapidly and unexpectedly produced DNA degradation products of two different lengths (indicated by the open and closed arrows in Fig. 2A). In contrast, DNA degradation by helicase-dead yDna2 K1080E was slower and more gradual and resulted in only the shorter ssDNA degradation products (Fig. 2A). We next analyzed the reaction products on denaturing gels to determine the length of the ssDNA products (Fig. 2B). Wild-type yDna2 gave rise to fragments ranging from ∼5 to ∼100 nt in length (Fig. 2B [lanes 2–4], C), while helicase-deficient yDna2 K1080E yielded only products between ∼5 and ∼12 nt in length (Fig. 2B [lanes 6–8], C). Kinetic experiments revealed that wild-type yDna2 first produced the longer fragments, which were further degraded at later time points (Fig. 2D,G). In contrast, the majority of the products generated by helicase-deficient yDna2 K1080E were <20 nt at any time point tested (Fig. 2E,G). In the absence of ATP, the rates of DNA degradation by wild-type and helicase-deficient yDna2 variants were indistinguishable, and both enzymes gave rise to only the short DNA degradation products (Fig. 2F; Supplemental Fig. S2A), confirming the involvement of the ATP-dependent motor activity in ssDNA degradation by yDna2. Changing the ATP:Mg2+ ratio influenced the product size distribution produced by wild-type but not by helicase-deficient yDna2 K1080E (Fig. 2H; Supplemental Fig. S2B). Specifically, higher ATP to Mg2+ ratios are known to favor the yDna2 helicase (Bae and Seo 2000; Budd et al. 2000; Masuda-Sasa et al. 2006; Fortini et al. 2011), which consequently stimulated the production of longer ssDNA fragments (Fig. 2H; Supplemental Fig. S2B). Elevated Mg2+ ions instead promote the yDna2 nuclease; hence, we observed a reduction of the long yDna2 translocase-dependent degradation fragments (Supplemental Fig. S2C). The yDna2 ΔN variant lacking the first 405 amino acids behaved similarly to the full-length protein and produced the long ssDNA degradation fragments in an ATP-dependent manner (Supplemental Fig. S2D). As the yDNA2/hDNA2 motor promotes the degradation of ssDNA but not dsDNA, these results collectively imply that the motor of DNA2 functions as a ssDNA translocase rather than a helicase to promote efficient degradation of ssDNA.
Figure 2.

Cleavage of ssDNA by yDna2 results in two groups of DNA degradation products. (A) Degradation kinetics of ssDNA randomly labeled with 32P by wild-type and helicase-dead K1080E yDna2 in the presence of 315 nM yRPA as analyzed by 1% agarose gel electrophoresis. The two main groups of DNA degradation products are indicated by the open and closed arrows. (B) Representative 20% polyacrylamide denaturing gels showing the degradation of ssDNA randomly labeled with 32P by wild-type and K1080E yDna2. The DNA substrate was incubated with various concentrations of yDna2 for 10 min in the presence of 315 nM yRPA. (C) Image analysis of the experiment shown in B showing optical density analysis of gel lanes 1–9. Red arrows indicate the position of ∼80-nt-long DNA fragments. (D,E) Same reactions as in A, but the products were separated on 20% polyacrylamide denaturing gels. (F) Degradation kinetics of randomly labeled ssDNA by wild-type yDna2 in the absence of ATP with 315 nM yRPA. (G) Quantitation of experiments shown in D and E. The relative proportion of DNA degradation products >20 nt in length was determined. Averages are shown for n = 2. Error bars indicate range. (H) The effect of ATP concentration on the ssDNA degradation product length by 0.5 nM wild-type and helicase-deficient K1080E yDna2 in the presence of 315 nM yRPA in standard Mg2+ concentration (2 mM). Quantitation of experiments that are shown in Supplemental Figure S2B. Averages are shown for n = 2. Error bars indicate range.
RPA is required for the DNA2 motor-dependent degradation of ssDNA
Cognate RPA is a critical regulator of nuclease and helicase activities of DNA2 in both yeast and human cells (Bae et al. 2001, 2003; Cejka et al. 2010; Niu et al. 2010; Nimonkar et al. 2011; Levikova et al. 2013). In accord with previous data, ssDNA degradation by yDna2 without yRPA was slow (cf. Figs. 3A and 2B). Surprisingly, in the absence of yRPA, wild-type yDna2 produced only the short DNA degradation fragments in a manner similar to the helicase-deficient yDna2 K1080E variant even in the presence of ATP (Fig. 3A,B). Thus, the motor activity of yDna2 facilitates ssDNA degradation only when both yRPA and ATP are present. Likewise, the long DNA degradation products can be observed only with wild-type yDna2 in the presence of both ATP and yRPA, implying that the long DNA degradation products result from fast ssDNA degradation (Figs. 2B, 3B). We failed to detect a similar variation of DNA degradation fragment lengths in reactions with human DNA2 (Supplemental Fig. S3). However, the motor of hDNA2, similar to that in the yeast system, accelerated the speed of ssDNA degradation only in the presence of hRPA (Fig. 3C,D; Supplemental Fig. S3). The results presented here suggest that DNA2 is using its motor activity to facilitate the degradation of RPA-coated ssDNA, which results from DNA unwinding by a cognate RecQ family helicase partner.
Figure 3.
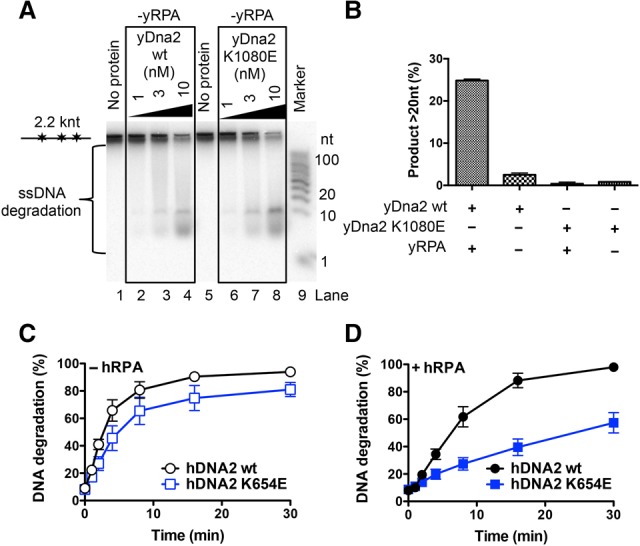
The motor of human and yeast DNA2 promotes degradation of RPA-coated ssDNA. (A) Degradation of ssDNA by wild-type and K1080E yDna2 as in Figure 2B but in the absence of yRPA, analyzed by 20% polyacrylamide denaturing gel electrophoresis. (B) Quantitation of DNA degradation product lengths by 1 nM wild-type and K1080E yDna2 variants from the experiments shown in A and Figure 2B. Averages are shown for n = 3. Error bars indicate SEM. (C,D) Degradation of randomly labeled ssDNA by 20 nM wild-type or helicase-deficient K654E hDNA2 variants. Quantitation of experiments that are shown in Supplemental Figure S3 without (C) or with (D) 176 nM hRPA. Averages are shown for n = 4. Error bars indicate SEM.
WRN or BLM stimulate the ssDNA nuclease activity of DNA2
Recently, we demonstrated that the helicase activity of nuclease-deficient hDNA2 was enhanced by physical interactions with BLM or WRN and vice versa: hDNA2 stimulated DNA unwinding by WRN/BLM in the presence of hRPA (Pinto et al. 2016). This suggested that hDNA2–BLM and hRPA as well as hDNA2–WRN and hRPA form functional complexes, with their subunits stimulating each other. Here we set out to test whether BLM or WRN could promote the degradation of long ssDNA by hDNA2 and hRPA. As above, wild-type hDNA2 was much more efficient in ssDNA degradation than the helicase-deficient enzyme (Fig. 4A [cf. lanes 3 and 4], B,D). Strikingly, helicase-deficient WRN or BLM proteins strongly stimulated ssDNA degradation by both wild-type and helicase-deficient hDNA2 (Fig. 4A–D). Helicase-deficient WRN or BLM preparations alone did not show any nuclease activity, confirming that the ssDNA degradation capacity was inherent to hDNA2 (Fig. 4A, lanes 9,10). Interestingly, wild-type WRN and BLM proteins promoted the degradative capacity of hDNA2 to a lesser extent than helicase-deficient polypeptides (Supplemental Fig. S4A, cf. lanes 4 and 5 and lanes 6 and 7), possibly due to being active translocases, with a fraction of molecules moving on the same DNA strand in the opposite direction from hDNA2 and thus blocking DNA degradation. The structural role of WRN/BLM in promoting DNA degradation by hDNA2 was apparent only on long ssDNA but not when hDNA2 was assayed with WRN on oligonucleotide-based 5′ overhang DNA (Supplemental Fig. S4B). As both wild-type and helicase-deficient hDNA2 variants were stimulated by WRN and BLM (Fig. 4), the acceleration of DNA degradation is a result of WRN/BLM stimulating the hDNA2 nuclease directly and not indirectly through promoting the hDNA2 motor activity. Together, these results indicate that BLM and WRN not only stimulate the hDNA2 motor as established previously (Pinto et al. 2016) but also have a structural role to promote the ssDNA-specific nuclease activity of hDNA2. This underpins the integrated nature of the hDNA2–WRN and hDNA2–BLM complexes.
Figure 4.
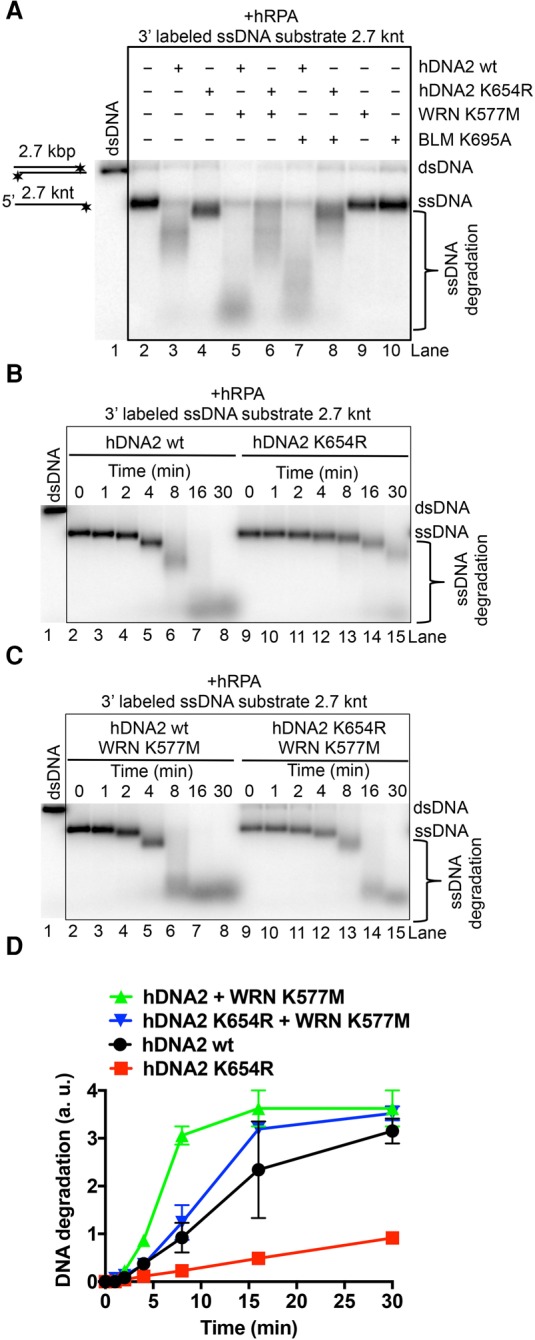
WRN or BLM enhances the ssDNA degradative capacity of hDNA2. (A) Representative 1% agarose gel showing degradation of 3′ 32P-labeled ssDNA (heat-denatured pUC19/HindIII) by 30 nM wild-type or helicase-deficient hDNA2 K654R variant with or without 30 nM helicase-deficient WRN K577M or 30 nM BLM K695A. The reactions were supplemented with 50 mM NaCl and 215 nM hRPA. (B) Representative 1% agarose gel showing the kinetics of ssDNA degradation (as in A) by 30 nM wild-type or helicase-deficient hDNA2 K654R. (C) Representative 1% agarose gel showing the kinetics of ssDNA degradation (as in A) by 30 nM wild-type or helicase-deficient hDNA2 K654R together with 30 nM helicase-deficient WRN K577M. (D) Quantitation of the experiments shown in B and C. The Y-axis shows the distance of the band midpoint from the position of intact ssDNA, relative to the distance between dsDNA and ssDNA. Averages are shown for n = 2. Error bars indicate range.
DNA end resection by Sgs1 and yDna2 is stimulated by yDna2 helicase in vitro and in vivo
Initial reports suggested that the nuclease but not the helicase activity of DNA2 was required for DNA end resection in yeast and humans (Zhu et al. 2008; Cejka et al. 2010; Nimonkar et al. 2011). These studies suggested that the DNA2 motor function may be dispensable in resection. Using limiting concentrations of recombinant proteins, we instead later observed that the helicase activity of hDNA2 promoted DNA end resection in conjunction with WRN or BLM helicases (Pinto et al. 2016). We could recapitulate this observation with yeast recombinant proteins: Helicase-deficient yDna2 K1080E was approximately twofold less efficient in DNA end resection than wild-type yDna2 in conjunction with Sgs1 (Supplemental Fig. S5A,B). Notably, the stimulatory effect of the yDna2 helicase was also apparent only at limiting yDna2 concentrations, suggesting that higher than physiological yDna2 levels might mask the involvement of the yDna2 helicase in DNA end resection in vivo (Zhu et al. 2008). Therefore, we set out to test for the effect of the yDna2 helicase in yeast cells where each yDna2 variant was expressed from its endogenous promoter on chromosomal DNA and thus was present at physiological levels. To this end, we monitored the resection of a single HO endonuclease-induced DSB by Southern blotting (White and Haber 1990). DNA end resection renders DNA single-stranded, which prevents cleavage by a restriction endonuclease and leads to the disappearance of the Southern blot signal. By using probes complementary to DNA at various distances from the break, the progression of DNA end resection can be monitored at various time points upon DSB induction, as used previously (Fig. 5A; Zhu et al. 2008). We constructed strains containing either wild-type DNA2 or helicase-deficient dna2 K1080E in an exo1Δ rad51Δ pif1-m2 background. The exo1Δ mutation eliminates long-range resection by the separate Exo1 pathway, making the relative contribution of the yDna2–Sgs1 pathway more apparent. The rad51Δ mutation renders the DSB unrepairable, which allows monitoring of resection of long DNA lengths over extended periods of time. The pif1-m2 mutation is a suppressor of yDna2 function in Okazaki fragment processing (Budd et al. 2006; Zhu et al. 2008). As the dna2Δ exo1Δ rad51Δ pif1-m2 mutant is not viable, we used sgs1Δ exo1Δ rad51Δ pif1-m2 cells as a reference strain for deficient long-range DNA end resection. We monitored resection 0, 3, 10, and 28 kb away from the HO-induced DSB at the indicated time points (Fig. 5A,B). The dna2 K1080E strain was slower than DNA2 at every distance measured but faster than the corresponding sgs1Δ strain in the exo1Δ rad51Δ pif1-m2 background (Fig. 5B,C). Collectively, these observations demonstrate that the yDna2 motor is required for efficient DNA end resection in conjunction with Sgs1 in vivo. This is apparent only when yDna2 is present at physiological levels, and our data thus also provide an explanation for why this effect was not detected when the yDna2 variants were expressed from a plasmid (Zhu et al. 2008).
Figure 5.

The helicase activity of wild-type yDna2 promotes resection of DSBs in vivo. (A) A scheme of the resection assay. DSB is induced by HO endonuclease. Resection past the indicated EcoRI sites (in black) leads to the disappearance of the signal obtained with site-specific probes (in red) by Southern blotting. Only those restriction sites that govern the appearance of the respective DNA fragments are shown. (B) Southern blot analysis of 5′ DNA end resection kinetics of HO-induced DSBs in DNA2, helicase-deficient dna2 K1080E, or sgs1Δ cells. All strains are exo1Δ rad51Δ pif1-m2. (C) Quantitation of B. Plots show the fraction of unresected 5′ strand (percentages) at each distance from the DSB. Averages are shown for n ≥ 2. Error bars indicate range.
Finally, we analyzed the length of the DNA degradation products as a footprint for the yDna2 motor activity in DNA end resection. To this end, we reconstituted an in vitro kinetic resection experiment of plasmid-length dsDNA with Sgs1, yDna2 (or its helicase-deficient yDna2 K1080E variant), and yRPA as well as Top3–Rmi1 and Mre11–Rad50–Xrs2 complexes, which stimulate Sgs1–yDna2 (Cejka et al. 2010; Cejka and Kowalczykowski 2010; Niu et al. 2010). As demonstrated in Supplemental Figure S5C,D, reactions with wild-type but not helicase-deficient yDna2 K1080E produced the extended ssDNA degradation products. This is consistent with the model that DNA2 motor activity functions in DNA end resection and further demonstrates that the motor of DNA2 engages even in the presence of other DNA end resection factors.
Discussion
Our results collectively demonstrate that the motors of both yeast and human DNA2 promote the degradation of long ssDNA. DNA2 is known to function together with a cognate RecQ family helicase in DNA end resection (Zhu et al. 2008). Our results support a model in which the RecQ family helicase—either Sgs1 or WRN/BLM—uses its motor activity to translocate in a 3′ → 5′ direction and unwinds duplex DNA. Helicase-deficient Sgs1/WRN/BLM variants in conjunction with the respective yDna2 or hDNA2 are completely inactive in DNA end resection in vitro and in vivo (Zhu et al. 2008; Cejka et al. 2010; Niu et al. 2010; Nimonkar et al. 2011), supporting the notion that the RecQ helicase provides the lead helicase activity (Fig. 6A). The motor of DNA2 then powers its translocation on the unwound 5′-terminated ssDNA strand in a 5′ → 3′ direction. As the motor exclusively accelerates the degradation of ssDNA, the most likely explanation is that the DNA2 motor functions as an ssDNA translocase rather than a helicase to facilitate movement along the unwound ssDNA behind Sgs1/BLM/WRN (Fig. 6A). DNA2 thus likely uses an active translocation mode on ssDNA to accelerate its degradation rather than relying on passive diffusion. RPA promotes DNA unwinding by Sgs1/BLM/WRN and additionally directs the nuclease activity of yDna2/hDNA2 to the 5′-terminated DNA strand in a species-specific manner (Cejka et al. 2010; Niu et al. 2010; Nimonkar et al. 2011; Pinto et al. 2016). Here we show that the motor of DNA2 facilitates DNA degradation in the presence of RPA. This further supports a model in which RPA is an integral component of the Sgs1–yDna2, WRN–hDNA2, and BLM–hDNA2 resection machineries (Fig. 6A).
Figure 6.
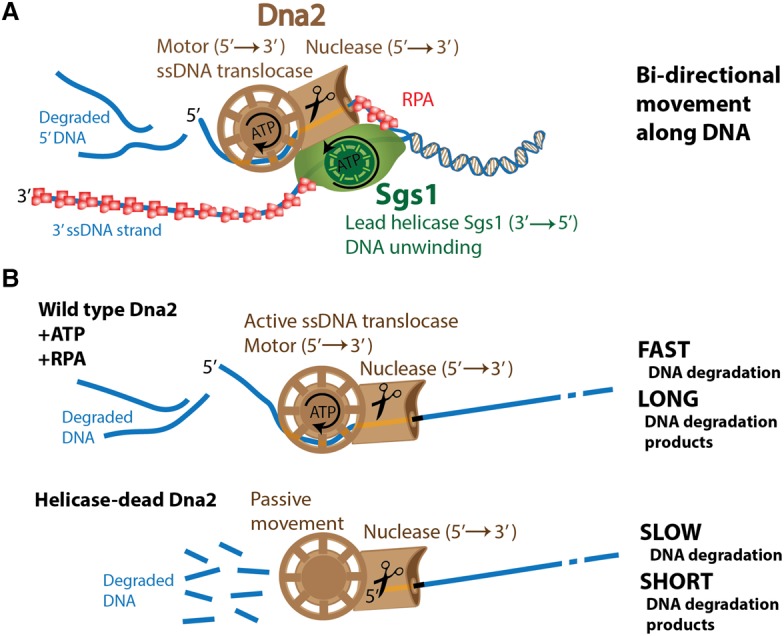
Model for the involvement of yDna2 helicase activity in DNA end resection. (A) Sgs1 translocates on the 3′-terminated DNA strand and functions as the lead helicase. Yeast Dna2 is using its motor activity to translocate on the unwound 5′-terminated DNA strand, which is degraded by its nuclease activity. (B, top) The motor of yDna2 accelerates degradation of ssDNA, which requires ATP hydrolysis and the presence of yRPA. This fast mode of DNA degradation results in long DNA degradation fragments. (Bottom) Without active translocation, the ssDNA degradation by yDna2 is slow, resulting in short DNA degradation fragments.
Previously, we observed that the nucleases of yeast and human DNA2 cleaved short 5′-terminated ssDNA overhangs of DNA duplexes, which prevented loading of DNA2 onto ssDNA and therefore duplex DNA unwinding (Levikova et al. 2013; Pinto et al. 2016). Paradoxically, the nuclease activity of DNA2 thus masks its helicase function in yeast and humans (Levikova et al. 2013; Pinto et al. 2016). The mouse DNA2 structure revealed that the DNA2 nuclease forms a narrow tunnel that accommodates only ssDNA (Zhou et al. 2015). Therefore, upon binding to a DNA end, wild-type DNA2 likely first moves along ssDNA in a passive diffusion-limited manner. DNA2 likely pauses once it encounters a junction between ssDNA and dsDNA, which represents an energy barrier. This provides time to the DNA2 nuclease active site within the tunnel to efficiently cleave the ssDNA overhang before it can reach the helicase domain located behind the tunnel. This prevents the engagement of the DNA2 motor activity and thus DNA unwinding of overhanging substrates by wild-type DNA2. The nuclease-deficient DNA2 instead does not cleave the ssDNA overhang within the tunnel. This allows time for the DNA2 variant to melt into the DNA duplex, possibly exploiting spontaneous “breathing” at the ssDNA and dsDNA junction or through a potential inherent dsDNA destabilization capacity of DNA2. This DNA melting then allows the nuclease-dead DNA2 variant to thread further onto the 5′-terminated DNA strand, which can reach the helicase domain to start the active ATP hydrolysis-dependent translocation and processive dsDNA unwinding observed previously (Levikova et al. 2013; Pinto et al. 2016).
The results presented here show that the nuclease of DNA2 instead does not prevent translocation on extended ssDNA. Translocation along ssDNA represents a lower-energy barrier than the unwinding of duplex DNA, which might allow the ssDNA strand to pass uncleaved with a higher frequency through the nuclease tunnel into the helicase domain, which would power the ssDNA translocation (Fig. 6B). This model is in accord with previous observations that DNA2 nuclease must load onto a free ssDNA end but then cleaves ssDNA endonucleolytically (Balakrishnan et al. 2010). This also implies that DNA2 may cleave ssDNA with a certain frequency, which might depend on the rate of ssDNA translocation. To this point, using yeast Dna2, we observed dramatic variations between the degradation fragment lengths: A passive mode of ssDNA translocation resulted in DNA degradation fragments of ∼5 to ∼10 nt in length, whereas active mode produced much longer fragments of ∼80 to ∼100 nt in length. The structure of mouse DNA2 revealed that 7 nt of ssDNA are bound to the helicase domain followed by a 2-nt-long linker and 6-nt-long fragment bound to the nuclease domain; therefore, the distance between the two active sites is larger than the “short” DNA fragments observed in our experiments. The presence of the “long” DNA degradation fragments is thus a clear indicator that the ssDNA passes intact through the nuclease tunnel of yDna2 to the helicase domain. Similar product fragment length variation was not observed with hDNA2, and thus it remains to be determined whether this was due to technical issues or whether the mode of translocation along ssDNA and its degradation by human DNA2 are different. Nevertheless, the ATP-dependent motor activity promoted ssDNA degradation by human DNA2 as well. It is possible that any “long” ssDNA fragments produced by hDNA2 were rapidly converted into shorter ones by other hDNA2 molecules present in solution, which prevented detection. The difference between the yeast and human enzymes could be explained by distinct DNA association versus degradation and translocation rates. Interestingly, ssDNA fragments produced during DNA end resection have been proposed to regulate checkpoint response (Jazayeri et al. 2008; Eapen et al. 2012); however, any function of the DNA2 motor activity in this process has yet to be demonstrated.
The Sgs1–yDna2 and WRN/BLM–hDNA2 complexes have been compared with AddAB, RecBCD, or other bacterial DNA end resection machines (Dillingham et al. 2003; Yeeles and Dillingham 2007; Dillingham and Kowalczykowski 2008; Yeeles et al. 2009; Cejka et al. 2010). Unlike in Escherichia coli’s RecBCD, where both RecB and RecD are per se DNA helicases capable of unwinding dsDNA, we propose that the motor of DNA2 is engaged only downstream from the RecQ family helicase partner. Despite this difference, Sgs1–yDna2, WRN/BLM–hDNA2, and RecBCD appear quite similar, as both complexes likely use a bidirectional DNA translocation mode and form a complex that is more than the sum of its parts (Dillingham and Kowalczykowski 2008). This bidirectional mode of DNA translocation is likely not used by Bacillus subtilis’s AddAB, which contains only one motor activity within the AddA subunit despite the nuclease of AddB being similar to the nuclease of DNA2 due to the presence of the iron–sulfur cluster (Yeeles et al. 2009). However, the DNA2 domain organization in which the nuclease of DNA2 precedes its motor is unique and not found in other prokaryotic resection enzymes characterized to date.
Many DNA helicases unwind dsDNA as a result of their ssDNA translocase activity, although not all ssDNA translocases are DNA helicases (Lohman et al. 2008). In fact, it has been demonstrated that several superfamily I DNA helicases, including E. coli UvrD, are ssDNA translocases as monomers and become capable of processive dsDNA unwinding only upon dimerization or interaction with a protein partner (Maluf et al. 2003; Fischer et al. 2004). Although direct DNA binding between DNA2 and the respective cognate RecQ family helicase has been demonstrated (Cejka et al. 2010; Nimonkar et al. 2011; Sturzenegger et al. 2014), there are no data on oligomerization of DNA2. DNA2 might thus be an unusual example of an enzyme in which the motor functions as a ssDNA translocase, and the nuclease domain prevents duplex DNA unwinding. The results presented here identify a conserved function for the DNA2 helicase to promote DNA end resection in conjunction with a cognate RecQ family helicase both in vitro and in vivo. The mechanism might be also relevant for the degradation of reversed replication forks that form upon replication stress, which might explain reduced viability and/or pronounced sensitivity of helicase-deficient yeast cells to alkylating agents and ionizing radiation, treatments that also lead to DSBs (Formosa and Nittis 1999; Budd and Campbell 2000; Thangavel et al. 2015).
Materials and methods
Recombinant proteins
Wild-type yDna2 as well yDna2 E675A and yDna2 K1080E variants were expressed and purified as described previously (Levikova et al. 2013). The construct for the expression of the yDna2 ΔN variant was prepared by cloning the yDNA2 sequence lacking the first 1215 residues (corresponding to 405 amino acids) between the 5′-terminal Flag tag and the 3′-terminal 6His tag into BamHI and EcoRI sites in a pYes2 vector (Invitrogen). The truncated protein was expressed and purified as the full-length protein. Wild-type hDNA2, hDNA2 K654E, and hDNA2 K654R were prepared as described previously (Pinto et al. 2016). No difference was observed between the hDNA2 K654E and hDNA2 K654R variants in the experiments presented in this report (data not shown). Wild-type BLM, WRN, BLM K695A, and WRN K577M were expressed and purified as described (Pinto et al. 2016). Sgs1, Top3–Rmi1, and Mre11–Rad50–Xrs2 were expressed and purified as described previously (Cejka and Kowalczykowski 2010; Cejka et al. 2010; Cannavo and Cejka 2014). The yRPA and hRPA proteins were expressed and purified as described (Henricksen et al. 1994; Kantake et al. 2003).
DNA substrates
The oligonucleotides X12-3 and X12-4NC were used for the preparation of the Y-structured DNA substrate, and oligonucleotides X12-3 and X12-4SC were used for the preparation of the 5′ tailed DNA, as described previously (Cejka and Kowalczykowski 2010; Levikova et al. 2013). Bacteriophage λ dsDNA (New England Biolabs) was digested by HindIII (New England Biolabs). The linearized dsDNA fragments were then labeled with [α-32P] dATP and a Klenow fragment of DNA polymerase I (New England Biolabs) at the 3′ end. Unincorporated nucleotides were removed using MicroSpin G25 columns (GE Healthcare). The substrate was denatured by heating for 5 min at 95°C prior to each experiment. The 6.4-kb-long ssDNA (M13mp18) was purchased from New England Biolabs. The 2686-base-pair-long pUC19 dsDNA was linearized with HindIII, purified by phenol-chloroform extraction and ethanol precipitation, and denatured as described above where necessary.
The randomly labeled 2200-nt-long substrate was prepared by amplification of the S. cerevisiae LIG1 gene by PCR from yeast genomic DNA (yWH436 strain) (see Supplemental Table S2) using Phusion high-fidelity DNA polymerase (New England Biolabs) and the following primers: forward, 5′-ACGCATTAGCTAGCGGATCCCTGGAAGTTCTGTTCCAGGGGCCCATGCGCAGATTACTGACCGGTTG-3′; and reverse, 5′-ACGCATTACTCGAGATTTTGCATGTGGGATTGGT-3′. In addition to the standard dNTP concentration in the PCR reaction (200 µM each), 66 nM [α-32P] dATP was added. The PCR reaction product was purified using Chroma Spin TE-400 columns (Clontech). The substrate was denatured by heating for 5 min at 95°C before the experiment to obtain ssDNA where necessary. The 10- to 100-nt low-molecular-weight marker (Affymetrix) was 32P-labeled at the 5′ terminus with [γ-32P] ATP and T4 polynucleotide kinase (New England Biolabs). An asterisk indicates the position of the radioactive label where indicated.
Nuclease and helicase assays
Unless indicated otherwise, the experiments were performed in a 15-µL volume in 25 mM Tris-acetate (pH 7.5), 2 mM magnesium acetate, 1 mM ATP, 1 mM dithiothreitol, 0.1 mg/mL bovine serum albumin (New England Biolabs), 1 mM phosphoenolpyruvate, 16 U/mL pyruvate kinase (Sigma), and 1 nM (in molecules) 32P-labeled oligonucleotide-based, pUC19-based, or PCR-based DNA substrate or 0.15 nM λ DNA-based substrate (corresponding to 2.4 nM 5′-terminated ssDNA ends upon denaturation). For reactions with unlabeled substrates, 100 ng of DNA was used. Where indicated, human and yeast RPA was included to saturate all ssDNA. Recombinant proteins were added on ice to the assembled reaction mixtures. The reactions were incubated for 30 min at 30°C for yeast and 37°C for human proteins unless indicated otherwise. Reactions were stopped by adding either 5 µL of 2% stop solution for native gels as described previously (Cejka and Kowalczykowski 2010) or 15 µL of formamide dye for denaturing gels (Cannavo and Cejka 2014). All gels with radioactive substrates were dried on DE81 chromatography paper (Whatman) and exposed to storage phosphor screens (GE Healthcare). The screens were scanned by a Typhoon 9400 phosphorimager (GE Healthcare). Where unlabeled DNA substrates were used, DNA was visualized by staining with ethidium bromide (Sigma) or GelRed (Biotium) as indicated.
Southern blot
Yeast cell growth, HO-break induction, and DNA isolation were performed as described previously (Zhu et al. 2008). The 0-h time point was collected immediately after adding galactose, before the HO cut occurred. DNA samples were separated on 1% agarose gel, and DNA was transferred onto a nylon membrane (GE Healthcare) and hybridized with DNA probes radioactively labeled by random primed DNA labeling kit (Roche) according to manufacturer's instructions. Primers used to prepare the 32P-labeled probes were used as described previously (Zhu et al. 2008) and are listed in Supplemental Table S1. Membranes were exposed to storage phosphor screens (GE Healthcare) that were scanned by a Typhoon 9400 phosphorimager (GE Healthcare).
Yeast strains used in this study are listed in Supplemental Table S2. Briefly, the dna2 K1080E mutation was introduced using the allele replacement strategy (Widlund and Davis 2005) by transforming the parental yWH436 strain with the pRS306 plasmid (Sikorski and Hieter 1989) containing the SacII/BamHI fragment of the point-mutagenized yDNA2 gene that was excised previously from the pGAL18 Dna2 K1080E plasmid (gift from J. Campbell). Gene deletions were performed by PCR-based substitution cassettes, as described previously (Janke et al. 2004; Hegemann et al. 2006). The EXO1 gene was deleted by using the PCR cassette amplified from pFA6a plasmid using the following primers: forward, 5′-ACCACATTAAAATAAAAGGAGCTCGAAAAAACTGAAAGGCGTAGAAAGGACAGCTGAAGCTTCGTACGCTGC-3′; and reverse, 5′-TTTTCATTTGAAAAATATACCTCCGATATGAAACGTGCAGTACTTAACTTCATAGGCCACTAGTGGATCTG-3′.
The SGS1 gene was deleted by using the PCR cassette amplified from pUG72 with the following primers: forward, 5′-ATTATTGTTGTATATATTTAAAAAATCATACACGTACACACAAGGCGGTA-3′; and reverse, 5′-TTGGCGAATGGTGTCGTAGTTATAAGTAACACTATTTATTTTTCTACTCTGCATAGGCCACTAGTGGATCTG-3′. The RAD51 gene was deleted by using the PCR cassette amplified from pUG6 using the following primers: forward, 5′-AAGAGCAGACGTAGTTATTTGTTAAAGGCCTACTAATTTGTTATCGTCATCAGCTGAAGCTTCGTACGCTGC-3′; and reverse, 5′-AGAATTGAAAGTAAACCTGTGTAAATAAATAGAGACAAGAGACCAAATACCATAGGCCACTAGTGGATCTG-3′.
Supplementary Material
Acknowledgments
We thank Grzegorz Ira and Alma Papusha (Baylor College of Medicine) for the yWH436 strain and help in establishing the in vivo resection assays. We thank members of the Cejka laboratory—including Elda Cannavo, Roopesh Anand, Lepakshi Ranjha, Vera Kissling, and Sean Howard—for comments on the manuscript, and Jim Haber (Brandeis University) for discussions. We thank Patrick Sung (Yale University) and colleagues for communicating their results before publication. This work has been supported by Swiss National Science Foundation professorship PP00P3 133636 and Swiss Cancer League grant KFS-3089-02-2013 to P.C.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.295196.116.
References
- Bae SH, Seo YS. 2000. Characterization of the enzymatic properties of the yeast dna2 Helicase/endonuclease suggests a new model for Okazaki fragment processing. J Biol Chem 275: 38022–38031. [DOI] [PubMed] [Google Scholar]
- Bae SH, Choi E, Lee KH, Park JS, Lee SH, Seo YS. 1998. Dna2 of Saccharomyces cerevisiae possesses a single-stranded DNA-specific endonuclease activity that is able to act on double-stranded DNA in the presence of ATP. J Biol Chem 273: 26880–26890. [DOI] [PubMed] [Google Scholar]
- Bae SH, Bae KH, Kim JA, Seo YS. 2001. RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature 412: 456–461. [DOI] [PubMed] [Google Scholar]
- Bae KH, Kim HS, Bae SH, Kang HY, Brill S, Seo YS. 2003. Bimodal interaction between replication-protein A and Dna2 is critical for Dna2 function both in vivo and in vitro. Nucleic Acids Res 31: 3006–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan L, Polaczek P, Pokharel S, Campbell JL, Bambara RA. 2010. Dna2 exhibits a unique strand end-dependent helicase function. J Biol Chem 285: 38861–38868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd ME, Campbell JL. 2000. The pattern of sensitivity of yeast dna2 mutants to DNA damaging agents suggests a role in DSB and postreplication repair pathways. Mutat Res 459: 173–186. [DOI] [PubMed] [Google Scholar]
- Budd ME, Choe WC, Campbell JL. 1995. DNA2 encodes a DNA helicase essential for replication of eukaryotic chromosomes. J Biol Chem 270: 26766–26769. [DOI] [PubMed] [Google Scholar]
- Budd ME, Choe W, Campbell JL. 2000. The nuclease activity of the yeast DNA2 protein, which is related to the RecB-like nucleases, is essential in vivo. J Biol Chem 275: 16518–16529. [DOI] [PubMed] [Google Scholar]
- Budd ME, Reis CC, Smith S, Myung K, Campbell JL. 2006. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase δ. Mol Cell Biol 26: 2490–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P. 2014. Sae2 promotes dsDNA endonuclease activity within Mre11–Rad50–Xrs2 to resect DNA breaks. Nature 514: 122–125. [DOI] [PubMed] [Google Scholar]
- Cannavo E, Cejka P, Kowalczykowski SC. 2013. Relationship of DNA degradation by Saccharomyces cerevisiae exonuclease 1 and its stimulation by RPA and Mre11–Rad50–Xrs2 to DNA end resection. Proc Natl Acad Sci 110: E1661–E1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P. 2015. DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J Biol Chem 290: 22931–22938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Kowalczykowski SC. 2010. The full-length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds holliday junctions. J Biol Chem 285: 8290–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, Kowalczykowski SC. 2010. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3–Rmi1 and Mre11–Rad50–Xrs2. Nature 467: 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham MS, Kowalczykowski SC. 2008. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev 72: 642–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham MS, Spies M, Kowalczykowski SC. 2003. RecBCD enzyme is a bipolar DNA helicase. Nature 423: 893–897. [DOI] [PubMed] [Google Scholar]
- Duxin JP, Moore HR, Sidorova J, Karanja K, Honaker Y, Dao B, Piwnica-Worms H, Campbell JL, Monnat RJ Jr, Stewart SA. 2012. Okazaki fragment processing-independent role for human Dna2 enzyme during DNA replication. J Biol Chem 287: 21980–21991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eapen VV, Sugawara N, Tsabar M, Wu WH, Haber JE. 2012. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol 32: 4727–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer CJ, Maluf NK, Lohman TM. 2004. Mechanism of ATP-dependent translocation of E. coli UvrD monomers along single-stranded DNA. J Mol Biol 344: 1287–1309. [DOI] [PubMed] [Google Scholar]
- Formosa T, Nittis T. 1999. Dna2 mutants reveal interactions with Dna polymerase α and Ctf4, a Pol α accessory factor, and show that full Dna2 helicase activity is not essential for growth. Genetics 151: 1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortini BK, Pokharel S, Polaczek P, Balakrishnan L, Bambara RA, Campbell JL. 2011. Characterization of the endonuclease and ATP-dependent flap endo/exonuclease of Dna2. J Biol Chem 286: 23763–23770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S, Chapman JR, Magill C, Jackson SP. 2008. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev 22: 2767–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegemann JH, Guldener U, Kohler GJ. 2006. Gene disruption in the budding yeast Saccharomyces cerevisiae. Methods Mol Biol 313: 129–144. [DOI] [PubMed] [Google Scholar]
- Henricksen LA, Umbricht CB, Wold MS. 1994. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem 269: 11121–11132. [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962. [DOI] [PubMed] [Google Scholar]
- Jazayeri A, Balestrini A, Garner E, Haber JE, Costanzo V. 2008. Mre11–Rad50–Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. EMBO J 27: 1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantake N, Sugiyama T, Kolodner RD, Kowalczykowski SC. 2003. The recombination-deficient mutant RPA (rfa1-t11) is displaced slowly from single-stranded DNA by Rad51 protein. J Biol Chem 278: 23410–23417. [DOI] [PubMed] [Google Scholar]
- Levikova M, Klaue D, Seidel R, Cejka P. 2013. Nuclease activity of Saccharomyces cerevisiae Dna2 inhibits its potent DNA helicase activity. Proc Natl Acad Sci 110: E1992–E2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman TM, Tomko EJ, Wu CG. 2008. Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat Rev Mol Cell Biol 9: 391–401. [DOI] [PubMed] [Google Scholar]
- Maluf NK, Fischer CJ, Lohman TM. 2003. A Dimer of Escherichia coli UvrD is the active form of the helicase in vitro. J Mol Biol 325: 913–935. [DOI] [PubMed] [Google Scholar]
- Masuda-Sasa T, Imamura O, Campbell JL. 2006. Biochemical analysis of human Dna2. Nucleic Acids Res 34: 1865–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455: 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. 2011. BLM–DNA2–RPA–MRN and EXO1–BLM–RPA–MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25: 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu H, Chung WH, Zhu Z, Kwon Y, Zhao W, Chi P, Prakash R, Seong C, Liu D, Lu L, et al. 2010. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 467: 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmezer G, Levikova M, Klein D, Falquet B, Fontana GA, Cejka P, Rass U. 2016. Replication intermediates that escape Dna2 activity are processed by Holliday junction resolvase Yen1. Nat Commun 7: 13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto C, Kasaciunaite K, Seidel R, Cejka P. 2016. Human DNA2 possesses a cryptic DNA unwinding activity that functionally integrates with BLM or WRN helicases. Elife 5: e18574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturzenegger A, Burdova K, Kanagaraj R, Levikova M, Pinto C, Cejka P, Janscak P. 2014. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J Biol Chem 289: 27314–27326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. 2015. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208: 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PT, Erdeniz N, Dudley S, Liskay RM. 2002. Characterization of nuclease-dependent functions of Exo1p in Saccharomyces cerevisiae. DNA Repair (Amst) 1: 895–912. [DOI] [PubMed] [Google Scholar]
- Wanrooij PH, Burgers PM. 2015. Yet another job for Dna2: checkpoint activation. DNA Repair (Amst) 32: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CI, Haber JE. 1990. Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. EMBO J 9: 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlund PO, Davis TN. 2005. A high-efficiency method to replace essential genes with mutant alleles in yeast. Yeast 22: 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JT, Dillingham MS. 2007. A dual-nuclease mechanism for DNA break processing by AddAB-type helicase-nucleases. J Mol Biol 371: 66–78. [DOI] [PubMed] [Google Scholar]
- Yeeles JT, Cammack R, Dillingham MS. 2009. An iron-sulfur cluster is essential for the binding of broken DNA by AddAB-type helicase-nucleases. J Biol Chem 284: 7746–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Pourmal S, Pavletich NP. 2015. Dna2 nuclease–helicase structure, mechanism and regulation by Rpa. Elife 4: e09832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134: 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.